
Arytmogenní kardiomyopatie levé komory
Arrhythmogenic left ventricular cardiomyopathy
Arrhythmogenic left ventricular cardiomyopathy (ALVC) is a rare condition characterised by progressive fibrofatty replacement of the myocardium of the left ventricle in combination with arrhythmias of left ventricular origin. ALVC has been linked to autosomal dominant mutations of genes encoding desmosomal proteins, similarly to the classic arrhythmogenic right ventricular cardiomyopathy with which it also shares pathological and prognostic features. It seems that isolated left or right ventricular abnormalities represent two extremes of the spectrum of clinical manifestations of a single disease: arrhythmogenic cardiomyopathy. In addition to arrhythmias originating from the left ventricle, the diagnosis of ALVC is based on identification of morphological changes of the left ventricle including late gadolinium enhancement with subepicardial to midwall distribution, corresponding to fibrous or fibrofatty replacement on histopathology. The diagnosis is confirmed by detection of a causal mutation. ALVC should be kept in mind in the differential diagnosis of ventricular tachycardia of non-ischemic origin.
Key words:
arrhythmogenic cardiomyopathy – cardiac magnetic resonance – late gadolinium enhancement – ventricular tachycardia
Autoři:
Štěpán Havránek 1; Tomáš Paleček 1; Petr Kuchynka 1; Ivana Vítková 2
Působiště autorů:
II. interní klinika kardiologie a angiologie 1. LF UK a VFN v Praze
1; Ústav patologie 1. LF UK a VFN v Praze
2
Vyšlo v časopise:
Vnitř Lék 2016; 62(9): 728-735
Kategorie:
Přehledné referáty
Souhrn
Arytmogenní kardiomyopatie levé komory (ALVC) představuje vzácné onemocnění charakterizované progresivní přestavbou myokardu levé komory fibrózní a tukovou tkání a manifestující se komorovými tachykardiemi levokomorového původu. ALVC lze pokládat za jednu z fenotypických variant arytmogenní kardiomyopatie, jejíž spektrum může nabývat celé škály projevů od levostranně dominantní, přes biventrikulární až po čistě pravostrannou variantu. Správný název tohoto onemocnění by tak měl znít arytmogenní kardiomyopatie, s následnou specifikací univentrikulárního či biventrikulárního postižení. V etiopatogenezi onemocnění je všeobecně přijímán koncept geneticky podmíněné jednotky vznikající v důsledku mutací v genech kódujících převážně desmosomální proteiny, v naprosté většině případů s autosomálně dominantním typem přenosu s neúplnou penetrancí. Mimo přítomnosti komorových tachykardií vycházejících z levé komory je diagnóza ALVC založena na typickém obraze subepikardiální fibrózy různého rozsahu při vyšetření magnetickou rezonancí, ideálně verifikovaném bioptickým vyšetřením myokardu a následným průkazem kauzální mutace. Na možnost ALVC je nutno myslet v diferenciálně diagnostické rozvaze etiologie neischemických komorových tachykardií.
Klíčová slova:
arytmogenní kardiomyopatie – komorová tachykardie – magnetická rezonance srdce – pozdní sycení gadoliniem
Úvod
Arytmogenní kardiomyopatie levé komory (Arrhythmogenic left ventricular cardiomyopathy – ALVC) je vzácné, geneticky podmíněné onemocnění charakterizované progresivní přestavbou myokardu levé komory (LK) tukovou a fibrózní tkání v kombinaci s komorovými tachykardiemi (KT) levokomorového původu, tj. s morfologií blokády pravého Tawarova raménka [1]. Patologicky je postižena exkluzivně či zcela dominantně LK. Patologická přestavba tkáně typicky počíná lokalizovaně subepikardiálně, tj. ve vnější třetině komorového myokardu [2–5]. S progresí choroby zasahuje fibrózní přestavba i hlouběji do nitra myokardu s možností až prakticky transmurálního postižení. Nejvíce vyjádřené formy ALVC pak mají postižení cirkumferenciální, po celém povrchu LK, včetně mezikomorové přepážky, postižena je strana septa přiléhající k pravé komoře (obr. 1).

Jako první byla ALVC popsána při autoptickém vyšetření obětí náhlé srdeční smrti, následně i u příbuzných náhle zemřelých jedinců, u nemocných s KT levokomorového původu a v rodinách postižených mutacemi v genech pro desmoplakin [6–10]. Později byla ALVC rozpoznána jako fenotypická varianta v rodinách s „klasickou“ arytmogenní kardiomyopatií pravé komory (ARVC) [11,12]. Spektrum postižení myokardu fibrózní či fibrolipomatózní přestavbou tak může nabývat celé škály projevů od levostranně dominantní, přes biventrikulární až po čistě pravostrannou variantu. Izolovaná levostranná a pravostranná varianta představují extrémní projevy jedné geneticky podmíněné nemoci [12]. Arytmogenní kardiomyopatie je pak nový termín, který sdružuje všechny varianty onemocnění (schéma). V kontrastu s klasickou variantou ARVC a biventrikulární formou arytmogenní kardiomyopatie zahrnující dohromady postižení obou komor, je ALVC charakterizována izolovaným či zcela dominantním postižením myokardu LK. Jelikož je ALVC v současnosti stále považována za vzácnou jednotku, jsou literární údaje omezeny hlavně na kazuistická sdělení nebo popis malých kohort nemocných. Dosud nejvíce studovaná kohorta nemocných s ALVC čítala 42 osob [1]. Je však téměř jisté, že s narůstající znalostí existence a s tím spojené zlepšené diagnostiky onemocnění bude prevalence ALVC stoupat.

Genetický podklad onemocnění
U většiny případů arytmogenní kardiomyopatie je předpokládán autosomálně dominantní typ přenosu s neúplnou penetrancí. Není tedy překvapivé, že je možné vystopovat srdeční postižení i u některých dalších členů rodiny nemocného. Avšak část postižených jedinců je jistě nositelem de novo vzniklé mutace v příslušných genech. Až u poloviny nemocných s ALVC lze v současné době identifikovat kauzální mutace v desmosomálních genech (desmoplakin, desmoglein 2, plakoglobin, plakophilin 2, desmocollin 2), tedy v genech, jejichž mutace jsou zodpovědné za vznik klasické ARVC [13–16]. Pro společný genetický podklad ALVC a ARVC svědčí i prokázaný výskyt obou forem onemocnění u příbuzných pacientů [12].
Autosomálně recesivní typy mutací genů jsou jako příčiny arytmogenní kardiomyopatie vzácné. Nicméně byly popsány mutace v homozygotní konstituci pro gen kódující desmocollin i u nemocných s ALVC. Srdeční postižení při autosomálně recesivní variantě může, ale nemusí být provázeno kožními projevy obdobnými Carvajalovu syndromu [16–20]. Carvajal (nebo také Carvajalův syndrom, popsaný poprvé Carvajal-Huertou v roce 1998) syndrom je jednotka vznikající na podkladě mutace v genu pro desmoplakin v homozygotní konstituci a je charakterizována trias klinických příznaků: palmoplantární keratodermie, vlněné vlasy a dilatační kardiomyopatie [21,22].
Je sice pravděpodobné, že dominantní genetickou příčinu arytmogenní kardiomyopatie představují mutace v genech kódující desmosomální proteiny, avšak relativně nedávno byly identifikovány i varianty arytmogenní kardiomyopatie vzniklé na podkladě mutací v genech pro jiné proteiny, konkrétně non-desmosomální fosfolamban [23].
Klinický obraz
ALVC se může manifestovat prakticky kdykoli od adolescence až po stáří. Ačkoliv průměrný věk pacientů s ALVC sledovaných Sen-Chowdhrym et al byl 44 let [1], je třeba si uvědomit, že část pacientů s ALVC může být delší dobu sledována pod jinou diagnózou a první manifestaci onemocnění je možné vystopovat v předchorobí již dříve. Typickými klinickými projevy onemocnění jsou u symptomatických jedinců obtíže vycházející z přítomnosti komorových arytmií: palpitace, presynkopální stavy a synkopa; nejzávažnějším projevem ALVC je pak náhlá smrt. Často uváděné obtíže představují i atypické bolesti na hrudi, které se vyskytují bez návaznosti na přítomnost ischemické choroby srdeční. Jelikož ALVC může být v určité fázi vývoje asociována i s vyššími plazmatickými hladinami srdečních troponinů a změnami na EKG, jsou pak nemocní s epizodou bolestí na hrudi často vyšetřováni koronarograficky pro podezření na akutní koronární syndrom s negativním výsledkem. Naopak vzácnějšími symptomy ALVC jsou projevy srdečního selhání v podobě progredující námahové dušnosti a otoků dolních končetin. Postižení jedinci mohou být však až ve třetině případů zcela asymptomatičtí [1]. I dosavadní zkušenosti autorů tohoto sdělení potvrzují, že dominujícími klinickými projevy ALVC jsou symptomy KT.
Je velmi důležité zdůraznit, že výskyt KT u ALVC neodráží v naprosté většině případů tíži systolické dysfunkce LK. I u nemocných s jen mírně sníženou, ba i normální ejekční frakcí (EF) LK mohou být přítomny významné komorové arytmie. Část nemocných má mimo levokomorových tachykardií i arytmie vycházející z pravé komory. Mimo náhlé srdeční smrti, u níž lze předpokládat podkladový mechanizmus v podobě setrvalé hemodynamicky závažné KT, jsou u nemocných s ALVC dokumentovány také komorové extrasystoly a běhy nesetrvalých KT. Manifestace onemocnění hemodynamicky tolerovanou, setrvalou monomorfní komorovou tachykardií se jeví spíše jako vzácnost, byť i s touto možností měli možnost se autoři sdělení setkat (obr. 2).

Diagnostika arytmogenní kardiomyopatie levé komory
EKG
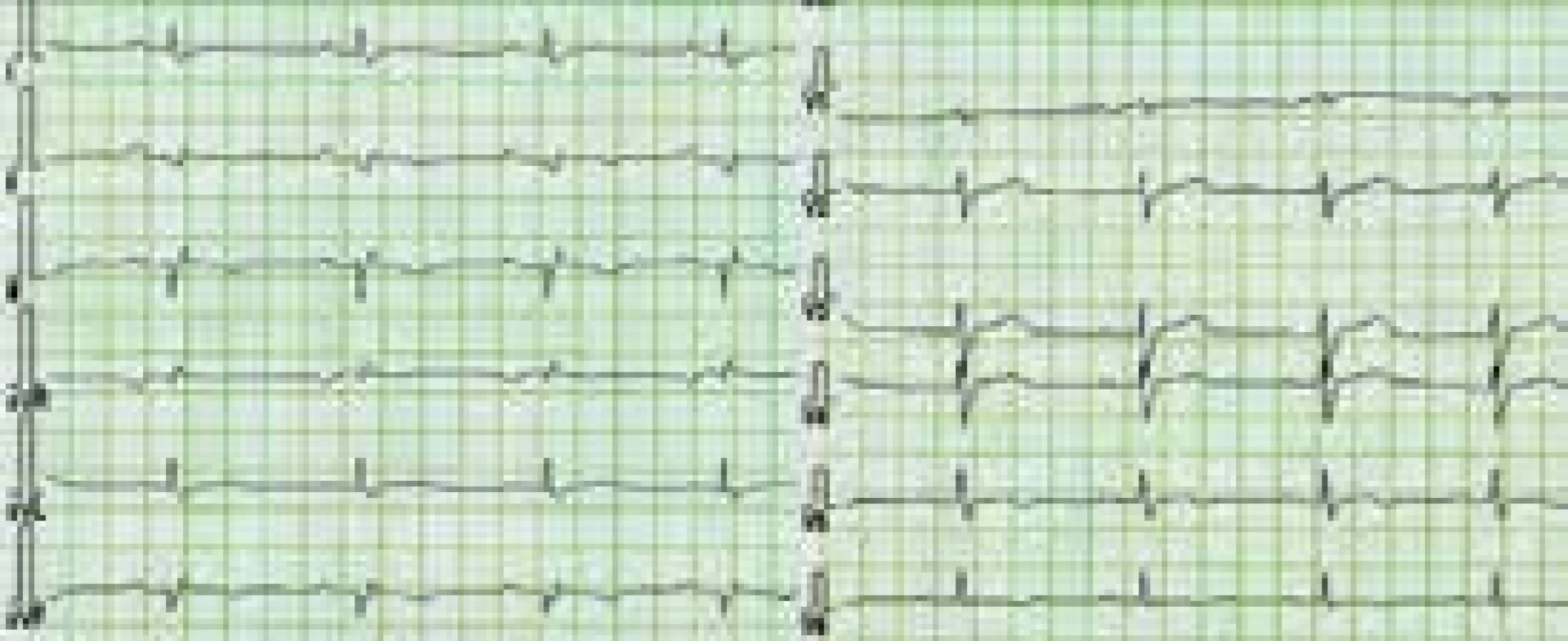
Charakteristickým nálezem na EKG je u ALVC přítomnost plochých nebo invertovaných T vln v laterálních nebo spodních svodech s možnou extenzí změn směrem k V4–V2 [1,3,8,10,12] (obr. 3). V běžné klinické praxi jsou tyto relativně časté změny považovány, při absenci jasné strukturální srdeční nemoci, za benigní a nespecifické. Méně obvyklá je přítomnost deviace srdeční osy doleva a pomalá progrese r kmitu v hrudních svodech [1,8]. Při vyšetření signálově zprůměrovaného EKG mohou být až u poloviny nemocných s ALVC zachyceny pozdní komorové potenciály [1]. Dle autorů tohoto sdělení a v kontextu již dříve publikovaných některých kazuistik s dostupným EKG [11] lze spekulovat, že do obrazu změn při ALVC patří i snížená amplituda QRS komplexů v končetinových svodech provázená nespecifickou poruchou nitrokomorového vedení (obr. 3 a 4). Tato pozorování však nebyla systematicky zpracována, a není tedy znám přesný výskyt a význam těchto nálezů u pacientů s ALVC.

Zobrazovací metody
Zobrazovací metody hrají v diagnostice ALVC klíčovou úlohu. Echokardiograficky či jinými zobrazovacími metodami, zejména magnetickou rezonancí (MRI), je možné detekovat regionální nebo difuzní poruchu kinetiky LK. Jak již bylo uvedeno výše, přítomnost normální systolické funkce LK však přítomnost ALVC nevylučuje. V kohortě nemocných s ALVC sledovaných Sen-Chowdhrym et al [1] byla snížená EF LK < 55 % zachycena jen u přibližně poloviny nemocných. Minimální echokardiograficky hodnocená EF LK přitom činila 40 %, neboli nebyla zaznamenána těžká systolická dysfunkce. Přibližně polovina nemocných měla dilatovanou LK, ale s maximální zachycenou hodnotou telediastolického rozměru pouze 61 mm [1]. Lze tedy vyvodit, že těžká dysfunkce nebo dilatace LK není pro ALVC typická, což může být velmi důležité v diferenciální diagnostice etiologie KT. Pokud jsou vyjádřeny regionální poruchy kinetiky LK, pak dle literárních údajů jsou nejčastěji detekovatelné v oblastech septa, srdečního hrotu, spodní a boční stěny [1]. U části nemocných splňujících kritéria pro ALVC může být též detekovatelná dilatace nebo abnormální kinetika stěn pravé komory [1]. Nicméně, aneuryzma pravé komory nebo snížené hodnoty EF pravé komory vykazuje sotva třetina postižených jedinců.
Zcela zásadní v diagnostice ALVC je v současnosti srdeční MRI, která prokazuje fibrózu myokardu a její extenzi na základě přítomnosti, rozsahu a lokalizace sycení kontrastní látkou na bázi gadolinia (late gadolinium enhancement – LGE). Pro ALVC je charakteristická subepikardiální lokalizace LGE, typicky vyjádřená v oblastech volných stěn LK, především posterolaterálně či v oblasti spodní stěny (obr. 1), s možností extenze midmyokardiálně, hlouběji do myokardu. V menším procentu lze LGE detekovat v mezikomorovém septu a na přední stěně LK. Asi u 25 % nemocných s ALVC je pak patrno kompletní cirkumferenciální subepikardiální LGE (obr. 1) [1]. Takto extenzivně vyjádřené LGE odpovídá původním autoptickým nálezům fibrózního kroužku popsaným u ALVC [2–5]. Subepikardiální lokalizace LGE ve stěnách LK je logickým „zrcadlovým obrazem“ strukturálních změn vznikajících při klasické ARVC, při níž ložiska fibrózy také iniciálně postihují subepikardiální oblasti volných stěn pravé komory. Rozsah LGE, tj. fibrózy myokardu, u ALVC vysoce koreluje s přítomností komorových tachykardií [1]. Z klinického hlediska je velmi důležité, že u zatím málo morfologicky vyjádřených forem ALVC je možné ložiska LGE dokumentovat i ve stěnách LK, které jinak nevykazují regionální či globální poruchu kontraktility. Je tedy zjevné, že echokardiografické vyšetření v plné detekci strukturálních abnormalit u jedinců s ALVC selhává. V neinvazivní diagnostice ALVC naopak napomáhá pouze vyšetření MRI prokazující charakteristický, byť ne zcela specifický typ subepikardiálního LGE.
U některých nemocných s ALVC je možné nalézt i lipomatózní infiltraci v T1 vážených sekvencích cílených k detekci tukové tkáně při vyšetření MRI (obr. 5). Pro diagnostiku ALVC však přesná senzitivita a specificita průkazu přítomnosti tukové tkáně v myokardu není známa, navíc určité množství tuku je fyziologicky nalézáno i v myokardu jinak zdravých jedinců. Absenci průkazu lipomatózní infiltrace při MRI vyšetření proto nelze považovat za nález vylučující ALVC; hlavní kritérium MRI tkáňové charakteristiky představuje v diagnostice ALVC přítomnost LGE, tj. fibrózy myokardu.

Genetická analýza
S rozvojem moderních metod genetického testování se nabízí i možnost detekce přítomnosti kauzální mutace v genech zodpovědných za rozvoj arytmogenní kardiomyopatie. Jasně pozitivní výsledek genetické analýzy může být jistě definitivním potvrzením přítomnosti ALVC a být nápomocný v rámci diferenciální diagnostiky arytmogenní kardiomyopatie a jejích fenokopií. Avšak diagnózu této jednotky čistě na výsledku genetického testování nelze v současnosti založit. V souboru Sen-Chowdryho et al byla kauzální mutace jasně potvrzena pouze zhruba ve třetině postižených rodin [1]. Absence průkazu mutace v genech kódujících klíčové proteiny tedy neznamená vyloučení přítomnosti ALVC. Dle konsenzu expertů z roku 2011 může být genetické testování přínosné u nemocných, kteří mají klinicky prokázanou diagnózu arytmogenní kardiomyopatie, a dále u příbuzných pacientů s jasně prokázanou kauzální mutací (tzv. kaskádový screening). Dále může být genetické testování zváženo u nemocných s klinickou suspekcí na možnou arytmogenní kardiomyopatii, u kterých definitivní diagnóza není ještě jistá [24]. Při diskusi o přínosu genetického testování je však nutné vzít do úvahy, zda případný průkaz dané mutace u příbuzných s doposud nemanifestním onemocněním povede ke změně způsobu sledování a léčby nemocných, popř. zda bude indikovat změnu postupů v prevenci náhlé srdeční smrti.
Histologické vyšetření
Vzhledem k subepikardiální lokalizaci strukturálních změn u nemocných s ALVC je patognomický histologický průkaz fibrózní či fibrolipomatózní náhrady myokardu při klasicky prováděné endomyokardiální biopsii myokardu, ať již z pravé či levé komory, obtížný. V souladu se zkušenostmi autorů sdělení, pouze u jedinců s MRI průkazem postižení interventrikulárního septa, které je typicky lokalizováno na jeho pravokomorové straně, lze očekávat validní výsledek takto provedeného bioptického vyšetření. V ostatních případech je nutné k potvrzení diagnózy ALVC, pokud ji nelze stanovit pouze na základě klinického obrazu, výsledku MRI a ev. i pozitivní genetické analýzy, provést biopsii myokardu epikardiálním přístupem, který je však technicky výrazně náročný. Detekované histopatologické změny jsou obdobné nálezům u ARVC (obr. 6) [25,26].

Diferenciální diagnostika
Nemocní s diagnostikovanou ALVC jsou zpočátku často vedeni pod různými jinými diagnózami. Nejčastější onemocnění, pro která jsou pacienti mylně sledováni, představují dilatační kardiomyopatie a myokarditida, levokomorová non-kompakce, hypertrofická kardiomyopatie, prolaps mitrální chlopně, idiopatická komorová tachykardie či ektopie [1].
Odlišení ALVC fenotypu od dilatační kardiomyopatie je klinicky patrně nejčastější a nesmírně důležité, zejména s ohledem na daleko vyšší riziko náhlé smrti u ALVC a její familiární výskyt. Základním rozdílem mezi oběma jednotkami je skutečnost, že vysoké riziko KT včetně maligních forem prakticky vždy předbíhá u ALVC morfologické abnormality a výraznou poruchu systolické funkce levé komory. Pro ALVC tak svědčí přítomnost subepikardiálního typu LGE, zvláště cirkumferenciálního, kontrastujícího s přítomností většinou jen mírně snížené globální systolické funkce LK. Dále na přítomnost arytmogenní KMP jako takové může pochopitelně ukazovat konkomitantní postižení pravé komory.
Význam zánětu v patogenezi ALVC byl opakovaně diskutován. Dle některých názorů je myokarditida možnou iniciální příčinou ALVC, a to jednak jako samostatného onemocnění, jednak v podobě biventrikulárního postižení [27]. Některá fakta opravdu svědčí pro podíl myokarditidy v etiopatogenezi ALVC. V kohortě nemocných, reportované Sen-Chowdrhym et al, udávala přibližně třetina pacientů symptomy suspektní z myokarditidy. Někteří jedinci s ALVC byli také původně sledováni, protože se u nich předpokládalo, že prodělali virovou myokarditidu [1]. Též ložisko LGE v subepikardiální oblasti LK může být zjištěno i u nemocných splňujících jiná kritéria myokarditidy včetně jejího bioptického průkazu [28,29]. Na druhou stranu byl u značné části nemocných s ALVC potvrzen familiární výskyt onemocnění s průkazem mutací v genech pro desmosomální proteiny [1]. Dále, histologický obraz u nemocných s ALVC se zdá být obdobný situaci u ARVC, při níž je nález fokální lymfocytární infiltrace a myokardiální nekrózy přítomen až u 2 třetin nemocných s jasně prokázaným onemocněním [24]. Z těchto důvodů se v současné době obraz zánětu myokardu považuje za nedílnou součást vývoje ALVC, ovšem infekce myokardu není základní příčinou onemocnění, ale zánětlivá celulizace je vysoce pravděpodobně nespecifickou reakcí vznikající v okolí myocytárních nekróz a předcházející reparaci tkáně fibrózou, tak jako je tomu i u jiných myokardiálních patologií. Nezdá se však, že by onemocnění bylo trvale pozvolna progredující. Jako pravděpodobnější se jeví – podobně jako u ARVC – progrese skokovitá a fáze relativního klidu se střídají s epizodami tzv. „horkých fází“, ve kterých dochází k nekróze myocytů a reaktivnímu zánětu [13]. Je pak nasnadě, že okamžik skokové progrese onemocnění může být provázen bolestmi na hrudi a popř. dokumentovaným zvýšením hladin kardiospecifických enzymů připomínajícím myokarditidu či akutní koronární syndrom. Zůstává otázkou, zda okamžik aktivní progrese onemocnění je provázen rovněž elektrickou nestabilitou a zvýšeným rizikem náhlé srdeční smrti v důsledku maligní arytmie.
Levokomorová non-kompakce je porucha struktury LK charakterizovaná přítomností prominující trabekularizace a hlubokými mezitrabekulárními recesy, které jsou překryty ztenčenou kompaktní epikardiální svalovou vrstvou. Tyto změny nemusí být nutně ohraničeny jen na levou komoru, ale mohou se vyskytovat i v pravé komoře. U většiny pacientů je non-kompakce provázena dilatací a systolickou dysfunkcí LK. Non-kompakce se může vyskytovat jak izolovaně, tak ve formě familiární (až 50 % postižených), s autosomálně dominantní dědičností při postižení genů pro sarkomerické proteiny, proteiny metabolizmu kalcia a laminin [30].
Idiopatická myokardiální fibróza (IMF) je méně známé onemocnění, které se některými znaky překrývá s arytmogenní kardiomyopatií. Autopticky je IMF identifikovatelná až u 3 % obětí náhlé srdeční smrti [31–33]. Po strukturální stránce je charakterizována heterogenní intersticiální fibrotizací predilekčně přítomnou v oblasti spodní stěny LK [31]. Ačkoli je náhrada myokardu fibrózní tkání nespecifickou reakcí vyskytující se i u jiných onemocnění, nelze vyloučit, že IMF je entitou blízkou k arytmogenní kardiomyopatii. Vyšetření pomocí MRI však u IMF na rozdíl od ALVC vykazuje více difuzní a midmyokardiálně lokalizované LGE. K dalšímu odlišení arytmogenní kardiomyopatie od IMF může sloužit přítomnost lipomatózní tkáně v myokardu. Na druhou stranu není tuková tkáň přítomna ani u kardiomyopatie blízké arytmogenní – u již zmíněného Carvajalova syndromu, který však pro svůj fenotypický obraz a genetický podklad připomíná spíše arytmogenní než dilatační kardiomyopatii [22].
Postižení levé komory u pacientů s pokročilou ARVC je přítomno u více než 50 % nemocných [7,26]. Je ovšem nutné znovu zdůraznit, že fenotyp ALVC je jinou fenotypovou formou manifestace arytmogenní kardiomyopatie než ARVC. Většina pacientů s ALVC má přítomny komorové tachykardie z LK, tj. morfologie blokády pravého Tawarova raménka, zatímco klasická forma ARVC je provázena komorovými tachykardiemi morfologie blokády levého Tawarova raménka a levokomorová morfologie je vzácná. V případě ALVC nejsou též vyjádřeny změny na EKG v pravém prekordiu charakteristické pro „čistou“ ARVC, naopak nacházíme repolarizační změny inferolaterálně. Myokard mezikomorového septa bývá u ARVC zpravidla až do pozdních stadií ušetřen. U nemocných s ALVC je naopak mezikomorové septum postiženo přibližně v polovině případů. U klasické formy ARVC je izolovaná globální dysfunkce pravé komory vyjádřena vždy dříve než změny LK [12]. Naopak u 30 % nemocných s ALVC je přítomna zachovalá funkce pravé komory [1]. U biventrikulární formy arytmogenní kardiomyopatie s konkomitantním a funkčně obdobným postižením obou komor je pak předpokládána paralelní progrese ventrikulárního postižení.
Terapie
V současné době neexistují žádná cílená doporučení pro léčbu nemocných s ALVC fenotypem. Pokud budeme aproximovat doporučení pro léčbu KT uváděná u ARVC [34] a přihlédneme k publikovaným, bohužel zejména kazuistickým datům u nemocných s ALVC, lze léčbu shrnout v následujících odstavcích.
Při stanovení diagnózy ALVC doporučujeme postiženým jedincům, aby neprovozovali výkonnostní sporty s cílem zmírnění rizika náhlého úmrtí v důsledku maligní arytmie.
K ovlivnění palpitačních obtíží při četných komorových extrasystolách či epizodách nesetrvalé KT lze jakožto prvou volbu využít empirickou terapii betablokátory v maximálně tolerované dávce. Pokud nemocný léčbu betablokátory netoleruje, má ji kontraindikovánu, nebo na ni nereaguje, přichází do úvahy jako další krok k potlačení symptomů léčba sotalolem či amiodaronem.
Po prodělané oběhové zástavě či po epizodě hemodynamicky netolerované KT je u nemocných s ALVC, tak jako v jiných případech, jasně indikovaná sekundárně preventivní implantace kardioverteru defibrilátoru (ICD). Implantace ICD by měla být jistě zvážena u i pacientů s hemodynamicky tolerovanými setrvalými KT. Zkušenosti našeho pracoviště totiž ukazují, že i u pacientů s ALVC prezentující se hemodynamicky tolerovanou KT se může následně vyvinout oběhově kompromitující arytmie.
V oblasti primární prevence náhlého úmrtí je situace u ALVC méně jasná, neboť indikace k zavedení ICD je stále diskutovaným tématem i u nemocných s klasickou ARVC. V současnosti se jako racionální jeví postup obdobný situaci u ARVC, tj. orientujeme se dle přítomnosti rizikových faktorů arytmické příhody: anamnézy náhlé srdeční smrti v rodině, přítomnosti nevysvětlitelných synkop, četnosti běhů nesetrvalých KT, rozsahu postižení jedné či obou komor, event. i dle výsledku programované stimulace komor [34].
Katetrizační ablaci arytmogenního substrátu je možné zvážit v případě vysoce symptomatických epizod KT u pacientů nereagujících na antiarytmickou terapii, popřípadě s četnými výboji ICD. Katetrizační ablaci bychom indikovali s cílem zlepšení symptomatologie a kvality života, základem prevence náhlé úmrtí je pouze implantace ICD. Při zvažování ablační léčby je třeba mít na paměti dominantně subepikardiální lokalizaci substrátu KT u ALVC. Tradiční endomyokardiální ablace je prakticky proveditelná jen v případě KT závislé na substrátu v oblasti mezikomorové přepážky, v ostatních případech je nutné provést ablaci epikardiálním přístupem, který je pro svůj charakter prováděn pouze ve vysoce specializovaných centrech [35]. Nicméně naše zkušenosti ukazují, že endokardiální katetrizační ablace pro recidivující KT v případě přítomného kritického substrátu v oblasti mezikomorové přepážky je proveditelná a účinná. Obecně je ale na ALVC třeba nahlížet jako na progredující onemocnění s jednoznačně přítomným rizikem rekurence různých morfologií KT.
Závěr
ALVC představuje fenotypickou variantu arytmogenní kardiomyopatie postihující izolovaně či zcela dominantně myokard LK. Toto doposud vzácně popisované a geneticky podmíněné onemocnění patří jednoznačně do diferenciální diagnostiky etiologie KT u nemocných bez významné ischemické choroby srdeční. Zcela zásadní roli při stanovení přítomnosti ALVC má srdeční MRI, resp. průkaz přítomnosti subepikardiální fibrózy. Lze očekávat, že počet popsaných případů bude s nárůstem dostupnosti a využíváním MRI stoupat.
Podpořeno: PRVOUK-P35/LF1/5 a projektem OPPK CZ.2.16/3.1.00/21565.
prof. MUDr. Tomáš Paleček, Ph.D.
tomas.palecek@lf1.cuni.cz
II. interní klinika kardiologie a angiologie 1. LF UK a VFN v Praze
www.vfn.cz
Doručeno do redakce 28. 3. 2016
Přijato po recenzi 31. 5. 2016
Zdroje
1. Sen-Chowdhry S, Syrris P, Prasad SK et al. Left-dominant arrhythmogenic cardiomyopathy: an under-recognized clinical entity. Dostupné z DOI: J Am Coll Cardiol 2008; 52(25): 2175–2187. <http://dx.doi.org/10.1016/j.jacc.2008.09.019>.
2. Collett BA, Davis GJ, Rohr WB. Extensive fibrofatty infiltration of the left ventricle in two cases of sudden cardiac death. J Forensic Sci 1994; 39(5): 1182–1187.
3. De Pasquale CG, Heddle WF. Left sided arrhythmogenic ventricular dysplasia in siblings. Heart 2001; 86(2): 128–130.
4. Gallo P, d‘Amati G, Pelliccia F. Pathologic evidence of extensive left ventricular involvement in arrhythmogenic right ventricular cardiomyopathy. Hum Pathol 1992; 23(8): 948–952.
5. Michalodimitrakis M, Papadomanolakis A, Stiakakis J et al. Left side right ventricular cardiomyopathy. Med Sci Law 2002; 42(4): 313–317.
6. Sen-Chowdhry S, Syrris P, McKenna WJ. Desmoplakin disease in arrhythmogenic right ventricular cardiomyopathy: early genotype-phenotype studies. Eur Heart J 2005; 26(16): 1582–1584.
7. Bauce B, Basso C, Rampazzo A et al. Clinical profile of four families with arrhythmogenic right ventricular cardiomyopathy caused by dominant desmoplakin mutations. Eur Heart J 2005; 26(16): 1666–1675.
8. Norman M, Simpson M, Mogensen J et al. Novel mutation in desmoplakin causes arrhythmogenic left ventricular cardiomyopathy. Circulation 2005; 112(5): 636–642.
9. Suzuki H, Sumiyoshi M, Kawai S et al. Arrhythmogenic right ventricular cardiomyopathy with an initial manifestation of severe left ventricular impairment and normal contraction of the right ventricle. Jpn Circ J 2000; 64(3): 209–213.
10. Okabe M, Fukuda K, Nakashima Y et al. An isolated left ventricular lesion associated with left ventricular tachycardia – arrhythmogenic “left” ventricular dysplasia? Jpn Circ J 1995; 59(1):49–54.
11. Coats CJ, Quarta G, Flett AS et al. Arrhythmogenic Left Ventricular Cardiomyopathy. Circulation 2009; 120(25): 2613–2614. Dostupné z DOI: <http://dx.doi.org/10.1161/CIRCULATIONAHA.109.874628>.
12. Sen-Chowdhry S, Syrris P, Ward D et al. Clinical and genetic characterization of families with arrhythmogenic right ventricular dysplasia/cardiomyopathy provides novel insights into patterns of disease expression. Circulation 2007; 115(3): 1710–1720.
13. Sen-Chowdhry S, Syrris P, McKenna WJ. Role of genetic analysis in the management of patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy. J Am Coll Cardiol 2007; 50(19): 1813–1821.
14. Gandjbakhch E, Vite A, Gary F et al. Screening of genes encoding junctional candidates in arrhythmogenic right ventricular cardiomyopathy/dysplasia. Europace 2013; 15(10): 1522–1525. Dostupné z DOI: <http://dx.doi.org/10.1093/europace/eut224>.
15. Calore M, Lorenzon A, De Bortoli M et al. Arrhythmogenic cardiomyopathy: a disease of intercalated discs. Cell Tissue Res 2015; 360(3): 491–500. Dostupné z DOI: <http://dx.doi.org/10.1007/s00441–014–2015–5>.
16. Cox MG, van der Zwaag PA, van der Werf C et al. Arrhythmogenic right ventricular dysplasia/cardiomyopathy: pathogenic desmosome mutations in index-patients predict outcome of family screening: Dutch arrhythmogenic right ventricular dysplasia/cardiomyopathy genotype-phenotype follow-up study. Circulation 2011; 123(23): 2690–2700. Dostupné z DOI: <http://dx.doi.org/10.1161/CIRCULATIONAHA.110.988287>.
17. Lorenzon A, Pilichou K, Rigato I et al. Homozygous Desmocollin-2 Mutations and Arrhythmogenic Cardiomyopathy. Am J Cardiol 2015; 116(8): 1245–1251. Dostupné z DOI: <http://dx.doi.org/10.1016/j.amjcard.2015.07.037>.
18. Al-Sabeq B, Krahn AD, Conacher S et al. Arrhythmogenic right ventricular cardiomyopathy with recessive inheritance related to a new homozygous desmocollin-2 mutation. Can J Cardiol 2014; 30(6): 696.e1-e3. Dostupné z DOI: <http://dx.doi.org/10.1016/j.cjca.2014.01.014>.
19. Gerull B, Kirchner F, Chong JX et al. Homozygous founder mutation in desmocollin-2 (DSC2) causes arrhythmogenic cardiomyopathy in the Hutterite population. Circ Cardiovasc Genet 2013; 6(4): 327–336. Dostupné z DOI: <http://dx.doi.org/10.1161/CIRCGENETICS.113.000097>.
20. Simpson MA, Mansour S, Ahnood D et al. Homozygous mutation of desmocollin-2 in arrhythmogenic right ventricular cardiomyopathy with mild palmoplantar keratoderma and woolly hair. Cardiology 2009; 113(1): 28–34. Dostupné z DOI: <http://dx.doi.org/10.1159/000165696>.
21. Carvajal-Huerta L. Epidermolytic palmoplantar keratoderma with woolly hair and dilated cardiomyopathy. J Am Acad Dermatol 1998; 39(3): 418–421.
22. Protonotarios N, Tsatsopoulou A. Naxos disease and Carvajal syndrome: cardiocutaneous disorders that highlight the pathogenesis and broaden the spectrum of arrhythmogenic right ventricular cardiomyopathy. Cardiovasc Pathol 2004; 13(4): 185–194.
23. Groeneweg JA, van der Zwaag PA, Jongbloed JD et al. Left-dominant arrhythmogenic cardiomyopathy in a large family: associated desmosomal or nondesmosomal genotype? Heart Rhythm 2013; 10(4): 548–559. Dostupné z DOI: <http://dx.doi.org/10.1016/j.hrthm.2012.12.020>.
24. Ackerman MJ, Priori SG, Willems S et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Heart Rhythm 2011; 8(8): 1308–1339. Dostupné z DOI: <http://dx.doi.org/10.1016/j.hrthm.2011.05.020>.
25. Basso C, Thiene G, Corrado D et al. Arrhythmogenic right ventricular cardiomyopathy. Dysplasia, dystrophy, or myocarditis? Circulation 1996; 94(5): 983–991.
26. Corrado D, Basso C, Thiene G et al. Spectrum of clinicopathologic manifestations of arrhythmogenic right ventricular cardiomyopathy/dysplasia: a multicenter study. J Am Coll Cardiol 1997; 30(6): 1512–1520.
27. Fontaine GH, Fornes P. Letter regarding article by Norman et al, “novel mutation in desmoplakin causes arrhythmogenic left ventricular cardiomyopathy”. Circulation 2006; 113(5): e68-e69.
28. Assomull RG, Lyne JC, Keenan N et al. The role of cardiovascular magnetic resonance in patients presenting with chest pain, raised troponin, and unobstructed coronary arteries. Eur Heart J 2007; 28(10): 1242–1249.
29. De Cobelli F, Pieroni M, Esposito A et al. Delayed gadolinium-enhanced cardiac magnetic resonance in patients with chronic myocarditis presenting with heart failure or recurrent arrhythmias. J Am Coll Cardiol 2006; 47(8): 1649–1654.
30. Jenni R, Oechslin E, Schneider J et al. Echocardiographic and pathoanatomical characteristics of isolated left ventricular non-compaction: a step towards classification as a distinct cardiomyopathy. Heart 2001; 86(6): 666–671.
31. John BT, Tamarappoo BK, Titus JL et al. Global remodeling of the ventricular interstitium in idiopathic myocardial fibrosis and sudden cardiac death. Heart Rhythm 2004; 1(2): 141–149.
32. Bowker TJ, Wood DA, Davies MJ et al. Sudden, unexpected cardiac or unexplained death in England: a national survey. QJM 2003; 96(4): 269–279.
33. Davies MJ. The investigation of sudden cardiac death. Histopathology 1999; 34(2): 93–98.
34. Priori SG, Blomstrom-Lundqvist C, Mazzanti A et al. [Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC)]. 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: The Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC) Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Europace 2015; 17(11): 1601–1687. Dostupné z DOI: <http://dx.doi.org/10.1093/europace/euv319>.
35. Havranek S, Palecek T, Kovarnik T et al. Arrhythmogenic substrate at the interventricular septum as a target site for radiofrequency catheter ablation of recurrent ventricular tachycardia in left dominant arrhythmogenic cardiomyopathy. BMC Cardiovasc Disord 2015; 15 : 18. Dostupné z DOI: <http://dx.doi.org/10.1186/s12872–015–0010–8>.
Štítky
Diabetologie Endokrinologie Interní lékařstvíČlánek vyšel v časopise
Vnitřní lékařství

2016 Číslo 9
- Diosmin a hesperidin: Co ukazuje farmakokinetika?
- Omeprazol a mechanismus hojení krvácení z peptického vředu
- Porovnání farmakologických vlastností mikronizovaného diosminu a hesperidinu v terapii chronické žilní insuficience a hemoroidů
- Srovnání vlivu omeprazolu a pantoprazolu na antiagregační účinek klopidogrelu
Nejčtenější v tomto čísle
- Arytmogenní kardiomyopatie levé komory
- Jak postupovat při podezření na sekundární arteriální hypertenzi
-
Schnitzlerové syndrom
Diferenciální diagnostika, přehled léčebných možností a popis 5 případů léčených anakinrou - Perorálne antikoagulanciá v primárnej a sekundárnej prevencii vénovej tromboembólie
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy