
Patogeneze inzulinové rezistence u vybraných endokrinopatií
Pathogenesis of insulin resistance in different endocrinopathies
Visceral obesity, insulin resistance and increased cardiovascular morbidity and mortality are strongly related. Development of visceral obesity and regulation of body fat distribution in general are influenced by many factors. This article is a summary of current knowledge about pathogenesis of insulin resistance in different endocrinopathies, particularly in the Cushingʼs syndrome and acromegaly, as they are both connected with the pathological conditions mentioned above.
Key words:
insulin signaling pathway – visceral obesity – cortisol – growth hormone
Autoři:
V. Ďurovcová; M. Kršek; M. Haluzík
![]()
Působiště autorů:
III. interní klinika 1. lékařské fakulty UK a VFN Praha, přednosta prof. MUDr. Štěpán Svačina, DrSc., MBA
Vyšlo v časopise:
Vnitř Lék 2008; 54(4): 368-376
Kategorie:
Přehledný referát
Souhrn
Spojitost mezi centrální (viscerální) obezitou, inzulinovou rezistencí a předčasnou mortalitou na kardiovaskulární onemocnění soustřeďuje pozornost na identifikaci faktorů, které regulují distribuci tuku v organizmu. U některých endokrinopatií (zejména Cushingova syndromu a akromegalie) jsou inzulinová rezistence, resp. viscerální obezita jedním z charakteristických rysů nemoci. Náš článek prezentuje aktuální přehled dostupných informací ohledně vzniku inzulinové rezistence u těchto endokrinopatií na různých úrovních, tkáňových a buněčných.
Klíčová slova:
inzulinová signální kaskáda – viscerální obezita – kortizol – růstový hormon
Úvod
Obezita je úzce spojena s metabolickým syndromem, který zahrnuje inzulinovou rezistenci, hyperglykemii, dyslipidemii, arteriální hypertenzi, protrombotický a proinflamatorní stav. Nejlepším prediktorem rozvoje těchto onemocnění není ani tak celkový objem tuku v organizmu, jako množství viscerálního, tj. omentálního a mezenterického tuku [1]. Tendence k rozvoji viscerální obezity je částečně vysvětlitelná změnami inzulinové senzitivity a působení inzulinu na jednotlivé metabolické pochody.
Inzulinová signalizace metabolických procesů a viscerální obezita
Konformační změny molekuly inzulinového receptoru (IR), vyvolané vazbou ligandu (inzulinu), vedou k aktivaci tyrozinkinázové domény intracytoplazmatické části β-podjednotky receptoru. To vede k autofosforylaci tyrozinových zbytků na několika místech β-podjednotky IR s další aktivací její tyrozinkinázy a fosforylací intermediárních proteinů inzulinové signalizace, jako jsou substráty inzulinového receptoru (IRS) a Shc proteiny (Src Homologous and Collagen like proteiny; Src - onkogen s názvem odvozeným od v-src - viral sarcoma).
Dle současných poznatků obsahuje skupina IRS proteinů u savců 4 členy, a to IRS-1 a IRS-2, které jsou nejprozkoumanější a exprimované téměř ve všech tkáních, IRS-3, který je přítomen pouze v tukové tkáni a β-buňkách pankreatu, a IRS-4, který je exprimovaný v tymu, mozku a ledvinách. Substráty inzulinového receptoru, Shc proteiny a IR s fosforylovanými tyrozinovými zbytky na sebe váží několik proteinů obsahujících Src homologní (SH2) doménu, které zprostředkují další přenos signálů v buňce. Jedním z SH2 doménu obsahujících proteinů je fosfatidylinositol-3-kináza (PI3K). Tento enzym se skládá ze 2 podjednotek - regulační p85 a katalytické p110: navázáním p85 podjednotky na fosforylované IRS proteiny dochází k aktivaci enzymu. PI3K je enzym nezbytný ke spuštění téměř všech inzulinem zprostředkovaných procesů včetně stimulace glukózového transportu, aktivace glykogensyntázy a inhibice jaterní glukoneogeneze [2,3]. Dalším důležitým signálním proteinem je proteinkináza B (PKB), známá též jako Akt/PKB, která je také začleněna do procesu inzulinem-stimulovaného vychytávání glukózy a aktivace glykogensyntázy [2,4,5] (obr. 1).
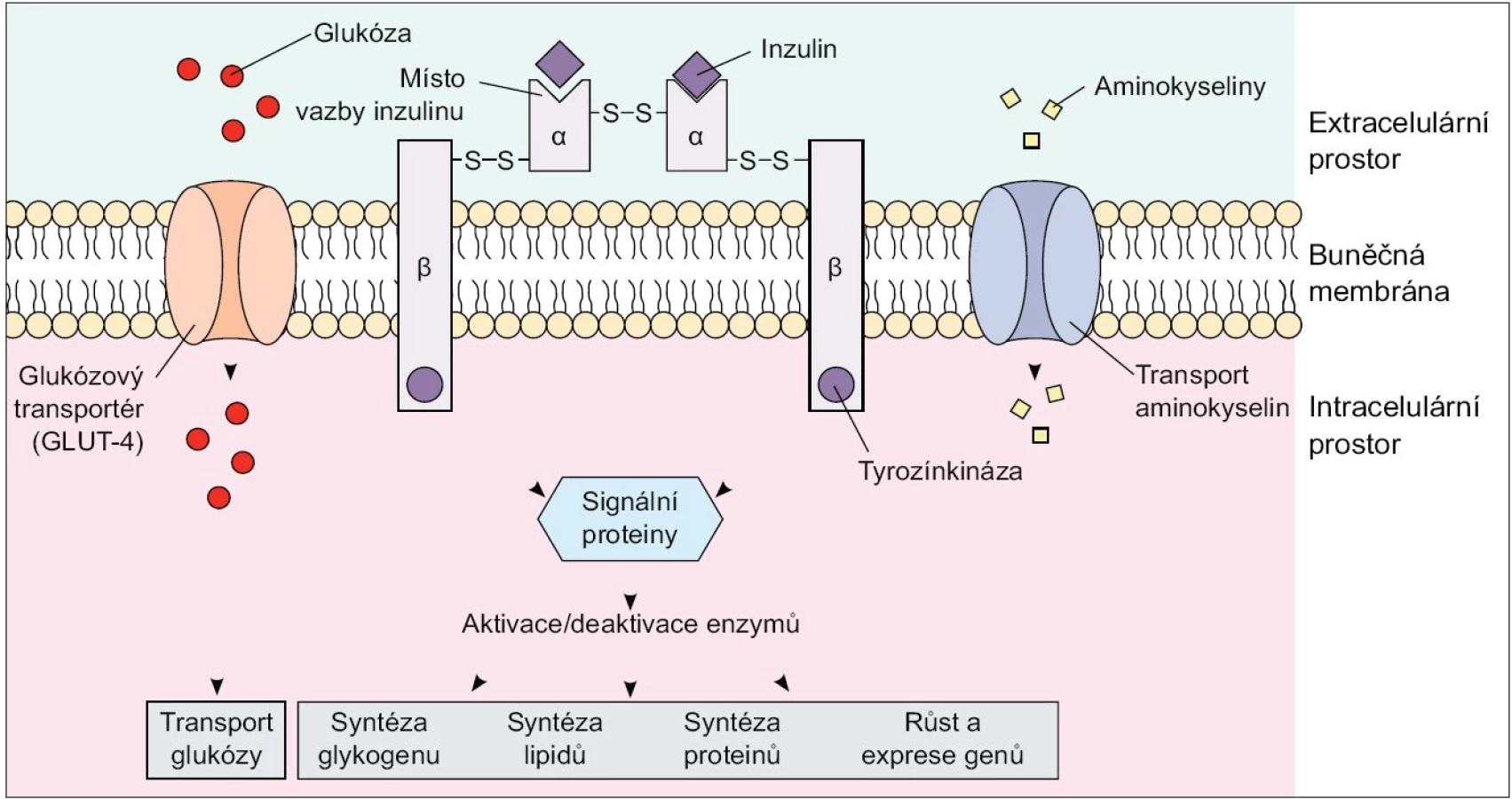
Inzulinem stimulované vychytávání glukózy z oběhu se děje prostřednictvím translokace tkáňově specifických glukózových transportérů (GLUT 4) z nitrobuněčných skladovacích míst do plazmatické membrány, a to především v tukové tkáni a kosterním svalstvu. Omentální adipocyty vykazují ve srovnání se subkutánním tukem 2krát vyšší inzulinem stimulované vychytávání glukózy z krve, a to díky vyššímu obsahu IRS 1, Akt/PKB a GLUT 4. Dá se tedy předpokládat, že vyšší příjem glukózy buňkou a následná lipogeneze s využitím glykolýzou vzniklého acetyl-koenzymu A vede k preferenčnímu hromadění triacylglycerolů ve viscerálním tuku. Výzkumy ale ukazují, že de novo syntéza volných mastných kyselin (VMK) z glukózy je v lidské tukové tkáni minimální a jen nepatrně stimulovaná i při vysokém kalorickém nebo glukózovém příjmu, jelikož absolutně převažuje vychytávání a esterifikace VMK z plazmatických lipoproteinů. Na druhou stranu zvýšená utilizace glukózy ve viscerálním tuku je doprovázená sníženou lipidovou oxidací a tak hromaděním triacylglycerolů v adipocytech [6].
Negativní metabolický efekt viscerální tukové tkáně
Podkožní i viscerální tuk jsou metabolicky vysoce aktivní tkáně s pestrým spektrem secernovaných produktů, podléhající komplexním hormonálním i jiným regulacím (tab. 1 a 2). Jednou z důležitých funkcí tukové tkáně je skladování triacylglycerolů, které jsou zdrojem energie v případech lačnění nebo zvýšené energetické potřeby v organizmu. Skladování, resp. použití této energie je regulované různými hormony, např. právě inzulinem a katecholaminy. Katecholaminy se váží na α - a/nebo β-adrenergní receptory na povrchu buněk. Stimulací β-receptorů dochází prostřednictvím elevace cAMP (cyklického adenozinmonofosfátu) a proteinkinázy A k aktivaci hormonsenzitivní lipázy (HSL). HSL hydrolyzuje triacylglyceroly na glycerol a VMK, které mohou být následně využity v energetickém metabolizmu jiných tkání. Dle některých hypotéz uvolňuje viscerální tuková tkáň větší množství VMK než subkutánní tuková tkáň, a to díky vyšší lipolytické aktivitě – většímu množství β-adrenergních receptorů a nižší senzitivitě k antilipolytickému účinku inzulinu. Nadbytek viscerální tukové tkáně tedy potencionálně vede k větší dodávce VMK do jater portálním oběhem, což zhoršuje clearance a účinek inzulinu a zvyšuje výdej glukózy a lipoproteinů s velmi nízkou denzitou (VLDL) z jater [6].
![Příklady receptorů exprimovaných v tukové tkáni; upraveno podle [40].](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/c72be509a888a1072e5350a1dbcceaa9.png)
![Příklady adipocyty secernovaných proteinů s endokrinními funkcemi; upraveno podle [40].](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/198f579206d4f12657f0bc99a9c066b0.png)
Hyperkortizolizmus a inzulinová rezistence
Glukokortikoidy (GK) hrají jednu z klíčových rolí v regulaci metabolizmu, funkce a distribuce tukové tkáně. S tím souvisí také fakt, že u pacientů s Cushingovým syndromem (onemocnění charakterizované systémovým nadbytkem GK vznikající na podkladě adrenálních, pituitárních, méně často i jiných tumorů) nebo pacientů na glukokortikoidní terapii se rozvíjí poměrně výrazná inzulinová rezistence a viscerální obezita [7,8].
GK jsou hormony syntetizované v kůře nadledvin pod kontrolou hypotalamo-hypofýzo-adrenokortikální osy (HPA). U lidí je hlavním glukokortikoidem kortizol. Negativní vliv GK na parametry metabolického syndromu je dán jejich působením v rámci regulací metabolizmu lipidů a glukózy. Negativní vliv na kardiovaskulární systém je zprostředkovaný také přítomností receptorů GK (GKR), i mineralokortikoidů (MKR) v srdečním svalu, cévních stěnách, nebo přítomností GKR v zánětlivých buňkách invadujících endotel, např. makrofázích [8].
Ovlivnění sérových koncentrací lipidů je způsobeno stimulací lipolýzy tukových zásob se zvýšenou dodávkou VMK do jater, stimulací činnosti HMG-CoA reduktázy (3-hydroxy-3-methyl-glutaryl-CoA reduktázy), klíčového enzymu syntézy cholesterolu, či snížením hladin lipoproteinové lipázy (LPL), enzymu štěpícího lipidy v lipoproteinech, zejména chylomikronech a VLDL s následnou hypertriglyceridemií. GK zároveň stimulují diferenciaci preadipocytů na adipocyty, což může vést k nárůstu objemu tukové tkáně. Hyperglykemizující účinek GK je způsoben zvýšením nitrobuněčné koncentrace enzymů (stimulací jejich transkripce) a substrátů (aminokyselin z proteolýzy, glycerolu z lipolýzy) pro glukoneogenezi, což vede ke zvýšené produkci glukózy játry. V pankreatu vedou GK působením na B-buňky k inhibici inzulinové sekrece, na úrovni kosterních svalů a tukové tkáně zhoršují vychytávání glukózy z oběhu (inhibicí translokace glukózových transportérů) a její utilizaci [9–12]. Hypertenzní účinek GK je dán jejich částečným mineralokortikoidním účinkem s retencí sodíku a chloridů, ale také přímým vlivem na tonus cév [8,11,12]. Za zmínku stojí i schopnost GK modifikovat zánětlivé, proliferativní a remodelační odezvy cév na poškození [8].
Význam glukokortikoidů v patogenezi lidské obezity zůstává nejasný, jelikož u většiny obézních nejsou jejich cirkulující hladiny zvýšené [13]. I když v rámci některých studií byla prokázaná korelace mezi hladinou glukokortikoidů v krvi a symptomy metabolického syndromu i v normálním rozmezí hodnot kortizolu [14]. Výsledek působení glukokortikoidů na cílové tkáně však nezávisí pouze na jejich hladinách v krvi, ale i na jejich intracelulárních koncentracích [1,15]. Ty jsou v úzké souvislosti s metabolizmem kortizolu.
11-β-hydroxysteroiddehydrogenáza (11-β-HSD) a inzulinová rezistence
Metabolizmus kortizolu je komplexní, tkáňově dependentní proces, jehož hlavní součástí je konverze aktivního kortizolu na inaktivní kortizon, respektive přeměna inaktivního kortizonu na kortizol, katalyzované enzymem 11-β-hydroxysteroiddehydrogenázou (11-β-HSD). V současné době jsou známy 2 izoformy tohoto enzymu.
11-β-HSD1 byla izolována původně z jater, ale je exprimována i v tukové tkáni, svalech, gonádách, kostech a očních tkáních. Působí in vivo jako reduktáza měnící inaktivní kortizon na aktivní kortizol a zároveň urychluje účinek kortizolu v tkáních.
Naproti tomu 11-β-HSD2 je exprimována v tkáních s vysokým obsahem mineralokortikoidních receptorů (MKR) – ledvinách, tlustém střevě a slinných žlázách, kde má protektivní účinek na MKR vůči přesycení kortizolem [11,15–17].
V případě metabolického syndromu jsou zvýšené intracelulární koncentrace kortizolu v tukové tkáni a kosterních svalech. Tento jev je důsledkem zvýšené exprese 11-β-HSD1 v tukových depech obézních jedinců.
Důkaz o důležitosti tohoto enzymu v patogenezi metabolického syndromu dokazují i pokusy, ve kterých u 11-β-HSD1 knockout myší nedošlo k vývoji viscerální obezity ani při krmení potravou s vysokým obsahem tuků. Naopak u myší se zvýšenou aktivitou enzymu na hladiny srovnatelné s hladinami u obézních lidí se rozvinula obezita s inzulinovou a leptinovou rezistencí, dyslipidemií a arteriální hypertenzí [1,11,16]. Syntetické selektivní 11-β-HSD1 inhibitory byly testovány na myších modelech diabetes mellitus 2. typu s obezitou a byly zjištěny jejich pozitivní vlivy na glukózovou toleranci, se schopností upravit i těžkou hyperglykemií. Nadto i antidiabetické preparáty typu PPARγ (peroxisome proliferator-activated receptor-γ) agonistů a LXRα (liver X receptor-α) agonistů významně redukují transkripci 11-β-HSD1 a její enzymatickou aktivitu, což rovněž naznačuje, že suprese 11-β-HSD1 v tukové tkáni může být jedním z mechanizmů, kterým tyto léky dosahují své příznivé metabolické účinky [1].
Výsledky klinických studií jsou ale v relativním rozporu s daným pozorováním, jelikož prokázaly celkovou inhibici a ne stimulaci 11-β-HSD1 u obezity. Redukované hladiny 11-β-HSD1 mRNA (messenger ribonucleic acid) v nepřímé úměře k narůstajícímu BMI (body mass index) byly nalezeny v tukových biopsiích jak subkutánního, tak i omentálního tuku. Předpokládá se ale spíš protektivní než patogenetický význam tohoto pozorování, kdy pokles aktivity 11-β-HSD1 způsobující redukci diferenciace adipocytů a jaterní glukoneogeneze má vést ke snížení negativních dopadů obezity na glukózovou toleranci [15].
Výzkumy na humánních i potkaních subjektech ukazují, že vliv glukokortikoidů se v souvislosti s inzulinovou rezistencí projevuje téměř výlučně ve viscerálním, ne v subkutánním tuku. 24hodinové podávání dexametazonu v různých dávkách vedlo k inhibici jak bazálního, tak i inzulinem stimulovaného vychytávání glukózy omentárními adipocyty. Došlo totiž ke snížení exprese inzulinových signálních proteinů IRS1 a PKB s následnou nedostatečnou translokací GLUT 4. Avšak ani jeden ze zmíněných jevů nebyl prokázán v subkutánním tuku [6]. Tyto procesy jsou významným vodítkem k pochopení úzké souvislosti GK s inzulinovou signální kaskádou a tak i s potencionálním rozvojem inzulinové rezistence.
Glukokortikoidní receptor a inzulinová rezistence
Glukokortikoidní receptor (GKR) patří do velké rodiny nukleárních receptorů, které se nacházejí v cytoplazmě buněk a působí jako transkripční faktory regulující genovou expresi. Po navázání GK na receptor dochází ke konformační změně GKR s odpojením od velkého komplexu proteinů, ze kterých je nejdůležitější tzv. protein tepelného šoku 90. GK-GKR komplex se následně přesouvá do jádra, kde může účinkovat několika způsoby. Jednak se může navázat na GK-responzivní elementy cílového genu s následnou iniciací jeho transkripce, nebo může pozitivně i negativně regulovat transkripci genů prostřednictvím interproteinových interakcí [18]. Takto je stimulována, resp. inhibována exprese enzymů zapojených do metabolických pochodů regulovaných GK.
Zajímavým pozorováním v souvislosti s GKR je fakt, že senzitivita k působení GK měřená pomocí dexametazonového supresního testu mezi jednotlivými, jinak zdravými jedinci, značně kolísá. Za jedno z možných vysvětlení tohoto pozorování se považuje existence polymorfizmů v genu kódujícím GKR [8,18].
Polymorfizmy jsou definované jako běžné variace DNA vyskytující se v normální populaci s frekvencí vyšší než 1 %. Existují minimálně 3 polymorfizmy GKR, které jsou spojované s alterovanou citlivostí GKR k působení GK a změnami v tělesném složení a metabolizmu.
Polymorfizmus N363S je asociovaný se zvýšenou citlivostí ke GK, zvýšenou inzulinovou odezvou na podání dexametazonu, s tendencí k nižší kostní denzitě a zvýšenému BMI. Dále je u něj prokázaná asociace s koronární aterosklerózou, nezávisle na přítomnosti obezity, ale také se zvýšenou hladinou celkového cholesterolu, triacylglycerolů a sníženou hodnotou HDL-cholesterolu [8,18,19]. Další polymorfizmus, označovaný jako polymorfizmus délky BclI restrikčního fragmentu, je rovněž spojený se zvýšenou citlivostí GKR k působení GK. U osob ve středním věku je asociovaný se zvýšenou kumulací abdominálního tuku a u osob ve věku vyšším spíš s nižším BMI, s tendencí k nižšímu obsahu tukuprosté tělesné hmoty [8,18]. Přítomnost tohoto polymorfizmu predisponuje k hyperinzulinemii, viscerální obezitě, arteriální hypertenzi a hypersenzitivitě hippokampu ke GK [8,20]. Třetí polymorfizmus vzniká v důsledku 2 propojených mutací nukleotidů v kodonech 22 a 23. Tento tzv. ER22/23EK polymorfizmus je asociovaný s relativní GK rezistencí. Jeho nositelé mají nižší celkový i LDL-cholesterol [8,18,21], nižší hladiny CRP (C-reaktivního proteinu), inzulinu nalačno a vyšší inzulinovou senzitivitu [8,18,22]. I když určitá hladina GK je nezbytná pro adekvátní mozkovou výkonnost, jejich nadměrné množství je spojené s negativním efektem na mozkovou morfologii a funkce. U nositelů polymorfizmu ER22/23EK bylo ve vyšším věku nalezeno nižší riziko demence a defektů bílé mozkové hmoty [8,18,23]. Zároveň jsou u nich patrné změny v rozložení tukové hmoty v těle. Mladí mužští nositelé daného polymorfizmu jsou vyššího vzrůstu, mají tendenci k většímu obsahu svalové hmoty a větší svalové síle než jedinci bez této odchylky, u žen je pozorován nižší obvod pasu a boků a nižší hmotnost [8,18,24].
Efekty polymorfizmů se mohou lišit mezi rasami vzhledem k různým kombinacím polymorfizmů několika genů, či vzhledem k působení rozdílných vlivů prostředí a socioekonomických faktorů i v rámci jedné rasy. Proto je obtížné interpretovat výsledky odlišných studií. Nicméně je pravděpodobné, že tyto polymorfizmy GKR mají vliv na patologické stavy, jako je např. ateroskleróza. Je známo, že někteří jedinci se dožívají vysokého věku i přes značnou hypercholesterolemii a právě u těch nelze vyloučit pozitivní vliv genetické odchylky, jakou je ER22/23EK. Na druhou stranu nositelé polymorfizmů N363S nebo BclI mohou mít kardiovaskulární riziko vyšší i přes uspokojivé metabolické ukazatele [18] (obr. 2).
![Gen glukokortikoidního receptoru (GKR) a jeho polymorfizmů; upraveno podle [18].](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/bfe8278b4029c01ab86381ea31a0227f.jpg)
Kortikosteroidy vážící protein a inzulinová rezistence
Kortikosteroidy vážící protein (corticosteroid-binding globulin – CBG) je hlavním transportním proteinem kortizolu v lidském séru. Jeho hladina negativně koreluje s volnou frakcí kortizolu a kortikosteronu, jelikož je na sebe váže. Nicméně výrazně nadměrná produkce CBG játry nebo jeho významný nedostatek ovlivňují celou HPA osu s nadprodukcí, resp. hypoprodukcí kortizolu [11]. O vztahu CBG a patogeneze obezity a inzulinové rezistence je známo několik zajímavých skutečností.
Předpokládá se, že mírný zánětlivý stav v organizmu je predisponujícím faktorem vzniku inzulinové rezistence, obezity a diabetes mellitus 2. typu. CBG negativně koreluje s hladinami interleukinu 6 (IL-6), solubilního receptoru TNF (tumor necrosis factor/tumor nekrotizující faktor) a CRP. Hladina CBG je negativně asociovaná s rizikovými parametry vzniku inzulinové rezistence jako zvýšeným BMI, WHR (waist-to-hip ratio, poměr obvodu pasu a boků), či HOMA indexem (homeostatic model assessment, počítán jako součin hladiny inzulinu v μU/ml a glykemie v mmol/l nalačno dělený konstantou 22,5, s hodnotou kolem 1 u zdravých mladých jedinců) [11,25,26].
CBG je zároveň členem rodiny tzv. SERPIN (serine protease inhibitors) proteinů, které jsou substráty pro elastázu exprimovanou na povrchu neutrofilů. Proto hladiny cirkulujícího CBG závisí i na míře štěpení CBG aktivovanými neutrofily - dochází k odloučení části proteinu vážícího kortizol. Leukocytóza a neutrofilie jsou přitom častým úkazem přítomným u obezity a inzulinové rezistence, čím lze vysvětlit negativní vztah těchto patologických stavů k množství CBG v krvi.
Jelikož CBG je exprimován tukovou tkání, pokles jeho množství znamená lokálně zvýšenou dostupnost kortizolu (bez změn cirkulujícího kortizolu) s následnou zvýšenou kumulací tuku s patřičnými metabolickými důsledky. Existují i pohlavně dané rozdíly v množství CBG. Muži mají slabší korelaci CBG a zánětlivých parametrů, ale také nižší hladiny CBG. Estrogeny totiž stimulují tvorbu CBG, a to buď indukcí zvýšené genové transkripce, nebo změny glykozylace molekuly CBG [11,25].
Růstový hormon a inzulinová rezistence
Růstový hormon (growth hormone – GH) je peptidový hormon produkovaný a secernovaný hypofýzou pod kontrolou hypotalamických hormonů a periferních zpětnovazebných působků, včetně inzulinu podobného růstového faktoru-1 (insulin-like growth factor-I - IGF-I). IGF-I, secernovaný zejména v játrech, ale i v jiných tkáních pod vlivem GH, je zároveň zprostředkovatelem některých účinků GH v cílových tkáních.
Význam GH v regulaci objemu viscerálního tuku je zjevný z pozorování, že u pacientů s akromegalií (nadprodukcí GH převážně na podkladě tumoru hypofýzy) dochází v této oblasti k redukci tukové tkáně. Naopak u dospělých jedinců s nedostatkem GH se vyvíjí stav podobný metabolickému syndromu spojený s hromaděním abdominálního tuku, inzulinovou rezistencí, hypertriglyceridemií a snížením HDL-cholesterolu [12]. Po substituci GH ve fyziologických dávkách dochází k úpravě stavu a snížení kardiovaskulárních rizik [27–29].
Z tohoto hlediska lze konstatovat, že mnoho účinků GH je soustředěno na prevenci akumulace tuku a stimulaci lipidové mobilizace. To vyžaduje synergizmus se steroidními hormony – např. LPL je výrazně inhibovaná působením GH, ale pouze v přítomnosti testosteronu nebo kortizolu. Na druhou stranu GH potencuje katecholaminy indukovanou lipolýzu přes β-adrenergní receptory, jednak prostřednictvím zvýšení denzity těchto receptorů, jednak stimulací aktivity adenylátcyklázy, proteinkinázy a hormonsenzitivní lipázy [30,31].
Z hlediska souvislosti s inzulinovým metabolizmem lze konstatovat existenci akutních a chronických účinků GH na metabolizmus tuků a uhlovodanů. Ty akutní jsou podobné účinkům inzulinu, tj. snížení glykemie, stimulace vychytávání glukózy kosterními svaly, nebo stimulace glukózového transportu a lipogeneze v izolovaných adipocytech. Fyziologický význam těchto účinků není dosud úplně objasněn a jsou pouze přechodné [32,33]. Již po několika hodinách stoupá inzulin antagonizující efekt GH. Dochází ke zvýšení koncentrace glukózy v séru, k vzestupu inzulinové rezistence, ke stimulaci lipolýzy a inhibici glukózového transportu. Vznik inzulinové rezistence působením GH je opět dobře dokumentovatelný na pacientech s diagnózou akromegalie a je vyvolatelný také exogenní aplikací GH [4].
Interakce GH a inzulinové signalizace
Dle recentních údajů je vliv GH na inzulinový metabolizmus daný jeho úzkou vazbou na postreceptorovou úroveň inzulinové signalizace. Receptor pro GH (GHR) na rozdíl od IR postrádá vnitřní tyrozinkinázovou aktivitu. Po navázání GH dochází k fosforylaci non-receptorové tyrozinkinázy Janus2 (JAK2) a k její aktivaci po dobu vazby s GHR. Následně jsou fosforylovány tyrozinové zbytky několika intracelulárních proteinů, včetně cytoplazmatické domény GHR a JAK2, což iniciuje spuštění signalizačních kaskád (schéma).
![Schéma. Konvergence mezi signalizačními kaskádami inzulinu a růstového hormonu (GH); upraveno podle [4].](https://pl-master.mdcdn.cz/media/image/f66bbf8773304b387b0ef5cd760790a4.jpg?version=1537797676)
Akutní, inzulinu-podobné, účinky GH jsou přechodné (jak už bylo zmíněno) a vznikají pouze v buňkách, které byly vystavené několikahodinové deprivaci GH. Jedná se o proces zprostředkovaný JAK2, která indukuje tyrozinovou fosforylaci GHR, ale i IRS-1 a IRS-2. Analogicky k inzulinové signalizaci dochází následně k navázání PI3K na IRS proteiny a k aktivaci enzymu. Působením Akt/PKB se pak aktivují glukózové transportéry GLUT 4 se zvýšením vychytávání glukózy buňkou. Zároveň se aktivuje fosfodiesteráza 3B a spouští hydrolýza cAMP, což vede k defosforylací HSL a inhibici lipolýzy. JAK2 současně aktivuje STAT-rodinu transkripčních faktorů, které se přesouvají do jádra a aktivují, mezi jiným, transkripci genů kódujících tzv. supresory cytokinové signalizace (SOCS) [32,34]. Jedná se o buněčné proteiny, které inhibují signalizaci IR. V přítomnosti SOCS JAK2 nadále aktivuje STAT proteiny, ale už nezpůsobuje fosforylaci IRS proteinů. Tak se přeruší inzulinu podobný účinek GH a nastupuje účinek diabetogenní [32,35].
Předpokládá se existence několika mechanizmů, jakými GH dosahuje svůj antagonistický účinek vůči inzulinu. Např. prostřednictvím další signalizační cesty vedoucí ke stimulaci glukózového transportu. Jedná se o cestu paralelní s PI3K, ve které dochází pod vlivem inzulinu k tyrozinové fosforylaci produktu c-Cbl protoonkogenu (Cbl), který je napojený na IR prostřednictvím adaptačního proteinu CAP. Po fosforylaci se komplex Cbl-CAP z IR odpojí a iniciuje aktivaci celé řady dalších signálních bílkovin. Míra exprese CAP dobře koreluje s inzulinovou senzitivitou a je stimulovaná např. thiazolidindiony, tedy PPARγ agonisty se současnou zvýšenou fosforylací Cbl. Tato cesta je dalším místem konvergence GH a inzulinového působení, při němž GH stimuluje tyrozinovou fosforylaci Cbl i jeho asociaci s jinými proteiny. Důsledkem je snížení citlivosti tkání k inzulinu, zejména jater a kosterního svalstva, jelikož buňky stimulované GH nejsou schopné dostatečně reagovat na inzulinový signál, který měl spustit již částečně aktivovanou signální kaskádu [4,5].
Pozornost na sebe soustřeďuje i vztah GH a PI3K. Jednak se předpokládá, že GH je schopen odpojit účinek PI3K od dalších signálních molekul [4], jednak je tady významný vliv GH na regulaci p85 podjednotky enzymu. Jak bylo popsáno v úvodu, PI3K se skládá ze 2 podjednotek – regulační, p85 a katalytické, p110. Za normálních okolností se regulační podjednotka nachází v stoichiometrickém nadbytku v poměru k podjednotce katalytické, z čehož vyplývá existence volné zásoby p85 monomerů, které nejsou vázané s p110. Tyto monomery se váží na fosforylované IRS proteiny blokujíc tak přístup heterodimerům p85-p110. Důležitá je rovnováha mezi monomery a heterodimery, přičemž právě heterodimery jsou odpovědny za aktivitu PI3K. V podmínkách nadbytku GH dochází k nadprodukci p85 podjednotky, čímž vzniká dysbalance mezi monomery a heterodimery se snížením funkce PI3K a z toho vyplývajícím zhoršením inzulinové senzitivity daných buněk, zejména v kosterním svalstvu. Podjednotka p85 je tak považovaná za potenciální léčebný cíl u inzulinové rezistence, resp. možný faktor určující tendenci ke vzniku diabetes mellitus 2. typu [3,36].
Dalším mechanizmem, jakým může GH způsobovat inzulinovou rezistenci, je stimulace exprese buněčných proteinů, které inhibují signalizaci IR. Jedná se o již zmíněné SH2-doménu obsahující supresory cytokinové signalizace (SOCS). Ne jenom působením GH, ale i za situací spojených s elevací cytokinů a s inzulinovou rezistencí, jako jsou stres, infekce, či zánět, dochází ke zvýšení zejména SOCS 1 a 3 v játrech, svalech a tukové tkáni. Je prokázáno, že jejich působením dochází k supresi tyrozinové fosforylace IRS, přičemž SOCS 3 inhibuje fosforylaci IRS-1 i IRS-2 a SOCS 1 zejména IRS-2. Ani jeden z nich neovlivňuje fosforylaci IR, nicméně oba se na IR vážou. SOCS 3 se váže na místo, kde dochází k rozpoznávání IRS-1 a IRS-2 a SOCS 1 na místo nezbytné k rozpoznání IRS-2, čímž je vysvětlen princip jejich inhibičního působení v signalizační kaskádě. Při nadměrné expresi SOCS 1 a 3 dochází ke snížení syntézy glykogenu a k sníženému vychytávání glukózy adipocyty. Na druhou stranu v případě redukce hladin SOCS za pomoci speciálně syntetizovaných antagonistů dochází k restituci TNF-α indukované snížené fosforylace IRS proteinů v adipocytech. Tyto výsledky dokládají význam SOCS jako negativních regulátorů inzulinové signalizace a spojovacích článků mezi inzulinovou rezistencí a cytokinovou signalizací [4,37,38].
Na regulaci inzulinové senzitivity se podílí i IGF-I, a to jednak přímým zlepšením inzulinové senzitivity, jednak nepřímo – udržováním rovnováhy mezi působením GH a inzulinu, jelikož redukuje cirkulující hladiny GH v krvi [39].
Závěr
Závěrem lze konstatovat, že distribuce tukové tkáně a její metabolické dopady (včetně inzulinové rezistence), resp. regulace inzulinového metabolizmu jako takového, jsou velmi komplexně hormonálně regulované procesy. Nejedná se při tom pouze o tolik diskutované adipokiny, či adipocytokiny, ale i o sympatoadrenální systém, pohlavní hormony, glukokortikoidy (GK) či růstový hormon (growth hormon - GH). V našem přehledovém článku jsme se zabývali mechanizmy vzniku viscerální obezity a inzulinové rezistence u nemocných s nadbytkem GK a GH. V případě hyperkortizolizmu se kauzálně jedná zejména o přímé účinky zvýšených tkáňových hladin kortizolu, které vznikají v důsledku jeho zvýšené aktivace z neaktivního kortizonu, prostřednictvím polymorfizmy způsobené hypersenzitivity glukokortikoidního receptoru, či nedostatečných hladin kortizol vážícího globulinu s excesivním množstvím volného kortizolu v buňkách. V případě nadbytku GH vzniká inzulinová rezistence nezávisle na přítomnosti viscerální obezity, jelikož GH podporuje redukci množství abdominálního tuku. Dochází tu k četným postreceptorovým interakcím mezi GH a inzulinovou signální kaskádou v cílových buňkách s následným zhoršením inzulinové senzitivity.
Práce byla podpořena grantem IGA MZ ČR No. NR/9438-3.
MUDr. Viktória Ďurovcová
www.vfn.cz
e-mail: viktoria.durovcova@gmail.com
Doručeno do redakce: 26. 11. 2007
Přijato po recenzi: 22. 1. 2008
Zdroje
1. Masuzaki H, Flier JS. Tissue-specific glucocorticoid reactivating enzyme, 11 β-hydroxysteroid dehydrogenase type 1 (11 β-HSD1) – a promising drug target for the treatment of metabolic syndrome. Curr Drug Targets Immune Endocr Metabol Disord 2003; 3 : 255–262.
2. Karlsson HK, Zierath JR. Insulin signaling and glucose transport in insulin resistant human skeletal muscle. Cell Biochem Biophys 2007; 48 : 103-113.
3. Del Rincon JP, Iida K, Gaylinn BD et al. Growth Hormone Regulation of p85 α Expression and Phosphoinositide 3-Kinase Activity in Adipose Tissue. Mechanism for Growth Hormone-Mediated Insulin Resistance. Diabetes 2007; 56 : 1638–1646.
4. Dominici FP, Turyn D. Growth Hormone-Induced Alterations in the Insulin-Signaling System. Exp Biol Med 2002; 227 : 149–157.
5. Dominici FP, Cifone D, Bartke A et al. Loss of sensitivity to insulin at early events of the insulin signaling pathway in the liver of growth hormone-transgenic mice. J Endocrinol 1999; 161 : 383–392.
6. Lundgren M, Burén J, Ruge T et al. Glucocorticoids Down-Regulate Glucose Uptake Capacity and Insulin-Signaling Proteins in Omental But Not Subcutaneous Human Adipocytes. J Clin Endocrinol Metab 2004; 89 : 2989–2997.
7. Pivonello R, Faggiano A, Lombardi G et al. The metabolic syndrome and cardiovascular risk in Cushingʼs syndrome. Endocrinol Metab Clin North Am 2005; 34 : 327–339.
8. Walker BR. Glucocorticoids and Cardiovascular Disease. Eur J Endocrinol 2007; 157 : 545–559.
9. Vondra K, Hampl R. Glukokortikoidy a diabetes mellitus. Vnitř Lék 2006; 52(5): 493–497.
10. Watts LM, Machem VP, Leedom TA et al. Reduction of Hepatic and Adipose Tissue Glucocorticoid Receptor Expression With Antisense Oligonucleotides Improves Hyperglycemia and Hyperlipidemia in Diabetic Rodents Without Causing Systemic Glucocorticoid Antagonism. Diabetes 2005; 54 : 1846–1853.
11. Wang M. The role of glucocorticoid action in the patophysiology of the metabolic syndrome. Nutr Metab 2005; 2 : 3–17.
12. Marek J, Hána V, Krsek M. How corticoids, growth hormone and oestrogens influence lipids and atherosclerosis. Vnitř Lék 2007; 53 : 386–390.
13. Walker BR, Soderberg S, Lindahl B et al. Independent effects of obesity and cortisol in predicting cardiovascular risk factors in men and women. J Intern Med 2000; 247 : 198–204.
14. Oltmanns KM, Dodt B, Schulte B et al. Cortisol correlates with metabolic disturbances in a population study of type 2 diabetic patients. Eur J Endocrinol 2006; 154 : 325–331.
15. Stewart PM. Tissue-specific Cushing’s syndrome, 11b-hydroxysteroid dehydrogenases and the redefinition of corticosteroid hormone action. Eur J Endocrinol 2003; 149 : 163–168.
16. Wake DJ, Walker BR. Inhibition of 11β-hydroxysteroid dehydrogenase type 1 in obesity. Endocrine 2006; 29 : 101–108.
17. Stewart PM, Boulton A, Kumar S et al. Cortisol Metabolism in Human Obesity: Impaired Cortisone-Cortisol Conversion in Subjects with Central Adiposity. J Clin Endocrinol Metab 1999; 84 : 1022–1027.
18. Van Rossum EFC, Lamberts SWJ. Polymorphisms in the Glucocorticoid Receptor Gene and Their Associations with Metabolic Parameters and Body Composition. Recent Prog Horm Res 2004; 59 : 333–357.
19. Lin RCI, Wang XL, Morris BJ Association of Coronary Artery Disease With Glucocorticoid Receptor N363S Variant. Hypertension 2003; 41 : 404–407.
20. Rosmond R. The Glucocorticoid Receptor Gene and Its Association to Metabolic Syndrome. Obes Res 2002; 10 : 1078–1086.
21. Van Rossum EF, Koper JW, Huizenga NA et al. A polymorphism in the glucocorticoid receptor gene, which decreases sensitivity to glucocorticoids in vivo, is associated with low insulin and cholesterol levels. Diabetes 2002; 51 : 3128–3134.
22. Van Rossum EFC, Feelders RA, Van den Beld AW et al. The ER22/23EK polymorphism in the glucocorticoid receptor gene is associated with better survival and low C-reactive protein levels in elderly men. Am J Med 2004; 117 : 158–162.
23. Van Rossum EFC, De Jong FJ, Den Heijer T et al. The ER22/23EK polymorphism in the glucocorticoid receptor gene protects against white matter lesions and dementia, 82. In: Program & Abstracts of the 85th Annual Meeting of the Endocrine Society, Philadelphia 2003.
24. Van Rossum EFC, Voorhoeve PG, Te Velde SJ et al. The ER22/23EK polymorphism in the glucocorticoid receptor gene is associated with a beneficial body composition and muscle strength in young adults. J Clin Endocrinol Metab 2004; 89 : 4004–4009.
25. Fernandez-Real JM, Puget M, Grasa M et al. Serum Corticosteroid-Binding Globulin Concentration and Insulin Resistance Syndrome: A Population Study. J Clin Endocrinol Metab 2002; 87 : 4686-4690.
26. Fernandez-Real JM, Pugeat M, López-Bermejo A et al. Corticosteroid-binding globulin affects the relationship between circulating adiponectin and cortisol in men and women. Metabolism 2005; 54 : 584–589.
27. Pasarica M, Zachwieja JJ, Dejonge L et al. Effect of growth hormone on body composition and visceral adiposity in middle-aged men with visceral obesity. J Clin Endocrinol Metab 2007; 92 : 4265–4270.
28. Ahmad AM, Hopkins MT, Thomas J et al. Body composition and quality of life in adults with growth hormone deficiency; effects of low-dose growth hormone replacement. Clin Endocrinol 2001; 54 : 709–717.
29. Vilar L, Naves LA, Costa SS et al. Increase of classic and nonclassic cardiovascular risk factors in patients with acromegaly. Endocr Pract 2007; 13 : 363–372.
30. Wajchenberg BL. Subcutaneous and Visceral Adipose Tissue: Their Relation to the Metabolic Syndrome. Endocr Rev 2000; 21 : 697–738.
31. Franco C, Brandberg J, Lönn L et al. Growth Hormone Treatment Reduces Abdominal Visceral Fat in Postmenopausal Women with Abdominal Obesity: A 12-Month Placebo-Controlled Trial. J Clin Endocrinol Metab 2004; 90 : 1466–1474.
32. Ridderstrale M. Signaling Mechanism for the Insulin-like Effects of Growth Hormone – Another Example of a Classical Hormonal Negative Feedback Loop. Curr Drug Targets Immune Endocr Metabol Disord 2005; 5 : 79–92.
33. Gaur S, Schwartz Y, Tai LR et al. Insulin Produces a Growth Hormone-Like Increase in Intracellular Free Calcium Concentration in Okadaic Acid-Treated Adipocytes. Endocrinology 1998; 139 : 4953–4961.
34. Piwien-Pilipuk G, Huo JS, Schwartz J. Growth hormone signal transduction. J Pediatr Endocrinol Metab 2002; 15 : 771–786.
35. Ridderstråle M, Amstrup J, Hilton DJ et al. SOCS-3 is involved in the downregulation of the acute insulin-like effects of growth hormone in rat adipocytes by inhibition of Jak2/IRS-1 signaling. Horm Metab Res 2003; 35 : 169–177.
36. Barbour LA, Rahman SM, Gurevich I et al. Increased P85-αIs a Potent Negative Regulator of Skeletal Muscle Insulin Signaling and Induces in Vivo Insulin Resistance Associated with Growth Hormone Excess. J Biol Chem 2005; 280 : 37489–37494.
37. Ueki K, Kondo T, Kahn CR. Suppressor of cytokine signaling R1 (SOCS-1) and SOCS-3 cause insulin resistance through inhibition of tyrosine phosphorylation of insulin receptor substrate proteins by discrete mechanisms. Mol Cell Biol 2004; 24 : 5434–5446.
38. Fasshauer M, Kralisch S, Klier M et al. Insulin resistance-inducing cytokines differentially regulate SOCS mRNA expression via growth factor – and Jak/Stat-signaling pathways in 3T3-L1 adipocytes. J Endocrinol 2004; 181 : 129–138.
39. Dominici FP, Argentino DP, Munoz MC et al. Influence of the crosstalk between growth hormone and insulin signalling on the modulation of insulin sensitivity. Growth Horm IGF Res 2005; 15 : 324–336.
40. Kershaw EE, Flier JS. Adipose Tissue as an Endocrine Organ. J Clin Endocrinol Metab 2004; 89 : 2548–2556.
Štítky
Diabetologie Endokrinologie Interní lékařstvíČlánek vyšel v časopise
Vnitřní lékařství

2008 Číslo 4
- Plicní hypertenze – syndrom mnoha tváří – vyžaduje přesnou diagnostiku a specializovanou léčbu
- Proces hojení ran krok za krokem a co ho může zkomplikovat
- Limity glykovaného hemoglobinu a význam dalších glykovaných proteinů
- Korelace dávky levothyroxinu s titrem autoimunitních protilátek u primární hypotyreózy
- Zeolit-jodový komplex pomáhá v péči o infikované rány
Nejčtenější v tomto čísle
- Perikardiocentéza po kardiologické operaci – naše zkušenosti
- Lymeská karditida – vzácná příčina dilatační kardiomyopatie a poruch srdečního rytmu: kazuistika
- EKG zmeny pri akútnej intoxikácii alkoholom
- Lidské leukocytární antigeny z hlediska CD klasifikace
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy