
Kongenitální adrenální hyperplazie na podkladě deficitu 3-β-hydroxysteroidní dehydrogenázy
Congenital adrenal hyperplasia due to 3-β-hydroxysteroid dehydrogenase deficiency
Deficiency of 3-ß-hydroxysteroid dehydrogenasis belongs to less frequent types of congenital adrenal hyperplasia. It is a deficiency of the enzyme which function is to change pregnenolon to progesteron, 17-OH-pregnenolon to 17-OH-progesteron and dehydroepiandrosteron to androstendion in the adrenal glands and gonads. No prevalence of forms with clinical symptoms is known; however, it is less frequent compared to deficiency of 21-hydroxylase. Clinical manifestations are very variable, from serious salt disorder, ambiguous genitalia and precocious puberty to oligosymptomatic forms with late effects or asymptomatic forms. No routine genetic analysis in Czech republic is conducted. Diagnosis can be suspected through increased levels of some steroid hormones in basal condition; the final determinent indicator is the concentration of steroid hormones after stimulation by adrenocorticotropic hormone.
Key words:
3-β-hydroxysteroid dehydrogenase – hypogonadism – congenital adrenal hyperplasia – cortisol
Autoři:
M. Marunová
Působiště autorů:
Endokrinologický ústav, Praha, ředitel doc. MUDr. Vojtěch Hainer, CSc.
Vyšlo v časopise:
Vnitř Lék 2006; 52(10): 957-962
Kategorie:
Přehledný referát
Souhrn
Deficit 3-β-hydroxysteroidní dehydrogenázy patří mezi vzácnější varianty kongenitální adrenální hyperplazie. Jedná se o deficit enzymu, který v nadledvinách a gonádách mění pregnenolon na progesteron, 17-OH-pregnenolon na 17-OH-progesteron a dehydroepiandrosteron na androstendion. Prevalence klinicky manifestní formy není známa, ale je výrazně nižší ve srovnání s deficiencí 21-hydroxylázy. Onemocnění má velmi variabilní klinickou manifestaci od těžké solné poruchy přes obojetný genitál a předčasnou pubarché až k oligosymptomatickým formám s pozdním nástupem účinků nebo formám zcela asymptomatickým. Rutinní genetická analýza této poruchy se v České republice neprovádí, na diagnózu mohou upozornit zvýšené bazální hladiny některých steroidních hormonů, ale rozhodující pro diagnózu je jejich koncentrace po stimulaci adrenokortikotropním hormonem.
Klíčová slova:
3-β-hydroxysteroidní dehydrogenáza - hypogonadizmus - kongenitální adrenální hyperplazie - kortizol
Úvod
Přeměna cholesterolu na steroidní hormony, ke které dochází v nadledvinách, testes, ovariích a v placentě, je katalyzována 5 enzymy ze „superrodiny“ cytochromu P450 (steroidní hydroxylázy) a skupinou 3 steroidních dehydrogenáz. Tři steroidní hydroxylázy jsou mitochondriální (P45011A1, P45011B1, P44011B2) a dvě mikrozomální (P450c17, P450c21). Mezi steroidní hydroxylázy patří 3βHSD (3-β-hydroxysteroidní dehydrogenáza), StAR (steroidogenic acute regulatory protein) a Adx (adrenodoxin). Geneticky podmíněná deficience syntézy jednotlivých enzymů adrenální steroidogeneze představuje molekulární základ pro skupinu onemocnění zahrnovaných pod pojem kongenitální adrenální hyperplazie (CAH).
CAH představuje skupinu autozomálně recesivních onemocnění. Každá choroba je charakterizována specifickým enzymovým deficitem, který narušuje tvorbu kortizolu a dalších hormonů v kůře nadledvin [9]. Více než 95 % případů CAH souvisí s deficiencí 21-hydroxylázy (P450c21) [7], vzácnější jsou deficity 11β-hydroxylázy, 3βHSD, 17α-hydroxylázy/17,20-lyázy (P450c17) a cholesterol desmolázy. Byly popsány i vzácné případy deficitu dalších enzymů účastnících se v adrenální steroidogenezi a rovněž lokalizovány nejčastější i méně časté mutace jednotlivých genů. Rutinní genetická analýza v ČR je zaměřena na nejčastější variantu, deficit 21-hydroxylázy. Klasická forma CAH na podkladě deficitu 21-hydroxylázy se vyskytuje s incidencí 1 : 10 000-1 : 15 000 novorozenců. Onemocnění se vyznačuje širokou škálou projevů od asymptomatického průběhu, přes prostou virilizaci (simple virilizing form) po těžkou solnou poruchu (salt wasting form). Nadbytek androgenů může vést k maskulinizaci zevního genitálu u novorozených dívek a k progresivní postnatální virilizaci u obou pohlaví. Vzácnější je deficit 11β-hydroxylázy, který se v rozvinuté podobě manifestuje obojetným genitálem u dívek a excesem androgenů a hypertenzí u obou pohlaví. Dvě vzácné poruchy 17α-hydroxylázy/17,20-lyázy a cholesterol desmolázy vedou k vývoji ženského zevního genitálu u obou pohlaví [17].
Enzym 3βHSD/Δ5-Δ4 izomeráza
3βHSD/Δ5-Δ4 izomeráza, jejíž deficit je příčinou jedné z méně častých forem CAH, je mikrozomální membránově vázaný enzym, NAD (nicotinamid adenin dinucleotid) dependentní. Katalyzuje oxidaci a izomerizaci Δ5-3β-hydroxysteroidních prekurzorů do podoby Δ4-ketosteroidů. Tento krok je klíčovým mezistupněm pro tvorbu všech tříd steroidních hormonů [21]. 3βHSD je esenciální pro tvorbu progesteronu, který se dále mění na aldosteron, a 17-OH-progesteronu, který je prekurzorem kortizolu v kůře nadledvin. Je rovněž nezbytný pro tvorbu androstendionu, testosteronu a estrogenů v nadledvinách a gonádách [10]. Ukotvení 3βHSD ve fosfolipidové dvojvrstvě je zásadní pro normální funkci enzymu, pro jeho prostorovou orientaci i stabilitu vůči vlivům prostředí. Aktivita 3βHSD optimální pro průběh steroidogeneze je udržována na transkripční úrovni působením ACTH, cAMP-dependentní cestou přes specifické cAMP-responsivní sekvence (CRS) v promotorovém úseku genu.
Nadledviny fetu exprimují 3βHSD od 12. týdne, tedy ve stejné době jako např. 21-hydroxylázu (P450c21). Exprese 3βHSD se od fetálního období do dospělosti zvyšuje 21násobně, což je ve srovnání se všemi ostatními enzymy adrenální steroidogeneze zdaleka nejvyšší poměr.
Klinická manifestace deficitu 3βHSD
Podobně jako u dalších typů CAH závisí i zde klinická manifestace na míře narušení enzymatické aktivity a rovněž na pohlaví. Nedostatečná syntéza kortizolu vede zpětnovazebně k vystupňované produkci CRH v hypotalamu a ACTH v adenohypofýze. Dlouhodobé stimulační působení ACTH vede u části pacientů k hyperplazii nadledvin a ke zvýšené syntéze steroidních prekurzorů před místem enzymatické poruchy. Klinický obraz je velice heterogenní, od těžké solné poruchy přes obojetný genitál, neúplný vývoj genitálu (inkompletní maskulinizaci) u genetických mužů, předčasné pubarche u chlapců i dívek [13], mírné hyperandrogenní projevy u žen až k asymptomatickým formám (tab. 1) [15].

Zvýšená syntéza CRH u pacientů s deficitní syntézou kortizolu je možnou příčinou častých neuropsychických změn, anxiety a depresivních poruch, které jsou u těchto pacientů popisovány.
Onemocnění postihuje obě pohlaví, přesto zhruba 2/3 zaznamenaných případů tvoří muži. Protože genetická prevalence poruchy v populaci je pro obě pohlaví stejná, znamená to, že část postižených žen není správně diagnostikována [1]. V posledních 20 letech jsou stále častěji dokumentovány méně závažné „neklasické“ varianty 3βHSD deficience projevující se např. hirzutizmem a menstruačními poruchami u řady adolescentních a mladých žen [10].
Genetický základ onemocnění
U člověka se nacházejí 2 izoenzymy 3βHSD s 93,5% homologií primární struktury. Jejich geny, které byly chronologicky označeny jako HSD3B1 a HSD3B2, jsou lokalizovány na krátkém raménku 1. chromozomu v těsné vazbě v pozici 1p13.1 [8,12]. Rodina genů 3βHSD obsahuje v těchto místech celkem 7 genů, z nichž aktivní jsou pouze zmíněné 2 a zbývajících 5 (označovaných HSD3Bpsi1 až 5) jsou nefunkční pseudogeny [14].
Gen pro izoenzym typu I (HSD3B1) je exprimován v placentě a v periferních tkáních. Gen pro izoenzym typu II (HSD3B2) je lokalizován v nadledvinách, ovariích a testes [5]. Z uvedených 2 izoenzymů je klinicky významnější HSD3B2, jehož mutace podmiňují vznik CAH. Do roku 2002 bylo popsáno 34 různých mutací HSD3B2, postihujících nekódující i kódující regiony genu [16,23]. Tato genetická variabilita vysvětluje rozdílnou enzymovou aktivitu izoenzymu typu II, a tím i značnou fenotypovou heterogenitu. Vedle změny enzymatické aktivity mění různé mutace výrazně i stabilitu proteinu [22]. Žádná z těchto 34 mutací HSD3B2 není u pacientů výrazně častější.
Genetická variabilita izoenzymu typu I (HSD3B1) zůstává zatím v pozadí a výsledky výzkumu jsou nekonzistentní. Podle Robertse et al [19] tato variabilita ovlivňuje riziko vzniku benigní hyperplazie prostaty ve vyšším věku - podle jeho zjištění mají heterozygoti HSD3B1 (c.1100 A/C) riziko vzniku hyperplazie nižší (HR = 0,7) ve srovnání s ostatní populací. Chang et al [4] řadí variabilitu genů HSD3B1 a HSDB2 mezi minoritní rizikové faktory vzniku karcinomu prostaty s tím, že dopad některých rizikových variant obou genů se neznámým mechanizmem vzájemně umocňuje.
Rosmond et al [20] zjistili, že záměna T→C v kodonu Leu338 ve 4. exonu genu HSD3B1 ovlivňuje krevní tlak. Homozygoti s alelou C měli signifikantně vyšší systolický i diastolický krevní tlak ve srovnání s heterozygoty a homozygoty s alelou T. U hypertoniků I. stupně WHO byla C alela přítomna signifikantně častěji ve srovnání s normotoniky.
Laboratorní kritéria
Genetická analýza nejčastějších mutací HSD3B2 se v České republice neprovádí. Diagnóza onemocnění se proto opírá o ukazatele laboratorní - bazální hladiny steroidních hormonů a jejich prekurzorů a zejména o stimulované hladiny po podání ACTH. U pacientů s klasickou formou 3βHSD deficience je obvykle zvýšený poměr Δ5/Δ4 steroidů, pro diagnostiku poruchy ale není dostačující [1]. Senzitivitu tohoto vyšetření snižuje skutečnost, že nedostatečnost 3βHSD typu II je částečně kompenzována izoenzymem 3βHSD typu I, který je exprimován v kůži, prsních žlázách, endometriu, myometriu, prostatě, tuku, játrech a v tlustém střevě [3].
Laboratorní kritéria se opírají o ACTH-stimulovanou hormonální odpověď. Kritéria podle Pangové [18] jsou nastavena tak, aby požadované hodnoty parametrů přesahovaly 2 směrodatné odchylky od průměru zdravé populace (tab. 2). Za rozhodující Pangová pokládá vysokou hladinu 17-OH-pregnenolonu po standardní stimulaci ACTH (v bolusové dávce 0,25 mg i.v.). Mezi další kritéria patří stimulovaná hodnota 17-OH-progesteronu a dehydroepiandrosteronu a poměr 17-OH-pregnenolonu k 17-OH-progesteronu a 17-OH-pregnenolonu ke kortizolu.

Zejména u lehčích, neklasických variant ale tyto hormonální ukazatele často selhávají a nezachytí pacienty s neúplně vyjádřeným deficitem 3βHSD. Diagnostika je tedy obtížnější ve srovnání s mnohem častější formou CAH na podkladě deficitu 21-hydroxylázy. U této choroby je 17-OH-progesteron pokládán za citlivý ukazatel i u mírných variant poruchy. V posledních 4 letech se objevily návrhy na úpravu užívaných hormonálních kritérií (tab. 3) [10,15]. Jejich autoři se již mohou opřít o porovnání biochemických ukazatelů s genetickou analýzou. Mermejo [15] přitom u řady dříve diskutovaných parametrů prokázal velký překryv mezi (geneticky definovanými) pacienty s deficitem 3βHSD a zdravou populací - týká se to plazmatického kortizolu a DHEA, poměru Δ5-17-pregnenolonu k 17-OH-progesteronu a poměru DHEA k androstendionu. Jak bazální, tak i stimulované hladiny těchto parametrů nedokázaly odlišit pacienty od geneticky normální zdravé populace.

Také Lutfallah et al [10] konfrontovali klinický obraz, standardně užívané laboratorní testy a genotyp pacientů s předpokládaným defektem 3βHSD. Na základě svých pozorování představili v roce 2002 modifikovanou podobu laboratorních kritérií pro deficit 3βHSD. Jejich kritéria respektují měnící se hormonální profil pacientů v různých věkových kategoriích (tab. 4).
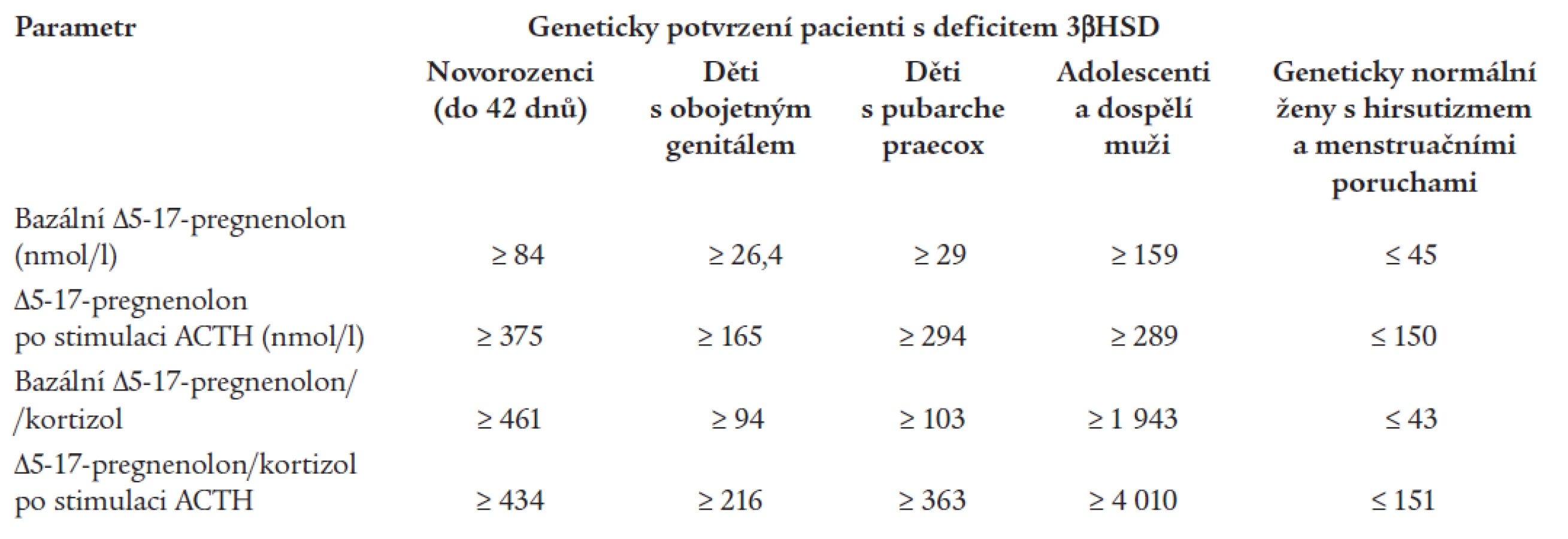
Diagnostika heterozygotů
Přenos onemocnění je autozomálně recesivní. Heterozygoti - nosiči mutované alely HSD3B2 jsou klinicky asymptomatičtí. Jestliže u nejčastější formy CAH, deficitu 21-hydroxylázy, je podle řady studií možný záchyt heterozygotů na základě abnormální elevace 17-OH-progesteronu po stimulaci ACTH, u této varianty CAH nic takového neplatí. Nepotvrdila se hypotéza, že heterozygoti s defektní alelou HSD3B2 mají sníženou aktivitu enzymu, podobně jako je to známo u nositelů genové mutace CYP21 ve vztahu k mírnému poklesu aktivity adrenální 21-hydroxylázy. Klinicky asymptomatiční heterozygoti v genu pro HSD3B2 mají normální bazální i stimulované hormonální hladiny. Jakým mechanizmem je u heterozygotů udržována normální enzymatická aktivita HSD3B2, přitom známo není. Ukazuje se ale, že heterodimery mutovaného a normálního enzymu mohou být stabilní a vykazovat podobnou enzymatickou aktivitu jako homodiméry normálního enzymu. Záchyt heterozygotů - nosičů mutované alely HSD3B2 je tak možný pouze genetickou analýzou.
Popis vybraných případů
Johannsen [6] uvádí případ 2 sester (věk 7 roků a 3 roky). První z nich byla hospitalizována ve věku 20 měsíců pod obrazem sepse a v této souvislosti byla zjištěna minerálová dysbalance (Na 124 mmol/l, K 5,5 mmol/l). Příčina infekce zjištěna nebyla a po přeléčení antibiotiky byla dívka propuštěna domů. V dalších letech byla bez obtíží až do věku 7 roků, kdy byla poslána k endokrinologovi pro pubarche praecox (Tanner 3-4), mírnou růstovou akceleraci a urychlení kostního věku. Druhá sestra, v té době ve věku 3 roků, měla za sebou rovněž septickou příhodu ve věku 19 měsíců s nálezem minerálové dysbalance (Na 124 mmol/l, K 6,8 mmol/l), CRP 1 602 mg/l a P-kortizolem 60 nmol/l. Vzhledem k evelaci 17-OH-progesteronu u starší z dívek bylo zpočátku vysloveno podezření na neklasickou formu deficitu 21-hydroxylázy. Vyšetřením CYP21 genu se ukázalo, že obě sestry jsou pouze heterozygotkami v mutaci V281L. Následným vyšetřením genu pro izoenzym II 3βHSD byla zjištěna deleční mutace 1105delA a potvrzena diagnóza 3βHSD insuficience.
Cavanah a Dons [2] publikovali případ mladého muže s normálním průběhem puberty, u kterého se ve věku 24 roků objevila gynekomastie. Pacient měl normální spermiogram a normální odezvu testosteronu v LHRH testu. Poté co byly vyloučeny běžné příčiny gynekomastie a nepodařilo se prokázat ani původně uvažovaný estrogeny-produkující tumor, následoval ACTH stimulační test. Ten ukázal normální produkci kortizolu, 11-deoxykortizolu, 17-OH-progesteronu a aldosteronu, ale současně signifikantní zvýšení pregnenolonu, 17-OH-pregnenolonu, DHEA a androstendionu odpovídající parciálnímu deficitu 3βHSD. Gynekomastie byla tedy vysvětlena jako výsledek alterace poměru mezi androgeny a estrogeny v důsledku tohoto defektu.
V naší ambulanci byl vyšetřen 23letý muž, u něhož byla dominujícím klinickým příznakem oboustranná gynekomastie [11]. Gynekomastie, bez bolestivosti a bez sekrece, se objevila v pubertě kolem 13. roku věku a v době záchytu onemocnění trvala 10 let. Průběh puberty byl jinak fyziologický. Pacient dále uváděl větší únavu, holení v intervalech 4-5 dní, ranní erekce jen občas. Ve fyzikálním nálezu byl konstatován normální vzrůst a hmotnost, normotenze, symetrické zvětšení prsů, bez rezistence, bez sekrece, palpačně nebolestivé. Ochlupení mužského typu, normálně rozvinuté sekundární pohlavní znaky. Testes palpačně menší, hypoplazie testes poté potvrzena sonograficky (vpravo 10,1 ml; vlevo 9,2 ml). Genetické vyšetření (46XY), sonografie nadledvin i spermiogram - vše v mezích normy. V bazálních hormonálních náběrech byl nižší testosteron a vyšší DHEA a následné výsledky ACTH testu (normální reakce kortizolu, zvýšená stimulace 17-OH pregnenolonu) odpovídaly diagnóze CAH na podkladě parciálního bloku 3β-HSD při použití kritérií dle Pangové. Genetická analýza tohoto deficitu se v ČR neprovádí a diagnóza byla proto založena jen na laboratorních nálezech.
Postpubertální klinická manifestace 3βHSD deficitu je typicky méně vyjádřená u dospělých mužů než u žen, které často přicházejí k lékaři pro hirzutizmus. Gynekomastie může být rovněž jediným projevem parciálního deficitu 3βHSD u muže.
Závěr
Kongenitální adrenální hyperplazie na podkladě deficitu 3βHSD je vzácným onemocněním s variabilní klinickou manifestací - od asymptomatických forem přes obojetný genitál až po těžkou solnou poruchu. Při nedostupnosti genetické analýzy postiženého genu HSD3B2 se diagnóza opírá o hormonální kritéria. V bazálních laboratorních hodnotách může na diagnózu upozornit zvýšení DHEA a 17-OH-pregnenolonu a snížení testosteronu. Rozhodující jsou ale hormonální hladiny po stimulaci ACTH (kritéria dle Pangové, nebo jejich novější modifikace). Jejich výtěžnost u neklasických případů onemocnění je ale omezená a část pacientů není správně diagnostikována.
Přehled zkratek
3βHSD 3-β-hydroxysteroidní dehydrogenáza
ACTH adrenokortikotropin
Adx adrenodoxin
CAH kongenitální adrenální hyperplazie
cAMP cyklický adenozinmonofosfát
CRS cAMP-responzivní sekvence
DHEA dehydroepiandrosteron
HSD3B1, HSD3B2 geny 3-β-hydroxysteroidní dehydrogenázy
NAD nicotinamid adenin dinucleotid
StAR steroidogenic acute regulatory protein
MUDr. Michaela Marunová
www.endo.cz
e-mail: mmarunova@endo.cz
Doručeno do redakce: 26. 6. 2006
Zdroje
1. Alos N, Moisan AM, Ward L et al. A novel A10E homozygous mutation in the HSD3B2 gene causing severe salt-wasting 3β-hydroxysteroid dehydrogenase deficiency in 46,XX and 46,XY French-Canadians: evaluation of gonadal function after puberty. J Clin Endocrinol Metab 2000; 85 : 1968-1974.
2. Cavanah SF, Dons RF. Partial 3-β-hydroxysteroid dehydrogenase deficiency presenting as new-onset gynecomastia in a eugonadal adult male. Metabolism 1993; 42 : 65-68.
3. Gingras S, Simard J. Induction of 3β-hydroxysteroid dehydrogenase type 1 expression by interleukin-4 in human normal prostate epithelial cells, immortalized keratinocytes, colon and cervix cancer cell lines. Endocrinology 1999; 140 : 4573-4584.
4. Chang BL, Zheng SL, Hawkins GA et al. Joint effect of HSD3B1 and HSD3B2 genes is associated with hereditary and sporadic prostate cancer susceptibility. Cancer Res 2002; 62 : 1784-1789.
5. Chang YT, Kappy MS, Iwamoto K et al. Mutations in the type II 3β-hydroxysteroid dehydrogenase gene in a patient with classic salt wasting 3β-hydroxysteroid dehydrogenase deficiency congenital adrenal hyperplasia. Pediatr Res 1993; 34 : 698-700.
6. Johannsen TH, Mallet D, Dige-Petersen H et al. Delayed diagnosis of congenital adrenal hyperplasia with salt wasting due to type II 3 β-hydroxysteroid dehydrogenase deficiency. J Clin Endocrinol Metab 2005; 90 : 2076-2080.
7. Kotaška K, Lisá L, Průša R. Common CYP21 gene mutations in Czech patients and statistical analysis of worldwide mutation distribution. Centr Eur J Public Health 2003; 11 : 124-128.
8. Labrie F, Simard J, Luu-The V et al. Structure, function and tissue-specific gene expression of 3β-hydroxysteroid dehydrogenase/5-ene-4-ene isomerase enzymes in classical and peripheral intracrine steroidogenic tissues. J Steroid Biochem Mol Biol 1992; 43 : 805-826.
9. Lisá L. Vrozená adrenální hyperplazie. Praha, Triton 2004.
10. Lutfallah C, Wang W, Mason JI et al. Newly proposed hormonal criteria via genotypic proof for type II 3β-hydroxysteroid dehydrogenase deficiency. J Clin Endocrinol Metab 2002; 87 : 2611-2622.
11. Marunová M. Kongenitální adrenální hyperplazie s neúplným deficitem 3-β hydroxysteroidní dehydrogenázy u dospělého muže. Kazuistiky v diabetologii 2006; 4 : 34-40.
12. Mason JI. The 3β-hydroxysteroid dehydrogenase gene family of enzymes. Trends Endocrinol Metab 1993; 4 : 199-202.
13. Mauri M, Castro A, Latronico C et al. Mutations in the type II 3β-hydroxysteroid dehydrogenase (HSD3B2) gene can cause premature pubarche in girls. Clin Endocrinol 2000; 52 : 67-75.
14. McBride MW, McVie AJ, Burridge SM et al. Cloning, expression, and physical mapping of the 3β-hydroxysteroid dehydrogenase gene cluster (HSD3BP1-HSD3BP5) in human. Genomics 1999; 61 : 277-284.
15. Mermejo LM, Elias LL, Marui S et al. Refining hormonal diagnosis of type II 3β-hydroxysteroid dehydrogenase deficiency in patients with premature pubarche and hirsutism based on HSD3B2 genotyping. J Clin Endocrinol Metab 2005; 90 : 1287-1293.
16. Moisan AM, Ricketts ML, Tardy V et al. New insight in mutations in the HSD3B2 gene in eleven patients from seven new families and comparison of the functional properties of twenty-five mutant enzymes. J Clin Endocrinol Metab 1999; 84 : 4410-4425.
17. New MI. Inborn errors of adrenal steroidogenesis. Mol Cell Endocrinol 2003; 211 : 75-83.
18. Pang S, Carbunaru G, Haider A et al. Carriers for type II 3β-hydroxysteroid dehydrogenase (HSD3B2) deficiency can only be identified by HSD3B2 genotype study and not by hormone test. Clin Endocrinol (Oxf) 2003; 58 : 323-331.
19. Roberts RO, Bergstralh EJ, Farmer SA et al. Polymorphisms in genes involved in sex hormone metabolism may increase risk of benign prostatic hyperplasia. Prostate 2006; 66 : 392-404.
20. Rosmond R, Chagnon M, Bouchard C et al. Polymorphism in exon 4 of the human 3β-hydroxysteroid dehydrogenase type I gene (HSD3B1) and blood pressure. Biochem Biophys Res Commun 2002; 293 : 629-632.
21. Simard J, Rheaume E, Sanchez R et al. Molecular basis of congenital adrenal hyperplasia due to 3β-hydroxysteroid dehydrogenase deficiency. Mol Endocrinol 1993; 7 : 716-728.
22. Simard J, Moisan AM, Morel Y. Congenital adrenal hyperplasia due to 3β-hydroxysteroid dehydrogenase/delta5-delta4 isomerase deficiency. Semin Reprod Med 2002; 20 : 255-276.
23. Zhang L, Mason I, Naiki Y et al. Characterization of two novel homozygous missense mutations involving codon 6 and 259 of type II 3β-hydroxysteroid dehydrogenase (3β-HSD) gene causing respectively, nonsalt wasting and salt-wasting 3βHSD deficiency disorder. J Clin Endocrinol Metab 2000; 85 : 1678-1685.
Štítky
Diabetologie Endokrinologie Interní lékařstvíČlánek vyšel v časopise
Vnitřní lékařství

2006 Číslo 10
- Plicní hypertenze – syndrom mnoha tváří – vyžaduje přesnou diagnostiku a specializovanou léčbu
- Proces hojení ran krok za krokem a co ho může zkomplikovat
- Limity glykovaného hemoglobinu a význam dalších glykovaných proteinů
- Korelace dávky levothyroxinu s titrem autoimunitních protilátek u primární hypotyreózy
- Zeolit-jodový komplex pomáhá v péči o infikované rány
Nejčtenější v tomto čísle
- Hashimotova encefalopatie
- Radionuklidové zobrazovací metody používané v endokrinologii
- Kongenitální adrenální hyperplazie na podkladě deficitu 3-β-hydroxysteroidní dehydrogenázy
- Autoimunitní tyreoiditida: vybrané etiopatogenetické mechanizmy
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy