
Léčba histiocytózy z Langerhansových buněk u dospělých osob
Treatment of Langerhans cell histiocytosis in adults
Langerhans cell histiocytosis has diverse clinical manifestations and the aggressiveness of its course varies. As in the case of other malignant diseases, involvement of other organs must be determined once the diagnosis has been made. Local treatment may suffice in the case of a single lesion (surgery, instillation of methylprednisolone within the lesion). Skin involvement responds to phototherapy (PUVA or electron beam irradiation). Remissions following the administration of thalidomide or methotrexate have been described in cases of isolated skin involvement. In adults, 2-cholorodeoxyadenosine (cladribine) in monotherapy and less often in combination with cytosine-arabinoside is the treatment of choice in the case of multi-system involvement. Extremely aggressive forms may be treated with combined chemotherapy regimens that include etoposide as in the case of lymphomas. In resistant cases, patients may be referred to transplant centres and undergo high-dose chemotherapy with autologous or allogeneic stem cell transplantation. Patients with the BRAF V600E gene mutation have been shown to respond to vemurafenib or dabrafenib, which are registered in the Czech Republic for the treatment of disseminated melanoma. On demonstrating this mutation and documenting resistance to prior therapy, it would be possible to request reimbursement of these agents from the health insurance company.
Keywords:
Langerhans cell histiocytosis – 2-cholorodeoxyadenosine (cladribine) – vemurafenib – dabrafenib
Autoři:
Z. Král 1; Z. Adam 1; M. Doubková 2; M. Krejčí 1; T. Horvát 3; M. Doubek 1; L. Pour 1; Z. Řehák 4,5; R. Koukalová 4
Působiště autorů:
Interní hematologická a onkologická klinika LF MU a FN Brno
1; Klinika nemocní plicních a TBC LF MU a FN Brno
2; Chirurgická klinika LF MU a FN Brno
3; Oddělení nukleární medicíny, Masarykův onkologický ústav, Brno
4; Regionální centrum aplikované molekulární onkologie (RECAMO), Masarykův onkologický ústav, Brno
5
Vyšlo v časopise:
Transfuze Hematol. dnes,26, 2020, No. 2, p. 101-112.
Kategorie:
Souhrnné/edukační práce
Souhrn
Histiocytóza z Langerhansových buněk má velmi pestré klinické projevy a u jednotlivých pacientů odlišnou míru agresivity. Po stanovení diagnózy je zásadní, stejně jako u ostatních maligních chorob, určit rozsah choroby, zjistit, které z orgánů jsou postiženy a v jaké míře. V případě jednoho ložiska lze použít lokální terapii (operační řešení, nebo instilaci metylprednisolonu do ložiska). Kožní postižení reaguje na světloléčbu (PUVA, případně electron beam irradiation). V případě izolovaného kožního postižení byly také popsány remise po léčbě thalidomidem nebo metotrexátem. V případně multisystémového postižení se u dospělých preferuje léčba 2-chlorodeoxyadenosinem (kladribinem) v monoterapii, méně často v kombinaci s cytosin-arabinosidem. Pro velmi agresivní formy lze použít chemoterapeutická schémata s etoposidem, které se jinak používají pro léčbu lymfomů. V případě rezistentních forem lze po domluvě s transplantačním centrem použít vysokodávkovanou chemoterapii s autologní či alogenní transplantací krvetvorných buněk. U pacientů s mutací genu BRAF V600E byla prokázána účinnost vemurafenibu či dabrafenibu, které jsou v ČR registrované pro léčbu diseminovaného melanomu, a bylo by možné při prokázání této mutace a při rezistenci na přechozí léčbu požádat plátce zdravotní péče o schválení úhrady i pro tuto diagnózu.
Klíčová slova:
histiocytóza z Langerhansových buněk – 2-chlorodeoxyadenosin (kladribin) – vemurafenib – dabrafenib
ÚVOD
Historii poznání, klinickým příznakům a projevům histiocytózy z Langerhansových buněk (LCH) byl věnován článek, který vyšel v říjnovém čísle časopisu Transfuze a hematologie dnes v roce 2019 [1]. Tento článek na něj navazuje a dominantně se věnuje léčbě této nemoci. Léčebné postupy se odvozují z publikovaných zkušeností ve formě popisu případů či malých souborů, případně závěrů z mezinárodního registru [2]. Vzácnost této nemoci je důvodem, proč je málo dokončených klinických studií. První mezinárodní doporučení pro léčbu u dospělých nemocných bylo zveřejněno v roce 2013, ale tato doporučení nebyla později revidována a znovu vydána, i když se objevily nové poznatky a léky [3].
Langerhansova histiocytóza (LCH) je velmi divergentní onemocnění s variabilním průběhem. Buňky LCH jsou klonální s výjimkou primární plicní formy LCH. Mutace genu BRAF V600E byla nalezena ve více než polovině případů, což svědčí pro klonální neoplastickou etiologii, nikoliv pro etiologii reaktivní [4]. V některých případech byla prokázána asociace LCH s některými maligními tumory, ale to jsou zcela výjimečné případy.
LCH může postihnout kterýkoliv orgán či systém, nejčastěji však bývají postiženy kosti, kůže a hypofýza. Méně často jsou pak postiženy lymfatické uzliny, játra, slezina, střevo a hemopoetický systém. Plíce mohou být postiženy současně či následně při postižení jiných orgánů. U dospělých se však často vyskytuje izolovaná plicní forma (PLCH) a postižení plic může předcházet postižení dalších orgánů.
Klinická manifestace je velmi pestrá, závisí na tom, který orgán je postižen. Průběh nemoci kolísá od spontánně se hojícího procesu po chronické recidivující onemocnění. Rychle progredující formy LCH, typické pro dětský věk, u dospělých většinou nebývají. Zcela výjimečně se může u dospělých vyskytnout sarkom z Langerhansových buněk, a to buď de novo, nebo transformací z klasické formy LCH.
V některých případech se může současně vyskytnout Erdheimova-Chesterova choroba, případně Rosaiova--Dorfmanova nemoc. V případně současného výskytu těchto chorob je léčba cílena na dominující onemocnění.
Léčba LCH se odvíjí od rozsahu nemoci a její závažnosti. Problémem je, že typ orgánového postižení určuje nejen příznaky, ale také odbornost lékaře, kterého pacient se svými problémy navštíví. To někdy svádí k tomu, že je léčba zahájena bez podrobného vyšetření.
Stanovení diagnózy
Stanovení diagnózy musí být vždy založeno na histologickém a imunofenotypizačním vyšetření suspektní tkáně. Normální Langerhansovy buňky vykazují pozitivitu antigenu CD1a a/nebo Langerinu [5]. Někdy je možná i falešně pozitivní diagnóza LCH, protože jak v kůži, tak v lymfatických uzlinách se může objevit zvýšený počet normální reaktivních Langerhansových buněk, obvykle jako nespecifická reakce na nějaký zánětlivý podnět. Z tohoto důvodu jsou definovány dvě úrovně diagnostické jistoty (tab. 1).
![Kritéria stanovení diagnózy [3]](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image_pdf/7dd9b6d58abde7a28e083d11ab4cba57.png)
Vyšetření při podezření na LCH
Pacienti s LCH jsou velmi často asymptomatičtí, nebo vykazují jen mírné symptomy.
Nejčastější symptomy LCH:
- dušnost a kašel při plicní formě,
- bolesti kostí a zduření nad kostním ložiskem,
- zvětšení břicha,
- necharakteristický kožní raš, svědění kůže,
- zvýšená žízeň (příznak diabetes insipidus),
- lymfadenopatie,
- dále mohou být přítomny celkové příznaky systémové zánětlivé reakce jako při jiných maligních chorobách: patologická únava (fatigue), celková slabost, úbytek hmotnosti, noční poty, nevolnost a subfebrilie či febrilie.
Je třeba v rámci pečlivé anamnézy zjistit, zda se nevyskytly nevysvětlené příznaky z dřívějška, typu idiopatického ekzému, diabetrs insipidus, plicních cyst a spontánních pneumotoraxů.
Klinické vyšetření musí zahrnovat vyšetření kůže a viditelných slizničních povrchů. V případě neuropatie, myopatie či kognitivního zhoršení se doporučuje též vyšetření neurologem či psychologem.
V rámci laboratorních vyšetření se provádějí běžná vyšetření, protože LCH nemá žádný specifický biochemický marker. Navíc je vhodné i vyšetření TSH a při podezření na postižení hypofýzy i dalších hypofyzárních hormonů včetně hormonů dalších endokrinních žláz, které jsou regulovány hypofýzou, tedy i hormonů pohlavních [3].
Zásadní je vždy získat informace o případném postižení skeletu. Dříve k tomuto účelu sloužily klasické snímky skeletu, dnes lze použít low dose CT skeletu případně FDG-PET/CT.
MR zobrazení může také velmi dobře odhalit kostní ložiska s přesahem infiltrátů do měkkých tkání, ale také infiltraci stopky hypofýzy, takže MR mozku je standardní metoda pro odhalení postižení hypofýzy a její stopky, případně pro odhalení ložisek v CNS.
Klasická scintigrafie skeletu pomocí Tc-pyrofosfátů se dnes nepovažuje za vhodnou metodu pro detekci kostního postižení LCH, protože kostní osteolytická ložiska nemusí akumulovat Tc-pyrofosfonát, podobně jako u mnohočetného myelomu.
Při podezření na postižení plic je třeba provést HRCT plic, které může odhalit typické nodularity a cystickou přestavbu, což na běžném snímku plic nemusí být zřetelné [6].
Sonografie břišní dutiny může odhalit ložiska v játrech.
Cílem těchto vyšetření je zjistit rozsah choroby a množství postižených orgánů či tkání.
Rizikové orgány (kostní dřeň, játra, slezina CNS), jejichž postižení signalizuje nepříznivý průběh
Postižení hematopoetického systému, které je u dospělých velmi vzácné, stejně jako postižení sleziny a jater nebo CNS, indikuje horší prognózu oproti průměru s možností úmrtí na tuto chorobu, pokud se nepodaří léčbou chorobu zastavit. Výrazně horší prognóza při postižení těchto rizikových orgánů byla jednoznačně prokázána u dětí, u dospělých statistický průkaz chybí pro velmi malý počet takových případů, ale není znám důvod, proč by tomu bylo u dospělých jinak.
Pokud jsou přítomny febrilie, noční poty a úbytek hmotnosti, tak jak jsou definovány B-symptomy u lymfomů, tak to také signalizuje velmi agresivní průběh podobný high grade lymfomu.
Speciální místa postižení signalizující vyšší riziko postižení CNS
Postižení obratlů s intraspinálním šířením infiltrátů anebo postižení kraniofaciálního skeletu se šířením do měkkých tkání (orbit, mastoideů, postižení sfenoidní či temporální kosti) je spojeno s akutním rizikem větších klinických komplikací vzhledem k lokalizaci. Lokální léčba v těchto oblastech je spojena s větším rizikem. Postižení těchto lokalizací vyžaduje vždy systémovou léčbu. Postižení kraniofaciálního skeletu je mimo jiné vždy spojeno s vyšším rizikem vzniku diabetes insipidus. Tento fakt byl opět jednoznačně prokázán v dětské populaci, ale panel expertů se domnívá, že tuto zkušenost lze extrapolovat i do dospělé populace [3].
Endokrinologické postižení a doporučená vyšetření
LCH má velkou afinitu k postižení hypotalamo-pituitární osy, vedoucí k permanentnímu deficitu hormonů zadní části hypofýzy a v některých případech k panhypopituarismu, který vyžaduje substituci.
Diabetes insipidus je nejčastější hormonální komplikací LCH, jeho manifestace může být prvním příznakem nemoci, anebo se může rozvinout v průběhu choroby. Diabetes insipidus bývá prokázán až u 30 % dospělých pacientů s LCH [2] a v případně multisystémového postižení se vyskytuje v 40 % [3]. Polyurie či polydypsie nebo strukturální abnormality v oblasti hypotalamu a hypofýzy jsou důvodem k vyšetření s cílem potvrdit či vyloučit diabetes insipidus.
Poškození funkce předního laloku hypofýzy je prokazováno asi u 20 % dospělých osob s LCH, téměř vždy společně s diabetes insipidus. Nejčastěji je postižena tvorba růstového hormonu (STH), jeho deficit bývá nalézán až u 50 % pacientů s diabetes insipidus. Dospělí muži nemají žádný specifický projev nedostatku STH, nedostatek se ale projeví u žen, které bez dostačující hladiny STH nejsou schopny donosit dítě. Proto je v případech gravidity nutná jeho substituce.
Deficit gonadotropinů je druhou nejčastější poruchou z oblasti adenohypofýzy. Projeví se poruchou menstruačního cyklu u žen a sníženým libidem u mužů [3].
Deficit ACTH může být parciální či kompletní a projeví se nespecifickými příznaky nebo akutním selháním nadledvin při zátěži.
Mírně zvýšená hladina prolaktinu může způsobit galaktoreu u žen a deficit gonadotropinů u všech osob.
Hormonální deficit, pokud již jednou vznikne, je trvalý, i když abnormality MR při zobrazení hypofýzy a hypotalamu po účinné léčbě regredují.
Postižení hypotalamu je méně časté než postižení hypofýzy, mimo hormonální poruchy způsobuje také neuropsychiatrické poruchy, poruchy termoregulace a spánku. Nejčastějším důsledkem infiltrace hypotalamu je těžká obezita na podkladě zvýšené chuti k jídlu. A to je spojeno s porušením glukózové tolerance a se vznikem diabetes mellitus.
LCH má také vliv na kostní metabolismus. Pacienti s aktivní LCH mají nižší kostní hustotu [7], což lze dokumentovat vyšetřením kostní denzity.
Za základní endokrinologické vyšetření pro všechny pacienty s LCH se proto považuje vyšetření osmolarity ranní moči, zda není přítomen diabetes insipidus. Pokud je podezření na snížení tvorby dalších hormonů, tak je vhodné požádat o komplexní endokrinologické vyšetření vyšetření osmolality plazmy, vyšetření sérového kortisolu, růstového faktoru, gonadálních steroidních hormonů a gonadotropinu [3].
Kožní a gingivální postižení a doporučená vyšetření
Kožní forma LCH může imitovat velmi různé kožní diagnózy a může být první známkou nemoci. Typické jsou papuly ve skalpu velikosti 1–2 mm růžově žluté barvy. Často tvoří krusty, a proto jsou často mylně interpretovány jako seboreická dermatitida.
Dále bývá časté intertriginózní postižení v axilách, inguinách a v anogenitální oblasti spojené s erytémem a erozemi, které jsou často špatně interpretovány jako ekzém a psoriáza, kandidové infekce nebo intertrigo.
Generalizované kožní erupce mohou být mylně interpretovány jako psoriasis guttata, prurigo nodularis nebo lichen planus. Bez odebrání kožní biopsie není možné kožní formu LCH rozpoznat.
Gingivální postižení je často spojeno s postižením alveolární části čelisti a ztrátou zubů. V případně uvolnění zubů se tyto zuby nemají extrahovat, protože pokud se však včas zahájí léčba, je možné opětovné zpevnění zubního lůžka.
Změny na nehtech zahrnují paronychia, onycholysis, subunguální hyperkeratózu a purpurové strie nehtového lůžka. Někdy jsou subunguálně viditelné tmavohnědé strie podobné, jaké způsobují některé medikamenty.
Postižení kožního povrchu LCH zahrnuje tedy značně pestré projevy. Z makroskopického pohledu nelze nikdy potvrdit, že se jedná o kožní formu LCH, a proto je vždy nutno provést bioptické a histologické ověření [3].
Postižení trávicího traktu
Postižení trávicího traktu je vzácné. Může se projevit jako solitární kolorektální polyp či mnohočetná granulomatózní ložiska v mukóze horní či dolní části trávicího traktu. Postižení trávicího traktu je často asymptomatické. Mnohočetné infiltráty jsou spojené s bolestmi břicha, průjmem a hypoalbuminemií.
Postižení jater je charakterizováno někdy infiltráty, které obsahují CD1a pozitivní buňky, ale mohou být i lymfocytární infiltráty kolem portálních žil, které nakonec vedou ke sklerotizující cholangitidě. Postižení pankreatu je extrémně vzácné [3].
Postižení plicního parenchymu
Primární pulmonální forma LCH postihuje převážně mladé osoby ve věkové skupině 20–40 let [5, 6]. Prevalence případů plicní formy LCH tvoří kolem 5 % ze všech intersticiálních plicních chorob.
Plicní forma LCH, podobně jako jiné plicní choroby, se může projevit kašlem, který mimo komplikující bronchitidy je neproduktivní, dále dušností a dalšími nespecifickými potížemi – známkami zánětlivé reakce organismu (úbytkem hmotnosti, nočním pocením a subfebriliemi či febriliemi s patologickou únavou – fatigue). Infekce dolních dýchacích cest je typickou komplikací plicní formy LCH.
Opakované záněty plic jsou častou příčinou zhoršení celkového stavu a jsou vždy indikací k intenzivní léčbě. Proto se také doporučuje vakcinace proti pneumokokovým infekcím a každoroční očkování proti chřipce [6]. Asi 20 % osob nemá žádné symptomy a prvním příznakem plicní formy LCH je spontánní pneumotorax. Obzvláště při cestování letadlem je vyšší riziko vzniku spontánního pneumotoraxu, a proto by na to měli být upozorněni všichni pacienti s plicní formou LCH. V případě kašle a/nebo bolestí na hrudníku by pacienti s plicní formou LCH měli konzultovat před letem svého plicního specialistu a probrat míru rizika spontánního pneumotoraxu v letadle [6, 7]. A naopak při každém spontánním pneumotoraxu by mělo být v rámci diferenciální diagnostiky pomýšleno i na možnost plicní formy LCH [6, 7].
Rentgenový snímek plic obvykle není velkým přínosem pro diagnostiku plicní formy LCH, jak ilustruje obrázek 1. Typické změny, noduly a mikrocystické změny, nejsou na snímku plic jasně patrné. Retikulární a mikronodulární přeměna struktury plíce je viditelná pouze při HRCT zobrazení hrudníku. Změny postihují obě plíce, dominantně střední a horní plicní pole, a chybí v kostofrenických oblastech [6]. HRCT hrudníku je nejlepší metodou pro detekci těchto změn (malé noduly, které přecházejí v kavitované noduly, a ty se postupně mění v tlustostěnné a posléze tenkostěnné cysty). S rozvojem nemoci přibývá cystických změn [6, 7], jak ilustruje obrázek 2.

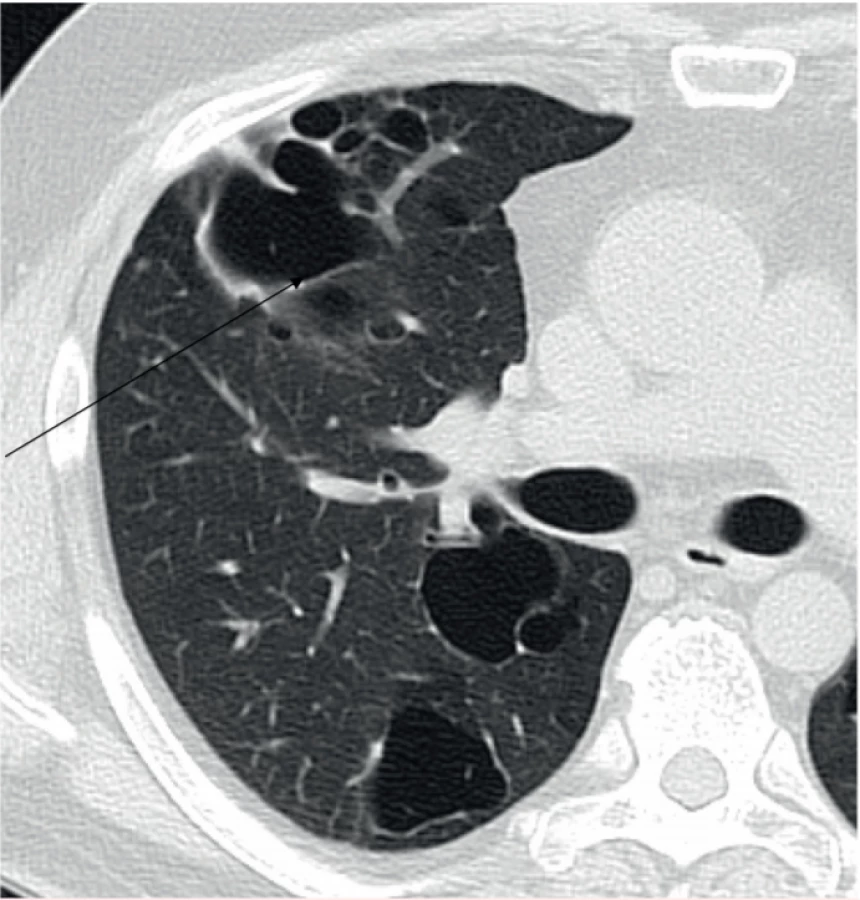
Vyšetření plicních funkcí prokazuje sníženou difuzní kapacitu asi u 70–90 % vyšetřených. Obvykle je snížená vitální kapacita a zvýšený reziduální objem. Celková plicní kapacita tak může být v normě. U většiny pacientů pozorujeme obstrukční ventilační poruchu. Zcela vzácně se může objevit i restriktivní komponenta [8].
Při bronchoskopii vidíme obvykle normální makroskopický nález. Bronchoalveolární laváž (výplach) obvykle provádíme z té části bronchiálního stromu, která ventiluje nejvíce postiženou část plic. V diferenciálním rozpočtu buněk z bronchoalveolární tekutiny (BAT) získané z bronchoalveolární laváže (BAL) převládají u LCH alveolární makrofágy, což odráží expozici cigaretovému kouři. Nález Langerhansových buněk (LB) zjištěný imunohistochemicky pomocí monoklonálních protilátek, především proti CD1a (CD1a je glykoprotein exprimovaný na antigen prezentujících buňkách), může vést k diagnóze. Za „diagnostický“ považujeme zvýšený počet LB v BAT nad 5 % v korelaci s klinickým a radiologickým nálezem (kavitované noduly, tenkostěnné cysty, mladý silný kuřák). Tento test vykazuje vysokou specificitu, ale nízkou senzitivitu, proto samotný průkaz LB v BAT diagnózu nedělá [9, 10]. Kromě CD1a jsou dalšími diagnostickými markery LB zjistitelné imunohistochemicky: protein S100, langerin (CD207; receptor na Langerhansových buňkách), Birbeckova granula.
Za standardní diagnostickou metodu volby se proto stále považuje plicní biopsie, kde místo odběru je určeno podle HRCT nálezu. U asymptomatických pacientů s makrofágovou alveolitidou a typickým nálezem na HRCT hrudníku (viz výše) se považuje průkaz CD1a+ LB v BAT za dostatečný pro diagnózu LCH [9, 10]. V případě, kdy nález z BAL není diagnostický a podle HRCT hrudníku je podezření na plicní formu LCH, je indikována plicní biopsie (transbronchiální, chirurgická plicní biopsie). U pacientů s velkými cystickými ložisky je nutno zvážit riziko a přínos plicní biopsie. Výhodou chirurgické plicní biopsie (videotorakoskopie) je možnost získat reprezentativní vzorek plicní tkáně s histologickým průkazem LCH a typických mutací genu BRAF i dalších, jejichž průkaz otevře cestu do budoucna pro biologickou léčbu.
Stratifikace nemoci
Po provedení všech výše uvedených vyšetření je možné pacienta zařadit do kategorie LCH s postižením jednoho orgánu nebo kategorie multisystémové LCH, jak uvádí tabulka 2.
![Stratifikace pacientů s LCH [3]](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image_pdf/04d296ebac096454f265568a87cae1e0.png)
Léčba histiocytózy z Langerhansových buněk
Léčba LCH se vždy odvíjí od místa postižení a rozsahu nemoci.
Léčba kostního postižení
V případě LCH s unifokálním postižením kosti, jejíž infiltrace není spojena s vyšším rizikem postižení CNS, je považována za dostačující lokální léčba a dále sledování.
Opět způsob léčby závisí na velikosti a uložení ložiska. Biopsie s kyretáží ložiska je dostačující pro histologické ověření, ale i pro zahájení procesu hojení. Kompletní excize kostního ložiska se nepovažuje za indikovanou, protože by způsobila velký a nehojící se kostní defekt a mutilovala by člověka.
Nitroložisková injekce kortikoidů může urychlit hojení. Doporučuje se metylprednisolon v dávce 40–160 mg [11].
Cílená radioterapie je indikována, pokud je riziko neurologického defektu nebo vyšší riziko operačního zákroku, například při lokalizaci v čelisti či kraniálních kostech, či na lební bázi.
V případě multifokálního postižení nebo při postižení speciálních kostních lokalizací je indikována systémová léčba.
Léčba izolovaného postižení uzlin
Izolované postižení uzlin je velmi vzácné a jsou možné i spontánní regrese. Proto by se neměly v těchto případech používat extenzivní operační výkony (disekce krčních uzlin) ani systémová léčba.
Léčba izolovaného postižení kůže
Kožní excize lze limitovat na solitární nevelká ložiska, ale neměla by se provádět v žádném případě mutilující operace. V případně multisystémového postižení reagují kožní morfy obvykle na systémovou léčbu.
V případě postižení omezeného na kůži nebo v případě, že kožní postižení nereaguje dobře na celkovou léčbu, existuje několik lokálních léčebných alternativ.
Lze použít fototerapii, psoralen a ultrafialové záření (PUVA) [12] nebo ozáření úzkým pásmem ultrafialového světla (UV) B [13]. Efekt této léčby potvrzují některé popisy případů. Obtížně se touto metodou léčí intertriginózní lokalizace či postižení vlasové části hlavy. Tuto léčbu nelze také použít při postižení penisu.
Při postižení kůže lze s úspěchem použít i ozáření nízce energetickými elektrony (electron beam irradiation) [14].
Je popsán i hojivý účinek thalidomidu na kožní postižení LCH (33), ale jeho efekt je nedostatečný při multisystémové formě nemoci. Thalidomid se používá v dávce 100 mg denně za bedlivého sledování jeho nežádoucích účinků [15]. Azathioprin je další lék, který je efektní u dětí, ale u dospělých nebyl až tolik testován. Dnes se doporučuje, aby u pacientů byla vyšetřena thiopurin metyltransferáza, a pokud je v normě, je možné otestovat léčebný potenciál azathioprinu v dávce 2 mg/kg/den. Lék se obvykle podává alespoň 6 týdnů. Až pak je možno hodnotit jeho efekt [3].
Jsou publikovány zprávy u účinnosti metotrexátu podávaného buď v monoterapii, nebo v kombinaci s azathioprinem nebo prednisolonem. Metotrexát se podává v dávce 20 mg 1× týdně [16].
Postižení sliznice úst se obvykle léčí systémovou léčbu a, pokud možno, nemají se extrahovat zuby [3].
Léčba plicní formy LCH
Prognózu izolované formy LCH není možné v době stanovení diagnózy dostupnými metodami stanovit. Asi u 40–50 % pacientů dojde k částečnému či úplnému vymizení abnormalit na HRCT vyšetření bez terapeutického zásahu. Spontánní vymizení plicní formy se však popisuje pouze v souvislosti s ukončením kouření [6, 7]. Obrázek 3 ukazuje regresi nálezu u silného kuřáka (abúzus 20 cigaret denně po dobu 20 let) po zanechání kouření.

Pacienty je potřeba komplexně sledovat včetně funkčních plicních vyšetření a myslet na možnost rozvoje plicní hypertenze.
Pokud nevede vyloučení cigaretového kouře ze života pacienta s plicní formou k regresi v průběhu několika měsíců, nemoc je stále aktivní a jsou zřetelné nové plicní noduly na HRCT plic, je nutno zvážit medikamentózní léčbu.
Za léčbu první linie u primární plicní formy LCH se stále považují glukokortikosteroidy. Účinnost glukokortikosteroidů je sice popisována, ale nebyla prokázána klinickými studiemi. Za standardní se považuje podávání prednisonu v dávce 1 mg/kg/den po dobu 1 měsíce s následující redukcí dávky v průběhu několika dalších měsíců [3, 17].
Onemocnění refrakterní k léčbě prednisonem nebo při intoleranci dlouhodobého podávání prednisonu je pak indikováno stejně jako u multisystémové formy k léčbě 2-chlorodeoxyadenosinem, synonymem kladribinem. Lék se podává v dávce 5 mg/m2 ve formě podkožní injekce 5 dní po sobě v 4–5týdenních intervalech. Obvykle se podávají 4 cykly. Ale podle klinické studie je u těchto pacientů přínosné i použití vinblastinu [18].
U pacientů s plicní formou LCH se řeší opakované spontánní pneumotoraxy hrudními drenážemi nebo s pomocí pleurodézy (iatrogenní srůst viscerální a parietální pleury za účelem snížení rizika pneumotoraxu, například chirurgickým ošetřením) [18].
Při průkazu odpovídajících cytogenetických změn lze použít i nové léky (inhibitory BRAF nebo MAPK) [19].
Transplantace plic je řešení pouze pro pacienty s respirační a ventilační insuficiencí anebo s pokročilou plicní hypertenzí. Recidiva plicní formy LCH je popisována u 20 % nemocných po provedení transplantace plic [20].
Pokud se v souvislosti s LCH prokáže plicní hypertenze, je to indikací ke specifické vazodilatační léčbě [21].
Systémová léčba
V případě dospělých pacientů není na rozdíl od pediatrických pacientů definována standardní léčba. Vinblastin a prednison jsou zmiňovány četnými chemoterapeutickými manuály, ale účinnost této léčby nebyla nikdy u dospělých prokázána v rámci prospektivní studie. U dětí se používají léčebné protokoly s opakovaným dlouhodobým podáváním vinblastinu a prednisonu. Dlouhodobé podávání vinblastinu a prednisonu je ale u dospělých spojeno s neurotoxicitou vinblastinu a s nežádoucími účinky dlouhodobého podávání prednisonu. Proto v současnosti experti na léčbu LCH preferují u dospělých pacientů monoterapii kladribinem, dále pak cytosin-arabinosidem či etoposidem, ale již nedoporučují opakované aplikace vinblastinu [3]. V případě izolovaného postižení CNS je podle některých autorů považován cytosin-arabinosid za účinnější než kladribin. Při diskusi o cytosin-arabinosidu a kladribinu na setkání Německé skupiny pro léčbu histiocytózy z Langerhansových buněk vyplynulo, že cytosin-arabinosid je používán častěji u dětských pacientů než u dospělých.
První zpráva o úspěšné léčbě LCH pomocí kladribinu se objevila v roce 1997 [22].
Pak následovala exploze dalších informací potvrzujících účinnost této léčby. Od roku 1993 do roku 2020 však vznikla pouze jedna klinická studie, v jejímž rámci byl kladribin podáván pacientům s LCH, a ta jeho účinnost také potvrdila [23].
V odborné literatuře lze najít četné popisy případů a malých souborů pacientů s LCH, kteří byli léčeni kladribinem, a všechny tyto publikace potvrdily jeho účinnost (viz databázi Medline PUBMED). V retrospektivní studii 58 pacientů s kostním postižením pozorovali autoři výrazně lepší léčebnou odpověď po cytarabinu než po vinblastinu a prednisonu, která byla srovnatelná s 2-chlorodeoxyadenosinem [24].
Intenzivní polychemoterapeutické režimy, používané pro lymfomy a obsahující etoposid, například CHOEP či MACOB B, jsou sice účinné, ale měly by se používat jen ve vzácných případech agresivních forem LCH, kde může léčba kladribinem selhat [3, 25].
Většina expertů zahajuje léčbu v případech s postižením rizikových orgánů nebo s postižením CNS pomocí kladribinu. Cytosin-arabinosid je považován za racionální alternativu [3].
Někteří autoři podávají v případě vícečetného kostního postižení bisfosfonáty, ale je nutno dbát na rizika vzniku osteonekrózy [26].
Změna léčby při selhání první linie
Při refrakteritě k jedné léčebné linii se testuje další alternativa. V případě postižení CNS se za vhodnou kombinaci považuje kombinace cytosin-arabinosidu a kladribinu, protože oba léky prostupují hematocefalickou bariéru [3].
Alternativou pro agresivní formy nemoci je také provedení vysokodávkované chemoterapie s autologní či alogenní transplantací krvetvorných buněk [27].
Radioterapie
Radioterapie je další léčebnou alternativou [28]. Nejčastěji se používá u kostního postižení. Podíl léčebných odpovědí se pohybuje kolem 79–100 % [28]. Není shoda ve velikosti doporučené dávky s poměrně značným rozptylem od 1,4 do 45 Gy. Obecně je považována za optimální dávku 10–20 Gy pro dospělé a 10 Gy pro děti [28].
Pokud LCH postihuje nervový systém, tak je to nejčastěji hypotalamus a hypofýza i s její stopkou. Parenchymální či meningeální postižení je výjimečné.
Pokud je postižena pouze hypotalamicko hypofyzární oblast, tak léčbou volby je kladribin případně s cytosin-arabinosidem. Izolované postižení parenchymu CNS v ostatních lokalizacích se léčí cílenou radioterapií. Vícečetnou ložiskovou infiltraci CNS pak již nelze léčit cílenou radioterapií, takže zbývá pouze systémová léčba.
Léčba neurodegenerativního postižení CNS
Neurodegenerativní postižení CNS se rozvijí po mnohaletém průběhu onemocnění. Ložiska neurodegenerativního postižení již neobsahují CD1a histiocyty, ale obsahující CD8+ pozitivní lymfocyty.
Neumíme zde definovat jednoznačné doporučení. Formou popisů případů byly popsány případy, kdy došlo ke stabilizaci po i. v. imunoglobulinech, ale i po rituximabu. Ale byly popsány i další léčebné postupy s pozitivním výsledkem, jak uvádí citované práce. Zásadní otázkou je vždy časový interval mezi počátkem neurodegenerativních změn a jejich diagnostikou pomocí MR či FDG-PET/CT či FDG-PET/MR. Informace v publikované literatuře jsou natolik divergentní, že není možné stanovit postup, který s vysokou pravděpodobností povede ke klinickému zlepšení [29].
V našem souboru 38 pacientů máme pouze 2 pacienty s touto pozdní komplikací, LCH byla diagnostikována a léčena v dětském věku a po dosažení dospělosti byli tito pacienti předáni k nám na další sledování.
Biologická léčba LCH – vemurafenib a dabrafenib
V roce 2010 byla prokázána mutace BRAF V600E u pacientů s LCH a bylo prokázáno, že proliferace těchto buněk je závislá na MAPK aktivační cestě. U 25–65 % případů LCH je nalézána patogenní varianta BRAF V600E. Protein BRAF hraje důležitou roli v MAPK signální dráze. Tato dráha ovlivňuje tvorbu transkripčních faktorů důležitých pro buněčný růst a proliferaci. Mutace BRAF V600E je spouštěcí mutací i řady jiných nádorových nemocí – melanomu, leukemie z vlasatých buněk a dalších. Toto poznání otevřelo cestu biologické léčbě pomocí preparátu vemurafenib.
Molekulárně biologické podrobnosti byly nedávno publikovány v časopise Klinická onkologie [30]. V první klinické studii bylo zařazeno 14 pacientů s LCH nebo s Erdheimovou-Chesterovou chorobou. Léčebná odpověď podle kritérií RECIST byla popsána u 41 % pacientů [31].
V další retrospektivní studii u dospělých pacientů s Erdheimovou-Chesterovou nemocí byla léčebná odpověď, tentokráte definovaná vymizením aktivity na FDG-PET/CT zobrazení, prokázána ve 100 % [32].
V klinické studii VE-BASKET byla prokázána metabolická léčebná odpověď u všech pacientů. Zda se ale tato metabolická odpověď promítne i do dlouhodobého přežití, zatím není jasné [33].
Pozitivní léčebný efekt vemurafenibu byl potvrzen také u pediatrických pacientů [34–39], a to i kolegy ze Slovenska [39].
Proto americká agentura Food and Drug Administration (FDA) schválila dva inhibitory BRAF V600E kinázy, vemurafenib a dabrafenib pro léčbu Erdheimovy-Chesterovy choroby, jejíž buňky obsahují uvedenou mutaci [40].
Podle současných názorů je jak vemurafenib, tak i dabrafenib účinný při léčbě BRAF V600E pozitivní multisystémové Erdheimovy-Chesterovy choroby, stejně jako u dalších BRAF V600E pozitivních malignit, včetně BRAF V600E pozitivně mutované Langerhansovy histiocytózy (LCH) s postižením CNS [40, 41]. Zatím však není zcela jasné, jak dlouho léčebná odpověď po této léčbě vydrží. Ve studii, která sledovala dlouhodobý efekt vemurafenibu u Erdheimovy-Chesterovy choroby (LOVE study), byly prokázány relapsy u 75 % pacientů poté, co byla léčba inhibitorem BRAF V600E ukončena [41]. V případě Erdheimovy-Chesterovy choroby je mutace BRAF V600E přítomna asi v 50 % případů. Zde je možné při neúspěchu léčby první linie, za kterou považují někteří odborníci interferon alfa či chlorodeoxyadenosin použít i dabrafenib či trametinib [42].
Nejčastější nežádoucí účinky vemurafenibu (> 30 %) zahrnují artralgii, únavu, vyrážku, fotosenzitivní reakce, nauzeu, alopecii a pruritus. Často (20 %) byl hlášen kožní spinocelulární karcinom, který byl léčen lokální excizí. Dále byly hlášeny závažné reakce přecitlivělosti včetně anafylaxe, dermatologické reakce, včetně vzácných případů Stevensova-Johnsonova syndromu a toxické epidermální nekrolýzy. Použítí vemurafebinu tedy není bez nežádoucích účinků a stejně tak je tomu u dabrafenibu [43].
Dabrafenib se používá v kombinaci s trametinibem pro léčbu kožních neoplazií a jsou také první zprávy o použití této kombinace pro léčbu LCH, i když s tímto lékem je podstatně méně publikovaných zkušeností než s vemurafenibem [43]. Podle pravidel úřadu SÚKL je vemurafenib je hrazen v monoterapii nebo v kombinaci s cobimetinibem k léčbě dospělých pacientů s neresekovatelným nebo metastatickým melanomem s mutací V600E genu BRAF. Stejně tak je hrazen dabrafenib v monoterapii nebo v kombinaci s trametinibem k léčbě dospělých pacientů s neresekovatelným nebo metastatickým melanomem s mutací V600E genu BRAF, kteří nebyli v minulosti léčeni systémovou terapií pro inoperabilní pokročilé či metastatické onemocnění. Terapie těmito léky je hrazena do progrese onemocnění.
Nicméně data o účinnosti vemurafenibu u LCH stejně jako u Erdheimovy-Chesterovy choroby jsou dostatečně průkazná k tomu, aby při prokázání této mutace a při neúspěchu kladribinu či cytosin-arabinosidu byla zvažována léčba vemurafeniben, samozřejmě jen po schválení úhrady a indikce revizním lékařem zdravotní pojišťovny.
Sledování po léčbě
LCH může přejít do chronického stadia, ale může také recidivovat i po dosažení remise nemoci. Proto je třeba pacienty po ukončené léčbě sledovat. A protože LCH je také spojena s vyšším rizikem dalších malignit ve srovnání s průměrnou populací, je cílem kontrol jak včasné podchycení recidivy této nemoci, tak i časná diagnostika jiných maligních onemocnění [3].
Po ukončení léčby by měly následovat první kontroly ve 2–3měsíčních intervalech. Posléze lze intervaly kontrol protáhnout na 3–12 měsíců.
Metody pro sledování plicní formy LCH jsou limitované. Vhodné je v rámci kontrol opakovaně provádět komplexní funkční plicní vyšetření včetně plicní difuze. Pro detekci nových nodulů je nutné HRCT zobrazení, protože běžný snímek je neodhalí, jak ilustruje obrázek 1.
Zásadním přínosem FDG-PET/CT zobrazení s metodou měření difuzní aktivity definovaného objemu plicního parenchymu a její vyjadřování jak ve formě SUVmax, ale také ve formě indexu SUVmax pulmo : SUVmax hepar. A právě tento index, poměr difuzní aktivity v definovaném objemu plíce k aktivitě v definovaném objemu v játrech, umožnil dlouhodobé sledování těchto pacientů, protože hodnota tohoto indexu není závislá na celkové nitrožilně aplikované aktivitě radiofarmaka. U našich pacientů jsme tuto metodu sledování opakovaně použili a korelovala s klinickým průběhem [44, 45]. Obrázek 4 ilustruje použití této metody sledování aktivity LCH v plicním parenchymu pomocí FDG-PET/CT. Posléze tuto metodu zmínili i další pracovníci z PET/CT pracovišť. Měření difuzní plicní aktivity a její vyjadřování indexem SUVmax pulmo : SUVmax hepar se nám jeví pro sledování jako nejlepší metoda. Zobrazení pomocí HRCT sice také velmi dobře ukáže plicní noduly, ale vyhodnocování, zda nodulů přibylo, či ubylo na HRCT obraze, je velmi obtížné a zdlouhavé a vyžaduje nadstandardní nasazení lékaře, který vyhodnocuje HRCT plic.

Ložiska LCH poměrně dobře vychytávají fluorodeoxyglukózu, a proto je FDG-PET/CT poměrně často používáno pro sledování osob s LCH [46].
Závěr
Léčba dospělých pacientů dříve zcela vycházela z pediatrických zkušeností, a tak byla doporučována i u dospělých léčba vinblastinem a prednisonem. Nicméně u dospělých nebyl efekt této léčby řádně doložen a léčba byla provázena výraznými komplikacemi (neurotoxicita). A tak v posledních letech je za lék první volby pro dospělé nemocné s LCH v případě systémové léčby považován 2-chlorodexyadenosin, synonymem kladribin. Průkaz mutace BRAF V600E asi u poloviny pacientů s LCH otevřel v případě neúspěchu léčby klasické cestu k použití biologické léčby preparátem vemurafenib nebo dabrafenib. Zatím však ještě není vyhodnocen vliv této nové biologické léčby na dlouhodobou morbiditu a mortalitu.
Poděkování
Publikace vznikla na podporu grant u MZ ČR RVO, FNBR, 65269705.
Podíl autorů na přípravě rukopisu
KZ – příprava první verze a revizí rukopisu
AZ, DM, KM, HT, DM, PL, ŘZ, KR – připomínkování rukopisu a schválení finální verze
Čestné prohlášení
Autoři práce prohlašují, že v souvislosti s tématem, vznikem a publikací tohoto článku nejsou ve střetu zájmů a vznik ani publikace článku nebyly podpořeny žádnou farmaceutickou firmou.
Do redakce doručeno dne 18. 10. 2019.
Přijato po recenzi dne 16. 12. 2019.
prof. MUDr. Zdeněk Adam, CSc.
Interní hematologická a onkologická klinika LF MU a FN Brno
Jihlavská 20
Brno-Bohunice
e-mail: adam.zdenek@fnbrno.cz
Zdroje
- Adam Z, Ježová M, Nebeský T, et al. Klinické obrazy histiocytózy z Langerhansových buněk v dospělosti. Transfuze Hematol Dnes. 2019;25(3):219–228.
- Arico M. Langerhans cell histiocytosis in adults. Report from the International Registry of the Histiocyte Society. Eur J Cancer. 2003;39(16):2341–2348.
- Girschikofsky M, Arico M, Castilo D, et al. Management of adult patients with Langerhans cell histiocytosis, recommendation from an expert panel on behalf of Euro-Histo Net. Orphanet J Rare Dis. 2013; 8 : 72–78.
- Badalian-Very G, Vergilio JA, Degar BA, et al. Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood. 2010;116(11):1919–1923.
- Swerdlow SH, Campo E, Harris NL, et al. WHO classification of tumours of haematopoietic and lymphoid tissues. International Agency for Research on Cancer (IARC), 2008; 439 pages.
- Lorillon G, Tazi A. How I manage pulmonary Langerhans cell histiocytosis. Eur Respir Rev. 2017;26(145).
- Doubková M, Tomíšková M, Skřičková J. Plicní histiocytóza z Langerhansových buněk – nemoc kuřáků. Stud Pneumol Phtiseol. 2014;74(5):158–161.
- Tazi A, Marc K, Dominique S, et al. Serial CT and lung function testing in pulmonary Langerhans cell histiocytosis. Eur Respir J. 2012;40(4):905–912.
- Auerswald U, Barth J, Magnussen H. Value of CD-1 positive cells in bronchoalveolar lavage fluid for the diagnosis of pulmonary histiocytosis X. Lung. 1991;169(6):305–309.
- Lommatzsch M, Bratke K, Stoll P, et al. Bronchoalveolar lavage for the diagnosis of pulmonary Langerhans cell histiocytosis. Respir Med. 2016; 119 : 168–174.
- Yasko AW, Fanning CV, Avala AG, et al. Percutaneous techniques for the diagnosis and treatment of localized Langerhans-cell histiocytosis (eosinophilic granuloma of bone). J Bone Joint Surg Am. 1998;80(2):219–228.
- Sakai H, Ibe M, Takahashi H, et al. Satisfactory remission achieved by PUVA therapy in Langerhans cell hisiocytosis in an elderly patient. J Dermatol. 1996;23(1):42–46.
- Imafuku S, Shibata S, Tashiro A, et al. Cutaneous Langerhans cell histiocytosis in an elderly man successfully treated with narrowband ultraviolet B. Br J Dermatol. 2007;157(6):1277–1279.
- Adam Z, Ježová M, Šlampa P, et al. Histiocytóza z indeterminovaných buněk – vymizení kožní infiltrace po ozáření elektronovým svazkem a aplikace 2-chlorodeoxyadenozinu: kazuistika. Vnitř Lék. 2017;63(4):284–288.
- McClain KL, Kozinetz CA. A phase II trial using thalidomide for Langerhans cell histiocytosis. Pediatr Blood Cancer. 2007;48(1):44–49.
- Steen AE, Steen KH, Bauer R, et al. Successful treatment of cutaneous Langerhans cell histiocytosis with low-dose methotrexate. Br J Dermatol. 2001;145(1):137–140.
- Vassallo R, Harari S, Tazi A. Current understanding and management of pulmonary Langerhans cell histiocytosis. Thorax. 2017;72(10):937–945.
- Tazi A, Lorillon G, Haroche J, et al. Vinblastine chemotherapy in adult patients with langerhans cell histiocytosis: a multicenter retrospective study. Orphanet J Rare Dis. 2017;12(1):95.
- Lorillon G, Jouenne F, Baroudjian B, et al. Response to trametinib of a pulmonary Langerhans cell histiocytosis harboring a MAP2K1 deletion. Am J Respir Crit Care Med. 2018;198(5):675–678.
- Dauriat G, Mal H, Thabut JF, et al. Lung transplantation for pulmonary Langerhans´ cell histiocytosis: a multicenter analysis. Transplantation. 2006;81(5):746–750.
- Karampitsakos T, Tzouvelekis A, Chrysikos S, et al. Pulmonary hypertension in patients with interstitial lung disease. Pulm Pharmacol Ther. 2018;50 : 38–46.
- Dimopoulos MA, Theodorakis M, Kostis E, et al. Treatment of Langerhans cell histiocytosis with 2 chlorodeoxyadenosine. Leuk Lymphoma. 1997;25(1–2):187–189.
- Donadieu J, Bernard F, van Noesel M for Salvage Group of the Histiocyte Society. Cladribine and cytarabine in refractory multisystem Langerhans cell histiocytosis: results of an international phase 2 study. Blood. 2015;126(12):1415–1423.
- Cantu MA, Lupo PJ, Bilgi M, et al. Optimal therapy for adults with Langerhans cell histiocytosis bone lesions. PLoS One. 2012;7(8): e43257.
- Derenzini E, Fina MP, Stefoni V, et al. MACOP-B regimen in the treatment of adult Langerhans cell histiocytosis: experience on seven patients. Ann Oncol. 2010;21(6):1173–1178.
- Montella L, Merola C, Merola G, et al. Zoledronic acid in treatment of bone lesions by Langerhans cell histiocytosis. J Bone Miner Metab. 2009;27(1):110–113.
- Pan Y, Xi R, Wang C, et al. Autologous hematopoietic stem cell transplantation for efficient treatment of multisystem, high-risk, BRAF V600E-negative Langerhans cell histiocytosis. J Int Med Res. 2019;47(9):4522–4529.
- Micke O, Seegenschmiedt MH. Consensus guidelines for radiation therapy of benign diseases: a multicenter approach in Germany. Int J Radiat Oncol Biol Phys. 2002;52(2):496–513.
- Yeh EA, Greenberg J, Abla O, et al. North American Consortium for Histiocytosis. Evaluation and treatment of Langerhans cell histiocytosis patients with central nervous system abnormalities: Current views and new vistas. Pediatr Blood Cancer. 2018;65(1):e26784.
- Novosad O, Skrypets T, Pastushenko Y, et al. MAPK/ERK signal pathway alterations in patients with Langerhans cell histiocytosis. Klin Onkol. 2018;31(2):130–134.
- Hyman DM, Puzanov I, Subbiah V, et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N Engl J Med. 2015;373(8):726–736.
- Haroche J, Cohen-Aubart F, Emile JF, et al. Dramatic efficacy of vemurafenib in both multisystemic and refractory Erdheim-Chester disease and Langerhans cell histiocytosis harboring the BRAF V600E mutation. Blood. 2013;121(9):1495–500.
- Diamond EL, Subbiah V, Lockhart AC, et al. Vemurafenib for BRAF V600-mutant Erdheim-Chester disease and Langerhans cell histiocytosis: analysis of data from the histology-independent, phase 2, open-label VE-BASKET study. JAMA Oncol. 2018;4(3):384–388.
- Heisig A, Sörensen J, Zimmermann SY, et al. Vemurafenib in Langerhans cell histiocytosis: report of a pediatric patient and review of the literature. Oncotarget. 2018;9(31):22236–22240.
- Charles J, Beani JC, Fiandrino G, et al. Major response to vemurafenib in patient with severe cutaneous Langerhans cell histiocytosis harboring BRAF V600E mutation. J Am Acad Dermatol. 2014;71(3):e97–e99.
- Váradi Z, Bánusz R, Csomor J, et al. Effective BRAF inhibitor vemurafenib therapy in a two-year-old patient with sequentially diagnosed Langerhans cell histiocytosis and Erdheim-Chester disease. Onco Targets Ther. 2017;10 : 521–526.
- Haroche J, Cohen-Aubart F, Emile JF, et al. Vemurafenib as first line therapy in BRAF-mutated Langerhans cell histiocytosis. J Am Acad Dermatol. 2015;73(1):e29–e30.
- Donadieu J, Larabi IA, Tardieu M, et al. Vemurafenib for refractory multisystem Langerhans cell histiocytosis in children: an international observational study. J Clin Oncol. 2019;37(31):2857–2865.
- Kolenová A, Bubanská E, Špotová A, et al. Cielená liečba závažnej multisystémovej histiocytózy z Langerhansových buniek. Pediatr Prax. 2018;19(1):27–31.
- Oneal PA, Kwitkowski V, Luo L, et al. FDA approval summary: vemurafenib for the treatment of patients with Erdheim-Chester disease with the BRAFV600 mutation. Oncologist. 2018;23(12):1520–1524.
- Cohen Aubart F, Emile JF, Carrat F, et al. Targeted therapy in 54 patients with Erdheim Chester disease, including follow up after interruption (the LOVE study). Blood. 2017;130 : 1377–1380.
- Al Bayati A, Plate T, Al Bayati M, et al. Dabrafenib and trametinib treatment for Erdheim-Chester disease with brain stem involvement. Mayo Clin Proc Innov Qual Outcomes. 2018;2(3):303–308.
- Stewart JR, Murzaku EC, Sode TT, Gordon KA. Cutaneous Langerhans cell histiocytosis with gastrointestinal involvement treated with dabrafenib. JAAD Case Rep. 2017;4(1):95–97.
- Szturz P, Řehák Z, Koukalová R. Measuring diffuse metabolic activity on FDG-PET/CT: new method for evaluating Langerhans cell histiocytosis activity in pulmonary parenchyma. Nucl Med Biol. 2012;39(3):429–436.
- Adam Z, Řehák Z, Koukalová R, et al. Plicní forma histiocytózy z Langerhansových buněk: hodnocení aktivity nemoci a léčebné odpovědi pomocí PETCT (indexu SUVmax Pulmo/ SUVmax Hepar). Popis vlastních zkušeností a přehled literatury. Vnitř Lék. 2010;56(12):1228–1250.
- Obert J, Vercellino L, Van Der Gucht A, et al. (18) F-fluoro-deoxyglucose positron emission tomography-computed tomography in the management of adult multisystem Langerhans cell histiocytosis. Eur J Nucl Med Mol Imaging. 2017;44(4):598–610.
Štítky
Hematologie a transfuzní lékařství Interní lékařství OnkologieČlánek vyšel v časopise
Transfuze a hematologie dnes

2020 Číslo 2
- Ivabradin zlepšuje kvalitu života starších pacientů se srdečním selháním
- Srovnání vlivu tamoxifenu a exemestanu na tloušťku endometria
- Mutace BRCA1/2 nezvyšuje vliv perorální antikoncepce na riziko karcinomu prsa a ovarií
- Limity glykovaného hemoglobinu a význam dalších glykovaných proteinů
- Plazmatický fibrinogen jako možný prognostický marker u nemetastatického renálního karcinomu
Nejčtenější v tomto čísle
- Castlemanova nemoc, jedna z příčin chronické systémové zánětlivé reakce, někdy i retence tekutin, vaskulitid a poruch imunity – Mezinárodní diagnostická kritéria z roku 2017
- Trombotické mikroangiopatie
- Léčba histiocytózy z Langerhansových buněk u dospělých osob
- Překvapivý nález v kostní dřeni: co za ním je?
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy