
Získaná aplastická anémie v dětském věku – dlouhodobé výsledky a rizika kombinované imunosupresivní léčby antithymocytárním globulinem a cyklosporinem A
Immunosuppressive therapy combining anti-thymocyte globulin and cyclosporine A in childhood acquired aplastic anaemia – long term-outcomes and risks
In patients with acquired aplastic anaemia (AA), autoimmunity has been shown to play an important role in the pathogenesis. Therefore immunosuppressive therapy combining cyclosporine A (CsA) and antithymocyte globulin (ATG) is currently a standard treatment regimen for paediatric patients with AA, if a matched sibling donor is not available. Response rate approximately 70% and a 10-20% risk of clonal evolution (paroxysmal nocturnal haemoglobinuria, myelodysplastic syndrome, acute myeloid leukaemia) are the main limitations of IST compared to allogeneic stem cell transplantation (MSD-SCT). Long-term experience with AA management is presented through a cohort of paediatric patients diagnosed with aplastic anaemia between 1996 and 2009 in the Czech Republic. A total of 72 children with a median age of 10.5 yrs (1.6-17.9 yrs) met the criteria of aplastic anaemia. First-line therapy followed HLA-identical sibling donor availability - 18 patients underwent MSD-SCT and 54 were given immunosuppressive therapy (IST). Overall survival (OS) at a median follow-up of 7 years was 96±3.5% in the SCT group and 92±3.8% in the IST group. There was a significant difference between 7-year EFS in the SCT group (92.3±7.4%) and in the IST group (58±7.1%). Overall response to IST was 76%. Cummulative relapse rate 22%, with 75% probability of response to repeated courses of IST. First-line IST failure and relapse treatment non-response in 12 patients was managed by stem cell transplantation from an alternative donor (MUD-SCT) followed by 5-year OS 80%. No MDS/AML clonal transformation was registered within a 7-year median follow-up after IST. PNH clonal evolution was registered in 7 out of 37 evaluated patients (19%) at a median interval of 108 months from start of IST. Five of these patients presented with clinical or laboratory symptoms of PNH including two thrombotic events. Only two patients were asymptomatic, with a subclinical course typical of PNH/AA syndrome. The cumulative risk of PNH evolution following immunosuppressive treatment for aplastic anaemia is 2.2%...4.6% ...7.2%...19.8% 2, 4, 6 and 10 years from start of treatment. IST should be considered an effective alternative first-line therapy for paediatric aplastic anaemia, with acceptable toxicity facilitating at least temporary disease control in patients lacking a matched sibling donor. From a long-term perspective not relapses, but PNH clonal evolution including symptomatic haemolytic and thrombotic forms are of a major concern in therapy consequencies.
Key words:
aplastic anaemia, immunosuppressive therapy, antithymocyte globulin, response, relapse, PNH, clonal evolution
Autoři:
M. Suková 1; E. Mejstříková 2; V. Campr 3; E. Vodičková 4; P. Smíšek 1; P. Keslová 1; P. Sedláček 1; L. Šrámková 1; V. Vávra 1; E. Pindurová 1; Z. Zemanová 5; J. Starý 1
Působiště autorů:
Klinika dětské hematologie a onkologie, 2. lékařská fakulta UK a FN v Motole, Praha
1; Laboratoř průtokové cytometrie CLIP, 2. lékařská fakulta UK a FN v Motole, Praha
2; Ústav lékařské patologie, 2. lékařská fakulta UK a FN v Motole, Praha
3; Oddělení klinické hematologie FN v Motole, Praha
4; Laboratoř nádorové cytogenetiky, 1. lékařská fakulta UK, Praha
5
Vyšlo v časopise:
Transfuze Hematol. dnes,18, 2012, No. 3, p. 112-123.
Kategorie:
Souhrnné práce, původní práce, kazuistiky
Souhrn
Získaná aplastická anémie (AA) je onemocnění, v jehož patogeneze hrají klíčovou roli autoagresivní mechanismy. Pro pacienty, kteří nenaleznou dárce pro prioritní transplantaci kostní dřeně od HLA identického sourozence, je kombinovaná léčba antithymocytárním globulinem (ATG) a cyklosporinem A (CsA) léčebnou alternativou první volby. Limitující je dosažení léčebné odpovědi jen v 60–70 % případů, cca 30 % výskyt relapsů a 10–20 % riziko klonální evoluce: paroxysmální noční hemoglobinurie (PNH) myelodysplastický syndrom, akutní myeloidní leukemie (MDS/ADL). Cílem naší práce je zhodnocení dlouhodobých výsledků léčby aplastické anémie u dětí se zaměřením na účinnost a komplikace imunosupresivní léčby (IST). V období 1996-2009 bylo v ČR diagnostikováno celkem 72 dětí s aplastickou anémií ve věku 1,6–17,9 (medián 10,5) roku, z nichž 18 bylo primárně indikováno k MSD-SCT, 54 podstoupilo imunosupresivní léčbu. Ve skupině léčených imunosupresí (IST) bylo dosaženo léčebné odpovědi u 76 %pacientů, kumulativní výskyt relapsů byl 22 % a pravděpodobnost odpovědi na opakovanou léčbu ATG/CsA – 75 %. Celkové přežití (7letý OS) ve skupimě IST (92 ± 3,8 %) je srovnatelné se skupinou primárně transplantovaných (96 ± 3,5 %). Dlouhodobé přežití bez události (7letý EFS) ve skupině IST (58 ± 7,1 %) je významně horší než ve skupině MSD-SCT (92,3 ± 7,4 %). Transplantace kmenových buněk krvetvorby od alternativního dárce jako záchranná léčba při primárním selhání IST nebo neúspěšné léčbě relapsu je v našem souboru efektivním postupem s 5letým přežitím 80 %. Během dlouhodobého sledování nebylo ve skupině IST zaznamenáno žádné klonální onemocnění typu MDS/AML. U 7 z 37 monitorovaných pacientů (19 %) byl zachycen klonální vývoj do PNH. U 5 z nich byla PNH symptomatická a to včetně hemolytické/trombotické formy netypické pro PNH/AA syndrom. Kumulativní incidence PNH po imunosupresivní léčbě je v našem souboru 2,2 % (2 roky), 4,6 % (4 roky) 7,2 % (6 let) a 19,8 % (10 let od jejího zahájení). Naše zkušenosti potvrzují význam ATG v první linii léčby dětské aplastické anémie, umožňující při nízké toxicitě dosažení léčebné odpovědi u tří čtvrtin pacientů. Z dlouhodobého pohledu není vážnějším rizikem výskyt relapsů, ale oproti předpokladům významné riziko klonálního vývoje PNH včetně symptomatické hemolytické i trombotické formy.
Klíčová slova:
aplastická anémie, imunosupresivní léčba, antithymocytární globulin, léčebná odpověď, relaps, PNH, klonální evoluce
Úvod
Získaná aplastická anémie (AA) je vzácné onemocnění kmenové hemopoetické buňky. V patogeneze hraje klíčovou roli autoagresivní působení oligoklonálně expandujících cytotoxických T lymfocytů namířené vůči CD34+ hematopoetickým prekurzorům, porucha opravy DNA a selhání funkce mikroprostředí kostní dřeně (1). V dětském věku se jedná o velmi vzácné onemocnění s incidencí 2/1 000 000/rok. Stejně jako u dospělých se v etiologii může uplatnit virová infekce (hepatitida) nebo toxické látky, většina případů přesto zůstává idiopatických. Diagnostika aplastické anémie je založena na průkazu periferní pancytopenie a hypoplazie kostní dřeně v trepanobiopsii. Podmínkou pro stanovení diagnózy je vyloučení vrozených selhání kostní dřeně (jmenovitě Fanconiho anémie, Shwachman-Diamondova syndromu a Dyskeratosis congenita) a klonálních onemocnění typu myelodysplastického syndromu (MDS). Vývoj útlumu kostní dřeně v čase a diskrétní morfologické rozdíly ve srovnání s refrakterní cytopenií (RCC) činí odlišení aplastické anémie a hypoplastické formy MDS často velmi obtížné, což se odráží v požadavku na opakované a v referenční laboratoři hodnocené morfologické a cytogenetické analýzy. Dříve nepříznivý osud dětských pacientů s aplastickou anémií (pravděpodobnost dlouhodobého přežití 32 %) (2) se zásadně změnil počátkem 90. let minulého století se zavedením transplantace kmenových buněk krvetvorby a kombinované imunosupresivní léčby (IST) jako léčebných modalit. I v současné době zůstává zlatým standardem pro dosažení plné rekonstituce krvetvorby transplantace kmenových buněk krvetvorby od HLA identického sourozeneckého dárce (MSD-SCT) s více než 90% pravděpodobností vyléčení (3). Alternativou při chybění HLA kompatibilního rodinného dárce je včasné zahájení kombinované imunosupresivní léčby antithymocytárním globulinem (ATG) a cyklosporinem A, případně v kombinaci s růstovými faktory (G-CSF). Účinnost různých režimů IST byla ověřena v několika multicentrických studiích (4–6) s obdobnými výsledky – primární odpovědi je dosaženo u cca 70 % pacientů, z dlouhodobého pohledu je limitující 20–30% výskyt relapsů a riziko klonální transformace (MDS/AML) 10–20 %. Úloha zkracování telomér, respektive porušená funkce telomerázového komplexu (TERT a TERC) v hematopoetických prekurzorech je diskutována jako příčina větší „zranitelnosti“ aplastické krvetvorby vysvětlující tendenci k relapsu a klonální evoluci (MDS/AML, PNH) (11, 12, 13). Paroxysmální noční hemoglobinurie jako klonální onemocnění na podkladě získané somatické mutace PIG-A genu (Xp.22.1) je v klasické podobě charakterizována triádou příznaků: intravaskulární hemolýza s intermitentní hemoglobinurií, trombózy v atypických lokalizacích a selhání kostní dřeně. Asociace PNH a AA je dlouhodobě známá, (14). Výskyt malých klonů buněk s deficitem glycosylphosphatidylinositol kotvy (GPI-AP) v kostní dřeni při diagnóze aplastické anémie nebo vývoj PNH jako komplikace imunosupresivní léčby je popsán v různých souborech u 10–40 % dospělých pacientů (14–16) a u 30 % dětí (16–19). Možnou evoluci PNH klonu v průběhu aplastické anémie vysvětluje teorie o selektivní proliferativní výhodě poskytované PNH klonu v terénu aplastické dřeně (20). Tento stav, označovaný termínem PNH/AA syndrom, je spojen s projevy selhávání krvetvorby. Jen vzácně je nárůst klonu provázen symptomatickou hemolýzou, typickou pro klasickou hemolytickou formu PNH. Typický asymptomatický obraz PNH/AA syndromu je označovaný také jako subklinická forma PNH (15, 16, 21, 22).
V ČR je diagnostika a léčba dětských pacientů s aplastickou anémií od roku 1991 centralizována a koordinována v návaznosti na evropské projekty (EWOG-MDS a EWOG-SAA). Dosud publikované výsledky léčby v souboru českých pacientů za období 1991–1996 (23), ukazující pravděpodobnost 5letého přežití po SCT – 91 %, po IST – 82 % a 25% výskyt relapsů, jsou srovnatelné s daty uváděnými na velkých pediatrických souborech (6, 10, 24). V tomto sdělení předkládáme aktualizovanou analýzu osudu dětských pacientů léčených pro získanou aplastickou anémii v ČR v letech 1996–2009, s důrazem na zhodnocení efektu a komplikací IST. Zvláštní zaměření je cíleno na výskyt relapsů a úspěšnost jejich léčby, výskyt klonálních transformací a PNH/AA syndromu. Hodnocení je prováděno na základě retrospektivní analýzy dat a prospektivního monitorování po IST.
Pacienti a metody
Definice
Aplastická anémie je charakterizovaná periferní cytopenií minimálně ve 2 liniích (granulocyty < 0,5 x 109/l, trombocyty < 20 x 109/l, retikulocyty < 20 x 109/l) a hypocelulární kostní dření (buněčnost v biopsii pod 30 %). Podmínkou stanovení diagnózy AA je absence chromozomálních abnormit a vyloučení vrozených selhání krvetvorby a MDS. Stratifikace podle tíže je založena na hloubce periferní neutropenie: těžká aplastická anémie (SAA) má počet granulocytů < 0,5 x 109/l, velmi těžká aplastická anémie (vSAA) < 0,2 x 109/l. Pokud nejsou splněna kritéria hlubokého útlumu podle parametrů periferního krevního obrazu, je stav označován jako středně těžká aplastická anémie (non-SAA), jež je považována za iniciální fázi selhání krvetvorby.
Vyšetření při diagnóze
U všech dětí byla provedena aspirace kostní dřeně minimálně ze dvou míst a biopsie kostní dřeně. Dřeň byla podrobena histologické, cytomorfologické a konvenční cytogenetické analýze a FISH screeningu na přítomnost monozomie nebo delece 7. a trizomie 8. chromozomu. U pacientů s klinickým podezřením na Fanconiho anémii bylo provedeno genetické vyšetření včetně testování spontánních a indukovaných chromozomálních zlomů (DEB test). Všichni pacienti byli sérologicky vyšetřeni k vyloučení infekce hepatitidou A, B, C, autoimunitní hepatitidou, cytomegalovirem a virem Epsteina-Barrové. Dynamika vývoje u non-SAA byla ověřena opakovanou biopsií kostní dřeně.
Léčebný protokol
Léčba dětských pacientů s aplastickou anémií v ČR byla ve sledovaném období vedena v souladu s postupem německo-rakousko-nizozemské skupiny (SAA-94) (5), vycházející se zkušeností s IST u dospělých pacientů (25, 26). Léčebnou alternativou první volby byla alogenní transplantace kostní dřeně od HLA identického sourozence jako dárce štěpu s použitím standardního přípravného režimu Cyklofosfamid (200 mg/kg) + rATG Fresenius (40 mg/kg) nebo imunosupresivní léčba kombinací ATG a CsA. Koňský antithymocytární globulin (hATG = ALG, Lymphoglobuline R, Genzyme) byl podáván v jednotlivé dávce 15 mg/kg, po dobu 8 následujících dnů (od roku 2003 5 dnů). Variantou při nedostupnosti koňského ATG bylo užití králičího antithymocytárního globulinu (rATG, Thymoglobuline R, Genzyme) v jednotlivé dávce 3,75 mg/kg po dobu 5 dnů. V obou případech jde o preparáty připravené ze séra zvířat imunizovaných lidskými thymocyty. ATG byl podáván v kombinaci s metylprednizolonem (MP) k prevenci sérové nemoci v dávce 2–1 mg/kg/den s redukcí k vysazení do dne 28. Cyklosporin A (CsA) byl podáván od prvního dne v dávce 5 mg/kg/den s cílovou hladinou 150–250 ng/ml. Po dobu neutropenie byly paralelně v režimu „on demand“ podávány růstové granulocyty stimulující faktory (G-CSF) v dávce 5 μg/kg/den. Celková doba léčby CsA byla závislá na dosažení léčebné odpovědi, minimální doba podávání byla 6 měsíců, maximální 24 měsíců. Vysazování CsA formou stupňovité redukce bylo vzhledem ke známému riziku relapsů velmi pomalé. Nedosažení léčebné odpovědi nebo relaps byly indikací ke změně léčby – buď opakování ALG/ATG, nebo transplantace kmenových buněk od alternativního dárce (MUD-SCT). Podpůrná léčba vyvíjející se během 15 let zkušeností zahrnovala semisterilní, nízkobakteriální režim, s prevencí infekce Pneumocystis carinii (trimetoprim 6 mg/kg/den 2 dny v týdnu) a antimykotickou profylaxí zohledňující hloubku neutropenie (itraconazol, posaconazol). Případná empirická antibakteriální léčba zohledňovala recentní kolonizace a výskyt nozokomiálních kmenů.
Hodnocení odpovědi na léčbu
Odpověď na IST byla hodnocena podle dosažených parametrů krevního obrazu a závislosti na transfuzích v časových bodech den 120, 180, 270, 365 od zahájení IST (hematologická kritéria jsou definována evropskou pracovní skupinou pro SAA). Hodnocení primární odpovědi bylo stanoveno na den 120 od zahájení imunosupresivní léčby. Kompletní odpověď (CR) předpokládá dosažení hodnot ANC > 1,5 x 109/l, trombocytů > 150 x 109/l, a hemoglobinu nad normu pro daný věk. Jako parciální odpověď bylo hodnoceno dosažení nezávislosti na transfuzích a určitý vzestup všech parametrů krevního obrazu (hemoglobin [Hgb] > 6,0 g/l, trombocyty >20 x 109/l, absolutní počet neutrofilů [ANC] >0,5 x 109/l). Za nedosažení léčebné odpovědi (nonresponse = NR) je označována trvající závislost na transfuzích a/nebo setrvalá hodnota ANC < 0,5 x 109/l. Relaps je definovaný jako recidiva potřeby transfuzí respektive pokles parametrů krevního obrazu o více než 50 % maximální dosažené hodnoty.
Pacienti
V období od 1. ledna 1996 do 31. prosince 2009 bylo v České republice diagnostikováno celkem 72 dětí se získanou AA, ve věku 1,6–17,9 roku (medián 10,5 roku), 40 chlapců a 32 dívek. Etiologicky byla aplastická anémie asociovaná se sérologicky negativní hepatitidou ve 14 případech (19 %) u ostatních 58 bylo onemocnění označeno jako idiopatické. Podle tíže onemocnění byli pacienti stratifikováni do skupin: těžká (n = 37) – 51 %, velmi těžká (n = 21) – 30 % a středně těžká AA (n = 14) – 19 %. Podle dostupnosti dárce bylo celkem 18 pacientů (25 %) primárně indikováno k MSD-SCT, 54 pacientů (75 %) bez HLA identického sourozence bylo léčeno IST. Detailní charakteristika skupin pacientů podle typu primární léčby je uvedena v tabulce 1.

Metody
V rámci celého souboru bylo hodnoceno celkové přežití, přežití bez události a srovnání obou parametrů podle typu primární léčby. Ve skupině pacientů léčených IST byl prospektivně sledován výskyt relapsů, klonálních onemocnění typu MDS/AML a PNH a úspěšnost jejich léčby. Prospektivní monitorování PNH klonu v periferní krvi je u pacientů po IST prováděno prospektivně od roku 2003. V roce 2007 byla navíc (na základě odhalení několika případů klinické PNH) provedena plošná klinická a laboratorní analýza PNH u všech dostupných nemonitorovaných pacientů po IST s cílem stanovit incidenci PNH/AA syndromu.
Laboratorní screening PNH je prováděn standardní metodou imunofenotypizace buněk periferní krve (erytrocytů, granulocytů/monocytů) panelem povrchových markerů GPI vázaných molekul (CD55, CD59, CD66c, CD14, CD16, CD24) (27). Výskyt PNH klonu v granulocytech a monocytech je navíc ověřován metodou FLAER (průkaz defektu pomocí bakteriálního toxinu se specifickou vazbou na GPI-AP kotvu). Metoda s citlivostí 10-3 dokáže zachytit a kvantifikovat PNH klon o velikosti < 1 %, včetně prognosticky významné specifikace typu I-III GPI-AP deficitních erytrocytů. Za významný klon je považován nález >1 % GPI-AP deficitních buněk v erytrocytech, granulocytech nebo monocytech, respektive kvantifikovatelný klon typu III v erytrocytech. U pacientů s klonem >5 % byla následně doplněna PCR sekvenační DNA analýza PIG-A genu (exonu 2-6) s cílem identifikace mutace (28). U všech pacientů s prokázaným signifikantním PNH klonem v granulocytech, monocytech a/nebo erytrocytech byla provedena retrospektivní klinická analýza ve smyslu symptomatické PNH a odpovědi na léčbu AA. Nález významného PNH klonu III. typu v erytrocytech a/nebo hemolytické projevy byly důvodem pro následné frekventní monitorování v periferní krvi.
Statistika
Výsledky léčby byly vyhodnoceny k 1. dubnu 2011. Data s nenominální distribucí byla vyjádřena v hodnotách mediánu. Odpověď na léčbu byla hodnocena podle hematologických kritérií den 120, den 180 a den 270 od zahájení IST, vyjádřena v procentech. Celkové přežití (OS) a přežití do události (EFS) jsou vyjádřeny jako pravděpodobnost události ve vztahu k datu diagnózy, k hodnocení byla použita metoda dle Kaplana-Meiera, s porovnáním rozdílů pomocí log-rank testu (29, 30). Jako událost pro stanovení celkového přežití (OS) bylo hodnoceno úmrtí z jakékoli příčiny. Pro stanovení přežití do události (EFS) úmrtí, nedosažení odpovědi, transplantace od alternativního dárce, relaps onemocnění, klonální transformace (MDS, AML), symptomatická PNH a sekundární malignita. Censorem pro dobu sledování byla smrt, transplantace od alternativního dárce a ztráta ze sledování. Hodnocení vzniku PNH v souvislosti s imunosupresivní léčbou (PNH/AA syndrom) bylo založeno na průkazu vzniku nového nebo symptomatické progrese původního PNH klonu. Pravděpodobnost výskytu PNH/AA syndromu v časovém vztahu k zahájení imunosupresivní léčby, je vyjádřena jako kumulativní incidence za přítomnosti konkurujících rizik, stanovena pomocí programu XLSTAT Version 2012.3.01 Addinsoft SARL (31).
Výsledky
Imunosupresivní léčba - léčebná odpověď
Z 54 dětí primárně léčených IST bylo léčebné odpovědi (hodnocené jako zlepšení parametrů periferního krevního obrazu a celularity kostní dřeně) den 120 od zahájení ALG dosaženo u 41 (76 %), z toho u 11 byla odpověď kompletní (CR - 20 %). 11 dětí nedosáhlo léčebné odpovědi den 120 (NR - 20 %). Odpověď den 180 byla hodnocena u 48 pacientů pokračujících v IST: léčebné odpovědi dosáhlo 40 z nich (85 %), 13 (28 %) mělo odpověď kompletní, 27 (57 %) parciální. 5 pacientů (11 %) zůstalo bez odpovědi v den 180 a 2 (4 %) prodělali časný relaps (podrobněji tab. 2). Z šesti dětí léčených druhou kúrou ATG tři zůstaly bez odpovědi a byly transplantovány od alternativního dárce a žijí v kompletní remisi, tři dosáhly postupně plné léčebné odpovědi, u jednoho z nich se ale v delším odstupu manifestovala PNH. U jednoho pacienta, který z důvodu absence vhodného dárce pro transplantaci pokračoval v prologované léčbě CsA, bylo zaznamenáno dosažení pozdní odpovědi po dni 180. Z primárně léčených IST zemřeli celkem 4 pacienti (7 %). 2 z nich na infekční komplikace do dne 100 (sepse Pseudomonas aeruginosa, respektive smíšená bakteriální a aspergilová infekce), 2 na komplikace při záchranné transplantaci od alternativního dárce. Detailní vyhodnocení léčebné odpovědi shrnuje schéma 1.

Relaps po imunosupresivní léčbě
Celkem 9 pacientů (22 % z responderů) prodělalo relaps po imunosupresivní léčbě v odstupu 5–32 měsíců (medián 19 měsíců) od diagnózy. Relaps byl zaznamenán ve 4 případech časně, 2x ještě na léčbě CsA a 2x v intervalu do 6 měsíců po jeho vysazení. Vznik relapsu se nezdál být v souvislosti s předchozí dosaženou odpovědí (6 pacientů původně dosáhlo parciální, 3 kompletní remise). Opakovaná IST (u 4 dětí kombinace ATG a CsA, u 4 pouze CsA) vedla k léčebné odpovědi u 6 pacientů (75 %) – 3 dosáhli druhé kompletní remise, 3 prodělali druhý relaps, který byl u dvou úspěšně léčen CsA. Recidivující průběh aplastické anémie byl příčinou dlouhodobé léčby CsA (45 a 47 měsíců) u 2 pacientů a byl spojen s PNH klonální evolucí. V současné době při mediánu sledování 10,2 roku žije 8 z 9 původně relabujících pacientů, 6 v kompletní remisi (4 z nich po MUD-SCT), 2 v parciální remisi na léčbě CsA. 2 pacienti s PNH manifestující se bez souvislosti s relapsem jsou léčeni kombinací kortikoidů a CsA (tab. 3).
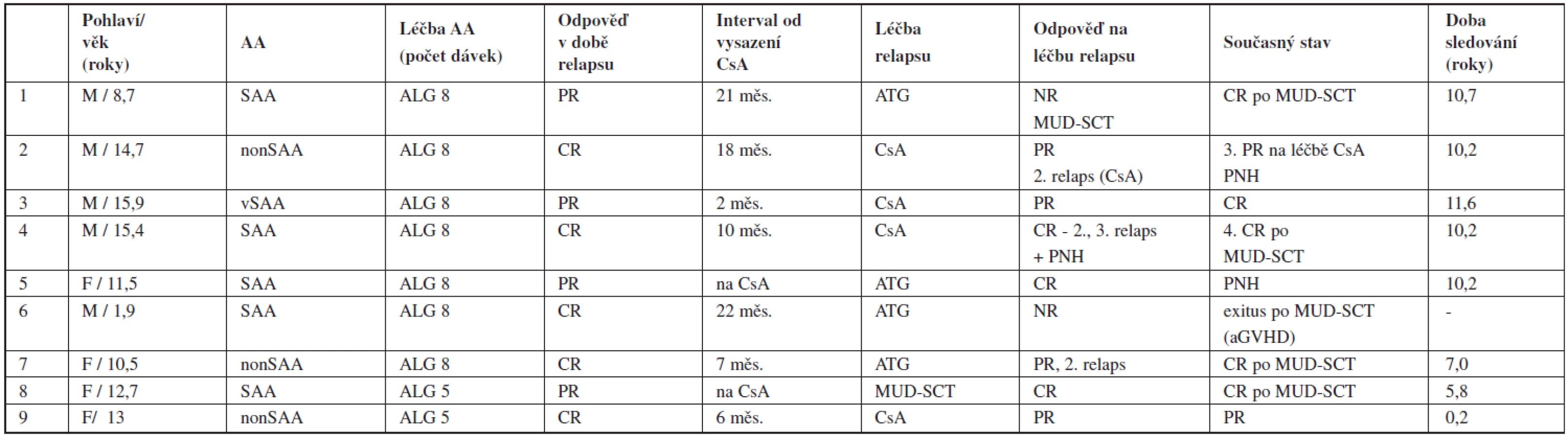
Osud pacientů při selhání imunosupresivní léčby
K záchranné MUD-SCT bylo indikováno 8 pacientů s absencí primární léčebné odpovědi (čtyři po 1. kúře ATG, tři po 2. kúře ATG a jeden pro postupné vyhasnutí parciální odpovědi) a čtyři pacienti s relapsem a selháním opakované IST. Jako štěp byla ve všech případech (n = 12) využita dárcovská kostní dřeň. Myeloablativní přípravné režimy byly převážně (n = 8) založeny na kombinaci cyklofosfamidu, ATG a celotělového ozáření (TBI), které bylo postupně redukováno z vysokých dávek 10–14,4 Gy na 6 Gy. U tří pacientů transplantovaných od roku 2008 byl jako příprava k MUD-SCT použit režim s redukovanou intenzitou (fludarabin + cyklofosfamid nebo fludarabin + thiotepa + threosulfan). Detailní analýza předtransplantačních režimů, komplikací a výsledků transplantace je uvedena v článku Pindurové, et al. (32). Celková úspěšnost záchranné léčby po selhání IST formou transplantace od alternativního dárce představuje v naší skupině 5letý OS 80 ± 12,6 % a 5letý EFS 70 ± 14,5 %.
Celkové přežití a přežití bez události – srovnání účinnosti IST a SCT
V celé skupině dětí léčených pro aplastickou anémii je při mediánu sledování 7 let dlouhodobé přežití 92 ± 3,4 %, přežití bez události 67 ± 5,8 %. Ve skupině léčených imunosupresí je pravděpodobnost dosažení primární léčebné odpovědi 76 %, dlouhodobé přežití (7letý OS) je 92 ± 3,8 %, přežití bez události (7letý EFS) 58 ± 7,1 % (graf 1). U žádného pacienta léčeného imunosupresí nebyl zaznamenán výskyt klonální transformace typu MDS/AML ani vznik chromozomální aberace. Léčebná odpověď po MSD-SCT, respektive přihojení štěpu s rekonstitucí krvetvorby je v našem souboru 100 %. Při nulovém výskytu úmrtí v souvislosti s transplantací (TRM) a nulovém výskytu relapsů je 7letý EFS ve skupině MSD-SCT 92,3 ± 7,4 % a 7letý OS 96 ± 3,5 % (32). Srovnání dlouhodobé účinnosti léčby u primárně léčených IST a transplantovaných od HLA-identického sourozence ukazuje graf 2. Přežití bez události je statisticky významně horší ve skupině IST (p = 0,0054).


Klonální evoluce po imunosupresivní léčbě, PNH/AA syndrom
Standardní screening PNH klonu metodou průtokové cytometrie byl při diagnóze proveden u 38/72 pacientů, z toho u 26 v IST skupině. Významný PNH klon byl iniciálně zachycen pouze u jedné pacientky. Klon s velikostí 5 % v erytrocytech a 8 % v granulocytech zůstal stabilní v průběhu IST, při dosažení kompletní odpovědi i při následném relapsu. Celkem 28 pacientů bylo monitorováno na vývoj PNH klonu po IST (24 a při diagnóze + 4 původně nevyšetření). Z 27 původně negativních byla evoluce klonu zachycena u 2 (7 %), v intervalu 24 a 108 měsíců po podání ATG. Nález PNH klonu o velikosti 40 % a 22 %, v granulocytech respektive 10 % a 3 % v erytrocytech periferní krve byl provázen subklinickou hemolýzou a určitým poklesem parametrů krevního obrazu při normální buněčnosti kostní dřeně. Kromě toho byl vznik PNH/AA syndromu zachycen u dalších pěti původně nevyšetřených pacientů. U tří z nich s velikostí klonu > 50 % byla PNH symptomatická, provázená hemolytickými/trombotickými projevy (Budd-Chiari syndrom, recidivující těžká hemolýza, refrakterní pancytopenie) zjištěnými 32, 60 a 134 měsíců od diagnózy AA v kompletní nebo parciální remisi. Další 2 případy asymptomatické PHN byly zachyceny na základě plošné analýzy provedené v IST skupině v roce 2007. Podrobná charakteristika jednotlivých případů PNH/AA syndromu je uvedena v tabulce 4. PCR sekvenační analýza PIG-A genu byla provedena u šesti pacientů, u pěti z nich byla potvrzena kauzální somatická mutace asociovaná s PNH. Výskyt PNH po IST je v našem souboru 19 %. V 5 případech (14 %) se jednalo o obraz PNH s klinickými a laboratorními známkami hemolýzy, eventuálně trombózy, pouze ve 2 případech o klasickou subklinickou formu PNH. Pravděpodobnost vzniku PNH/AA syndromu po IST vyjádřená jako kumulativní incidence je v našem souboru 2,2 % (2 roky), 4,6 % (4 roky) 7,2 % (6 let) a 19,8 % (10 let) od jejího zahájení (graf 3).



Shrnutí léčebných výsledků IST
Z celé skupiny 54 dětí léčených IST žije v současné době 51, bez léčby 45, z toho 40 v kompletní remisi (10 z nich po MUD-SCT) a 5 v parciální remisi. Sedm z těchto dětí je ztraceno ze sledování. Šest pacientů je dosud, z důvodu relabujícího průběhu nebo PNH, léčeno CsA v kontinuální délce podávání 23–48 měsíců. Kvalita života všech pacientů po ukončení IST je velmi dobrá. Dosud se, kromě sekundárního NHL u pacientky po komplikované MUD-SCT, nevyskytly žádné pozdní následky léčby.
Diskuse
První podmínkou úspěšné IST léčby AA je rychlá a přesná diagnostika, respektive vyloučení vrozených selhání kostní dřeně a hypoplastického MDS - refrakterní cytopenie (RCC), které v dětském věku obraz AA mohou imitovat. Zatímco evropská spolupráce má v diagnostice a léčbě MDS u dětí tradici od první poloviny devadesátých let, kdy byla založena evropská pracovní skupina EWOG-MDS, mezinárodní studie diagnostiky a léčby AA (EWOG-SAA) byla zahájena až v roce 2011. Jednotný systém referenčních cytomorfologických, cytogenetických a výzkumných laboratoří zabývajících se současně MDS i SAA povede k hlubšímu poznání etiologie a patogeneze aplastické anémie a refrakterní cytopenie. V rámci studie EWOG-SAA jsou doporučeny před zahájením léčby dvě biopsie kostní dřeně v intervalu 2 týdnů stejně jako je tomu i u refrakterní cytopenie. Tento přístup mírně oddálí zahájení IST či transplantace, přispěje ale k lepšímu odlišení AA a RCC. Není vyloučeno, že někteří pacienti se středně těžkou anémií v našem souboru by za dnešního stavu poznání mohli být diagnostikováni jako RCC (WHO klasifikace 2008) s nižší pravděpodobností odpovědi na IST (26). Rutinní cytogenetické vyšetření a FISH screening při diagnóze jistě přispěly k odlišení hypoplastického MDS s nálezem klonálních změn, kterými jsou v dětském věku prakticky výlučně monozomie 7 a/nebo nebo trizomie 8.
Prvotní zkušenosti s léčbou AA v éře MSD-SCT a IST v České republice jsou shrnuty v článku v Čs Pediatrii 1998 (23), potvrzujícím vysokou úspěšnost obou léčebných postupů (OS MSD-SCT: 91 %, OS IST: 82 %) jako výsledek časného zahájení léčby a centralizace pacientů. Naše práce hodnotí výsledky léčby dětské AA od roku 1996, v následném 14letém období stabilně nastaveného léčebného postupu, za postupně se zdokonalující transplantační a podpůrné péče. Ve srovnání s historickými soubory je rychlost diagnostiky, vyjádřená jako interval k zahájení léčby, srovnatelná (tab. 1). MSD-SCT je nadále vzhledem k nízké toxicitě a vysoké pravděpodobnosti vyléčení preferována jako léčba první volby. V našem souboru mělo HLA identického sourozence 18 (25 %) pacientů, z nich až na jedno dítě, které zemřelo tragicky na následky dopravní nehody, ostatní žijí, vyléčeni, nikdo neprodělal relaps nemoci. Karnofského/Lanského skóre pacientů je 100 %. Až na jednoho pacienta nikdo netrpí extenzivní chronickou reakcí štěpu proti hostiteli. Alternativou pro pacienty postrádající HLA identického sourozence je IST. Účinnost Lymphoglobulinu byla ověřena v četných studiích i u dětských pacientů s aplastickou anémií, alternativní použití Thymoglobulinu při další nedostupnosti ALG je podloženo studiemi u dospělých pacientů s nízce rizikovým MDS (26). IST v našem souboru podstoupilo 54 dětí. Významné snížení časné mortality (4 %) ve srovnání s historickým souborem z 80. let (32 %) (2) je výsledkem standardního užití G-CSF v časné fázi neutropenie, umožňující zkrácení fáze agranulocytózy. Na dalším zlepšení ve srovnání se souborem z 90. let (9 %) (23) se nepochybně podílí zlepšující se podpůrná péče. Recentně publikovaná randomizovaná studie evropské pracovní skupiny pro AA EBMT srovnávající IST léčbu s a bez současného podání G-CSF neprokázala rozdíl v EFS i OS v obou skupinách (v celém souboru zastoupeno 16 % dětí). Podání G-CSF snížilo výskyt časných infekcí a zkrátilo dobu hospitalizace (33). Současným doporučením v rámci studie EWOG-SAA je nepodávat G-CSF déle než 60 dní i při chybějícím vzestupu neutrofilů. Důvodem je zvýšené riziko vývoje klonálního onemocnění (MDS, AML) s aberacemi 7. chromozomu spojené s dlouhodobou léčbou G-CSF (9, 34). Klinická pozorování jsou podložena hypotézou o expanzi preexistujících malých klonů s monozomií 7 pod „tlakem“ G-CSF (35). Významný podíl na vzniku této komplikace má jistě ale i dlouhodobá imunosuprese. Sami jsme G-CSF podávali intermitentně v závislosti na periferním krevním obrazu s cílem držet absolutní počet neutrofilů nad 0,5 x 109/l. Délka léčby byla individuální v závislosti na léčebné odpovědi a nepřesahovala u žádného pacienta 120 dnů. Tato relativně krátká doba podávání G-CSF spolu s rutinně prováděným cytogenetickým vyšetřením při diagnóze a centralizovanou cytomorfologickou diagnostikou může být vysvětlením skutečnosti, že se v našem souboru i při dostatečně dlouhé době sledování nevyskytl žádný případ MDS či AML.
Výsledky hodnotící primární odpověď na IST (76 %), dlouhodobé přežití (7letý OS – 92 %) a přežití bez události (7letý EFS – 58 %) jsou jasně lepší ve srovnání se situací v 80. letech (dosažení dočasné odpovědi u 15/22 pacientů, OS 32 %) (2) a jsou srovnatelné s velkými zahraničními pediatrickými soubory (6, 10, 36). Ale zdá se, že IST kombinací ATG+CsA dosáhla svého maxima. Výskyt non-responderů na tuto léčbu je 30 %. V současné době panují obavy, že se výsledky IST naopak mohou zhoršovat. Po ukončení výroby koňského Lymphoglobulinu (Genzyme) jsou v Evropě všichni pacienti léčeni králičím ATG (Thymoglobulin, Genzyme). Předběžné výsledky této léčby vykazují zhoršení léčebných odpovědí i přežití ve srovnání s léčbou koňským ALG (37). Za prediktivní faktory léčebné odpovědi se považují absolutní počet retikulocytů před léčbou ≥ 25x109/l, nález malého PNH klonu a dětský věk (38). Naopak nedostatečný vzestup granulocytů (< 0,5x109/l) při podání G-GSF 30 dní od zahájení IST je nepříznivým prognostickým faktorem (33). Klíčovou roli v odpovědi na IST tak hraje reziduální funkce kostní dřeně (počet zbylých kmenových buněk).
Pravděpodobnost dosažení opožděné odpovědi u nonresponderů den 120 je při pokračování léčby cyklosporinem A 10–20 %. V našem souboru takto reagovalo 8 % dětí, v souboru Führerové 13 % pacientů (8). Protože se nedaří tuto malou skupinu pozdních responderů na IST predikovat, byla v našem souboru při nedosažení léčebné odpovědi 120 dní od začátku IST podána 2. kúra ATG. 50 % dětí (3 ze 6) na tuto léčbu odpovědělo a žije v remisi, i když jeden z pacientů pozdně vyvinul PNH.
Vzhledem ke zlepšujícím se výsledkům transplantace od nepříbuzného dárce (díky optimalizaci vyhledávání dárců při aplikaci molekulárních metod do HLA analýz a snížení toxicity předtransplantační přípravy zařazením fludarabinu a redukcí celotělového ozáření) je v posledních letech MUD-SCT od HLA identického alternativního dárce metodou první volby při nedosažení léčebné odpovědi den 120. Bacigalupo dosahuje při tomto postupu 5letého přežití 92 % v souboru transplantovaných po roce 2004 (39). V našem souboru byli jako nonrespondeři den 120 transplantováni 4 pacienti, z nichž 3 žijí v kompletní remisi, jeden zemřel na komplikace v souvislosti s transplantací. V celém souboru transplantovaných od alternativního dárce (n = 12) je úspěšnost přihojení štěpu (HLA shoda 8/-10/10) 100 % s jedním případem časné rejekce, 5letý OS je 80 % a 5letý EFS 70 %. Smrt na transplantační komplikace dosahuje 18 % (32). Podmínkou zařazení MUD-SCT do druhé linie léčby je zahájení hledání dárce v registrech ihned po zahájení IST. Není-li nalezen HLA identický dárce (shoda 9-10/10), je u nonresponderů léčebnou metodou volby druhá kúra ATG, pokračování v léčbě CsA s nadějí na pozdní odpověď.
V souladu s publikovanými daty (5, 6) je i v našem souboru srovnatelné dlouhodobé přežití ve skupině primárně transplantovaných od MSD (7letý OS 96 ± 3,5 %) a primárně léčených IST (7letý OS 92 ± 3,8 %). Nepříznivou informaci Frickhofena o dlouhodobých výsledcích IST léčby u dospělých pacientů (10leté přežití 60 %) zmírnila Monica Führer analýzou dlouhodobých výsledků SAA-94 studie. Deset let od diagnózy žilo 81 % dětí léčených IST a 89 % pacientů léčených MSD-SCT (ústní sdělení). V našem souboru žije deset let od diagnózy 88 % dětí po imunosupresivní léčbě a 93 % dětí po MSD-SCT. Přežití bez události (7letý EFS) je ve skupině IST (58 ± 7,1 %) signifikantně horší než ve skupině MSD-SCT 92,3 ± 7,4 %) (p = 0,0054) (graf 3). Hlavní podíl na horších dlouhodobých výsledcích ve skupině IST má výskyt relapsů (22 %), který je ale nižší než v historicky uváděné skupině do roku 1996 (34 %) (23). Vysvětlením je již výše zmiňovaná časnější indikce k MUD-SCT při selhávání léčby CsA. Opakování IST v případě relapsu (2. cyklus ATG nebo pouze CsA podle stupně a dynamiky nástupu selhání dřeně) vedlo v našem souboru k alespoň určité léčebné odpovědi u tří čtvrtin pacientů, což potvrzuje opodstatněnost tohoto postupu ve snaze navození druhé remise (40). Literárně udávaný prognostický význam předchozí délky léčby CsA pro vznik relapsu (10, 24) v našem souboru nebyl potvrzen: délka původní léčby CsA ve skupině relabujících má medián 9 měsíců (7–18) měsíců, ve skupině nerelabujících: 11 měsíců (6–27 měsíců), výsledek je pravděpodobně ovlivněn malým počtem pacientů. V souladu s literárními údaji třetina relabujících pacientů prodělá recidivy opakovaně, zřejmě jako důsledek imunosupresí nepostižitelného poškození mikroprostředí stromatu kostní dřeně (1). Opakované relapsy jsou přesto úspěšně ovlivnitelné IST s rizikem vzniku závislosti na CsA v 15–25 % (10, 24). V naší skupině responderů na imunosupresivní léčbu jsme zaznamenali závislost na CsA (podávání déle než 36 měsíců) pouze u 2 pacientů (5 %). Opakovaně relabující průběh AA je v našem souboru asociován s vývojem PNH klonu, nicméně vzhledem k absenci diagnostického vyšetření PNH klonu je prediktivní význam tohoto vztahu nehodnotitelný. Stejně tak, vzhledem k malému počtu pacientů není validně proveditelná ani prediktivní analýza dalších parametrů vzhledem k relapsu. Selhání IST v léčbě relapsu je indikací k MUD-SCT, která v našich podmínkách má 75% pravděpodobnost dosažení trvalé remise.
PNH jako pozdní komplikace IST (PNH/AA syndrom) je v souborech dospělých pacientů dokumentována v 10–40 % případů. V detailně hodnocených souborech je zdůrazňována častá spontánní regrese preexistujícího klonu během IST, evoluce klonu cca u 20 % pacientů a typická subklinická forma PNH. Progrese PNH klonu je méně obvyklá (15–25 %) výskyt symptomatické hemolytické/trombotické PNH asociované s velikostí klonu > 50 % je vzácný (16, 17, 21). U dětí je dosud publikováno pouze několik desítek případů PNH, převážně formou kazuistik nebo malých souborů (17–19). Jediná retrospektivní analýza hodnotící asociaci PNH a AA na podsouboru dětských pacientů byla publikována v roce 2010 Scheinbergem (16). Z celkem 207 pacientů 40 % mělo prokazatelný PNH klon při diagnóze, který u 25 % progredoval v průběhu IST, ale jen u 7 (3 %) do symptomatické formy vyžadující léčbu PNH. Ze 47 dětí mělo 14 (31 %) preexistující PNH klon s mediánem velikosti 16 %, který u všech v průběhu IST regredoval, nevyskytl se ani jeden případ symptomatické PNH. Scheinberg uzavírá, že výskyt ani průběh PNH asociované s AA se u dětí neliší od dospělých, hemolytické a trombotické projevy jsou považovány za raritní. V doporučeních pro diagnostiku AA se objevuje požadavek na vyšetření a monitorování PNH klonu v posledním desetiletí, u dětských pacientů v ČR byla analýza PNH klonu systematicky zavedena spolu se zavedením moderních metodik průtokové cytometrie od roku 2003. Do té doby byli pacienti vyšetřováni pouze selektivně, ne zcela senzitivní metodou Hamova testu. Významný PNH klon (dle FC >1 %) v erytrocytech/granulocytech – byl při diagnóze získané AA zachycen pouze u 1 z 38 vyšetřených (3 %). Přesto incidence PNH/AA syndromu, respektive vývoje PNH v průběhu imunosupresivní léčby je 19 % (7/37 sledovaných pacientů). Soubor je hodnocen jednak retrospektivně (n = 37), jednak (od roku 2003) prospektivně (n = 29). Medián vzniku PNH je 9 let (2,7–11,4 roku) od zahájení IST. Na rozdíl od literárních údajů se u tří z našich pacientů (43 % ze všech s nálezem významného PNH klonu) PNH manifestovala jako symptomatická, hemolytická/trombotická forma, 2 měli projevy intermitentní hemolýzy s cytopenií a pouze 2 pacienti splňovali kritéria pro PNH/AA syndrom typické formy subklinické. Stupeň hemolytických/trombotických projevů PNH se zdá dle původních předpokladů (16, 41) korelovat s velikostí klonu. Neobvykle vysoký výskyt hemolytické/trombotické formy PNH je částečně vysvětlitelný faktem, že žádný ze symptomatických pacientů nebyl (vzhledem k datu diagnózy) primárně vyšetřen na přítomnost PNH klonu ani na jeho vývoj monitorován. Symptomatologie PNH byla prvním projevem existence již významného klonu, který se pravděpodobně vyvíjel delší dobu. Na druhou stranu, o rychlosti vývoje klonu a její asociaci s klinickými projevy PNH lze jen spekulovat, jak dokládá případ pacientů č. 2 a 5 se subklinickými projevy, kteří jsou asymptomatičtí i při trvalé přítomnosti velmi významného klonu (38 % a 40 % v granulocytech a 13 % a 22 % v erytrocytech) a případ pacientky č. 7 se symptomatickou hemoglobinurií v přítomnosti „nevýznamného“ klonu (3 % v erytrocytech). Pacienti se symptomatickou PNH byli léčeni dle současných doporučení (41, 42) vzhledem k věku převážně na pracovištích pro dospělé hematologie, většinou kombinací kortikoidů, CsA a antikoagulancií. Pacient č. 3, kde byl vývoj PNH klonu spojen s relabujícím průběhem AA a progresí do rezistentního selhání kostní dřeně, podstoupil komplikovanou MUD-SCT se selháním štěpu následovanou rekonstitucí autologní krvetvorby s překvapivou obnovou trilineární hematopoézy a vymizením PNH klonu. Naše pozorování potvrzují významné riziko vývoje PNH jako pozdní komplikace imunosupresivní léčby i u dětí. Kumulativní incidence vzniku PNH po IST se významně zvyšuje po 10. roce od jejího zahájení (graf 3). Za závažný považujeme fakt, že evoluce do obrazu PNH/AA syndromu může trvat i déle než 10 let a že nelze vyloučit ani výskyt dosud nepředpokládané závažné formy PNH. Vztah PNH/AA syndromu k původní preexistenci PNH klonu v terénu selhávající dřeně a rychlost jeho evoluce nelze zatím z důvodu malého počtu systematicky monitorovaných pacientů (n = 23) a nedostatečné doby sledování (medián 4,1 roku) spolehlivě hodnotit. Prospektivní monitorování stávající skupiny pacientů pokračuje s perspektivou přehodnocení po dosažení mediánu follow-up minimálně 5 let, což u řady pacientů bude vyžadovat spolupráci s pracovišti hematologie pro dospělé.
Závěr
Získaná AA v dětském věku je vzácné onemocnění se závažnou prognózou, v dnešní době kauzálně ovlivnitelné MSD-SCT s perspektivou dosažení dlouhodobé léčebné odpovědi u více než 95 % pacientů. Pro skupinu pacientů postrádajících HLA identického sourozeneckého dárce je alternativou první volby IST, kterou lze i dle našich zkušeností dosáhnout více než 70% primární léčebné odpovědi, dlouhodobého přežití srovnatelného s MSD-SCT a dlouhodobého přežití bez události u více než poloviny pacientů při akceptovatelné toxicitě. Selhání IST nebo relaps onemocnění jsou s vysokou úspěšností (70–80 %) ovlivnitelné transplantací od alternativního dárce, jejíž indikace se díky stále se zlepšující metodice vyhledávání dárců a snižující se morbiditě spojené s MUD-SCT při trendu ke snížení toxicity přípravných režimů dostává do časnějších stadií terapeutického plánu. Riziko vývoje klonální transformace do MDS/AML se z našeho pohledu, díky cílenému vyřazení pacientů s možnými preexistujícími chromozomálními aberacemi, nejeví jako významné. Naopak riziko vývoje PNH klonu včetně symptomatické hemolytické formy PNH i ve velmi dlouhém intervalu od IST (>10 let) vyžaduje důsledné sledování pacientů v dlouhodobé remisi a pravidelnou monitorování PNH přesahující období dětského věku. Podmínkou pro dosažení výsledků, které jsou plně srovnatelné s velkými světovými pediatrickými soubory, je centralizace diagnostiky SAA, návaznost na mezinárodní výzkumné projekty (EWOG-SAA 2010) a léčba na specializovaném pracovišti se zkušenostmi s imunosupresivní léčbou a alogenní SCT.
Seznam zkratek
AA aplastická anémie
ANC absolutní počet neutrofilů
ATG antithymocytární globulin
ALG antilymfocytární globulin
BMF selhání kostní dřeně
CsA cyklosporin A
G-CSF růstové granulocyty stimulující faktory
IST imunosupresivní léčba
MDS/AML myelodysplastický syndrom/ akutní myeloidní leukemie
NR non-response – bez odpovědi
PNH paroxysmální noční hemoglobinurie
PNH klon vyjádřeno jako % CD55,59 negativních buněk v granulocytech/erytrocytech
SAA těžká aplastická anémie
vSAA velmi těžká aplastická anémie
nonSAA středně těžká aplastická anémie
SCT alogenní transplantace kmenových buněk krvetvorby
MSD-SCT transplantace krvetvorných buněk od HLA-identického sourozence
MUD-SCT transplantace kmenových buněk od alternativního dárce
Podíl autorů na rukopisu
M. S. - napsání rukopisu, vedení databáze, léčba nemocných; E. M. – imunologická diagnostika, vyšetření PNH klonu, revize rukopisu; V. C. - histologická diagnostika; E. V. – cytologická diagnostika; P. S. – léčba nemocných; P. K. – léčba nemocných; P. S. – léčba nemocných, vedoucí transplantačního programu; L. Š. – léčba nemocných; V. V. – léčba nemocných; E. P. – léčba nemocných; Z. Z. – cytogenetická diagnostika; J. S. – vedoucí pracoviště, koordinátor studie, revize rukopisu.
Poděkování
Podmínkou dosažených příznivých výsledků je nepochybně důsledkem centralizované péče, která by nebyla možná bez velmi úzké a flexibilní spolupráce všech center v rámci Pracovní skupiny pro dětskou hematologii (PSDH) – poděkování jmenovitě patří za jednotivá centra: Brno (prof. MUDr. J. Štěrba, MUDr. J. Blatný, MUDr. O. Zapletal), České Budějovice (MUDr. Y. Jabali, MUDr. P. Timr), Hradec Králové (MUDr. J. Hak, MUDr. K. Toušovská), Olomouc (prof. MUDr. V. Mihál, doc. MUDr. D. Pospíšilová), Ostrava (MUDr. B. Blažek, MUDr. H. Ptoszková), Plzeň (MUDr. Z. Černá, MUDr. T. Votava), Ústí nad Labem (MUDr. D. Procházková). Zvláštní poděkování patří všem lékařům a sestrám hematologického oddělení a transplantační jednotky KDHO a hematologům přebírajícím péči o dospívající pacienty (jmenovitě doc. MUDr. J. Čermákovi, z Ústavu hematologie a krevní transfuze Praha a MUDr. Y. Brychtové z Interní hematoonkologické kliniky FN Brno). Za vyšetření mutací PIG-A genu u pacientů s PNH děkujeme MUDr. S. Pekové z Laboratoře DNA diagnostiky Chambon v Praze. Konečně za významnou spolupráci umožňující rychlou indikaci SCT děkujme pracovníkům NRL pro DNA diagnostiku ÚHKT (RNDr. M. Loudová, CSc., RNDr. M. Dobrovolná – HLA analýza) a pracovníkům Českého registru dárců krvetvorných buněk (MUDr. E. Ivašková, Ing. L. Kupková, Mgr. M. Kuříková) a Českého národního registru dárců dřeně (MUDr. H. Pittrová a MUDr. J. Navrátilová).
Zvláštní poděkování za zásadní podíl na statistickém zpracování dat patří Mgr. I. Janotové z KDHO.
Podpora: podpořeno projektem koncepčního rozvoje výzkumné organizace 00064203.
MUDr. Martina Suková
Klinika dětské hematologie a onkologie
2. LF UK a FN Motol
V úvalu 84
150 06 Praha 5
Doručeno do redakce: 23. 4. 2012
Přijato po recenzi: 31. 5. 2012
Zdroje
1. Young NS, Calado RT, Scheinberg P, et al. Current concepts in the patophysiology and treatment of aplastic anemia. Blood 2006; 108 : 2509-2519.
2. Hrodek O, Hyniová H, Chudomel V. Dřeňové útlumy u dětí. Čs Pediatrie 1982; 37 : 562-570.
3. Davies JK, Guinan EC. An update on the management of severe idiopathic aplastic anaemia in children. Br J Haematol 2007; 136 : 549-564.
4. Frickhofen N, Heimpel H, Kaltwasser JP, Schrezenmeier H. Antithymocyte globulin with or without cyclosporin A: 11-year follow-up of a randomized trial comparing treatments of aplastic anemia. Blood 2003; 101 : 1236-1242.
5. Fuhrer M, Burdech S, Ebell W, et al. Relapse and clonal disease in children with aplastic anemia (AA) after immunosuppressive therapy (IST): the SAA 94 experience. German/Austrian Pediatric Aplastic Anemia Working Group. Klin Pediatr 1998; 210 : 173-179.
6. Kojima S, Horine K, Inaba J, et al. Long term outcome of acquired aplastic anaemia in children: comparison between immunosuppressive therapy and bone marrow transplantation. Br J Haematol 2000; 111 : 321-328.
7. Tichelli A, Gratwohl A, Nissen C, et al. Late clonal complications in severe aplastic anemia. Leuk Lymphoma 1994; 12 : 167-175.
8. Socie G, Rosenfeld S, Frickhofen N, et al. Late clonal diseases of treated aplastic anemia. Semin Hematol 2000; 37 : 91-101.
9. Kojima S, Ohara A, Tsuchida M, et al. Risk factors for evolution of acquired aplastic anaemia into myelodysplastic syndrome and acute myeloid leukemia after immunosuppressive therapy in children. Blood 2002; 100 : 766-790.
10. Fuhrer M, Rampf U, Baumann I, et al. Immunosuppressive therapy for aplastic anemia in children: a more severe disease predicts better survival. Blood 2005; 106 : 2102-2104.
11. Lee JJ, Kook H, Chung IJ, et al. Telomere length changes in patients with aplastic anaemia. Br J Haematol 2001; 112 : 1026-1030.
12. Calado RT, Young NS. Telomere maintenance and human bone marrow failure. Blood 2008; 111 : 4446-4455.
13. Scheinberg P, Cooper JN, Sloand EM, et al. Association of telomerase length of peripheral blood leukocytes with hematopoietic relapse, malignant transformation, and survival in severe aplastic anemia. JAMA 2010; 304 : 1358-1364.
14. Young NS, Maciejewski JP, Sloand E, et al. The ralationship of aplastic anemia and PNH: Int J Hematol 2002; 76 (suppl 2): 168-172.
15. Maciejewski JP, Rivera C, Kook H, Dunn D, Young NS. Relationship between bone marrow failure syndromes and the presence of glycophosphatidyl inositol-anchored protein-deficient clonek. Br J Haematol 2001; 115 : 1015-1022.
16. Scheinberg P, Male M, Nunez O, Young NS. PNH clones in severe aplastic anemia patients treated with horse antithymocyte globuline plus cyclosporine A. Haematologica 2010; 95 : 1075-1080.
17. Ware RE, Hall SE, Rosse WF. Paroxysmal nocturnal hemoglobinuria with onset in childhood and adolescence. N Engl J Med 1991; 325 : 991-996.
18. van den Heuvel-Eibrink M, Bredius RG, Winkel MI, et al. Childhood paroxysmal nocturnal haemoglobinuria, a report of 11 cases in the Netherlands. Br J Haematol 2005; 128 : 571-577.
19. Timeus F, Crescenzi O, Saracco P, et al. PNH clones in children wth acquired aplastic anemia - a prospective single centre study. Br J Haematol 2010; 150 : 483-485.
20. Rosenfeld S, Fellmann D, Nunez O, Young NS. Antithymocyte globulin and cyclosporine for severe aplastic anemia – association between hematologic response and long-term outcome. JAMA 2003; 289 : 1130-1135.
21. Young NS. Paroxysmal nocturnal hemoglobinuria and myelodysplastic syndromes: clonal expansion of PIG-A mutant hematopoietic cells in bone marrow faiilure. Haematologica 2009; 94 : 3-7.
22. de Latour Peffault R, Mary JY, Socie G, et al. Paroxysmal nocturnal hemoglobinuria: natural history of disease subcategories. Blood 2008; 118 : 3099-3106.
23. Starý J, Sedláček P, Kobylka P, et al. Významný pokrok v léčbě získané aplastické anémie u dětí v 90. letech. Čs Pediatrie 1998; 53 : 260-266.
24. Saracco P, Quarello P, Zecca M, et al. Cyclosporine A response and dependence in children with acquired aplastic anemia - a multicentre retrospective study with long-term follow-up. Br J Haematol 2008; 140 : 197-205.
25. Young NS, Barrett AJ. The treatment of severe acquired aplastic anemia. Blood 1995; 85 : 3367-3377.
26. Stadler M, Germing U, Kliche KO, et al. A prospective, randomised, phase II study of horse antithymocyte globulin vs rabbit antithymocyte globulin as immune-modulating therapy in patiens with low-risk myelodysplastic syndromes. Leukemia 2004; 18 : 460-465.
27. Hall SE, Rosse WF. The use of monoclonal antibodies and flow cytometry in the diagnosis of paroxysmal nocturnal hemoglobinuria. Blood 1996; 87 : 5332-5340.
28. Mortazavi Y, Merk B, Macintosh J, et al. The spectrum of PIG-A gene mutations in AA/PNH: a high incidence of multiple mutations and evidence of a mutational hot spot. Blood 2003; 101 : 2833-2841.
29. Kaplan E, Meier P. Nonparametric estimation for incomplete observations. JASA 1958; 53 : 457-481.
30. Mantel N. Evaluation of survival data and two new rank order statistics arising in its consideration. Cancer Chemother Rep 1966; 50 : 163-170.
31. Gooley TA, Leisenring W, Crowley J, et al. Estimation of failure probabilities in the presence of competing risks: new representations of old estimators. Stat Ass 1999; 18 : 695-706.
32. Pindurová E, Sedláček P, Keslová P, et al. Úloha alogenní transplantace buněk krvetvorby v léčbě získané aplastické anémie u dětí – zkušenost v České republice v letech 1991–2007. Transfuze hematol dnes 2011; 17 : 122-129.
33. Tichelli A, Schrezenmeier H, Socié G, et al. A randomized controlled study in patients with newly diagnosed severe aplastic anemia receiving antithymocyte globulin (ATG), cyclosporine, with or without G-CSF: a study of the SAA Working Party of the European Group for Blood and Marrow Transplantation. Blood 2011; 117 : 4434-4441.
34. Maciejewski JP, Selleri C. Evolution of clonal cytogenetic abnormalities in aplastic anemia. Leuk Lymphoma 2004; 45 : 433-440.
35. Sloand EM, Kim S, Fuhrer M, et al. Fas-mediated apoptosis is important in regulative cell replication and death in trisomy 8 hematopoietic cells but not in cells with other cytogenetic abnormalities. Blood 2002; 100 : 4427-4432.
35. Locasciulli A, Oneto R, Bacigalupo A, et al. Outcome of patients with acquired aplastic anemia given first line bone marrow transplantation or immunosuppressive treatment in the last decade: a report from the European Group for Bone and Marrow Transplantation (EBMT). Haematologica 2007; 92 : 11-18.
36. Scheinberg P, Nunez O, Young NS. Horse versus rabbit antithymocyte globulin in acquired aplastic anemia. N Engl J Med 2011; 365 : 430-438.
37. Scheinberg P, Wu CO, Nunez O, Young NS. Predicting response to immunosuppressive therapy and survival in severe aplastic anaemia. Br J Haematol 2009; 144 : 206-216.
38. Bacigalupo A, Socie G, Lanino E, et al. Fludarabine, cyclophosphamide, antithymocyte globulin, with or without low dose total body irradiation, for alternative donor transplants in acquired severe aplastic anemia: a retrospective study from the EBMT-SAA Working Party. Haematologica 2010; 95 : 976-982.
39. Tichelli A, Passweg J, Issen C, et al. Repeated treatment with horse antithymocyte globuline for severe aplastic anemia. Br J Haematol 1998; 100 : 393-400.
40. Hill A, Richards SJ, Hillmen P. Recent developments in the understanding and management of paroxysmal nocturnal hemoglobinuria. Br J Haematol 2007; 137 : 181-192.
41. Parker Ch, Omine M, Richards S, et al. Diagnosis and management of paroxysmal nocturnal hemoglobinuria, Blood 2001; 106 : 3699-3709.
Štítky
Hematologie a transfuzní lékařství Interní lékařství OnkologieČlánek vyšel v časopise
Transfuze a hematologie dnes

2012 Číslo 3
- Srovnání vlivu omeprazolu a pantoprazolu na antiagregační účinek klopidogrelu
- Fixní kombinace kandesartan/amlodipin v terapii arteriální hypertenze
- Současné postavení a přínos sartanů v klinické praxi
- Prognostický význam hladiny natriuretických peptidů při léčbě empagliflozinem
- Hypertrofická obstrukční kardiomyopatie ve světle moderní farmakoterapie – kazuistika
Nejčtenější v tomto čísle
- Získaná aplastická anémie v dětském věku – dlouhodobé výsledky a rizika kombinované imunosupresivní léčby antithymocytárním globulinem a cyklosporinem A
- Prof. MUDr. Mikuláš Hrubiško, DrSc., 95. výročí narození
- Stanovení mimodřeňové leukemické infiltrace u dětské akutní lymfoblastické leukemie a jeho klinické využití. Přehledný článek a vlastní výsledky
- Vzácny typ intestinálneho krvácania u pacienta s kolorektálnym karcinómom na antikoagulačnej liečbe
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy