
Nitrolební hypertenze u akutního jaterního selhání a možnosti mikrodialýzy
Intracranial Hypertension in Acute Liver Failure and Microdialysis
Acute liver failure is a severe condition with a very unfavourable prognosis. One of the common complications and limiting factors for the outcome is the development of the intracranial hypertension. The etiology and the pathogenetic pathways leading to the development of this fatal complication of acute liver failure is still not completely understood. The aim of this review is to inform about the actual knowledge of pathogenesis of the intracranial hypertenson in acute liver failure. Finally, a new method of tissue metabolism monitoring – microdialysis – is shortly discussed as a perspective method in monitoring and research on acute liver failure.
Key words:
intracranial hypertension – acute liver failure – microdialysis
Autoři:
J. Pražák 1; E. Lásziková 2; O. Ryska 3; M. Ryska 4
Působiště autorů:
Klinika anesteziologie, resuscitace a intenzivní péče 2. LF UK
1; Anesteziologicko-resuscitační oddělení ÚVN Praha
2; Chirurgická klinika 1. LF UK Praha, Nemocnice na Bulovce, Praha
3; Chirurgická klinika 2. LF UK a ÚVN Praha
4
Vyšlo v časopise:
Rozhl. Chir., 2010, roč. 89, č. 10, s. 642-646.
Kategorie:
Monotematický speciál - Původní práce
Souhrn
Akutní jaterní selhání (AJS) je závažné onemocnění s nepříznivou prognózou. Jedním z hlavních limitujících faktorů přežití AJS je vznik nitrolební hypertenze, jejíž etiologie a patogeneze stále není uspokojivě vysvětlena. Autoři v předkládané přehledné práci shrnují stav aktuálního poznání patofyziologických dějů vedoucích ke vzniku nitrolební hypertenze provázející vývoj akutního jaterního selhání. V druhé části přehledu je diskutována intracerebrální mikrodialýza jako nová potencionální technika monitorace při vzniku a průběhu nitrolební hypertenze u AJS.
Klíčová slova:
nitrolební hypertenze – akutní jaterní selhání – mikrodialýza
ÚVOD
Akutní jaterní selhání (AJS) je poměrně vzácně se vyskytující avšak velmi závažné onemocnění s nepříznivou prognózou. Ať už je příčina vzniku AJS jakákoliv, dochází u takto postižených pacientů velmi záhy k elektrolytovému a metabolickému rozvratu s poruchou koagulace [1] a k postižení mnoha orgánových systémů, mnohdy pod obrazem multiorgánového selhání (Multi Organ Failure – MOF). Nástup AJS je provázen jaterní encefalopatií a mozkovým edémem, vedoucím v později k vzestupu nitrolebního tlaku (ICP – intracranial pressure), což může vést k cerebrální herniaci, která je vedle MOF nebo společně s ním hlavní příčinou smrti u pacientů s ALF (acute liver failure). Navzdory pokrokům v klinickém i experimentálním výzkumu nejsou doposud uspokojivě a vyčerpávajícím způsobem objasněny všechny patofyziologické pochody vedoucí ke vzniku intrakraniální hypertenze (ICH) u AJS. Tento přehledový článek si klade za cíl seznámit čtenáře s aktuálním stavem poznání problematiky vzniku ICH u AJS a dále stručně informovat o relativně nové metodě – intracerebrální mikrodialýze – a její možnosti při výzkumu a sledování patofyziologických dějů v průběhu AJS, včetně jejího potenciálu v monitoraci terapeutických konsekvencí stejně jako možného indikátoru časné léčebné intervence.
NITROLEBNÍ HYPERTENZE
Mozkový otok je u jaterního selhání popisován a sledován relativně krátkou dobu. Dříve byly nejčastější příčinou smrti u AJS časné důsledky insuficience hepatocytů jako vykrvácení nebo nezvladatelná sepse [2], Teprve v roce 1944 popsal Lucké změny na mozku pitvaných amerických vojáků, kteří zemřeli na hepatitidu (etiologicky podmíněnou pravděpodobně očkováním proti žluté zimnici látkou obsahující lidské sérum), a jako první vyslovil domněnku, že se může jednat o změny způsobené špatnou detoxifikační funkcí jater. První práce, publikované v sedmdesátých letech dvacátého století popisující mozkový edém a herniaci jako komplikaci AJS, byly ve své době spíše kritizovány a širší odbornou veřejností byly tyto symptomy přijaty jako komplikace AJS vyžadující intenzivní terapii až v průběhu osmdesátých let [3]. Přítomnost fatálního mozkového edému jako zásadního symptomu, respektive příčiny smrti u tohoto onemocnění popisována až teprve v relativně nedávné době, souvisí pochopitelně především s výrazným zlepšením a novými možnostmi a pokrokem intenzivní péče konce minulého století. Přestože se v posledních 20 letech – opět díky optimalizaci péče o pacienty s AJS na ICU – snížila u těchto pacientů incidence smrti mozku, stále umírá na následky intrakraniální hypertenze 25–30 % pacientů s AJS [4].
PATOFYZIOLOGIE VZNIKU NITROLEBNÍ HYPERTENZE
Normální nitrolební tlak je u dospělého člověka mezi 5 a 10 mm Hg a o intrakraniální hypertenzi, která vyžaduje terapeutický zásah, hovoříme, pokud nitrolební tlak přesáhne 20 mm Hg [5]. Nitrolební tlak nad 15 mm Hg bývá označován jako zvýšený a v některých indikacích, nikoliv však u ALF, je momentem pro zahájení symptomatické léčby. Hlavním důsledkem maligní nitrolební hypertenze u pacientů s ALF je herniace v oblasti tentorium cerebeli. Tato transtentoriální herniace může vést jednak k útlaku a ischemii v povodí arteria cerebri posterior, jednak k útlaku aquaeductus mesencephali a subarachnoidálního prostoru vedoucí ke vzniku obstrukčního hydrocefalu a také k útlaku mozkového kmene vedoucí k jeho ischemii a smrti [6]. Zvýšení nitrolebního tlaku vede ke snížení mozkového perfuzního tlaku (CPP – cerebral perfusion presure), který je definován jako rozdíl středního arteriálního tlaku – MAP (mean arterial pressure) a intrakraniálního tlaku. CPP = MAP – ICP. Byť je optimální hodnota CPP při léčbě intrakraniální hypertenze obzvlášť v oblasti problematiky traumatického postižení mozku TBI (traumatic brain insury) stále předmětem diskusí [7], je jasné, že snížení CPP a tím i průtoku krve mozkem CBB (cerebral blood flow) povede k mozkové ischemii, jejímž důsledkem je u pacientů přeživších AJS různě těžký neurologický deficit.
Obecně je vzrůst ICP důsledkem vzrůstu objemu jednoho ze tří intrakranálních kompartmentů – cerebrospinální liquor, mozková tkáň a krev – uzavřených rigidní a u normálních dospělých jedinců neroztažitelnou lebkou. Protože cerebrospinální liquor u AJS svůj objem primárně nemění, lze shrnout, že příčinou vzniku intrakraniální hypertenze u AJS je mozkový edém, a změna v regulaci průtoku krve mozkem, které vedou k cerebrální hyperemii.
MOZKOVÝ EDÉM U AJS
Na vzniku mozkového edému u AJS se podílí mechanismus pravděpodobně jak vazogenní, tak cytotoxický. Hlavní roli hraje cytotoxický edém a k postižení funkce hematoencefalické bariéry a vzniku edému na podkladě vazogenním tak dochází až sekundárně. Pro vliv primárně vazogenního edému hovoří některé experimentální práce dokládající přestup za normálních okolností hematoencefalickou bariérou neprostupných látek [8], Je však třeba konstatovat, že se jedná o modely AJS na malých laboratorních zvířatech. Naproti tomu schopnost manitolu snížit ICP u pacientů s AJS hovoří pro to, že hematoencefalická bariéra zůstává nepoškozená. Stejně tak analýza mozkové tkáně elektronovým mikroskopem u pacientů, kteří podlehli AJS, potvrdila celistvost tight-junctions v hematoencefalické bariéře a otok perivaskulárních astrocytů [9]. Zobrazení magnetickou rezonancí u pacientů s ALF také potvrzuje převážně cytotoxický podklad mozkového edému u AJS. V případě nepoškozené hematoencefalické bariéry dochází při cytotoxickém edému k přestupu vody, zodpovědné za vlastní buněčný otok, pomocí tří základních mechanismů. Jedná se o prostup difuzí skrz cytoplazmatickou membránu a kotransport s organickými a anoganickými ionty a také o přestup díky speciálním vodním kanálům – aquaporinům. Hlavní aquaporin v mozku, který patrně hraje důležitou roli při vzniku cytotoxického edému u AJS, je aquaporin-4, jenž je nejčastěji lokalizován právě na endoteliálních buňkách v mozku a astrocytárních výběžcích obklopujících mozkové kapiláry. Zajímavé je, že u myší s experimentálně navozeným deficitem pro aquaporin-4 je mozkový otok u AJS zřetelně menší [10].
ASTROCYTY
Příčina mozkového otoku je multifaktoriální a zásadní roli v patofyziologických pochodech vedoucích k jeho vzniku hraje amoniak [11] a jeho vliv na otok astrocytů. Astrocyty jsou nejpočetnějším buněčným typem v mozkové kůře a mají zásadní význam pro metabolické pochody mozku včetně udržování a regulace stavu extracelulárního prostředí. Podílejí se na vodním hospodářství a hrají klíčovou roli v detoxifikaci amoniaku i udržování normálních hladin excitačních aminokyselin včetně glutamátu. Pokud hovoříme o cytotoxickém otoku u AJS, hovoříme de facto o astrocytárním otoku.
AMONIAK U AJS
Amoniak vzniká činností enterocytů při metabolismu glutaminu, při metabolických pochodech v ledvinách a dále činností ureázy střevní flory. Je také produktem při odbourávání aminokyselin ve svalech. Vylučován je ledvinami, a je využíván k syntéze glutaminu ve svalech. Nejdůležitější úlohu v jeho metabolismu mají játra, kde je amoniak spotřebováván při syntéze močoviny. V průběhu AJS pak dochází k několikanásobnému vzestupu hladiny amoniaku v krvi, což je vysvětlováno právě poruchou syntézy močoviny v játrech. Amoniak ze střeva je s krví nesen mimo játra portosystémovým kolaterálním oběhem. Arteriální hyperamonemie (hladina amoniaku nad 50 mmol/l) vede k jeho akumulaci v mozku a v důsledku poškození astrocytů ke vzniku mozkového otoku. Přestup amoniaku přes hematoencefalickou bariéru probíhá pasivní difuzí molekul NH3 a transportem skrz kationtové kanály, které mohou být identické s kanály pro draslíkové ionty [12]. Samostatný specifický transportér pro amonné ionty obdobný jako přenašeč amonných iontů v ledvinných tubulech zatím nebyl v mozku nalezen, ale jeho přítomnost je pravděpodobná. Amoniak je sám o sobě pro neurony toxický a ovlivňuje přímo i nepřímo neurotransmisi, aktivuje excitační NMDA (N-methyl D-aspartát) receptory, a ovlivňuje i GABA (gama-amino-butyric acid) receptory [13]. Byl však popsán i jeho vliv na serotoninergní a dopaminergní transmisi [14]. Tato amoniakem podmíněná neuromodulace vede ve svém důsledku až k neuronální apoptóze.
ASTROCYTÁRNÍ OTOK
Po přestupu amoniaku přes hematoencefalickou bariéru dochází k jeho detoxifikaci reakcí s glutamátem za vzniku glutaminu. Reakce je katalyzována enzymem glutamin-synthetazou, který se nachází v cytoplazmě astrocytů. Glutamin jako konečný produkt detoxifikace amoniaku v astrocytech je elektrofyziologicky inertní a dlouhou dobu byla syntéza glutaminu vysvětlována jako efektivní obrana neuronů proti toxickým účinkům amoniaku. Opakovaně však bylo jak v klinických tak i v experimentálních studiích ukázáno, že množství glutaminu v mozku koreluje se vznikem mozkového edému [15] podmíněného astrocytárním otokem u AJS. Toto tvrzení lze poměrně jednoduše hypoteticky vysvětlit osmotickým efektem intraplazmaticky nakumulovaného glutaminu – jeho zvýšená koncentrace vede k většímu influxu molekul vody do buněk. Tomuto vysvětlení nahrává nepřímo i skutečnost, že v pokusech in vitro hyperamonemie zvyšuje astrocytární expresi aquaporinu-4 [16]. V poslední době se ale ukazuje, že situace je pravděpodobně složitější a přímý osmotický vliv glutaminu není správným vysvětlením astrocytárního otoku. V roce 2006 formulovali Albrecht a Norenberg hypotézu tzv. „Trojského koně“, podepřenou výsledky experimentálních prací [17]. Podle této hypotézy hlavní mechanismus toxického účinku glutaminu spočívá ve funkci glutaminu jako přenašeče toxického amoniaku z cytoplazmy do mitochondrií. Tento transport je navíc ještě potencován právě zvýšenou koncentrací amoniaku. Uvnitř mitochondrie je glutamin hydrolyzován fosfát aktivovanou glutamázou (PAG). Uvolněný amoniak pak v relativně malém množství mitochondriálního prostoru dosahuje vysokých koncentrací. Intramitochondriální volný amoniak tvorbou volných radikálů způsobuje oxidativní stres, což vede k alteraci permeability mitochodriální membrány – MPT (Mitochondrial Permeability Transition) – a tím k mitochondriální dysfunkci a zahájení apoptotického procesu. Tuto myšlenku potvrzují i některé nejnovější studie in vivo, dokládající korelaci sníženého oxidativního metabolismu mozkové tkáně s vyššími koncentracemi glutaminu [18].
ZVĚTŠENÍ INTRAKRANIÁLNÍHO KREVNÍHO KOMPARTMENTU U AJS
Druhou složkou podílející se na vzniku nitrolební hypertenze u AJS je změna v regulaci krevního průtoku mozkem, která ASJ provází a vede k mozkové hyperémii. Za normálních okolností je krevní průtok mozkem – CBF (Cerebral Blood Flow) – do velké míry nezávislý na změnách krevního tlaku díky velmi dobrým mechanismům autoregulace, schopným přizpůsobit i regionální krevní průtok aktuálním metabolickým potřebám jednotlivých oblastí mozkové tkáně. U jaterního selhání však přestávají tyto mechanismy fungovat [19] a dochází ke globálnímu vzrůstu CBF a hyperemii. Jedním z možných vasodilatátorů, zodpovědných za generelní mozkovou vasodilataci, by mohl být adenosin, který se v mozkové tkáni kumuluje ve stavu snížené produkce ATP v průběhu AJS [20]. Důležitější roli však pravděpodobně hraje systémová zánětlivá odpověď – SIRS, která je pravidelným průvodcem rozvoje AJS. Zánětlivá odpověď tak působením prozánětlivých mediátorů způsobuje v mozkové tkáni vazodilataci a tím přispívá ke zvětšení CBF. Synergickým působením vazodilatace a zvýšení CBF společně s výše diskutovanými mechanismy vzniku vlastního otoku mozkové tkáně tak dochází ke vzniku fatální intrakraniální hypertenze.
MIKRODIALÝZA – PRINCIP
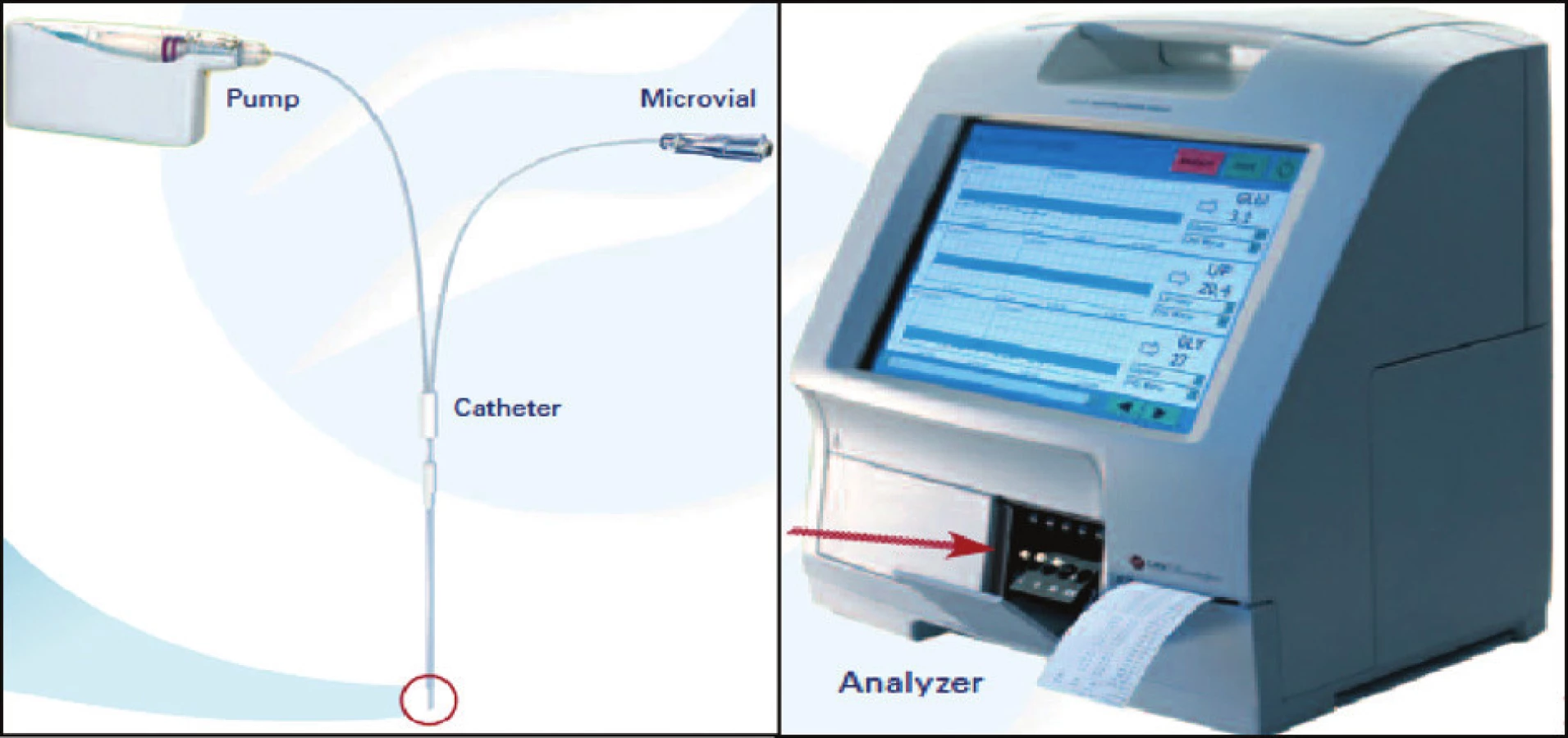
Mikrodialýza, přesněji intersticiální mikrodialýza, je jedinečná metoda umožňující téměř kontinuální monitoraci změn extracelulárního prostředí takřka všech tkání (21). Metoda funguje na principu napodobení funkce cévní kapiláry (Obr. 1).

Mikrodialyzační sonda, která má na svém konci membránu obsahující póry o definované velikosti, je zavedena jako umělá kapilára do tkáně.
Membrána mikrodializačního čidla velikostí pórů definuje skupinu látek, které mohou difundovat na základě koncentračního spádu mezi intersticiální tekutinou a mikrodialyzační tekutinou, jíž je artificiální mikrodialyzační céva promývána. Mikrodialyzační roztok je přiváděn do sondy speciální pumpou velmi pomalu, takže dochází téměř ke stoprocentnímu vyrovnání koncentrací látek v intersticiu a mikrodialyzátu, který velmi přesně odráží složení mezibuněčného prostředí v místě tkáně nebo orgánu, které je v kontaktu se zevní stranou mikrodialyzační membrány.
MIKRODIALÝZA PŘI ALF
Na klinické i experimentální úrovni je mikrodialýza uplatňována v širokém spektru indikací. Nejčastěji bývá metoda používána v oblasti neurointenzivní péče, především v problematice mozkové ischémie u cévních mozkových příhod nebo u závažných mozkových poranění [22] – obrázek 2.
Zmínky o jejím použití na poli akutního jaterního selhání, ať už v oblasti experimentální nebo v klinických studiích, se objevily teprve v několika posledních letech a její přínos jako nové metody monitorace zatím není jednoznačně prokázán. Pomineme-li studie monitorující pomocí mikrodialýzy viabilitu jaterního štěpu v případě transplantace jater u AJS, týkají se práce většinou monitorace cerebrálního metabolismu. Analýzou mikrodialyzátu byla klinicky prokázána souvislost mezi koncentrací glutaminu a koncentrací amoniaku, uvedená v první části tohoto přehledu [23]. Podobně ukazuje na výsledcích měření mikrodialýzy nedávno publikovaná práce skupiny F. S. Larsena souvislost mezi zhoršeným cerebrálním metabolismem, koncentrací glutaminu a intrakraniálním tlakem [24]. Na úrovni experimentu může sloužit mikrodialýza k doložení efektivity biologických nebo arteficiálních náhrad jater u AJS. Pro výhody analýzy mikrodialyzátu u pacientů s AJS hovoří i případová studie, dokumentující záchyt patologického snížení hladiny glukózy v mozkové tkáni bez korelace s patologickou hodnotou glykemie [25].

ZÁVĚR
Na vzniku nitrolební hypertenze má svůj podíl jednak astrocytární otok, jehož vznik souvisí s metabolismem amoniaku a glutaminu v mozkové tkáni a jednak tkáňová hyperémie, která provází vznik a vývoj akutního jaterního selhání. Otázka vzniku nitrolební hypertenze však stále není úplně zodpovězena a pro uspokojivé vysvětlení všech průvodních patofyziologických dějů je třeba dalších výsledků na všech úrovních preklinického i klinického výzkumu. Mikrodialýza, jakožto nově zaváděná efektivní metoda monitorace aktuálního stavu vnitřního prostředí konkrétních tkání organismu, by mohla být nadějnou pomůckou pro lepší pochopení metabolismu v průběhu akutního jaterního selhání. Mohla by v budoucnu sloužit jako časný marker efektivity klinické léčby tohoto závažného onemocnění.
Práce je podpořena grantem IGA MZ - NT 11463, výzkumným centrem LN00A065 a rezortní podporou S8120.
MUDr. Josef Pražák
Klinika anesteziologie, resuscitace a intenzivní péče 2. LF UK
FN Motol
V Úvalu 84
150 00 Praha 5
e-mail: jop6@centrum.cz
Zdroje
1. Detry, O., Honore, P., Meurisse, M., Jacquet, N. Management of fulminant hepatic failure. Acta Chir. Belg. 1998; 98 : 235–240.
2. Lucké, B. The pathology of fatal epidemic hepatitis. Am. J. Pathol., 1944; 20 : 471–525.
3. Ede, R. J., Gimson, A. E., Bihari, D., Williams, R. Controlled hyperventilation in the prevention of cerebral oedema in fulminant hepatic failure. J. Hepatol., 1986; 2 : 43–51.
4. Auzinger, G., Wendon, J. Intensive care management of acute liver failure. Curr. Opin. Crit. Care, 2008; 14 : 179–188.
5. Balestreri, M., Czosnyka, M., Hutchinson, P., et al. Impact of intracranial pressure and cerebral perfusion pressure on severe disability and mortality after head injury. Neurocrit. Care, 2006; 4 : 8–13.
6. Bingaman, W. E., Frank, J. I. Malignant cerebral edema and intracranial hypertension. Neurol. Clin., 1995; 13 : 479–509.
7. Grande, P. O. The ‘Lund Concept’ for the treatment of severe head trauma – physiological principles and clinical application. Intensive Care Med., 2006; 32 : 1475–1484.
8. Horowitz, M. E., Schafer, D. F., Molnar, P., Jones, E. A., Blasberg, R. G., Patlak, C. S., Waggoner, J., Fenstermacher, J. D. Increased blood-brain transfer in a rabbit model of acute liver failure. Gastroenterology, 1983; 84 : 1003–1011.
9. Kato, M., Hughes, R. D., Kraus, R. T., Williams, R. Electron microscopic study of brain capillaries in cerebral edema from fulminant hepatic failure. Hepatology, 1992; 15 : 1060–1066.
10. Manley, G. T., Binder, D. K., Papadopoulos, M. C., Verkman, A. S. New nsights into water transpot and edema in teh central nervous system from phenotype analysis of aquaporin-4 null mice. Neuroscience, 2004; 129 : 983–991.
11. Butterworth, R. F. Pathophysiology of hepatic encephalopathy: a new look at ammonia. Metab. Brain Dis., 2002; 7 : 221–227.
12. Nagaraja, T. N., Brookes, N. Intracellular acidification induced by passive and active transport of ammonium ions in astrocytes. Am. J. Physiol., 1998; 274: C883–C891.
13. Albrecht, J., Bender, A. S., Norenberg, M. D. Potassium-stimulated GABA release is a chloridedependent but sodium - and calcium-independent process in cultured astrocytes. Acta Neurobiol. Exp. (Wars), 1998; 58 : 169–175.
14. Lozeva, V., Montgomery, J. A., Tuomisto, L., Rocheleau, B., Pannunzio, M., Huet, P. M., Butterworth, R. F. Increased brain serotonin turnover correlates with the degree of shunting and hyperammonemia in rats following variable portal vein stenosis. J. Hepatol., 2004; 40 : 742–748.
15. Tofteng, F., Hauerberg, J., Hansen, B. A., Pedersen, C. B., Jorgensen, L., Larsen, F. S. Persistent arterial hyperammonemia increases the concentration of glutamine and alanine in the brain and correlates with intracranial pressure in patients with fulminant hepatic failure. J. Cereb. Blood Flow Metab., 2006; 26 : 21–27.
16. Rama Rao, K. V., Chen, M., Simard, J. M., Norenberg, M. D. Increased aquaporin-4 expression in ammonia trated cultured astrocytes. Neuroreport, 2003;14 : 2379–2382.
17. Albrecht, J., Norenberg, M. D. Glutamine: a Trojan horse in ammonia neurotoxicity. Hepatology, 2006; 44 : 788–794.
18. Bjerring, P. N., Hauerberg, J., Frederiksen, H. J., Jorgensen, L., Hansen, B. A., Tofteng, F., Larsen, F. S. Cerebral Glutamine Concentration and lactate-Pyruvate Ratio in Patients with Acute Liver Failure. Neurocrit. Care, 2008; 9 : 3–7.
19. Dethloff, T. J., Knudsen, G. M., Larsen, F. S. Cerebral blood flow autoregulation in experimental liver failure. J. Cereb. Blood Flow Metab., 2008; 28 : 916–926.
20. Bjerring, P. N., Eefsen, M., Hansen, B. A., Larsen, F. S. The brain in acute liver failure. A tortuous path from hyperammonemia to cerebral edema. Metab. Brain Dis., 2009; 24 : 5–14.
21. Ungerstedt, U. Microdialysis – principles and applications for studies in animals and man. J. Intern. Med., 1991; 230 : 365–373.
22. Goodman, J. C., Valadka, A. B., Gopinath, S. P., Uzura, M., Robertson, C. S. Extracelulare lactate and glucose alteration in the brain after head injury measured by mcrodialysis. Crit. Care Med., 1999; 27 : 1965–1973.
23. Tofteng, F., Hauerberg, J., Hansen, B. A., Pedersen, C. B., Jorgensen, L., Larsen, F. S. Persistent arterial hyperammonemia increases the concentration of glutamine and alanine in the brain and correlates with intracranial pressure in patients with fulminant hepatic failure. J. Cereb. Blood Flow Metab., 2006; 26 : 21–27.
24. Bjerring, P. N., Hauerberg, J., Frederiksen, H. J., Jorgensen, L., Hansen, B. A., Tofteng, F., Larsen, F. S. Cerebral glutamine concentration and lactate-pyruvate ratio in patients with acute liver failure. Neurocrit. Care, 2008; 9 : 3–7.
25. Hutchinson, P. J., Gimson, A., Al-Rawi, P. G., O’Connell, M. T., Czosnyka, M., Menon, D. K. Microdialysis in the management of hepatic encephalopathy. Neurocrit. Care, 2006; 5 : 202–205.
Štítky
Chirurgie všeobecná Ortopedie Urgentní medicínaČlánek vyšel v časopise
Rozhledy v chirurgii

2010 Číslo 10
- Aktuální evropská doporučení pro léčbu renální koliky v důsledku urolitiázy
- Rána vizitkou (nejen) chirurga
- Patogeneze vzniku keloidní jizvy
- Klinické studie neprokázaly vyšší účinnost obvazů s obsahem stříbra nebo medu při hojení bércových vředů
Nejčtenější v tomto čísle
- Gastrointestinální metastázy maligního melanomu
- Lokoregionální recidivy po konzervativních výkonech u časného karcinomu prsu
- Chirurgická léčba solidních nádorů v České republice
- Karcinom prsu u mužů – naše zkušenosti
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy