
Hepcidin and Host Defense against Infectious Diseases
Hepcidin is the master regulator of iron homeostasis in vertebrates. The synthesis of hepcidin is induced by systemic iron levels and by inflammatory stimuli. While the role of hepcidin in iron regulation is well established, its contribution to host defense is emerging as complex and multifaceted. In this review, we summarize the literature on the role of hepcidin as a mediator of antimicrobial immunity. Hepcidin induction during infection causes depletion of extracellular iron, which is thought to be a general defense mechanism against many infections by withholding iron from invading pathogens. Conversely, by promoting iron sequestration in macrophages, hepcidin may be detrimental to cellular defense against certain intracellular infections, although critical in vivo studies are needed to confirm this concept. It is not yet clear whether hepcidin exerts any iron-independent effects on host defenses.
Published in the journal:
. PLoS Pathog 11(8): e32767. doi:10.1371/journal.ppat.1004998
Category:
Review
doi:
https://doi.org/10.1371/journal.ppat.1004998
Summary
Hepcidin is the master regulator of iron homeostasis in vertebrates. The synthesis of hepcidin is induced by systemic iron levels and by inflammatory stimuli. While the role of hepcidin in iron regulation is well established, its contribution to host defense is emerging as complex and multifaceted. In this review, we summarize the literature on the role of hepcidin as a mediator of antimicrobial immunity. Hepcidin induction during infection causes depletion of extracellular iron, which is thought to be a general defense mechanism against many infections by withholding iron from invading pathogens. Conversely, by promoting iron sequestration in macrophages, hepcidin may be detrimental to cellular defense against certain intracellular infections, although critical in vivo studies are needed to confirm this concept. It is not yet clear whether hepcidin exerts any iron-independent effects on host defenses.
Introduction
Iron is necessary for the function of many proteins, including hemoglobin, myoglobin, and enzymes involved in oxidative phosphorylation, and is therefore essential to the survival of virtually all organisms. On the other hand, ionic iron is toxic because of its reactivity with oxygen in the so-called Fenton reaction, which generates the hydroxyl radical and other reactive oxygen species. As an element that is both essential and dangerous, iron availability is tightly regulated. The vast majority of iron is associated with proteins in biological systems, with free iron ions present in extremely low concentrations [1]. In multicellular organisms, this low availability of iron imposes a severe nutritional restriction on invading microbes, and hosts have evolved methods to further limit iron availability in the context of infection. Conversely, professional pathogens have evolved strategies to scavenge iron from the iron-binding proteins of the hosts [2]. Multiple lines of evidence suggest that this battle for iron is a critical component of antimicrobial defenses in many infections [3].
The peptide hepcidin is the master regulator of iron homeostasis in vertebrates [4–6]. Hepcidin was first described as a cationic antimicrobial peptide with microbicidal properties against many microorganisms in vitro [5,7]. Hepcidin is strongly induced during inflammation [8], and emerging data support its role in the pathogenesis of a number of infections. In this article, we review the literature on the role of hepcidin in the resistance and susceptibility to infectious diseases.
Hepcidin, inflammation, and the regulation of systemic iron availability
The global distribution of iron in mammalian hosts is depicted in Fig 1. The majority of the iron is in hemoglobin of circulating erythrocytes and bone marrow erythroid precursors and in splenic and hepatic macrophages that process senescent red blood cells. Hepatocytes serve as an important reservoir that stores or releases iron to maintain homeostasis. Extracellular iron, which constitutes a small proportion of total body iron, is transported in association with the protein transferrin. Stores of intracellular iron are maintained in association with ferritin. During steady state, little iron is absorbed from the diet or lost, with most of the iron requirement being met by recycling iron from red blood cell turnover.

The hepcidin–ferroportin axis controls both extracellular iron concentrations and total body iron levels. Ferroportin is a membrane protein that is the major exporter of iron from mammalian cells, including macrophages that recycle iron, duodenal enterocytes that absorb iron, and hepatocytes that store iron. Hepcidin limits the pool of extracellular iron by binding ferroportin and mediating its degradation, thus preventing iron release from intracellular sources (Fig 2) [9]. Sustained elevations of hepcidin result in insufficient iron availability for erythropoiesis, causing an iron-restricted anemia [10]. In contrast, inability to produce or respond to hepcidin causes hereditary hemochromatosis, a group of iron overload disorders resulting from excessive dietary iron absorption and inability to sequester iron in macrophages [4].
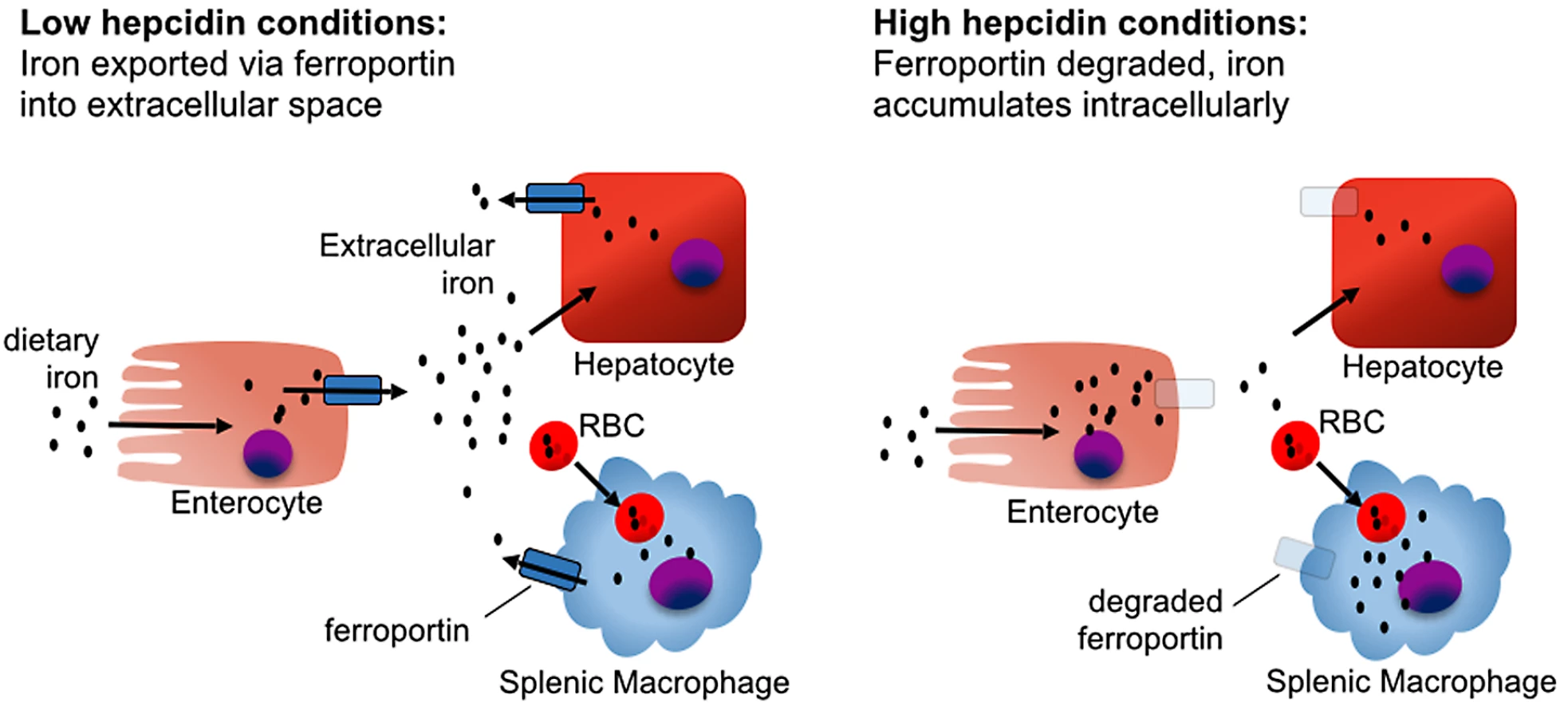
Most hepcidin is synthesized by hepatocytes. Hepatocyte hepcidin expression is stimulated by elevated extracellular or stored iron and, independently, also by inflammatory stimuli. Conversely, hepcidin expression is inhibited by hypoxia and erythropoiesis [11,12]. Regulation of hepcidin by iron depends on the hemojuvelin (HJV) and bone morphogenetic protein receptor (BMPR) complex activating the SMAD signaling pathway (Fig 3) [13–15]. The study of hepcidin regulation in mice housed under standard conditions is confounded by the high iron content of most commercial mouse chow blends. For mice, an iron-sufficient diet contains approximately 35 parts per million (ppm) iron, while commercial feed contains 150–300 ppm iron, resulting in high basal hepcidin expression [16].
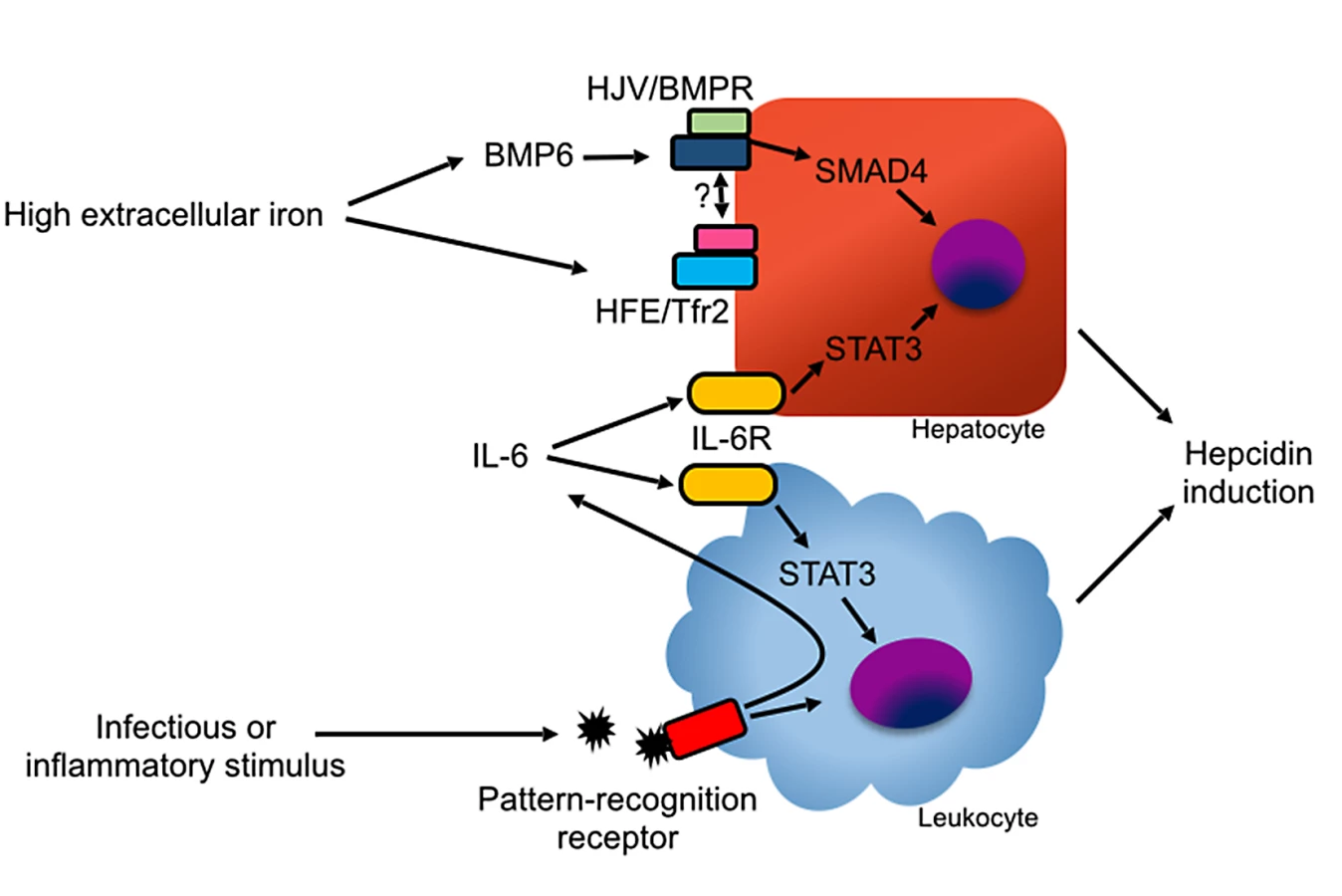
During inflammatory states, hepcidin expression is induced by the cytokine IL-6 via the Janus kinase (JAK) signal transducer and activator of transcription (STAT) 3 pathway [17]. Bone morphogenetic protein (BMP) signaling is also necessary for hepcidin induction during inflammation [18]. In animal models, diverse inflammatory and infectious stimuli, including Streptococcus species, Pseudomonas aeruginosa, Aspergillus fumigatus, influenza, turpentine, and lipopolysaccharide (LPS) robustly induce hepcidin in the liver via the induction of IL-6, resulting in rapid reduction in serum iron levels [16,19–21]. Sustained elevation of hepcidin expression, as occurs in many inflammatory states, results in anemia due to reduced availability of iron for erythropoiesis, a condition previously known as “anemia of chronic disease” and more accurately renamed as “anemia of inflammation” [16,22,23].
In addition to hepatocytes, many cell types, including myeloid leukocytes, express low levels of hepcidin [24,25]. Phagocyte hepcidin expression can be induced by autocrine and paracrine production of IL-6 and, at least in vitro, also by direct engagement of pathogen recognition receptors [24,26–29]. Hepatocyte-specific deletion of hepcidin recapitulates the hemochromatosis phenotype of global hepcidin-deficient animals, indicating that hepatocyte-derived hepcidin is necessary for iron homeostasis in steady state [30]. The role of leukocyte-derived hepcidin has not been formally examined but is hypothesized to contribute to host defense [31,32].
The Role of Hepcidin in Specific Infections
Intracellular infections
Hepcidin causes accumulation of iron within cells of the mononuclear phagocyte system, potentially benefiting pathogens that occupy this niche. Type I hemochromatosis, the most common form of hereditary hemochromatosis, is caused by loss-of-function mutations in the Hfe gene. HFE protein regulates hepcidin expression in response to increased extracellular iron. Loss of Hfe decreases hepcidin expression, increases iron absorption and extracellular iron concentrations, and releases iron from macrophages. Hfe mutations have arisen independently in several populations [33,34], and the most common mutation, C282Y, has a heterozygous prevalence of up to 10% in northern European populations [35]. Although difficult to test, it is hypothesized that Hfe mutations became prevalent by conferring a survival benefit during population bottlenecks caused by host-adapted pathogens that reside within macrophages as part of their life cycle [36–38].
Salmonellosis
Although in vitro data generally support the role of hepcidin in promoting Salmonella growth in macrophages, in vivo studies have provided conflicting results. In macrophages experimentally infected with Salmonella enterica serovar Typhimurium in vitro, hepcidin expression is induced via autocrine or paracrine mechanisms, causing intracellular iron accumulation and allowing for greater bacterial growth. In vitro infection of murine macrophages with Salmonella also resulted in lower intracellular bacterial burdens when macrophages were transfected to overexpress ferroportin [39,40]. In addition, hepcidin treatment increased bacterial burden in cells expressing wild-type, but not hepcidin-resistant, ferroportin [39]. Similarly, elicited peritoneal macrophages from Hfe-deficient mice had lower iron content and, when infected with S. Typhimurium in vitro, yielded fewer bacteria than wild-type cells [40].
In vivo, mice develop increased hepcidin levels and hypoferremia after oral infection with S. Typhimurium [41]. This induction was proposed to be detrimental to the host because prevention of hepcidin induction by an inverse agonist of estrogen-related receptor gamma was associated with improved mouse survival after S. Typhimurium infection. In a different study, however, opposite results were reported. Intravenous infection with S. Typhimurium did not increase hepcidin mRNA levels in the liver, and hepcidin-deficient mice were more susceptible to infection than their wild-type counterparts [42]. The literature on the susceptibility of Hfe-deficient mice to salmonellosis is also contradictory. Hfe-deficient mice have been reported to have attenuated intestinal inflammation but higher fecal and systemic bacterial burdens after oral infection in studies that used streptomycin-pretreated mice [43]. In other studies, Hfe-deficient mice were reported to have reduced death and bacterial burdens after intraperitoneal inoculation of S. Typhimurium that was ascribed to the greater production of lipocalin-2 in Hfe-/- macrophages [40].
The reasons for the discrepancies between in vivo studies of salmonellosis are unclear, but may relate to factors such as the age and diet of the experimental animals, which could influence the extent of iron overload at the time of infection and potentially alter the pathogenicity of bacteria. Furthermore, extrapolation of data from Hfe-deficient animals to the role of hepcidin is not straightforward, since hepcidin expression in Hfe-deficient mice is not abrogated, but only attenuated, relative to degree of iron overload [44]. In addition, there are conflicting reports as to whether Hfe influences hepcidin induction to inflammatory stimuli [21,44–46]. Finally, outcomes may be affected by variations in the extracellular portion of the bacterial life cycle, depending on the route of infection.
Mycobacterial infections
African iron overload, a condition caused by a combination of high dietary iron intake and non-Hfe hereditary hemochromatosis, is strongly associated with death from tuberculosis [47,48], potentially implicating iron homeostasis in host defense against mycobacteria. Consistent with this, macrophages from humans with type I hemochromatosis have lower iron accumulation and lower bacterial burden when infected with Mycobacterium tuberculosis as compared to cells from healthy donors; conversely, mycobacteria acquire iron more efficiently from macrophages of healthy subjects as compared to cells from patients with hemochromatosis [49,50]. These data suggest that hepcidin-mediated increase in intracellular iron may be harmful to the host during mycobacterial infections, but this hypothesis has not been addressed directly in vivo. On the other hand, Hfe-deficient mice with iron overload had increased tissue bacterial burden after intravenous infection with M. avium compared to wild-type animals [51], an unexpected result for animals predicted to have iron-depleted macrophages. As with studies in salmonellosis, however, it is difficult to extrapolate data from the Hfe mouse model to the role of hepcidin during the infection.
The direct roles of hepcidin and ferroportin in mycobacteria have only been assessed in vitro. Ferroportin overexpressing macrophages have lower mycobacterial burden as compared to normal macrophages when infected with M. tuberculosis in vitro [52]. Interestingly, ferroportin overexpressing macrophages also have reduced inducible nitric oxide synthase (iNOS) production and phagocytic ability, suggesting that depletion of intracellular iron stores may interfere with macrophage effector functions [52]. Exposure of macrophages to mycobacteria and IFN-γ synergistically induce the expression of hepcidin in vitro, and macrophage-derived hepcidin colocalizes with M. tuberculosis in coincubation studies [32,53], but the relevance of macrophage-derived hepcidin to in vivo host defense has not been evaluated. Similarly, very high concentrations of hepcidin have been shown to inhibit M. tuberculosis growth in vitro [32], but it is unknown whether these concentrations are relevant to in vivo infections.
Other intracellular pathogens
An in vitro study of murine bone marrow-derived macrophages infected with Leishmania amazonensis showed that they up-regulate hepcidin in a TLR4-dependent manner, reduce cell surface ferroportin, and accumulate intracellular iron. Macrophages from hepcidin-deficient mice had lower intracellular burden of parasites as compared to wild-type macrophages after in vitro infection, and hepcidin treatment of wild-type macrophages results in higher parasitic burdens [54]. Similar data have been published with macrophages infected with Chlamydia and Legionella species [55].
Infections caused by siderophilic bacteria
Individuals with hereditary hemochromatosis are notably susceptible to sepsis caused by specific microorganisms whose pathogenicity is augmented in the presence of free iron. These so-called siderophilic bacteria include some Vibrio and Yersinia species, and possibly other Gram-negative bacteria, including Salmonella and Escherichia species [56,57]. Susceptibility to siderophilic infections in hemochromatosis is thought to be mediated by increased availability of extracellular iron, but in observational studies it is difficult to exclude the contribution of other mechanisms, such as possible immune dysfunction as a result of iron overload and chronic liver disease [58].
Vibrio infection
Wild-type mice rapidly up-regulate hepcidin and develop hypoferremia after subcutaneous infection with Vibrio vulnificus, whereas hepcidin-deficient mice exhibited only mild hypoferremia and are much more susceptible to the infection [59]. The susceptibility of hepcidin-deficient mice was associated with high blood and tissue burden of V. vulnificus and was specifically attributable to high levels of extracellular iron, since hepcidin-deficient mice with low iron stores (but high serum iron) were also susceptible to infection. Treatment of iron-overloaded hepcidin-deficient mice with a hepcidin agonist was sufficient to restore hypoferremia (without changing iron stores) and to protect hepcidin-deficient mice from infection. Ex vivo studies of bacterial growth in sera from treated animals showed that hepcidin agonists prevented the growth of V. vulnificus by restricting iron availability rather than by direct antimicrobial activity. Hepcidin agonists did not provide any further protection from V. vulnificus infection in wild-type mice, again arguing against a direct microbicidal effect of these peptides. Overall, these studies point to hepcidin acting by lowering the concentration of iron species that are not bound to transferrin, and therefore available to V. vulnificus as nutrients and signals for rapid growth.
Sepsis
Patients presenting with sepsis syndromes have elevated hepcidin levels that fall during recovery, consistent with activation of the acute phase response during sepsis [60]. Whether hepcidin plays any role in modulating the course of sepsis is unknown, although several mouse studies suggested a protective effect. Hepcidin-deficient mice are more susceptible to death from lethal challenge with LPS as compared to wild-type controls, and administration of an unvalidated synthetic hepcidin protected both wild-type and hepcidin-deficient mice against LPS [61,62]. In murine cecal ligation and puncture model of polymicrobial intra-abdominal sepsis, mice treated intravenously with an adenovirus carrying anti-hepcidin shRNA had higher mortality and bacterial burden associated with increased serum iron levels, whereas conditioning mice on a low-iron diet or treating with an iron chelator resulted in protection following anti-hepcidin shRNA treatment [63]. Similarly, mice that received inhaled anti-hepcidin shRNA adenovirus had higher mortality and more severe lung injury after cecal ligation and puncture compared to mice that received control adenovirus [64]. These studies suggest that expression of hepcidin may be protective during sepsis caused by resident gut flora.
Malaria
In human studies, iron supplementation is associated with increased incidence and severity of malaria in some [65–67], but not all [68,69], studies, whereas dietary iron deficiency is associated with reduced malaria parasitemia and death [70–72]. Iron deficiency also promotes protection against infection with Plasmodium berghei in mice [73,74], suggesting a role for hepcidin in this infection. As expected, humans and mice infected with malaria have elevated plasma hepcidin levels, which correlate positively with parasitemia and plasma IL-6 [75–78], but patients with the most severe anemia in the context of P. falciparum infection have reduced hepcidin levels, perhaps suggesting negative feedback, such as the effect of compensatory erythropoiesis, on hepcidin expression [75]. In vitro, P. falciparum-infected erythrocytes induce hepcidin mRNA expression in human peripheral blood monocytes and monocyte-derived macrophages in an IL-6 independent but IL-10 dependent manner [79,80], but the relevance of leukocyte-derived hepcidin for host defense is unknown.
Hepcidin has complex effects in malaria [81]. On one hand, by causing iron restriction, elevated hepcidin likely contributes to anemia. On the other hand, hepcidin may have protective effects in mice during experimental malaria [77,78]. Immunoneutralization of hepcidin results in increased parasitemia and death in P. berghei infection, whereas pretreatment of animals with a hepcidin-expressing lentivirus protected against parasitemia and death as compared to mice treated with a control lentivirus [78]. Increased hepcidin protected hosts with parasitemia against a second malaria infection [77]; such super-infections are associated with increased mortality in endemic areas. Hepcidin was thought to act by causing the movement of iron from hepatocytes that could host superinfecting parasites to macrophages that cannot. In support of this mechanism, transgenic over-expression or administration of hepcidin to infected mice provided protection by reducing the burden of the parasites in the liver. These data suggest that hepcidin protects against malaria by reducing iron availability to parasites.
Viral infections
While the induction of serum hepcidin has been documented in several human and murine viral infections [19,26,82], relatively little is known about the contribution of hepcidin to pathogenesis of most viral infections. In an interesting study of serial samples, plasma hepcidin induction during early HIV infection correlated with subsequent viral load set point [83], although the mechanism underlying this correlation is unclear.
Hepatitis C
Hepatic iron accumulation is common in hepatitis C virus (HCV) infection and contributes both to liver fibrosis and to increased risk of hepatocellular carcinoma [84]. Patients with hepatitis C who have Hfe mutations are further predisposed to hepatic iron overload and worse liver fibrosis [85,86]. Although circulating hepcidin levels correlate positively with the severity of iron overload in chronic HCV infection, patients with HCV infection have a relative deficiency of hepcidin: compared to uninfected controls, HCV patients had lower hepcidin levels for given serum ferritin level, suggesting that hepcidin expression may be blunted in infected patients [87–89]. Similarly, hepcidin was not induced even during acute phases of HCV infection [83], unlike most other infections. Consistent with this, in vitro infection of a hepatocellular carcinoma cell line with HCV resulted in suppression of hepcidin transcription that was associated with higher production of reactive oxygen species [90,91]. This effect was attributable to reduced binding of the transcription factor CCAAT/enhancer-binding protein alpha (C/EBPa) to the hepcidin promoter, and both binding and hepcidin expression could be restored by treatment with antioxidants [91]. In the context of animal models, transgenic mice expressing HCV core protein develop increased serum and hepatic iron, but reduced splenic iron over time as compared to wild-type control animals [92]. This phenomenon was associated with suppressed hepcidin expression and higher ferroportin protein in liver, spleen, and duodenum. Primary hepatocytes from transgenic mice that were transfected with a hepcidin promoter and luciferase reporter construct showed lower luciferase activity compared to hepatocytes cultured from control mice, again associated with reduced binding of C/EBPa to the hepcidin promoter and increased levels of reactive oxygen species [92]. Taken together, these experiments suggest that HCV-mediated hepcidin suppression contributes to iron overload and disease pathology in HCV infection. Whether decreased hepcidin levels and consequent tissue iron loading play any role in viral replication is still unknown.
Iron-Independent Functions of Hepcidin
Some studies have proposed that hepcidin can influence immune responses independent of its role in iron homeostasis, but it remains to be demonstrated whether iron-independent functions of hepcidin have pathophysiological relevance. One study proposed that hepcidin regulates the production of cytokines and dampens inflammatory responses by activation of the Jak2 pathway [61]. However, it was subsequently shown that Jak2 is not activated by hepcidin and does not interact with ferroportin [93], challenging the proposed mechanism.
Another study showed that iron-deficient macrophages had a more pronounced inflammatory response to LPS treatment, and the inflammatory responses could be dampened with hepcidin treatment alone [94]. To confirm the role of hepcidin, mice deficient in the serine protease TMPRSS6 were used, as these animals have iron deficiency due to high constitutive expression of hepcidin [95]. Tmprss6-deficient mice demonstrated a blunted inflammatory response to LPS despite their iron-deficient status [94,96], suggesting that hepcidin may modulate inflammatory responses independently of iron. Other studies, however, reached opposite conclusions: iron-depleted macrophages from Hfe-deficient mice, which also have low hepcidin, had attenuated inflammatory response to LPS and Salmonella [43]. Thus, studies that dissect the separate effects of hepcidin and macrophage iron on inflammation are needed.
Although defensins and hepcidins are structurally distinct, the hepcidin molecule, like defensins, is an amphipathic peptide with a net cationic charge, is rich in cysteine bonds, and has a β-sheet structure [97]. Consistent with this, hepcidin has microbicidal activity against many classes of microbes in vitro, leading to the hypothesis that such a direct antimicrobial effect may be relevant in vivo during infection [5,7,98]. Serum hepcidin concentrations, however, are 1–2 orders of magnitude lower than those required for antimicrobial effects, making it unlikely that hepcidin directly kills pathogens in the bloodstream [59,98]. While local hepcidin production in infected tissue, and by leukocytes in particular, has been reported, it is unknown if local hepcidin concentrations reach antimicrobial concentrations [24,32]. There is currently little evidence in the literature that hepcidin plays a directly antimicrobial role in vivo in mammalian infections.
Conclusions and Areas for Further Study
Hepcidin is potently increased by inflammation, but the role of hepcidin in innate immunity is only beginning to be understood, as summarized in Table 1. Hepcidin restricts access to extracellular iron, and this form of “nutritional immunity” is important in at least some extracellular bacterial infections. In contrast, hepcidin induces iron accumulation in macrophages and may be detrimental in defense against pathogens that occupy this intracellular niche. This effect has been demonstrated convincingly in vitro but is not supported by in vivo data [32,39–43,49–55]. Interrogating the role of hepcidin in animal models of intracellular infections should further clarify the complex relationship between iron distribution and pathogenesis of such infections in humans.

In the absence of in vivo data, any iron-independent role of hepcidin in host defense remains speculative. In particular, there is little evidence to support a direct microbicidal role for hepcidin in mammalian infections. The current literature suggests that hepcidin may dampen inflammatory cytokines through a mechanism that is not well understood. As excessive inflammation is damaging in many infections, the potential role of hepcidin as a mediator of the innate immune response is a new and unexpected area of study.
The role of hepcidin remains undefined in most infections and awaits further investigation. For example, although hepcidin is induced in response to several viral and fungal pathogens [20,99], its contribution to host defenses against these infections is largely unknown. With the exception of malaria and Leishmaniasis, hepcidin has not been investigated in parasitic infections.
Zdroje
1. Hentze MW, Muckenthaler MU, Andrews NC. Balancing Acts: Molecular Control of Mammalian Iron Metabolism. Cell. 2004;117(3):285–97. 15109490
2. Ratledge C, Dover LG. Iron metabolism in pathogenic bacteria. Annual reviews in microbiology. 2000;54(1):881–941.
3. Skaar EP. The battle for iron between bacterial pathogens and their vertebrate hosts. PLoS pathogens. 2013;6(8):e1000949.
4. Nicolas Gl, Bennoun M, Devaux I, Beaumont C, Grandchamp B, Kahn A, et al. Lack of hepcidin gene expression and severe tissue iron overload in upstream stimulatory factor 2 (USF2) knockout mice. Proceedings of the National Academy of Sciences. 2001;98(15):8780–5.
5. Park CH, Valore EV, Waring AJ, Ganz T. Hepcidin, a urinary antimicrobial peptide synthesized in the liver. Journal of biological chemistry. 2001;276(11):7806–10. 11113131
6. Pigeon C, Ilyin G, Courselaud B, Leroyer P, Turlin B, Brissot P, et al. A new mouse liver-specific gene, encoding a protein homologous to human antimicrobial peptide hepcidin, is overexpressed during iron overload. Journal of biological chemistry. 2001;276(11):7811–9. 11113132
7. Krause A, Neitz S, Mägert H-J, Schulz A, Forssmann W-G, Schulz-Knappe P, et al. LEAP-1, a novel highly disulfide-bonded human peptide, exhibits antimicrobial activity. FEBS Letters. 2000;480(2):147–50.
8. Nemeth E, Valore EV, Territo M, Schiller G, Lichtenstein A, Ganz T. Hepcidin, a putative mediator of anemia of inflammation, is a type II acute-phase protein. Blood. 2003;101(7):2461–3. 12433676
9. Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306(5704):2090–3. 15514116
10. Nicolas Gl, Bennoun M, Porteu A, Mativet S, Beaumont C, Grandchamp B, et al. Severe iron deficiency anemia in transgenic mice expressing liver hepcidin. Proceedings of the National Academy of Sciences. 2002;99(7):4596–601.
11. Nicolas G, Chauvet C, Viatte L, Danan JL, Bigard X, Devaux I, et al. The gene encoding the iron regulatory peptide hepcidin is regulated by anemia, hypoxia, and inflammation. The Journal of Clinical Investigation. 2002;110(7):1037–44. 12370282
12. Kautz Lo, Jung G, Valore EV, Rivella S, Nemeth E, Ganz T. Identification of erythroferrone as an erythroid regulator of iron metabolism. Nature genetics. 2014. doi: 10.1038/ng.2996 24880340
13. Babitt JL, Huang FW, Wrighting DM, Xia Y, Sidis Y, Samad TA, et al. Bone morphogenetic protein signaling by hemojuvelin regulates hepcidin expression. Nature genetics. 2006;38(5):531–9. 16604073
14. Casanovas G, Mleczko-Sanecka K, Altamura S, Hentze MW, Muckenthaler MU. Bone morphogenetic protein (BMP)-responsive elements located in the proximal and distal hepcidin promoter are critical for its response to HJV/BMP/SMAD. Journal of molecular medicine. 2009;87(5):471–80. doi: 10.1007/s00109-009-0447-2 19229506
15. Andriopoulos B Jr, Corradini E, Xia Y, Faasse SA, Chen S, Grgurevic L, et al. BMP6 is a key endogenous regulator of hepcidin expression and iron metabolism. Nature genetics. 2009;41(4):482–7. doi: 10.1038/ng.335 19252486
16. Nemeth E, Rivera S, Gabayan V, Keller C, Taudorf S, Pedersen BK, et al. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. Journal of Clinical Investigation. 2004;113(9):1271–6. 15124018
17. Wrighting DM, Andrews NC. Interleukin-6 induces hepcidin expression through STAT3. Blood. 2006;108(9):3204–9. 16835372
18. Mayeur C, Leyton PA, Kolodziej SA, Yu B, Bloch KD. BMP type II receptors have redundant roles in the regulation of hepatic hepcidin gene expression and iron metabolism. Blood. 2014;124(13):2116–23. doi: 10.1182/blood-2014-04-572644 25075125
19. Rodriguez R, Jung C-L, Gabayan V, Deng JC, Ganz T, Nemeth E, et al. Hepcidin induction by pathogens and pathogen-derived molecules is strongly dependent on interleukin-6. Infection and Immunity. 2014;82(2):745–52. doi: 10.1128/IAI.00983-13 24478088
20. Leal SM Jr, Roy S, Vareechon C, deJesus Carrion S, Clark H, Lopez-Berges MS, et al. Targeting iron acquisition blocks infection with the fungal pathogens Aspergillus fumigatus and Fusarium oxysporum. PLoS pathogens. 2013;9(7):e1003436. doi: 10.1371/journal.ppat.1003436 23853581
21. Lee P, Peng H, Gelbart T, Beutler E. The IL-6-and lipopolysaccharide-induced transcription of hepcidin in HFE-, transferrin receptor 2-, and β2-microglobulin-deficient hepatocytes. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(25):9263–5. 15192150
22. Kemna E, Pickkers P, Nemeth E, van der Hoeven H, Swinkels D. Time-course analysis of hepcidin, serum iron, and plasma cytokine levels in humans injected with LPS. Blood. 2005;106(5):1864–6. 15886319
23. Kim A, Fung E, Parikh SG, Valore EV, Gabayan V, Nemeth E, et al. A mouse model of anemia of inflammation: complex pathogenesis with partial dependence on hepcidin2014. 1129–36 p.
24. Peyssonnaux C, Zinkernagel AS, Datta V, Lauth X, Johnson RS, Nizet V. TLR4-dependent hepcidin expression by myeloid cells in response to bacterial pathogens. Blood. 2006;107(9):3727–32. doi: 10.1182/blood-2005-06-2259 16391018
25. Ripley DA, Morris RH, Maddocks SE. Dual stimulation with bacterial and viral components increases the expression of hepcidin in human monocytes. FEMS microbiology letters. 2014;359(2):161–5. doi: 10.1111/1574-6968.12553 25145495
26. Armitage AE, Eddowes LA, Gileadi U, Cole S, Spottiswoode N, Selvakumar TA, et al. Hepcidin regulations by innate immune and infectious stimuli. Blood. 2011;118(15):4129–39. doi: 10.1182/blood-2011-04-351957 21873546
27. Nguyen N-B, Callaghan KD, Ghio AJ, Haile DJ, Yang F. Hepcidin expression and iron transport in alveolar macrophages. American Journal of Physiology—Lung Cellular and Molecular Physiology. 2006;291(3):L417–L25. 16648237
28. Layoun A, Santos MM. Bacterial cell wall constituents induce hepcidin expression in macrophages through MyD88 signaling. Inflammation. 2012;35(4):1500–6. doi: 10.1007/s10753-012-9463-4 22544439
29. Wu X, Yung L-M, Cheng W-H, Paul BY, Babitt JL, Lin HY, et al. Hepcidin regulation by BMP signaling in macrophages is lipopolysaccharide dependent. PloS one. 2012;7(9):e44622. doi: 10.1371/journal.pone.0044622 23028567
30. Zumerle S, Mathieu JRR, Delga Sp, Heinis Mn, Viatte L, Vaulont S, et al. Targeted disruption of hepcidin in the liver recapitulates the hemochromatotic phenotype. Blood. 2014;123(23):3646–50. doi: 10.1182/blood-2014-01-550467 24646470
31. Zhang X, Rovin BH. Beyond anemia: hepcidin, monocytes and inflammation. Biological Chemistry. 2013;394(2):231–8. doi: 10.1515/hsz-2012-0217 23314535
32. Sow FB, Florence WC, Satoskar AR, Schlesinger LS, Zwilling BS, Lafuse WP. Expression and localization of hepcidin in macrophages: a role in host defense against tuberculosis. Journal of Leukocyte Biology. 2007;82(4):934–45. 17609338
33. Rochette J, Pointon JJ, Fisher CA, Perera G, Arambepola M, Arichchi DSK, et al. Multicentric origin of hemochromatosis gene (HFE) mutations. The American Journal of Human Genetics. 1999;64(4):1056–62. 10090890
34. Distante S, Robson KJH, Graham-Campbell J, Arnaiz-Villena A, Brissot P, Worwood M. The origin and spread of the HFE-C282Y haemochromatosis mutation. Human genetics. 2004;115(4):269–79. 15290237
35. Hanson EH, Imperatore G, Burke W. HFE gene and hereditary hemochromatosis: a HuGE review. American Journal of Epidemiology. 2001;154(3):193–206. 11479183
36. Weinberg ED. Survival advantage of the hemochromatosis C282Y mutation. Perspectives in biology and medicine. 2008;51(1):98–102.
37. Olsson KS, Ritter B, Hansson N, Chowdhury RR. HLA haplotype map of river valley populations with hemochromatosis traced through five centuries in Central Sweden. European journal of haematology. 2008;81(1):36–46. doi: 10.1111/j.1600-0609.2008.01078.x 18363869
38. Moalem S, Percy ME, Kruck TPA, Gelbart RR. Epidemic pathogenic selection: an explanation for hereditary hemochromatosis? Medical Hypotheses. 2002;59(3):325–9. 12208162
39. Chlosta S, Fishman DS, Harrington L, Johnson EE, Knutson MD, Wessling-Resnick M, et al. The Iron Efflux Protein Ferroportin Regulates the Intracellular Growth of Salmonella enterica. Infection and Immunity. 2006;74(5):3065–7. 16622252
40. Nairz M, Theurl I, Schroll A, Theurl M, Fritsche G, Lindner E, et al. Absence of functional Hfe protects mice from invasive Salmonella enterica Serovar Typhimurium infection via induction of lipocalin-2. Blood. 2009;114 : 3642–51. doi: 10.1182/blood-2009-05-223354 19700664
41. Kim D-K, Jeong J-H, Lee J-M, Kim KS, Park S-H, Kim YD, et al. Inverse agonist of estrogen-related receptor [gamma] controls Salmonella typhimurium infection by modulating host iron homeostasis. Nat Med. 2014;20(4):419–24. doi: 10.1038/nm.3483 24658075
42. Yuki KE, Eva MM, Richer E, Chung D, Paquet Mn, Cellier M, et al. Suppression of Hepcidin Expression and Iron Overload Mediate Salmonella Susceptibility in Ankyrin 1 ENU-Induced Mutant. PloS one. 2013;8(2):e55331. doi: 10.1371/journal.pone.0055331 23390527
43. Wang L, Johnson EE, Shi HN, Walker WA, Wessling-Resnick M, Cherayil BJ. Attenuated inflammatory responses in hemochromatosis reveal a role for iron in the regulation of macrophage cytokine translation. The Journal of Immunology. 2008;181(4):2723–31. 18684963
44. Wallace DF, McDonald CJ, Ostini L, Subramaniam VN. Blunted hepcidin response to inflammation in the absence of Hfe and transferrin receptor 2. Blood. 2011;117(10):2960–6. doi: 10.1182/blood-2010-08-303859 21245482
45. Frazer DM, Wilkins SJ, Millard KN, McKie AT, Vulpe CD, Anderson GJ. Increased hepcidin expression and hypoferraemia associated with an acute phase response are not affected by inactivation of HFE. British Journal of Haematology. 2004;126(3):434–6. 15257718
46. Constante M, Jiang W, Wang D, Raymond V-A, Bilodeau M, Santos MM. Distinct requirements for Hfe in basal and induced hepcidin levels in iron overload and inflammation. American Journal of Physiology-Gastrointestinal and Liver Physiology. 2006;291(2):G229–G37. 16565419
47. Gordeuk VR, McLaren CE, MacPhail AP, Deichsel G, Bothwell TH. Associations of iron overload in Africa with hepatocellular carcinoma and tuberculosis: Strachan's 1929 thesis revisited. Blood. 1996;87(8):3470–6. 8605366
48. Boelaert JR, Vandecasteele SJ, Appelberg R, Gordeuk VR. The effect of the host's iron status on tuberculosis. Journal of Infectious Diseases. 2007;195(12):1745–53. 17492589
49. Olakanmi O, Schlesinger LS, Britigan BE. Hereditary hemochromatosis results in decreased iron acquisition and growth by Mycobacterium tuberculosis within human macrophages. Journal of Leukocyte Biology. 2007;81(1):195–204. 17038583
50. Olakanmi O, Schlesinger LS, Ahmed A, Britigan BE. Intraphagosomal Mycobacterium tuberculosis Acquires Iron from Both Extracellular Transferrin and Intracellular Iron Pools: IMPACT OF INTERFERON-γ AND HEMOCHROMATOSIS. Journal of Biological Chemistry. 2002;277(51):49727–34. 12399453
51. Gomes-Pereira S, Rodrigues PN, Appelberg R, Gomes MS. Increased Susceptibility to Mycobacterium avium in Hemochromatosis Protein HFE-Deficient Mice. Infection and Immunity. 2008;76(10):4713–9. doi: 10.1128/IAI.00612-08 18694968
52. Johnson EE, Sandgren A, Cherayil BJ, Murray M, Wessling-Resnick M. Role of Ferroportin in Macrophage-Mediated Immunity. Infection and Immunity. 2010;78(12):5099–106. doi: 10.1128/IAI.00498-10 20837712
53. Van Zandt KE, Sow FB, Florence WC, Zwilling BS, Satoskar AR, Schlesinger LS, et al. The iron export protein ferroportin 1 is differentially expressed in mouse macrophage populations and is present in the mycobacterial-containing phagosome. Journal of Leukocyte Biology. 2008;84(3):689–700. doi: 10.1189/jlb.1107781 18586980
54. Ben-Othman R, Flannery AR, Miguel DC, Ward DM, Kaplan J, Andrews NW. Leishmania-Mediated Inhibition of Iron Export Promotes Parasite Replication in Macrophages. PLoS Pathog. 2014;10(1):e1003901. doi: 10.1371/journal.ppat.1003901 24497831
55. Paradkar PN, De Domenico I, Durchfort N, Zohn I, Kaplan J, Ward DM. Iron depletion limits intracellular bacterial growth in macrophages. Blood. 2008;112(3):866–74. doi: 10.1182/blood-2007-12-126854 18369153
56. Khan FA, Fisher MA, Khakoo RA. Association of hemochromatosis with infectious diseases: expanding spectrum. International Journal of Infectious Diseases. 2007;11(6):482–7. 17600748
57. Weinberg ED. Iron availability and infection. Biochimica et Biophysica Acta (BBA)-General Subjects. 2009;1790(7):600–5.
58. Walker EM, Walker SM. Effects of iron overload on the immune system. Annals of Clinical & Laboratory Science. 2000;30(4):354–65.
59. Arezes Jo, Jung G, Gabayan V, Valore E, Ruchala P, Gulig PA, et al. Hepcidin-Induced Hypoferremia Is a Critical Host Defense Mechanism against the Siderophilic Bacterium Vibrio vulnificus. Cell host & microbe. 2015;17(1):47–57.
60. Van Eijk LT, Kroot JJ, Tromp M, van der Hoeven JG, Swinkels DW, Pickkers P. Inflammation-induced hepcidin-25 is associated with the development of anemia in septic patients: an observational study. Critical Care. 2011;15(1):R9. doi: 10.1186/cc9408 21219610
61. De Domenico I, Zhang TY, Koening CL, Branch RW, London N, Lo E, et al. Hepcidin mediates transcriptional changes that modulate acute cytokine-induced inflammatory responses in mice. The Journal of Clinical Investigation. 2010;120(7):2395–405. doi: 10.1172/JCI42011 20530874
62. Huang Y-H, Yang Y-L, Tiao M-M, Kuo H-C, Huang L-T, Chuang J-H. Hepcidin protects against lipopolysaccharide-induced liver injury in a mouse model of obstructive jaundice. Peptides. 2012;35(2):212–7. doi: 10.1016/j.peptides.2012.03.032 22504010
63. Zeng C, Chen Q, Zhang K, Chen Q, Song S, Fang X. Hepatic Hepcidin Protects against Polymicrobial Sepsis in Mice by Regulating Host Iron Status. Anesthesiology. 2014;122(2):374–86.
64. Chen Q, Song S, Chen Q, Zeng C, Zheng X, Wang J, et al. Silencing airway epithelial cell-derived hepcidin exacerbates sepsis-induced acute lung injury. Critical Care. 2014;18(4):470. doi: 10.1186/s13054-014-0470-8 25096529
65. Sazawal S, Black RE, Ramsan M, Chwaya HM, Stoltzfus RJ, Dutta A, et al. Effects of routine prophylactic supplementation with iron and folic acid on admission to hospital and mortality in preschool children in a high malaria transmission setting: community-based, randomised, placebo-controlled trial. The Lancet. 2006;367(9505):133–43. S0140-6736(06)67962-2.
66. Smith AW, Hendrickse RG, Harrison C, Hayes RJ, Greenwood BM. The effects on malaria of treatment of iron-deficiency anaemia with oral iron in Gambian children. Annals of tropical paediatrics. 1989;9(1):17–23. 2471438
67. Oppenheimer SJ, Gibson FD, Macfarlane SB, Moody JB, Harrison C, Spencer A, et al. Iron supplementation increases prevalence and effects of malaria: report on clinical studies in Papua New Guinea. Transactions of the Royal Society of Tropical Medicine and Hygiene. 1986;80(4):603–12. 3101243
68. Mebrahtu T, Stoltzfus RJ, Chwaya HM, Jape JK, Savioli L, Montresor A, et al. Low-dose daily iron supplementation for 12 months does not increase the prevalence of malarial infection or density of parasites in young Zanzibari children. The Journal of nutrition. 2004;134(11):3037–41. 15514272
69. Menendez C, Kahigwa E, Hirt R, Vounatsou P, Aponte JJ, Font F, et al. Randomised placebo-controlled trial of iron supplementation and malaria chemoprophylaxis for prevention of severe anaemia and malaria in Tanzanian infants. The Lancet. 1997;350(9081):844–50.
70. Gwamaka M, Kurtis JD, Sorensen BE, Holte S, Morrison R, Mutabingwa TK, et al. Iron Deficiency Protects Against Severe Plasmodium falciparum Malaria and Death in Young Children. Clinical Infectious Diseases. 2012;54(8):1137–44. doi: 10.1093/cid/cis010 22354919
71. Kabyemela ER, Fried M, Kurtis JD, Mutabingwa TK, Duffy PE. Decreased susceptibility to Plasmodium falciparum infection in pregnant women with iron deficiency. Journal of Infectious Diseases. 2008;198(2):163–6. doi: 10.1086/589512 18500927
72. Nyakeriga AM, Troye-Blomberg M, Dorfman JR, Alexander ND, Bäck R, Kortok M, et al. Iron deficiency and malaria among children living on the coast of Kenya. Journal of Infectious Diseases. 2004;190(3):439–44. 15243915
73. Koka S, Fοller M, Lamprecht G, Boini KM, Lang C, Huber SM, et al. Iron deficiency influences the course of malaria in Plasmodium berghei infected mice. Biochemical and Biophysical Research Communications. 2007;357(3):608–14. 17445762
74. Matsuzaki-Moriya C, Tu L, Ishida H, Imai T, Suzue K, Hirai M, et al. A critical role for phagocytosis in resistance to malaria in iron-deficient mice. European Journal of Immunology. 2011;41(5):1365–75. doi: 10.1002/eji.201040942 21469097
75. Casals-Pascual C, Huang H, Lakhal-Littleton S, Thezenas ML, Kai O, Newton CRJC, et al. Hepcidin demonstrates a biphasic association with anemia in acute Plasmodium falciparum malaria. haematologica. 2012;97(11):1695–8. doi: 10.3324/haematol.2012.065854 22689680
76. de Mast Q, Nadjm B, Reyburn H, Kemna EHJM, Amos B, Laarakkers CMM, et al. Assessment of Urinary Concentrations of Hepcidin Provides Novel Insight into Disturbances in Iron Homeostasis during Malarial Infection. Journal of Infectious Diseases. 2009;199(2):253–62. doi: 10.1086/595790 19032104
77. Portugal S, Carret C, Recker M, Armitage AE, Gonçalves LA, Epiphanio S, et al. Host mediated regulation of superinfection in malaria. Nature medicine. 2011;17(6):732–7. doi: 10.1038/nm.2368 21572427
78. Wang H-Z, He Y-X, Yang C-J, Zhou W, Zou C-G. Hepcidin is regulated during blood-stage malaria and plays a protective role in malaria infection. The Journal of Immunology. 2011;187(12):6410–6. doi: 10.4049/jimmunol.1101436 22084434
79. Armitage AE, Pinches R, Eddowes LA, Newbold CI, Drakesmith H. Plasmodium falciparum infected erythrocytes induce hepcidin (HAMP) mRNA synthesis by peripheral blood mononuclear cells. British Journal of Haematology. 2009;147(5):769–71. doi: 10.1111/j.1365-2141.2009.07880.x 19747361
80. Huang H, Lamikanra AA, Alkaitis MS, Thézénas ML, Ramaprasad A, Moussa E, et al. Interleukin-10 Regulates Hepcidin in Plasmodium falciparum Malaria. PloS one. 2014;9(2):e88408. doi: 10.1371/journal.pone.0088408 24520384
81. Spottiswoode N, Duffy PE, Drakesmith H. Iron, anemia and hepcidin in malaria. Frontiers in Pharmacology. 2014;5. doi: 10.3389/fphar.2014.00125 24910614
82. Wang X-h, Cheng P-P, Jiang F, Jiao X-Y. The Effect of Hepatitis B Virus Infection on Hepcidin Expression in Hepatitis B Patients. Annals of Clinical & Laboratory Science. 2013;43(2):126–34.
83. Armitage AE, Stacey AR, Giannoulatou E, Marshall E, Sturges P, Chatha K, et al. Distinct patterns of hepcidin and iron regulation during HIV-1, HBV, and HCV infections. Proceedings of the National Academy of Sciences. 2014;111(33):12187–92.
84. Lambrecht RW, Sterling RK, Naishadham D, Stoddard AM, Rogers T, Morishima C, et al. Iron levels in hepatocytes and portal tract cells predict progression and outcomes of patients with advanced chronic hepatitis C. Gastroenterology. 2011;140(5):1490–500. e3. doi: 10.1053/j.gastro.2011.01.053 21335007
85. Tung BY, Emond MJ, Bronner MP, Raaka SD, Cotler SJ, Kowdley KV. Hepatitis C, iron status, and disease severity: relationship with HFE mutations. Gastroenterology. 2003;124(2):318–26. 12557137
86. Lal P, Fernandes H, Koneru B, Albanese E, Hameed M. C282Y mutation and hepatic iron status in hepatitis C and cryptogenic cirrhosis. Archives of pathology & laboratory medicine. 2000;124(11):1632–5.
87. Fujita N, Sugimoto R, Takeo M, Urawa N, Mifuji R, Tanaka H, et al. Hepcidin Expression in the Liver: Relatively Low Level in Patients with Chronic Hepatitis C. Molecular Medicine. 2007;13(1–2):97–104. 17515961
88. Aoki CA, Rossaro L, Ramsamooj R, Brandhagen D, Burritt MF, Bowlus CL. Liver hepcidin mRNA correlates with iron stores, but not inflammation, in patients with chronic hepatitis C. Journal of clinical gastroenterology. 2005;39(1):71–4. 15599216
89. Girelli D, Pasino M, Goodnough JB, Nemeth E, Guido M, Castagna A, et al. Reduced serum hepcidin levels in patients with chronic hepatitis C. Journal of hepatology. 2009;51(5):845–52. doi: 10.1016/j.jhep.2009.06.027 19729219
90. Moriya K, Miyoshi H, Shinzawa S, Tsutsumi T, Fujie H, Goto K, et al. Hepatitis C virus core protein compromises iron-induced activation of antioxidants in mice and HepG2 cells. Journal of Medical Virology. 2010;82(5):776–92. doi: 10.1002/jmv.21661 20336713
91. Miura K, Taura K, Kodama Y, Schnabl B, Brenner DA. Hepatitis C virus–induced oxidative stress suppresses hepcidin expression through increased histone deacetylase activity. Hepatology. 2008;48(5):1420–9. doi: 10.1002/hep.22486 18671304
92. Nishina S, Hino K, Korenaga M, Vecchi C, Pietrangelo A, Mizukami Y, et al. Hepatitis C virus-induced reactive oxygen species raise hepatic iron level in mice by reducing hepcidin transcription. Gastroenterology. 2008;134(1):226–38. doi: 10.1053/j.gastro.2007.10.011 18166355
93. Ross SL, Tran L, Winters A, Lee K-J, Plewa C, Foltz I, et al. Molecular mechanism of hepcidin-mediated ferroportin internalization requires ferroportin lysines, not tyrosines or JAK-STAT. Cell Metabolism. 2012;15(6):905–17. doi: 10.1016/j.cmet.2012.03.017 22682226
94. Pagani A, Nai A, Corna G, Bosurgi L, Rovere-Querini P, Camaschella C, et al. Low hepcidin accounts for the proinflammatory status associated with iron deficiency. Blood. 2011;118(3):736–46. doi: 10.1182/blood-2011-02-337212 21628413
95. Du X, She E, Gelbart T, Truksa J, Lee P, Xia Y, et al. The serine protease TMPRSS6 is required to sense iron deficiency. Science. 2008;320(5879):1088–92. doi: 10.1126/science.1157121 18451267
96. Riba M, Rausa M, Sorosina M, Cittaro D, Manteiga JMG, Nai A, et al. A Strong Anti-Inflammatory Signature Revealed by Liver Transcription Profiling of Tmprss6-/ - Mice. PloS one. 2013;8(7):e69694. doi: 10.1371/journal.pone.0069694 23922777
97. Hunter HN, Fulton DB, Ganz T, Vogel HJ. The solution structure of human hepcidin, a peptide hormone with antimicrobial activity that is involved in iron uptake and hereditary hemochromatosis. Journal of Biological Chemistry. 2002;277(40):37597–603. 12138110
98. Maisetta G, Petruzzelli R, Brancatisano FL, Esin S, Vitali A, Campa M, et al. Antimicrobial activity of human hepcidin 20 and 25 against clinically relevant bacterial strains: effect of copper and acidic pH. Peptides. 2010;31(11):1995–2002. doi: 10.1016/j.peptides.2010.08.007 20713108
99. Potrykus J, Stead D, MacCallum DM, Urgast DS, Raab A, van Rooijen N, et al. Fungal iron availability during deep seated candidiasis is defined by a complex interplay involving systemic and local events. PLoS pathogens. 2013;9(10):e1003676. doi: 10.1371/journal.ppat.1003676 24146619
100. D'Alessio F, Hentze MW, Muckenthaler MU. The hemochromatosis proteins HFE, TfR2, and HJV form a membrane-associated protein complex for hepcidin regulation. Journal of hepatology. 2012;57(5):1052–60. doi: 10.1016/j.jhep.2012.06.015 22728873
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2015 Číslo 8
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Human Non-neutralizing HIV-1 Envelope Monoclonal Antibodies Limit the Number of Founder Viruses during SHIV Mucosal Infection in Rhesus Macaques
- Type VI Secretion System Toxins Horizontally Shared between Marine Bacteria
- Illuminating Targets of Bacterial Secretion
- Are Human Intestinal Eukaryotes Beneficial or Commensals?
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy