
Goblet Cell Derived RELM-β Recruits CD4 T Cells during Infectious Colitis to Promote Protective Intestinal Epithelial Cell Proliferation
Food and water-borne bacterial pathogens such as enterohemorrhagic Escherichia coli (EHEC) target the epithelial cells that line the inner surface of their host’s intestines, causing inflammation and diarrhea. While professional immune cells including T lymphocytes are well known for promoting host defense, we hypothesized that as the cells in closest contact with these bacterial pathogens, intestinal epithelial cells also play an active and essential role in protecting the host during infection. Infecting mice with Citrobacter rodentium, a mouse specific relative of EHEC, we noted a dramatic upregulation in the expression and secretion of the mediator RELM-β by a subset of epithelial cells called goblet cells. Compared to wildtype mice, mice lacking RELM-β showed less epithelial cell proliferation and suffered significantly more intestinal damage during infection. Rather than directly causing epithelial cell proliferation, we found RELM-β instead recruited T lymphocytes to the infected intestine. Upon reaching the intestine, the T lymphocytes produced the cytokine interleukin-22, which directly increased epithelial cell proliferation. Taken together, these findings indicate that epithelial/goblet cells play a critical role in orchestrating the host response to an intestinal pathogen, by recruiting T lymphocytes and by promoting epithelial proliferation to limit the intestinal damage suffered during infection.
Published in the journal:
. PLoS Pathog 11(8): e32767. doi:10.1371/journal.ppat.1005108
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1005108
Summary
Food and water-borne bacterial pathogens such as enterohemorrhagic Escherichia coli (EHEC) target the epithelial cells that line the inner surface of their host’s intestines, causing inflammation and diarrhea. While professional immune cells including T lymphocytes are well known for promoting host defense, we hypothesized that as the cells in closest contact with these bacterial pathogens, intestinal epithelial cells also play an active and essential role in protecting the host during infection. Infecting mice with Citrobacter rodentium, a mouse specific relative of EHEC, we noted a dramatic upregulation in the expression and secretion of the mediator RELM-β by a subset of epithelial cells called goblet cells. Compared to wildtype mice, mice lacking RELM-β showed less epithelial cell proliferation and suffered significantly more intestinal damage during infection. Rather than directly causing epithelial cell proliferation, we found RELM-β instead recruited T lymphocytes to the infected intestine. Upon reaching the intestine, the T lymphocytes produced the cytokine interleukin-22, which directly increased epithelial cell proliferation. Taken together, these findings indicate that epithelial/goblet cells play a critical role in orchestrating the host response to an intestinal pathogen, by recruiting T lymphocytes and by promoting epithelial proliferation to limit the intestinal damage suffered during infection.
Introduction
The enteric bacterial pathogens enterohemorrhagic Escherichia coli (EHEC) and enteropathogenic E. coli (EPEC) are important causes of infectious diarrhea. These food and waterborne pathogens infect intestinal epithelial cells (IEC) using a Type III secretion system (T3SS) [1]. Their infection leads to characteristic attaching and effacing (A/E) lesions on IEC, as well as diarrhea and transient enteritis or colitis in humans [1]. Exactly how the host defends against these A/E pathogens is poorly understood, largely because their luminal location segregates and protects them from most inflammatory and immune effector mechanisms. Instead, we and others have hypothesized that host defense against these microbes relies largely on immune mediated changes in the intestinal epithelium. In fact, several in vitro studies have shown that IEC actively promote “host resistance” to A/E pathogens by producing factors that recruit inflammatory/immune cells to the infected intestine, and by upregulating their expression of antimicrobial peptides to directly kill A/E bacteria [2–5]. However the efficacy of IEC-driven responses in clearing these pathogens is unclear, raising the question of whether infected hosts also promote IEC responses that help the host “tolerate” these infections, by limiting intestinal tissue damage to ensure the host’s survival.
Unfortunately the human specificity of EPEC and EHEC has limited our ability to study host responses against these microbes. Citrobacter rodentium, a natural A/E pathogen of mice has been widely used to model EPEC and EHEC infections, as well as study the host immune responses that develop against these pathogens [6, 7]. We and others have shown that CD4+ T cells are recruited to the infected intestine, where they drive a mixed Th1/Th17/Th22 immune response that promotes host defense, by limiting C. rodentium burdens [8–11]. Moreover C. rodentium infection leads to significant increases in IEC-based expression of antimicrobial proteins and chemokines, as well as dramatic elongation (hyperplasia) of colonic crypts due to increased IEC proliferation. We recently showed these hyperplastic crypts appear less susceptible to infection by C. rodentium [12] whereas the highly susceptible Rag1 deficient (-/-) mice (lacking T and B cells) are severely impaired in developing infection-induced IEC hyper-proliferation. Notably, reconstitution of Rag1-/- mice with CD4+, but not CD8+ T cells largely restored the IEC hyper-proliferative response during infection [13]. While IEC hyper-proliferation has been primarily seen as a characteristic pathology of C. rodentium infection, it may also reflect one mechanism by which CD4+ T cells promote host defense against A/E pathogens, although exactly how this response benefits the infected host remains controversial.
Increased proliferation and shedding of infected IEC are unlikely to clear the intestine of invading microbes, and also have detrimental effects on the host, such as limiting the maturation and differentiation of IEC including goblet cells, thereby limiting mucin production as well as ion transport in the colon [12–16]. Even so, increasing IEC turnover likely limits the potential for lumen dwelling microbes to escape the intestine and go systemic, as well as ensuring the replacement of IEC damaged by the pathogen, or by the host’s own inflammatory response. Thus the immune-mediated increase in IEC proliferation may fall under the new designation of “tolerance responses” that limit the pathology suffered by the host during infection [17]. Other potential tolerance responses described during C. rodentium infection include TLR2-dependent signaling, which rather than impacting C. rodentium burdens, was shown to limit mucosal damage as well as protect IEC barrier function during infection [18, 19]. In fact, tolerance responses may be selected for when dealing with intestinal pathogens since resistance responses aimed at killing pathogens may inadvertently deplete commensal microbes. We recently demonstrated this effect in mice lacking the negative regulator of TLR/IL-1R signaling termed SIGIRR [20]. Sigirr-/- mice proved highly susceptible to C. rodentium infection despite developing an exaggerated antimicrobial response, because rather than killing the pathogen, their host response caused a rapid depletion of commensal microbes, thus reducing colonization resistance against C. rodentium [20].
Aside from undergoing increased proliferation, secretory IEC such as goblet cells can also release mediators that promote host defense. For example, goblet cells produce and release the polymeric gel-forming mucin Muc2 into the intestinal lumen, where it hydrates and forms the protective mucus layer that overlies the IEC [21, 22]. Suspecting that Muc2 would play a protective role in this model, we previously infected wildtype mice as well as mice lacking intestinal mucus (Muc2-/-). The mucus barrier not only delayed C. rodentium infection, but mucin production increased during infection, acting to “flush” loosely adherent pathogens away from the mucosal surface [23]. Intestinal goblet cells also produce other mediators, including Resistin-like molecule (RELM)-β [24, 25]. RELM-β belongs to a family of cysteine-rich secretory molecules initially described to control insulin resistance in rodents [26, 27]. Interestingly, RELM proteins have been shown to modulate inflammation and wound healing processes [24, 28, 29]. RELM-β is produced solely by goblet cells, and is induced in the intestines of germfree mice following their colonization by commensal bacteria [30]. RELM-β expression is also strongly induced in mouse models of spontaneous ileitis [31] and during dextran-sodium sulfate driven colitis [24, 32], where it appears to worsen intestinal inflammation by stimulating macrophage production of pro-inflammatory cytokines such as TNFα, IL-6, and RANTES. Moreover, RELM-β has been shown to modulate host defense during helminth parasite infections (Trichuris muris, Nippostrongylus brasiliensis), by impacting on parasite viability and on CD4+ T cell cytokine responses during infection [33, 34].
Despite these findings, the actions of RELM-β within the GI tract remain controversial, and are largely unexplored during enteric bacterial infections. To better define its function, we tested whether RELM-β contributes to the host response to C. rodentium. Infection dramatically increased RELM-β levels within colonic goblet cells as well as in the stool and sera of mice. Although mice lacking the RELM-β gene (Retnlb-/-) carried roughly similar intestinal and systemic pathogen burdens to wildtype mice, they proved highly susceptible to C. rodentium, suffering exaggerated mucosal injury, in concert with impaired proliferation and replacement of infected IEC. Considering that CD4+ T cells are required for the increased IEC proliferative response during infection [13], we tested if Rag1-/- mice were impaired in RELM-β production, but instead found it was strongly expressed in their colons during infection. Instead we determined that RELM-β functions as a chemoattractant for CD4+ T cells, and that infected Retnlb-/- mice suffered a significant delay in CD4+ T cell recruitment to the intestine, along with reduced levels of interleukin (IL)-22, a cytokine that can directly increase IEC proliferation. Moreover, enema delivery of RELM-β to Retnlb-/- mice restored CD4+ T cell recruitment, elevating IL-22 levels and IEC proliferation, while reducing mucosal pathology. These results demonstrate that RELM-β and goblet cells play a novel host protective role during infectious colitis, accelerating the recruitment of CD4+ T cells and the promotion of IEC proliferation within the infected intestine, thereby limiting infection-associated tissue pathology.
Results
RELM-β expression is strongly induced during C. rodentium infection
Recent studies have shown that infection of the murine intestine by C. rodentium induces the resident goblet cells to strongly express RELM-β [30, 35], however the duration of this response and whether RELM-β was secreted by the goblet cells was not examined. To test this, C57BL/6 mice were infected with C. rodentium and colonic tissues, stool and serum were analyzed. Between 6 and 10 DPI when bacterial burdens were sustained at CFU levels up to 108/gram tissue (Fig 1A), we noted a dramatic increase in Retnlb gene transcript levels in the distal colon that remained elevated until the infection was cleared (21–28 DPI) (Fig 1A and 1B). When other goblet cell-specific mediators were assessed over this time course, Muc2 gene transcript levels remained fairly stable whereas trefoil factor (TFF) 3 transcripts decreased at 10 and 14 DPI as described previously [12], returning to near baseline levels by 28 DPI (Fig 1B). We also assessed RELM-β protein levels, revealing a corresponding increase in RELM-β expression in colon tissues at 6 DPI (Fig 1C). In addition, significant levels of RELM-β protein were also detected in stool samples isolated at 6 and 10 DPI (Fig 1C) as well as in the sera (Fig 1D) indicating that RELM-β is released from the goblet cells during infection. At later time points, RELM-β protein levels decreased from their peak (Fig 1C), likely reflecting increasing inflammatory damage in the colon, as well as accelerated IEC/goblet cell turnover rates and immaturity at these later stages of C. rodentium infection [13]. These data thus reveal a highly dynamic goblet cell response to C. rodentium, with RELM-β showing a distinct induction during infection.

To ensure the dramatic induction of RELM-β seen during infection was not simply due to the large oral dose of C. rodentium, we also tested a natural infection model by co-housing previously unexposed C57BL/6 mice with C. rodentium-infected mice. At day 7 post-exposure (DPE) when Citrobacter infection is well established in the previously naive mice (S1A Fig), we noted a significant ((40-fold) P < 0.0001) induction in Retnlb gene expression (S1B Fig). Western blotting of stool samples revealed a major increase in secreted RELM-β protein levels at 7 DPE (Fig 1E), while immunostaining confirmed both RELM-β induction and its specificity to goblet cells (Fig 1F).
RELM-β deficiency leads to increased mortality and mucosal damage during infection
We next examined mice genetically deficient in RELM-β (Retnlb-/- mice), and consistent with previous reports [24, 32] found no evidence of overt or spontaneous disease development under uninfected conditions. Similarly, upon examining the intestinal tissues of uninfected C57BL/6 and Retnlb-/- mice, we noted no overt pathology or other baseline differences between strains. In contrast, when we infected Retnlb-/- mice, we found they suffered a significant drop (15–20%) from their initial body weights that lasted until at least 16 DPI, whereas infected wildtype mice maintained their weight throughout the infection (Fig 2A). Furthermore, during each round of infections, on average 30–40% of Retnlb-/-mice exhibited such significant morbidity (became moribund) that they required euthanization (Fig 2B). While infected C57BL/6 mice suffered widespread but modest intestinal macroscopic pathology (as typical for this strain) at 8 and 10 DPI, infected Retnlb-/- mice displayed grossly swollen colons completely devoid of stool contents, as well as overt mucosal bleeding and patchy ulceration in both the cecum and colon (Fig 2C, arrows). Histologic analysis and pathology scoring of the colons of Retnlb-/- mice revealed marked epithelial disruption as well as significant but patchy influx of inflammatory cells in the infected mucosa, with some regions also showing significant submucosal edema and altered mucosal architecture (Fig 2D and 2F). The most severe phenotype seen in the Retnlb-/- mice involved pan-ulceration and massive dropout of crypts, leaving the entire tissue cross-section severely necrotic (Fig 2D and 2E). The intestinal pathology seen in Retnlb-/- mice at 8 DPI (S2A and S2B Fig) was similar to that seen at 10 DPI (Fig 2D), and exaggerated pathology was also observed in the ceca of infected Retnlb-/- mice (S2C and S2D Fig). In contrast, infected C57BL/6 mice suffered significantly (P < 0.0001) less colonic pathology, characterized by modest but widespread inflammatory cell infiltration and crypt hyperplasia (Fig 2D and 2F). Notably, the exaggerated pathology scores suffered by infected Retnlb-/- mice compared to C57BL/6 mice were also observed following their co-housing suggesting their susceptibility was due to their genotype rather than an aberrant microbiome. These results thus show that loss of RELM-β renders mice significantly more susceptible to C. rodentium-induced colitis.

RELM-β deficiency is associated with deeper penetration of crypts by C. rodentium
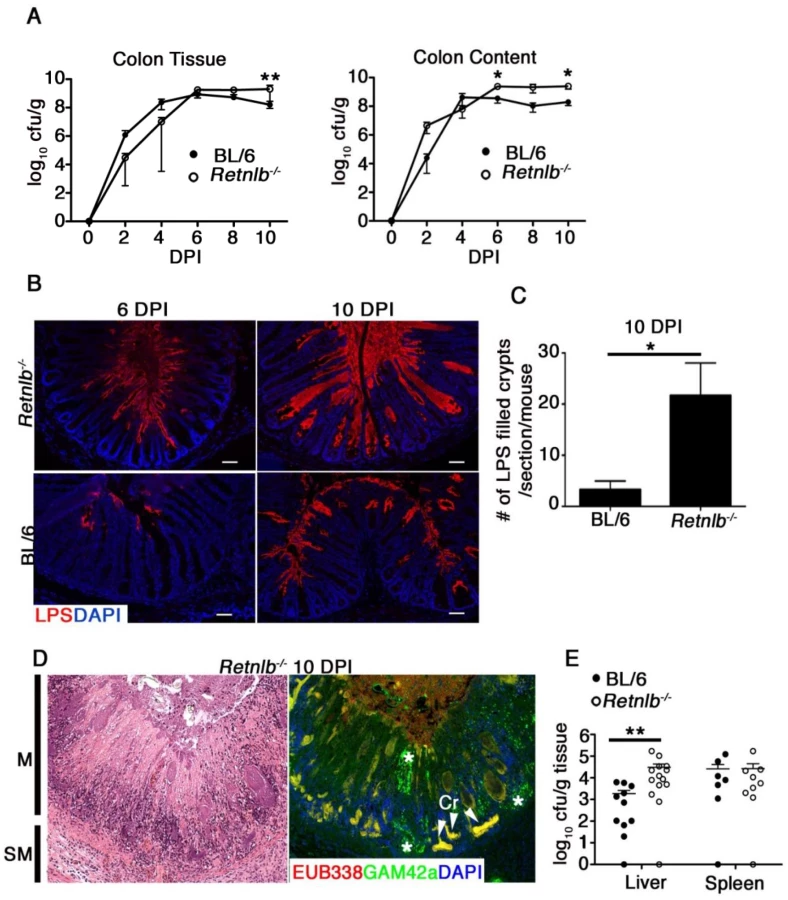
To test whether the severe damage suffered by Retnlb-/- mice reflected heavier pathogen burdens, we quantified C. rodentium within the colons and ceca of Retnlb-/- and C57BL/6 mice up to 10 DPI. C. rodentium burdens adherent to the colonic and cecal tissues of Retnlb-/- mice were not significantly different than those recovered from C57BL/6 mice from 2 to 8 DPI, but were significantly (5 fold) higher at 10 DPI (Fig 3A and S2E Fig). Similarly, pathogen burdens found in the colonic and cecal stool contents of Retnlb-/- mice were modestly (but significantly) elevated over those burdens recovered from C57BL/6 mice at 6 and 10 DPI (Fig 3A and S2E Fig). Considering the exaggerated morbidity and pathology suffered by infected Retnlb-/- mice, the observed differences in overall pathogen burdens seemed relatively unimpressive, so we next examined whether C. rodentium localization differed between the mouse strains by staining tissues for C. rodentium LPS. Strikingly, C. rodentium was found to deeply penetrate the crypts of Retnlb-/- mice at 6 DPI, (Fig 3B). At 10 DPI, C. rodentium filled the lumens of many Retnlb-/- colonic and cecal crypts, from the surface epithelia down to crypt bases (Fig 3B and 3C and S2F Fig) likely representing the source of the modestly increased burdens we recovered from these mice. In contrast, C. rodentium predominantly localized to surface epithelia in C57BL/6 mice, and only rarely penetrated their crypts (Fig 3B and 3C and S2F Fig). This difference in crypt penetration and evidence that C. rodentium directly infected epithelial cells near the base of Retnlb-/- colonic crypts was confirmed by immunostaining for the bacterial T3SS effector Tir (S2G Fig). FISH staining of ulcerated colonic regions revealed clusters of exclusively C. rodentium (yellow) and not commensal bacteria (red) localized in patterns resembling crypts, suggesting ulcers formed at sites where C. rodentium had been deeply penetrating crypt bases (Fig 3D). Consistent with this deep tissue invasion and ulceration, significantly higher pathogen burdens (P < 0.005) were found in the livers of Retnlb-/- mice (Fig 3E). Interestingly, when we infected Retnlb-/- mice with a C. rodentium strain lacking EspF (ΔespF), a T3SS effector linked to IEC damage and barrier disruption, most disease parameters including weight loss and colonic ulceration were abrogated, showing that the exaggerated tissue damage that developed in infected Retnlb-/- mice required the full pathogenic properties of C. rodentium (S3A–S3D Fig).
Infection-induced antimicrobial and inflammatory responses are intact in Retnlb-/- mice
The deep penetration of C. rodentium into Retnlb-/- mouse tissues led us to test whether RELM-β deficiency caused any defects in antimicrobial responses. Several antimicrobial genes (iNOS, mCRAMP, and RegIIIγ) primarily expressed by IEC were assessed. No defects in their expression were noted, and there was in fact a trend for elevated levels of RegIIIγ in the Retnlb-/- mice (S4A Fig). Moreover no defects were detected in the antimicrobial capacity of the colonic crypts themselves (S4B Fig). We also tested whether recombinant RELM-β itself possessed antimicrobial activity, and found that rRELM-β did not exhibit any C. rodentium killing capacity (S4C Fig).
We next examined whether inflammatory responses were impaired in the Retnlb-/- mice. Analysis of pro-inflammatory cytokine genes revealed heightened mRNA transcript levels for TNF-α, IL-1β, and IL-6 in the colons (S4D Fig) and ceca (S2B Fig) of infected Retnlb-/- vs C57BL/6 mice. We also found roughly similar numbers of macrophages and neutrophils infiltrating the colons of the two mouse strains by F4/80 and myeloperoxidase (MPO) staining respectively (S4E Fig), aside from areas of ulceration in the infected Retnlb-/- mice where neutrophils were found in much greater abundance, even by H&E staining (Fig 2D). Collectively, these studies indicate that loss of RELM-β does not cause any overt defects in launching antimicrobial or inflammatory responses to C. rodentium infection that could explain the deep crypt penetration seen in Retnlb-/- mice.
Retnlb-/- mice exhibit reduced IEC proliferation during infection
While exploring whether other host defenses might be compromised in Retnlb-/- mice, we observed through H&E staining that their colonic crypt structures did not change during infection in the same manner seen in C57BL/6 mice. This was most notable at 8 and 10 DPI, when the Retnlb-/- crypts showed less cellularity, wider lumens, and more mature goblet cells than those in C57BL/6 mice (Fig 4A). Since C. rodentium infection is known to dramatically increase IEC proliferation, leading to mature goblet cell depletion [12, 13], we examined IEC proliferation in the two mouse strains by staining for the proliferation marker Ki-67 [36]. While no baseline differences were noted between strains (S5A and S5B Fig) (9.3 ± 0.4 versus 10.2 ± 0.7 Ki67 +ve cells/crypt); at 8 DPI we saw significantly increased numbers of Ki67+ IEC in the colons of C57BL/6 mice (30.8 ± 1.5) whereas this IEC hyper-proliferative response was significantly impaired (16.8 ± 1.7, P < 0.005) in the Retnlb-/- mice (Fig 4B). Over the next six days, IEC proliferation gradually increased such that by 14 DPI, the reduction in IEC proliferation in Retnlb-/- mice that survived to that point (versus C57BL/6 mice) was no longer observed (both > 70 +ve cells/ crypt) suggesting that while RELM-β was not essential for IEC proliferation, the absence of RELM-β caused a major delay in IEC proliferative responses to C. rodentium infection.

Based on this data, we sought to determine whether the induction of IEC hyper-proliferation was related to the ability of C. rodentium to deeply penetrate colonic crypts. We analyzed IEC proliferation using 5-bromodeoxyuridine (BrdU) incorporation, and performed dual labeling for BrdU and C. rodentium LPS. The immunostaining revealed C. rodentium colonization in C57BL/6 mice was limited to the surface epithelium of crypts displaying abundant BrdU+ IEC extending from the crypt base to the surface (Fig 4C). However, in Retnlb-/- mice, C. rodentium deeply invaded many of the colonic crypts showing only sparse numbers of BrdU+ IEC limited to the crypt bases (Fig 4C). Overall, consistent with Ki67 staining, BrdU labeling was significantly reduced (P < 0.01) by approximately 4-fold in Retnlb-/- mice vs. C57BL/6 mice at 8 DPI (Fig 4C). As shown in Fig 3B, the number of LPS-filled crypts was increased 5-fold in Retnlb-/- mice, suggesting that their impaired IEC proliferative responses coincided with their crypts being highly susceptible to deep C. rodentium penetration. Notably, we previously identified deep C. rodentium colonization of colonic crypts exhibiting limited IEC proliferation in Rag1-/- mice [13].
CD4+ T cells are sufficient to induce IEC proliferative responses during infection
Crypt hyperplasia and increased IEC proliferation are common features of many infectious as well as non-infectious GI diseases, including tropical enteropathy and celiac disease [37, 38]. While the underlying mechanisms are unclear, T cell activation has been repeatedly linked to these pathologies. Moreover, we recently showed that adoptive transfer of CD4+ T cells into Rag1-/- mice increases IEC proliferation during C. rodentium infection [13]. Based on our finding of impaired IEC proliferation in Retnlb-/- mice, we examined whether CD4+ T cells might control IEC proliferation by modulating intestinal RELM-β levels. We therefore tested how reconstituting CD4+ T cells into Rag1-/- mice affected both IEC proliferation, as well as RELM-β expression. At 10 DPI, colon tissues were immunostained for CD4+ cells, proliferative responses (Ki67) and pathogen colonization (LPS), as well as RELM-β. As expected, CD4+ cells were virtually absent in non-reconstituted Rag1-/- mice, but were abundant in the mucosa of CD4+ T cell-reconstituted Rag1-/- mice (Fig 5A), confirming successful reconstitution. Analysis of IEC proliferation at 10 DPI found that infected CD4+ reconstituted Rag1-/- mice showed a dramatic increase in Ki67+ IEC compared to non-reconstituted Rag1-/- mice (Fig 5B and 5B1). Importantly, LPS staining revealed C. rodentium deeply penetrating the crypts of non-reconstituted Rag1-/- mice, whereas it was limited to the surface epithelium of CD4+ reconstituted Rag1-/- mice (Fig 5C). When tissues were stained for RELM-β, abundant positively staining goblet cells were noted in both the non-reconstituted Rag1-/- mice as well as the CD4+-reconstituted Rag1-/- mice (S5C Fig). These results indicate not only that induction of RELM-β expression does not require CD4+ T cells, but more importantly, that RELM-β is insufficient, in the absence of CD4+ T cells, to drive IEC hyper-proliferation during C. rodentium infection.

Infected Retnlb-/- mice display reduced numbers of colonic CD4+ T cells
The above results suggested that the impaired IEC proliferation seen in Retnlb-/- mice might reflect some defect in their CD4+ T cells, however baseline CD4+ T cell numbers within the intestine and spleen are known to be similar in Retnlb-/- vs C57BL/6 mice [24, 33]. Alternatively, we hypothesized that Retnlb-/- mice might suffer an impaired ability to recruit CD4+ T cells to the colon upon infection, since an inability to recruit these cells could potentially explain their impaired IEC proliferation during infection. We therefore immunostained colonic tissues from uninfected and infected C57BL/6 and Retnlb-/- mice, for the marker CD4. While CD4+ cells were sparse in both strains under uninfected conditions (Fig 5D), at 10 DPI, numerous intensely stained CD4+ cells with distinct lymphocyte morphology were seen in the submucosa and mucosa of C57BL/6 mice (Fig 5D). In contrast, CD4+ cell numbers were significantly reduced (P < 0.05) in Retnlb-/- mice, at roughly 15–25% the number found in C57BL/6 colons at both 8 and 10 DPI (Fig 5D and 5E). These results were confirmed by FACs analysis of cells isolated from the colonic lamina propria from the two mouse strains (Fig 5F). Furthermore, FACs analysis of CD8+ve T cell populations showed no significant differences between C57BL/6 and Retnlb-/- mice, indicating that loss of RELM-β did not affect all T cell populations. These data suggest that the paucity of CD4+ T cells in the mucosa of Retnlb-/- mice is likely the basis for their stunted IEC proliferative responses to C. rodentium infection.
Adaptive immune responses to C. rodentium are not compromised in Retnlb-/- mice
While CD4+ T cells are sufficient to drive increased IEC proliferation, it was unclear whether the reduced CD4+ T cell numbers found in infected Retnlb-/- mice reflected a defect in their priming, or in their recruitment to the site of infection. However, stimulation with C. rodentium antigen revealed similar antigen-specific secretion of IL-17A and IFNγ from splenocytes isolated from infected C57BL/6 and Retnlb-/- mice (S6A and S6B Fig). These results suggest that priming of adaptive immune responses to C. rodentium is not compromised in the absence of RELM-β.
RELM-β exhibits direct chemotactic activity for CD4+ T-cells in vitro
We next tested whether Retnlb-/- mice suffered defects in recruiting CD4+ T-cells to the colon, by measuring colonic gene expression of the T cell chemokines CCL8, CXCL9, and CCL25 [39]. We found significantly enhanced gene expression of CCL8 at 6 DPI, while both CCL8 and CCL25 were increased (but not significantly) at 10 DPI in Retnlb-/- mice, compared to that found in infected C57BL/6 mice (Fig 6A). While CXCL9 expression was mildly impaired at 6 DPI, it was not significant, and did not seem at a level that would have such a major effect on CD4+ T-cell recruitment. With no overt defects identified in these key chemokine genes, we noted previous studies showing that RELM-β could directly chemoattract stromal cells to sites of lung injury [40]. We therefore examined whether RELM-β could itself recruit CD4+ T cells using a Boyden Chamber assay. Interestingly, rRELM-β caused a dose-dependent increase in CD4+ T cell migration, similar to that seen using the T-cell chemokine CCL4 (Fig 6B), but the chemoattraction was abrogated when rRELM-β was heat inactivated. Therefore the impaired T cell recruitment seen in Retnlb-/- mice likely reflects the loss of RELM-β and its ability to chemoattract CD4+ T cells to the infected colon.

IL-22 increases IEC proliferation and is significantly reduced in Retnlb-/- mice
To determine the mechanism through which CD4+ T cells drive IEC proliferation during infection, we next examined levels of IL-22; a cytokine produced by CD4+ T cells during enteric infection that interacts with its receptors expressed by IEC [41]. IL-22 is unique amongst the interleukins in that its receptors are exclusively expressed on tissue resident non-hematopoietic cells such as IEC, inducing proliferative and antimicrobial responses in these cells [9, 41]. In fact, in separate studies, we found that neutralizing IL-22 in mice during C. rodentium infection caused a major impairment in IEC proliferation. C57BL/6 mice treated with an anti-IL-22 antibody showed less colonic IEC proliferation (19.1±1.7 Ki67+ve IEC/crypt) than infected mice receiving PBS alone (30.4 ±1.7) (P = 0.009) or mice receiving control antibodies (32.0 ±4.3) (P = 0.058). Interestingly, gene transcript and protein analysis revealed significantly impaired (P < 0.05) induction of IL-22 in Retnlb-/- mice during infection as compared to C57BL/6 mice (Fig 7A and 7B). Further, in vitro application of rIL-22 to the CMT93 colonic epithelial cell line significantly increased (P < 0.005) proliferation (Ki67+ cells) (Fig 7C), confirming the potential for its direct proliferative effects on IECs. In contrast, direct application of rRELM-β to this IEC line did not significantly increase Ki67+ cell numbers (S7A Fig). Since IL-22 can be produced by several cell types, including T lymphocytes and innate lymphoid cells, we compared its expression during infection in mice lacking T cells (Tcr-β-/-) and C57BL/6 mice. We found that Il-22 gene transcription, as well as IL-22 protein levels were dramatically reduced in the colons of Tcr-β-/- mice indicating that CD4+ T cells are the primary source of IL-22 at this time point in the infection (S7B and S7C Fig). These results thus suggest that IL-22 production by CD4+ T cells recruited to the infected intestine in response to RELM-β drives the protective IEC proliferative responses seen during infection.

RELM-β enemas restore CD4+ T cells, IL-22 levels and IEC proliferation in infected Retnlb-/- mice
To clarify whether restoring RELM-β could protect Retnlb-/- mice during infection, the effects of delivering rRELM-β into Retnlb-/- mice were tested. Repeated intraperitoneal (i.p) injection of murine rRELM-β had no protective effect on infected Retnlb-/- mice (S8 Fig). We next tried enema delivery, with infected Retnlb-/- mice given 400 ng doses of rRELM-β in 1% methyl propyl cellulose in PBS, or the vehicle alone (control), by enema each day between 5 to 7 DPI, and the mice were euthanized at 8 DPI. Once euthanized, the protective effects of rRELM-β were readily apparent as the macroscopic intestinal injury and bleeding seen in the Retnlb-/- mice was dramatically attenuated following rRELM-β treatment (Fig 8A). Moreover the colonic pathological scores as well as levels of crypt hyperplasia in the Retnlb-/- mice given rRELM-β enemas were found to be similar to that seen in C57BL/6 mice, and significantly different from those seen in vehicle treated Retnlb-/- mice (Fig 8B and 8B1). As assessed by FACs analysis, rRELM-β treatment led to a marked increase in CD4+ T cell numbers populating the colonic mucosa (Fig 8C). Significantly increased numbers of Ki67+ proliferating IEC (P < 0.0001) were also observed in rRELM-β treated mice (Fig 8D and 8E). Moreover significantly increased IL-22 production (P < 0.05) was observed at both gene transcript and protein levels in rRELM-β treated mice (Fig 8F). These results thus indicate that luminal delivery of rRELM-β to Retnlb-/- mice recruits CD4+ T cells to the infected colon; protecting the host by boosting IL-22 levels as well as proliferative IEC responses to replace infected and damaged cells.

Discussion
Intestinal goblet cells are considered important contributors to intestinal barrier function, through the release of the mucin Muc2 and their critical role in the generation of the intestinal mucus layer. While production of intestinal mucus is generally considered a passive means of host defense, herein we report that goblet cells can also play an active and critical role in driving inflammation and intestinal remodeling, through the production of RELM-β. In contrast to other goblet cell mediators (Muc2 and TFF3), RELM-β expression was strongly induced during C. rodentium infection. Moreover RELM-β production proved protective, since infected Retnlb-/- mice suffered exaggerated mucosal damage compared to C57BL/6 mice. Loss of RELM-β resulted in deeper pathogen penetration of colonic crypts, patchy ulceration, as well as heightened morbidity and mortality. The protective actions of RELM-β did not appear to involve modulation of antimicrobial defenses, but rather reflected its role in recruiting CD4+ T cells to the infected colon, where they drove a protective increase in IEC proliferation through the production of IL-22.
Our previous studies demonstrated that goblet cell-derived Muc2 protected mice during C. rodentium infection, by limiting the pathogen’s ability to reach the underlying IEC [23]. However once pathogens successfully cross the intestinal mucus layer, the protective actions of Muc2 are overshadowed by the host’s immune response. Thus a major impetus for the current study was to examine whether goblet cells respond to a successful infection by changing the mediators they release. We focused our attention on RELM-β since it is strongly induced in several models of colitis, yet its function is poorly defined. In fact, whether RELM-β plays a pro - or anti-inflammatory role during colitis remains controversial. Reports have shown that RELM-β aggravates pathology during DSS-induced colitis, by activating macrophages and by increasing their production of TNF-α [32]. Another study suggested that RELM-β promotes the chronicity of T. muris infections as well as the severity of colitis by increasing IFNγ production by CD4+ T cells [33]. In contrast, in the TNBS colitis mouse model, RELM-β deficiency was shown to worsen disease via unknown mechanisms and correspondingly, enema delivery of rRELM-β was shown to ameliorate TNBS colitis [42]. Collectively, these results indicate the biological function of RELM-β may be context dependent, while our findings suggest its impact may reflect whether recruitment of CD4+ T cells to the intestine provides protection within the specific model tested, or alternatively, simply worsens colitis.
In our model, we noted a rapid increase in RELM-β expression by intestinal goblet cells in response to C. rodentium infection. While RELM-β gene transcript levels remained elevated throughout the course of infection, RELM-β protein levels peaked at 6 DPI and declined by 10 to 14 DPI, in keeping with the increasingly immature state of goblet cells at these later time points [12]. Aside from goblet cells being strongly immunoreactive for RELM-β, the protein was also detected in large quantities within the stool. This suggests RELM-β is released apically into the intestinal lumen, however consistent with a role for RELM-β in recruiting T cells to the infected intestine, it was also found elevated within the sera of infected mice. At present it is unclear whether this reflects basolateral release of RELM-β by the goblet cells, or instead leakage of the luminally secreted molecule across a disrupted epithelial barrier. Recent studies have shown that goblet cells can act as a passageway for luminal antigens to cross the epithelium and reach underlying immune cells [43]. Presumably such a role would also facilitate the passage of luminal RELM-β into the underlying lamina propria and beyond.
Consistent with its strong induction in our model, RELM-β played an important protective role, as mice lacking the protein suffered more severe colitis than WT mice. The exaggerated tissue damage reflected the deep penetration of Retnlb-/- crypts by C. rodentium, resulting in direct pathogen damage to the epithelium as well as host immune-mediated loss of crypts and patchy ulceration. Considering that C. rodentium typically colonizes IEC at the surface of crypts, its presence at the crypt bases suggested the Retnlb-/- mice were impaired in host defense. While no overt defects in antimicrobial or inflammatory responses were detected, the Retnlb-/- mice did suffer a striking delay in their ability to induce the IEC hyper-proliferation typically seen during C. rodentium infection. We and others have shown this process increases the turnover and replacement of infected IEC and is driven by the host’s immune system [12–13, 44]. This process, previously described as an “epithelial escalator” has been highlighted as a protective response during Trichuris muris infection [45], and our data suggests this process also benefits the host during infection by a mucosal adherent bacterial pathogen.
While increasing IEC proliferation and ultimately sloughing IEC into the intestinal lumen may help keep mucosal adherent pathogens away from the base of intestinal crypts, an equally important protective role may be the replacement of damaged epithelial cells, as a form of regeneration and restitution of the epithelial layer. We propose this is a key aspect of host defense against C. rodentium infection, promoting tissue tolerance (the ability of the host to survive the infection), by limiting the extent of damage suffered during infection. While tissue tolerance has been primarily highlighted as a strategy by plants to survive infection [46], recent studies have begun to address its relevance in mammalian hosts [17]. We have also shown that TLR2 mediated maintenance of IEC barrier function during C. rodentium infection plays a critical role in limiting tissue damage and mortality, without impacting on pathogen burdens [18]. This suggests that promoting tissue tolerance is an important goal of the host immune system in this model. Notably, while increased IEC proliferation may be a requirement for host survival during C. rodentium infection, it is not on its own sufficient to overcome severe genetic susceptibilities in other aspects of host defense. For example, mice deficient in TNF-α as well as Muc2-/- mice exhibit significant susceptibility to C. rodentium infection, despite undergoing IEC hyper-proliferation. Thus IEC hyper-proliferation appears to work with other mechanisms of host defense to provide optimal protection against C. rodentium infection.
RELM-β’s role in driving IEC proliferation appears to be indirect, as exposing IEC to RELM-β in culture did not lead to increased proliferation. Moreover, although RELM-β is strongly induced in infected Rag1-/- mice, these mice develop only modest increases in IEC proliferation in response to C. rodentium. Instead RELM-β appears to require an intact immune system, and specifically CD4+ T cells to drive crypt IEC proliferation and hyperplasia. Retnlb-/- mice showed a selective reduction in CD4+ T cell numbers within the large bowel during infection, as compared to wildtype mice. We determined that RELM-β itself has direct chemoattractive activity for CD4+ T cells. While previous studies have shown that RELM-β can modulate cytokine production by CD4+ T cells [33], this is the first study to show it recruits these cells to sites of injury/infection. While there are several other chemokines known to recruit CD4+ T cells, a previous study showed that reduced production of IEC-derived chemokines (such as CCL25) during C. rodentium infection had little impact on CD4+ T cell recruitment at early stages (7 DPI) but only impacted CD4+ T cell numbers at later stages of infection (14 DPI) [47]. Why traditional chemokines have such a delayed effect in this model is unclear, however this delay in their actions may explain why the very rapid upregulation and release of RELM-β plays such an important role in protecting the host during the early stages of C. rodentium infection.
While CD4+ T cells can produce a number of cytokines, depending on their functional phenotype, C. rodentium infection is noted for the localized recruitment of Th22 CD4+ T cells to the colon, where they produce large quantities of the cytokine IL-22. A multifunctional cytokine, IL-22 has been shown to induce antimicrobial responses as well as promote cellular proliferation [9, 41], and we confirmed that IL-22 directly increases IEC proliferation in culture. While several studies have implicated innate lymphoid cells (ILC) as important producers of IL-22 during C. rodentium infection [48], the fact that Rag1-/- mice possess ILC in their intestines, but develop little increase in IEC proliferation during infection suggests ILC are not primarily involved in the IEC hyper-proliferative response. This may reflect the overall levels of IL-22 produced by ILC versus Th22 cells, or alternatively the localization of these cells during the course of infection. Similarly, while IL-22 has been shown to be required for the full induction of antimicrobial responses during C. rodentium infection [41], we noted no overt defect in antimicrobial defenses in Retnlb-/- mice, suggesting that the levels of IL-22 found in their tissues are sufficient to drive these responses, but insufficient to promote IEC proliferation.
RELM-β’s ability to drive CD4+ T cell recruitment to the gut may in part explain its complicated effects during parasite infections [25, 33–34], as well as its worsening effect on several models of colitis [31, 32]. Interestingly, RELM-β is not the only RELM family member to play a role during C. rodentium infection. RELM-α is produced primarily by macrophages, and was recently shown to promote a Th17 immune response by priming naive T-cells through increased MHCII expression and by increasing IL-23 expression by macrophages [49]. In contrast, we did not observe overt defects in T cell priming in the absence of RELM-β, suggesting distinct non-redundant functions of specific RELM family members in immunity to C. rodentium infection. Overall, our findings expand the role attributed to goblet cells in providing host defense against enteric bacteria. While intestinal mucus provides an important barrier against intestinal microbes, once that barrier is bypassed, the host’s immune system must be recruited to deal with invading pathogens. The induction of RELM-β within goblet cells, and its subsequent release may reflect a generalized but effective host approach to rapidly recruit CD4+ T cells to the intestine to deal with noxious stimuli threatening intestinal integrity. Considering the array of colitic stimuli shown to upregulate RELM-β expression, it will be interesting to define whether RELM-β promotes T cell recruitment in other models of colitis.
Material and Methods
Mice
Six to twelve-week-old Retnlb-/- and Tcr-β-/- mice (both generated on a C57BL/6 genetic background) and wildtype (C57BL/6) mice were bred in the animal facilities at the Child and Family Research Institute (CFRI), whereas Rag1-/- mice were purchased from The Jackson Laboratory. Mice were kept in sterilized, filter-topped cages, handled in tissue culture hoods and fed autoclaved food and water under specific pathogen free (SPF) conditions. Sentinel animals were routinely tested for common pathogens.
Ethics statement
All experiments were performed according to a protocol (A11-0290) approved by the University of British Columbia's Animal Care Committee and in direct accordance with The Canadian Council on Animal Care (CCAC) guidelines. Mice were monitored for mortality and morbidity throughout their infection and euthanized if they showed signs of extreme distress or >15% body weight loss.
Bacterial strains, infection and rRELM-β injections and enemas
Mice were infected by oral gavage with 0.1 ml of an overnight culture of Luria-Bertani (LB) broth grown at 37°C with shaking (200 rpm) containing 2.5 x 108 cfu of C. rodentium (strain DBS100) [23, 50]. For natural infection studies, the protocol was modified from Wiles et al. [51]; briefly, C57BL/6 mice were infected by oral gavage as described above. After colonization was confirmed by plating of stool contents (described below), a single mouse harboring over 108 cfu/g of stool was placed in a cage of uninfected BL/6 mice (n = 3–4). Infected and non-infected mice remained co-housed, and stool was taken at 4 and 7 PI to determine colonization. For RELM-β injections, 10μg recombinant (r)Relmβ in sterile PBS (100μL of 100μg/mL), or PBS alone as vehicle control, was administered intraperitoneally to Retnlb-/- mice 2 days prior to infection and every second day following infection. Mice were euthanized at 10 DPI and analyzed as above. For RELM-β enemas, mice were injected with 200μl of 2μg/ml rRELM-β in 1% methyl propyl cellulose in phospho-buffered saline (PBS), while control mice received just 1% methyl propyl cellulose in PBS. At 5 days post-infection (DPI), two doses of RELM-β were given, followed by single doses at 6 and 7 DPI. Mice were euthanized at 8 DPI to carry out tissue analysis.
Tissue collection
Tissues for histology, mRNA and cryosectioning were prepared as previously described [23, 50]. In brief, mice were anesthetized with halothane, euthanized by cervical dislocation, and their large bowel resected and regionally divided. Cecal and distal colonic tissues were immediately placed in 10% neutral buffered formalin (Fisher) (48 hrs, 4°C) or ice cold fresh Carnoy’s Fixative (2 hrs, 4°C) or 4% paraformaldehyde (PFA) (1 hr, room temp) for histological studies, or placed in RNAlater (Qiagen) and stored at -86°C for subsequent RNA and protein extraction. For histology, tissues collected in formalin or Carnoy’s were transferred into 70% or 100% ethanol, respectively (after they underwent the appropriate fixation time), embedded in paraffin and cut into 5-μm sections. PFA fixed tissues were embedded in Optimal Cutting Temperature (OCT) compound and sectioned with a microtome-cryostat.
Bacterial counts
For enumeration of C. rodentium within intestinal tissues and luminal compartments, whole mouse colons and ceca were opened longitudinally, and their luminal contents collected in pre-weighed 2.0 ml microtubes containing 1.0 ml of PBS and a 5.0 mm steel bead (Qiagen). Intestinal tissues were washed vigorously in PBS (pH 7.4), cut into several pieces, and also placed in a tube as above. Tissue and lumen contents were weighed, and then homogenized in a MixerMill 301 bead miller (Retche) for a total of 6 mins at 30 Hz at room temperature. Tissue homogenates were serially diluted in PBS and plated onto luria broth (LB) agar plates containing 100 mg/ml streptomycin, incubated overnight at 37°C, and bacterial colonies were enumerated the following day, normalizing them to the tissue or stool weight (per gram). A similar method was used to enumerate C. rodentium within the spleens and livers of infected mice.
For fecal bacterial burden analysis, stool was collected from live mice at various times post-infection (described in text) and processed as described for luminal contents. For some studies with non-antibiotic resistant C. rodentium, plating was performed on MacConkey Agar (Difco), C. rodentium colonies were clearly identified by their unique characteristic of being round with red centre and a thin white rim. Colonies were confirmed to be C. rodentium by PCR for the C. rodentium T3SS translocator gene escN.
Histology scoring and immunofluorescence staining
Preparation of paraffin embedded slides for histological analysis (H&E) and immunostaining was carried out as previously described [9, 21]. H&E stained tissues were assessed for the following parameters by two blinded observers to determine extent of histological damage: (i) submucosal edema (0, no change; 1, mild; 2, moderate; 3, severe); (ii) goblet cell depletion (0, no change; 1, mild depletion; 2, severe depletion; 3, absence of goblet cells); (iii) hyperplasia (0, no change; 1, 1 to 50%; 2, 51 to 100%; 3, >100%); (iv) epithelial integrity (0, no pathological changes detectable; 1, epithelial desquamation [a few cells sloughed, surface rippled]; 2, erosion of epithelial surface [epithelial surface rippled, damaged]; 3, epithelial surface severely disrupted/damaged, large amounts of cell sloughing; 4, ulceration [with an additional score of 1 added for each 25% of tissue in the cross-section affected, e.g., a large ulcer affecting 70% of the tissue section would be scored 4 + 3]); (v) inflammatory cell infiltration (per ×400 magnification field) (0, no change; 1, <20; 2, 20 to 50; 3, >50 cells/field); the maximum possible pathology score using this scheme was 20. When ulcers were specifically assayed (see Fig 2E), small ulcers were defined as < 25% of tissue ulcerated in the cross-section; medium ulcers involved >25 to <50% of the cross section and large extensive ulcers involved >50% of the cross section.
For immunofluorescence staining 5-μm sections were deparaffinized by heating to 60°C for 15 min, cleared with xylene, rehydrated through an ethanol gradient to water, steamed for 30 min in citrate buffer for antigen retrieval, and blocked using blocking buffer (goat or donkey serum in PBS containing 1% bovine serum albumin [BSA], 0.1% Triton X-100, 0.05% Tween 20, and 0.05% sodium azide). The rabbit derived primary antibodies - RELM-β (Peprotech), MPO (Thermo Scientific), LPS (Biotec) and Ki67 (Thermo Scientific) were used at dilutions 1 : 400, 1 : 200, 1 : 500 and 1 : 200 respectively. Rat anti-murine antibodies F4/80 (AbD Serotec), CD4 (e-Bioscience), BrdU (AbD Serotec) and Tir (gift of Dr. Wanyin Deng) were used at dilutions 1 : 200, 1 : 100, 1 : 200 and 1 : 2000 respectively. Secondary goat anti-rabbit and anti-rat antibodies conjugated to AlexaFluor 488 and 568 were used at 1 : 2000 dilution and ProLong gold antifade reagent with 4′,6-diamidino-2-phenylindole (DAPI; Invitrogen) to stain DNA was used to mount tissues. Tissues were viewed on a Zeiss Axio Imager microscope, and images were taken using AxioVision software and an AxioCam HRm camera.
RNA extraction and quantitative RT-PCR
Total RNA was extracted using an RNeasy kit (Qiagen) according to manufacturer’s instructions and quantified using a NanoDrop spectrophotometer. cDNA was synthesized using 1 μg of RNA reverse transcribed using an Omniscript RT kit (Qiagen). Quantitative PCR (qPCR) was performed on reactions containing 5 μL of 1 : 5 diluted cDNA in 10 μL of BioRad SYBR green mix with primers (300 nM final concentration) using an Opticon 2 (Bio-Rad) machine, as described previously [9]. Quantitation of data was carried out using Gene Expression Macro OM 3.0 software (Bio-Rad). Primer sequences and reaction conditions for all genes analyzed are given in S1 Table.
Colonic crypt isolation and antimicrobial assays
Colon tissue was harvested and the distal 3 cm was washed 3x in sterile cold PBS, cut into 0.5 mm pieces and placed in a 2 ml microtube with1 ml crypt isolation buffer (30 mM EDTA, 2 mM DTT in PBS, pH 7.2), and gently rocked at 4°C for 20 minutes. The supernatant was collected, stored on ice and fresh isolation buffer was added to the tissues. This was repeated until 4 separate fractions were collected. On collection of the 4th fraction, tissues were pulsed for 5s in a vortexor. The fractions were then centrifuged at 2000 rpm for 10 mins, 4°C, washed in 1 x PBS, pelleted again as above, the supernatant aspirated off, and samples were lysed in lysis buffer (1 x PBS + 0.1% TritonX-100 with 1x complete protease inhibitors (Roche)) and quantified. For the antimicrobial assay, an overnight culture of nalidixic acid-resistant C. rodentium grown in LB (+ nalidixic acid) was diluted 1 : 1000 in Tryptic Soy Broth (TSB) and grown to mid log phase (OD620 0.6–1.0). The bacteria was washed by centrifugation (3000 rpm, 4°C, 10 mins), the supernatant removed, and the pellet resuspended in ice cold 10mM sodium phosphate buffer (SPB) (pH 7.4). This step was repeated once. The washed sample was diluted to a final OD620 of 0.7, diluted 1000x, and 40 μL of this dilution (containing ≈ 8 x104 bacteria) were added to a microwell containing 100 μl 30% TSB, 25 μl 10mM SPB + 25 μl containing 100 μg of crypt lysate (fraction 4) or lysis buffer as a negative control. The total reaction volume was 200 μl. Cultures were incubated in a Wallace Victor plate reader (Perkin-Elmer Life Sciences, Boston, MA) at 37°C and O.D. 600 was measured and recorded every 15 minutes for 12 hours, and graphed. To evaluate the potential antimicrobial effects of recombinant mouse RELM-β, C. rodentium were serially diluted to 1000 bacteria and were then incubated with 10μl of either varying concentrations of RELM - or PBS as a control. Bacteria were allowed to grow overnight on LB-agar plates at 37°C after which the colony forming units were counted.
CD4+ T cell reconstitution of Rag1-/- mice
Positively selected CD4+ T cells were purified from spleens and mesenteric lymph nodes of C57BL/6 mice using the Miltenyi MiniMACs purification apparatus (Miltenyi Biotec). In brief, spleens and MLNs were homogenized with a 1.0-mL syringe plunger and filtered and washed into a single-cells suspension. Cells were incubated with biotinylated anti-CD4 antibody (1 : 200, UBC) followed by incubation with streptavidin coated magnetic beads (1 : 30, Miltenyi) and run through magnetic columns. 2 x 105 CD4+ cells were then injected into Rag1-/- mice via tail vein injection and left for 8 weeks, as previously described [13].
FACS analysis
Colons were resected, longitudinally opened and washed in PBS containing 50ug/ml gentamycin. The colons were cut into 0.5-1cm pieces and incubated in HBSS solution containing 5%FBS, 2mM EDTA (Sigma), 1mM DTT (Sigma) and 10 mM Hepes (Sigma) for 30 min at 37°C. After vortexing briefly, the intestines were passed through a 100μm cell strainer to remove intraepithelial lymphocytes. The sections were then incubated in 5ml of RPMI medium containing 5% FBS, 1.5mg/ml of collagenase VIII (Sigma) and 0.1mg/ml DNAse (Sigma) for 1h at 37°C in a shaker. After vortexing for 1min, supernatants were filtered in a 70μm cell strainer and then resuspended in a medium. Further enrichment of lamina propria lymphocytes was done by the Percoll gradient method where the pelleted cells were resuspended in 8ml of 40% Percoll (Sigma) made in PBS which was layered on top of 4ml of 80% Percoll prepared in DMEM medium. Purified lymphocytes were collected at the interphase ring visible after centrifuging at 2000 rpm for 20min (with no braking). Subsequent FACS analysis was performed on freshly isolated lamina propria lymphocytes to identify the CD4+ve T cell populations. Briefly, 5x105cells/well were fixed and permeabilized for 30min at room temperature using fixation/permeabilization concentrate (Ebioscience). Cells were then labelled with FITC and PE conjugated to anti-mouse CD4 - (Ebioscience) and CD8 (Ablab) antibodies respectively. Cells were analyzed by using BD FACSDiva machine and data were analyzed by using FlowJo (Tree star) software. At least 30,000 events were counted for each sample.
Transwell migration assays
4x105 purified CD4+ T cells in 50μl of RPMI (Gibco) complete medium (10% FBS, 10nM non-essential amino acids, 10nM sodium pyruvate, 50μM β-mercaptoethanol and 50U/ml Penicillin-streptomycin) were placed in upper chamber of Transwells containing permeable polyester membrane (0.4 μm pores). In the lower chamber, 150μl of RPMI complete medium containing different concentrations of mouse recombinant RELM-β, 100 μg/ml BSA, 100ng/ml CCL4 or 100ng/ml denatured RELM-β was placed and incubated at 37°C for 4h. The number of cells transmigrated into the bottom chamber were counted using hematocytometer for each treatment condition.
In vitro IEC proliferation
Mouse intestinal carcinogenic epithelial cells (CMT93) obtained from ATCC were cultured in high-glucose DMEM medium (Gibco) containing 10% FBS, 1x MEM and 5mM HEPES and 50U/ml penicillin-streptomycin. 5x104 cells/well were grown on sterile glass coverslips in a 12-well plate (Corning) in a humid incubator at 37°C and 5% CO2. After reaching 50% confluency, the cells were treated for 24hr with 100ng/ml recombinant mouse IL-22 (R&D Biosystems) or RELM-β (Peprotech). Cells were subsequently fixed in ice-cold paraformaldehyde for 15 min followed by blocking with 1% BSA for 30min and incubation with rabbit derived anti-Ki67 (Thermo Scientific) for 2hr. After washing 3X with PBS, cells were incubated in dark for 1hr with secondary antibody conjugated to Alexafluor 568. After removal of secondary antibodies, cells were washed 3X with PBS followed by mounting in Vectashield (Vector Laboratories, Burlington, ON, Canada) on glass slides and screened with a Zeiss microscope.
IL-22 neutralization studies and ELISA
IL-22 neutralization studies were carried out in C. rodentium infected C57BL/6 mice by injecting them with either neutralizing IL-22 antibodies (R&D systems), PBS or with an isotype matched control antibody (R&D systems) given on 0 and 6 DPI (100μg/mouse). Mice were euthanized at 8 DPI to collect colon tissues and then analyzed as above (including measuring IEC proliferation by Ki67 staining). For IL-22 ELISAs, colon and cecal tissues obtained from C. rodentium infected C57BL/6, Retnlb-/- and Tcrβ-/- mice either treated with rRELM-β or left untreated were extracted and the luminal contents were removed by washing with PBS. The pre-weighed tissues were cut into 0.5-1cm sections and incubated in DMEM containing 10% FBS supplemented with 50U/ml Penicillin-streptomycin and 50μg/ml gentamicin for 18 hours at 37°C in 5% CO2. Supernatants were collected by centrifugation at 12,000g for 10 min and the amount of IL-22 secreted into the supernatant was measured using the mouse IL-22 ELISA MAX (BioLegend, San Diego, CA) according to the manufacturer’s protocol.
Statistical analysis
Statistics was carried out using GraphPad Prism Software Version 5 (GraphPad Software, San Diego, California, USA). Significance was calculated using the unpaired two-tailed Student’s t-test, Mann-Whitney test and One-way ANOVA followed by Bonferroni or Tukey-post hoc test as appropriate. P < 0.05 was considered significant and results expressed as the mean ± standard error (SEM).
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Croxen MA & Finlay BB. Molecular mechanisms of Escherichia coli pathogenicity. Nat Rev Microbiol 2013;8 : 26–38.
2. Khan MA, Bouzari S, Ma C, Rosenberger CM, Bergstrom KS, Gibson DL, et al. Flagellin-dependent and-independent inflammatory responses following infection by Enteropathogenic Escherichia coli and Citrobacer rodentium. Infect Immun 2008;76 : 1410–1422. doi: 10.1128/IAI.01141-07 18227166
3. Michail SK, Halm DR & Abernathy F.. Enteropathogenic Escherichia coli: stimulating neutrophil migration across a cultured intestinal epithelium without altering transpetihelial conductance. J Pediatr Gastroenterol Nutr 2003;36 : 253–260. 12548063
4. Savkovic SD, Koutsouris A & Hecht G. Activation of NF-κB in intestinal epithelial cells by enteropathogenic Escherichia coli. Am J Physiol 1997;273:C1160–1167. 9357759
5. Savkovic SD, Koutsouris A & Hecht G. Attachment of a noninvasive enteric pathogen, enteropathogenic Escherichia coli, to cultured human intestinal epithelial monolayers induces transmigration of neutrophils. Infect Immun 1996;64 : 4480–4487 8890195
6. Mundy R, MacDonald TT, Dougan G, Frankel G & Wiles S. Citrobacter rodentium of mice and man. Cell Microbiol 2005;7 : 1697–1706. 16309456
7. Mundy R, Girard F, FitzGerald AJ & Frankel G. Comparison of colonization dynamics and pathology of mice infected with enteropathogenic Escherichia coli, enterohaemorrhagic E. coli and Citrobacter rodentium. FEMS Microbiol Lett 2006;265 : 126–132. 17034412
8. Ryz NR, Patterson S, Zhang Y, Ma C, Huang T, Bhinder G, et al. Active vitamin D increases host susceptibility to Citrobacter rodentium induced colitis by suppressing mucosal Th17 responses. Am J Physiol Gastrointest Liver Physiol 2012;303:G1299–311. doi: 10.1152/ajpgi.00320.2012 23019194
9. Basu R, O'Quinn DB, Silberger DJ, Schoeb TR, Fouser L, Ouyang W, et al. Th22 cells are an important source of IL-22 for host protection against enteropathogenic bacteria. Immunity 2012;37 : 1061–75. doi: 10.1016/j.immuni.2012.08.024 23200827
10. Mangan PR, Harrington LE, O'Quinn DB, Helms WS, Bullard DC, Elson CO et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature 2006;441 : 231–234. 16648837
11. Simmons CP, Clare S, Ghaem-Maghami M, Uren TK, Rankin J, Huett A, et al. Central role for B lymphocytes and CD4+ T cells in immunity to infection by the attaching and effacing pathogen Citrobacter rodentium. Infect Immun 2003;71 : 5077–86. 12933850
12. Bergstrom KS, Guttman JA, Rumi M, Ma C, Bouzari S, Khan MA et al. Modulation of intestinal goblet cell function during infection by an attaching and effacing bacterial pathogen. Infect Immun 2008;76 : 796–811. 17984203
13. Chan JM, Bhinder G, Sham HP, Ryz N, Huang T, Bergstrom KS, et al. CD4+ T cells drive goblet cell depletion during Citrobacter rodentium infection. Infect Immun 2013;81 : 4649–58. doi: 10.1128/IAI.00655-13 24101690
14. Paul G, Marchelletta RR, McCole DF & Barrett KE. Interferon-γ alters downstream signaling originating from epidermal growth factor receptor in intestinal epithelial cells: functional consequences for ion transport. J Biol Chem 2012;287 : 2144–2155. doi: 10.1074/jbc.M111.318139 22069319
15. O'Loughlin EV, Pang GP, Noltorp R, Koina C, Batey R & Clancy R. Interleukin 2 modulates ion secretion and cell proliferation in cultured human small intestinal enterocytes. Gut 2001;49 : 636–643. 11600465
16. Papapietro O, Teatero S, Thanabalasuriar A, Yuki KE, Diez E, Zhu L, et al. R-spondin 2 signalling mediates susceptiblity to fatal infecitous diarrhoea. Nat commun. 2013;4 : 1898. doi: 10.1038/ncomms2816 23695692
17. Medzhitov R. Damage control in host-pathogen interactions. Proc Natl Acad Sci USA 2009;106 : 15525–15526. doi: 10.1073/pnas.0908451106 19805209
18. Gibson DL, Ma C, Rosenberger CM, Bergstrom KS, Valdez Y, Huang JT, et al. Toll-like receptor 2 plays a critical role in maintaining mucosal integrity during Citrobacter rodentium-induced colitis. Cell Microbiol 2008;10 : 388–403. 17910742
19. Gibson DL, Montero M, Ropeleski MJ, Bergstrom KS, Ma C, Ghosh S, et al. Interleukin-11 reduces TLR4-induced colitis in TLR2-deficient mice and restores intestinal STAT3 signalling. Gastroenterology. 2010;139 : 1277–1288. doi: 10.1053/j.gastro.2010.06.057 20600022
20. Sham HP, Yu EY, Gulen MF, Bhinder G, Stahl M, Chan JM, et al. SIGIRR, a negative regulator of TLR/IL-1R signalling promotes Microbiota dependent resistance to colonization by enteric bacterial pathogens. PLoS Pathog 2013;9:e1003539. doi: 10.1371/journal.ppat.1003539 23950714
21. Kim YS & Ho SB. Intestinal Goblet Cells and Mucins in Health and Disease: Recent Insights and Progress. Curr Gastroenterol Rep. 2010;12 : 319–30. doi: 10.1007/s11894-010-0131-2 20703838
22. Johansson ME, Holmen Larsson JM & Hansson GC. The two mucus layers of colon are organized by the MUC2 mucin, whereas the outer layer is a legislator of host-microbial interactions. Proc Natl Acad Sci USA 2011;108 Suppl1 : 4659–4665.
23. Bergstrom KS, Kissoon-Singh V, Gibson DL, Ma C, Montero M, Sham HP, et al. Muc2 protects against lethal infectious colitis by disassociating pathogenic and commensal bacteria from the colonic mucosa. PLoS Pathog 2010;6:e1000902. doi: 10.1371/journal.ppat.1000902 20485566
24. Hogan SP, Seidu L, Blanchard C, Groschwitz K, Mishra A, Karow ML, et al. Resistin-like molecule beta regulates innate colonic function: barrier integrity and inflammation susceptibility. J Allergy Clin Immunol 2006;118 : 257–268. 16815164
25. Artis D, Wang ML, Keilbaugh SA, He W, Brenes M, Swain GP, et al. RELMbeta/FIZZ2 is a goblet cell-specific immune-effector molecule in the gastrointestinal tract. Proc Natl Acad Sci USA 2004;101 : 13596–13600. 15340149
26. Rajala MW, Obici S, Scherer PE & Rossetti L. Adipose-derived resistin and gut-derived resistin-like molecule-beta selectively impair insulin action on glucose production. J Clin Invest 2003;111 : 225–230. 12531878
27. Steppan CM, Brown EJ, Wright CM, Bhat S, Banerjee RR, Dai CY, et al. A family of tissue-specific resistin-like molecules. Proc Natl Acad Sci USA 2001;98 : 502–506. 11209052
28. Hergert B, Grambow E, Butschkau A & Vollmar B. Effects of systemic pretreatment with CpG oligodeoxynucleotides on skin wound healing in mice. Wound Repair Regen 2013;21 : 723–9. doi: 10.1111/wrr.12084 23927054
29. Jang JC, Chen G, Wang SH, Barnes MA, Chung JI, Camberis M, et al. Macrophage-derived human resistin is induced in multiple helminth infections and promotes inflammatory monocytes and increased parasite burden. PLoS Pathog 2015;11:e1004579. doi: 10.1371/journal.ppat.1004579 25568944
30. He W, Wang ML, Jiang HQ, Steppan CM, Shin ME, Thurnheer MC, et al. Bacterial colonization leads to the colonic secretion of RELMbeta/FIZZ2, a novel goblet cell-specific protein. Gastroenterology 2003;125 : 1388–1397. 14598255
31. Barnes SL, Vidrich A, Wang ML, Wu GD, Cominelli F, Rivera-Nieves J, et al. Resistin-like molecule beta (RELMbeta/FIZZ2) is highly expressed in the ileum of SAMP1/YitFc mice and is associated with initiation of ileitis. J Immunol 2007;179 : 7012–7020. 17982092
32. McVay LD, Keilbaugh SA, Wong TMH, Kierstein S, Shin ME, Lehrke M, et al. Absence of bacterially induced RELMbeta reduces injury in the dextran sodium sulfate model of colitis. The Journal of Clinical Investigation 2006;116 : 2914–2923. 17024245
33. Nair MG, Guild KJ, Du Y, Zaph C, Yancopoulos GD, Valenzuela DM, et al. Goblet cell-derived resistin-like molecule beta augments CD4+ T cell production of IFN-gamma and infection-induced intestinal inflammation. J Immunol 2008;181 : 4709–4715. 18802073
34. Herbert DR, Yang JQ, Hogan SP, Groschwitz K, Khodoun M, Munitz A, et al. Intestinal epithelial cell secretion of RELM-beta protects against gastrointestinal worm infection. J Exp Med 2009;206 : 2947–2957. doi: 10.1084/jem.20091268 19995957
35. Bhinder G, Stahl M, Sham HP, Crowley SM, Morampudi V, Dalwadi U, et al. Intestinal epithelium-specific MyD88 signaling impacts host susceptibility to infectious colitis by promoting protective goblet cell and antimicrobial responses. Infect Immun 2014;82 : 3753–63. doi: 10.1128/IAI.02045-14 24958710
36. Scholzen T & Gerdes J. The Ki-67 protein: from the known and the unknown. J Cell Physiol 2000;182 : 311–322. 10653597
37. Veitch AM, Kelly P, Zulu IS, Segal I & Farthing MJ. Tropical enteropathy: a T-cell-mediated crypt hyperplastic enteropathy. Eur J Gastroenterol Hepatol 2001;13 : 1175–1181. 11711773
38. Dickson BC, Streutker CJ & Chetty R. Coeliac disease: an update for pathologists. J Clin Pathol 2006;59 : 1008–1016. 17021129
39. Kunkel EJ & Butcher EC. Chemokines and the tissue-specific migration of lymphocytes. Immunity 2002;16 : 1–4. 11825560
40. Mishra A, Wang M, Schlotman J, Nikolaidis NM, DeBrosse CW, Karow ML, et al. Resistin-like molecule-beta is an allergen-induced cytokine with inflammatory and remodeling activity in the murine lung. Am J Physiol Lung Cell Mol Physiol 2007;293:L305–313. 17545488
41. Zheng Y, Valdez PA, Danilenko DM, Hu Y, Sa SM, Gong Q, et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med. 2008;14 : 282–9. doi: 10.1038/nm1720 18264109
42. Krimi RB, Kotelevets L, Dubuquoy L, Plaisancié P, Walker F, Lehy T, et al. Resistin-like molecule β regulates intestinal mucous secretion and curtails TNBS-induced colitis in mice. Inflam Bowel Dis 2008;14 : 931–941.
43. McDole JR, Wheeler LW, McDonald KG, Wang B, Konjufca V, Knoop KA, et al. Goblet cells deliver luminal antigen to CD103+ dendritic cells in the small intestine. Nature 2012;483 : 345–9. doi: 10.1038/nature10863 22422267
44. Vallance BA, Deng W, Knodler LA & Finlay BB. Mice lacking T and B Lymphocytes develop transient colitis and crypt hyperplasia yet suffer impaired bacterial clearance during Citrobacter rodentium infection. Infect Immun 2002;70 : 2070–2081. 11895973
45. Cliffe LJ, Humphreys NE, Lane TE, Potten CS, Booth C & Grencis RK. Accelerated intestinal epithelial cell turnover: a new mechanism of parasite expulsion. Science 2005;308 : 1463–5. 15933199
46. Schneider DS & Ayres JS. Two ways to survive infection: what resistance and tolerance can teach us about treating infectious diseases. Nat Rev Immunol 2008;8 : 889–895. doi: 10.1038/nri2432 18927577
47. Shim E, Bang BR, Kang S, Ma J, Otsuka M, Kang J, et al. Activation of p38α in T cells regulates the intestinal host defense against attaching and effacing bacterial infections. J Immunol 2013;191 : 2764–2770. doi: 10.4049/jimmunol.1300908 23918973
48. Sonnenberg GF, Monticelli LA, Elloso MM, Fouser LA & Artis D. CD4+ lymphoid tissue inducer cells promote innate immunity in the gut. Immunity 2011;34 : 122–134. doi: 10.1016/j.immuni.2010.12.009 21194981
49. Osborne LC, Joyce KL, Alenghat T, Sonnenberg GF, Giacomin PR, Du Y, et al. Resistin-like molecule α promotes pathogenic Th17 cell responses and bacterial-induced intestinal inflammation. J Immunol 2013;190 : 2292–300. doi: 10.4049/jimmunol.1200706 23355735
50. Gibson DL, Ma C, Bergstrom KS, Huan JT, Man C & Vallance BA. MyD88 signalling plays a critical role in host defence by controlling pathogen burden and promoting epithelial cell homeostasis during Citrobacter rodentium-induced colitis. Cell Microbiol 2008;10 : 618–631. 17979981
51. Wiles S, Dougan G & Frankel G. Emergence of a 'hyperinfectious' bacterial state after passage of Citrobacter rodentium through the host gastrointestinal tract. Cell Microbiol 2005;7 : 1163–1172. 16008583
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2015 Číslo 8
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Human Non-neutralizing HIV-1 Envelope Monoclonal Antibodies Limit the Number of Founder Viruses during SHIV Mucosal Infection in Rhesus Macaques
- Type VI Secretion System Toxins Horizontally Shared between Marine Bacteria
- Illuminating Targets of Bacterial Secretion
- Are Human Intestinal Eukaryotes Beneficial or Commensals?
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy