
The GAP Activity of Type III Effector YopE Triggers Killing of in Macrophages
The type III secretion system (T3SS) is a macromolecular protein export pathway found in gram-negative bacteria. It delivers bacterial toxins into eukaryotic cells to promote pathogenic infection. T3SSs and the bacterial toxins delivered are critical arsenals for many bacterial pathogens of clinical significance, such as Yersinia, Salmonella and Shigella. On the other hand, the mammalian immune system may recognize the T3SS as a danger signal to signify pathogenic infection, and to stimulate appropriate defense against pathogens. Here, we show that the innate immune system recognizes the activity of YopE delivered by the Yersinia T3SS. Modulation of host Rho GTPases by YopE elicits a defensive response, which results in killing of bacteria in host cells. Inhibition of host Rho GTPases by Clostridium difficile Toxin B, another bacterial toxin, mimics the YopE-triggered killing effect. Our study demonstrates that host cells sense manipulation of Rho GTPases by bacterial toxins as a surveillance mechanism, revealing new insights into innate immune recognition of pathogenic infections.
Published in the journal:
. PLoS Pathog 10(8): e32767. doi:10.1371/journal.ppat.1004346
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004346
Summary
The type III secretion system (T3SS) is a macromolecular protein export pathway found in gram-negative bacteria. It delivers bacterial toxins into eukaryotic cells to promote pathogenic infection. T3SSs and the bacterial toxins delivered are critical arsenals for many bacterial pathogens of clinical significance, such as Yersinia, Salmonella and Shigella. On the other hand, the mammalian immune system may recognize the T3SS as a danger signal to signify pathogenic infection, and to stimulate appropriate defense against pathogens. Here, we show that the innate immune system recognizes the activity of YopE delivered by the Yersinia T3SS. Modulation of host Rho GTPases by YopE elicits a defensive response, which results in killing of bacteria in host cells. Inhibition of host Rho GTPases by Clostridium difficile Toxin B, another bacterial toxin, mimics the YopE-triggered killing effect. Our study demonstrates that host cells sense manipulation of Rho GTPases by bacterial toxins as a surveillance mechanism, revealing new insights into innate immune recognition of pathogenic infections.
Introduction
Innate immunity provides an early and critical protection against pathogenic infection. In the dominant paradigm of innate immunity, host cells detect pathogens by recognition of “microorganism-associated molecular patterns” (MAMPs) via pattern recognition receptors (PRRs) [1]. However, MAMPs, such as flagellin or lipopolysaccharide (LPS), are conserved microbial structures found in both pathogenic and nonpathogenic bacteria. How then do host cells distinguish pathogens from innocuous microbes? Alternate theories propose that, in addition to MAMPs, host cells also respond to distinct pathogen-induced signals, termed “patterns of pathogenesis” [2]–[4]. Several recent studies have demonstrated that host cells sense the activities of bacterial effectors, such as inhibition of host protein synthesis, activation of host Rho GTPases or pore forming activity, resulting in an active response against the pathogenic attack [5]–[10]. The protective immune response that is triggered by the detection of microbial effectors is defined as an “effector-triggered immune response” (ETIR).
In the genus of Yersinia, three species are pathogenic for humans: Yersinia pestis, Yersinia pseudotuberculosis and Yersinia enterocolitica. Y. pestis is the causative agent of plague and is typically transmitted by fleabites or aerosols [11], [12]. Y. pseudotuberculosis and Y. enterocolitica are associated with self-limiting gastroenteritis acquired from contaminated food or water [11]. The virulence of pathogenic Yersinia requires a plasmid (pYV in Y. pseudotuberculosis), which encodes a T3SS and a suite of effectors named Yersinia outer proteins (Yops) [13]. Upon Yersinia infection, Yop effectors are translocated into host cells by the T3SS to modulate host signaling pathways [13]. Four Yop effectors act to target Rho GTPases by distinct mechanisms: YopE mimics the eukaryotic GTPase activating protein (GAP) and promotes GTP hydrolysis to inhibit Rho GTPase activation; YopH, a protein tyrosine phosphatase, impacts Rho GTPase activation by interrupting activating signals for guanine exchange factors (GEFs); YopT, a cysteine protease, proteolytically removes the C-terminal isoprenoid moiety of Rho GTPases, therefore releasing their membrane anchors; YpkA can bind to Rho GTPases with a guanine dissociation inhibitor (GDI) domain [13]. By disturbing Rho GTPase activity, YopE, YopH, YopT and YpkA exert a negative effect on cytoskeleton dynamics, thus contributing to the anti-phagocytic activity of the Yersinia T3SS. In addition, YopJ inhibits NF-κB and MAPK signaling pathways, while YopK regulates effector delivery as well as host responses [10]. Translocators YopB and YopD are required for the formation of the T3SS channel and delivery of effector Yops.
The prototypical bacterial effector YopE is a 219 amino acid protein containing a Rho GAP domain (residues 96 to 219) [14]. YopEGAP shares homology with SptPGAP from Salmonella Typhimurium and ExoSGAP from Pseudomonas aeruginosa. YopE introduces an “arginine finger” into the GTPases catalytic site, which results in efficient GTP hydrolysis and deactivation of GTPases. Exchanging Arg144 in the “arginine finger” with an alanine residue abolishes YopE GAP activity [14]. In mammalian cells, YopE localizes to plasma membrane and unidentified perinuclear compartments, which requires a hydrophobic leucine-rich motif within its membrane localization domain (MLD, residues 53 to 79) [15]–[18]. Stability of YopE in host cells is influenced by allelic variations of residues 62 and 75, as found in different Yersinia strains. The presence of lysine residues at position 62 or 75 can mediate YopE ubiquitination and degradation by the host cell proteasome pathway [19]. Both subcellular membrane localization and stability of YopE are important for its GAP activity [16], [19]. YopE is equally effective on Rac1, RhoA and Cdc42 in vitro [14], whereas it is preferably active on Rac1 and RhoA, but not Cdc42, in vivo [20]. Unlike YopE, YopT seems to be primarily effective on RhoA, but not Rac1 or Cdc42 in vivo [21]. However, overexpressed YopT also acts on Rac1 in Yersinia-infected epithelial cells [22]. Interestingly, under the latter condition, YopT competes with YopE for the same pool of membrane-associated Rac1, promotes translocation of cleaved Rac1 into the nucleus, and therefore interferes with the ability of YopE to inactivate Rac1 [22].
Yersinia is generally considered as an extracellular pathogen, as the bacteria grow primarily in an extracellular form in vivo; however, these bacteria can survive and grow inside phagocytic cells, which may be important for the early stages of colonization [23]. It is suggested that macrophages might serve as permissive sites for bacterial replication or even as transport vehicles from the initial site of infection to deeper lymph tissues [24]. Interestingly, T3SS function decreases survival of Y. pseudotuberculosis in murine macrophages. Under experimental conditions in which T3SS expression is pre-induced, macrophages restrict intracellular survival of wild-type Y. pseudotuberculosis, but not a yopB− mutant (deficient in Yops translocation) or a pYV− mutant (missing the entire T3SS) [25]. Thus, some T3SS-dependent factor encoded in the wild-type strain triggers a bactericidal response in macrophages, the mechanism of which remains unclear. It has been shown that upon internalization of Y. pseudotuberculosis, the T3SS stimulates Ca2+-dependent phagolysosome fusion in macrophages, mediated by the Ca2+ sensor SytVII, leading to increased killing of intracellular bacteria [26]. Also, it has been reported that the Y. pseudotuberculosis T3SS stimulates Ca2+ - and caspase-1-dependent lysosome exocytosis, releasing antimicrobial factors [27]. Yet, further studies are needed to determine the molecular basis of innate immune recognition of the Yersinia T3SS, and the role of this process in determining the fate of the bacteria in macrophages.
Here we hypothesize that the activities of the Yersinia T3SS effectors are sensed by host cells as patterns of pathogenesis, which stimulate an intracellular killing response against Yersinia as a type of ETIR. We show that macrophages recognize pathogenic Y. pseudotuberculosis through T3SS functions and elicit an intracellular killing response to counteract infection. We provide evidence that YopE GAP activity is a critical factor sensed by macrophages, with YopH playing a minor role. Overexpression of YopT counteracts the YopE-triggered killing effect possibly by competing for the Rho GTPase target and by reducing YopE translocation. Also, this YopE-triggered intracellular killing response can be mimicked by other bacterial derived toxins like Clostridium difficile Toxin B, indicating that host cells sense manipulation of Rho GTPases as a conserved surveillance pathway to detect pathogens. Thus, our data provide another example of a protective host response induced by pathogenic bacteria through recognition of bacterial effector activities on Rho GTPases, revealing a novel mode of innate immune recognition towards pathogenic infection.
Results
YopE and YopH restrict survival of Yersinia inside macrophages
Previous studies have shown that T3SS decreases survival of Y. pseudotuberculosis in murine macrophages [25], [26]. To determine if specific Yop effectors might contribute to decreased survival of Y. pseudotuberculosis in macrophages, the wild-type strain IP2666 and several yop deletion mutants were studied. Initially, the survival of IP2666 (wild-type), IP17 (yopEH−), IP27 (yopEHJ−) and IP37 (yopEHJMKYpkA−) (Table 1) in murine bone marrow-derived macrophages (BMDMs) was compared. Naïve BMDMs were infected with the indicated strains, followed by gentamicin treatment to eliminate extracellular bacteria. At 1 h and 23 h post infection, infected BMDMs were lysed and spread on LB plates to enumerate viable bacteria. CFU at 1 h post infection was considered as the initial intracellular bacterial count. The ratio of CFU between 23 h and 1 h post infection was calculated for each strain. At 1 h post infection, IP2666 showed lower CFU as compared to IP17, IP27 and IP37 (Figure S1A). This is expected because IP2666 expresses Yops with anti-phagocytic functions (YopE and YopH); however the other strains are yopEH− mutants. At 23 h post infection, IP17 displayed significantly higher level of CFU as compared to IP2666, but similar level as compared to IP27 and IP37 (Figure S1B). Consistently, for the ratio of CFU at 23 h/1 h, the level of IP17 was significantly higher than IP2666, but similar to IP27 and IP37 (Figure 1A). To rule out a threshold effect due to the differences in the initial bacterial uptake, BMDMs were infected with IP2666 or IP17 at different MOIs (Figure S2). Even with higher CFU at 1 h post infection, IP2666 (MOI = 10) still showed decreased survival in comparison to IP17 (MOI = 5 or 2.5) at 23 h post infection (Figure S2AB). Therefore, IP2666 shows reduced intracellular survival, in contrast to IPI7, which shows an intracellular growth phenotype, indicating that deletion of yopE and yopH promotes Yersinia survival inside macrophages.


To further elucidate the effects of YopE and YopH on intracellular survival of Yersinia, IP2666 (wild-type), IP6 (yopE−), IP15 (yopH−) and IP17 (yopEH−) (Table 1) were compared by CFU assay (Figure 1B). IP6 showed an intracellular growth phenotype similar to IP17, while IP15 had an intermediate phenotype (Figure 1B). The results were further confirmed by fluorescence microscopy. IP2666, IP6 and IP17 encoding GFP were used to infect BMDMs for different lengths of time. One hour before fixation and examination of the samples by fluorescence microscopy, IPTG was added to induce de novo expression of GFP from viable intracellular bacteria (Figure 2). At 23 h post infection, IP6 and IP17 showed greater survival compared to IP2666 (Figure 2). These results demonstrate that YopE is required for reduced survival of Yersinia in macrophages, and YopH cooperates with YopE in this process.

To determine whether SytVII-mediated phagolysosome fusion contributes to YopE-dependent intracellular killing, SytVII−/− BMDMs were compared to wild-type BMDMs for their ability to restrict intracellular survival of IP2666, IP17 or IP40 (yopB mutant, Table 1). The SytVII−/− genotype was verified by PCR using mouse-tail genomic DNA, in comparison to wild-type mice (Figure S3A). No significant difference was observed by CFU assay for IP2666 survival inside wild-type or SytVII−/− BMDMs (Figure S3B), suggesting that SytVII-mediated phagolysosome fusion does not contribute to the YopE-dependent killing of Yersinia in macrophages.
Under our experimental conditions, Yersinia infection does not cause significant cell death of macrophages (below 2% LDH release after 23 h from IP2666 or IP6 infected macrophages, data not shown). Accordingly, reduced intracellular survival of IP2666 is not due to enhanced Yersinia-induced macrophage cell death. We also investigated the possibility that IP2666 infection induces gentamicin uptake and leads to enhanced bacterial killing by gentamicin. If this is true, with increasing amount of gentamicin, intracellular IP2666 would be more sensitive than IP17, due to more gentamicin uptake. To analyze this possibility, the survival of IP2666 and IP17 in macrophages was compared with increasing amount of gentamicin. IP2666 and IP17 responded similarly to increasing amounts of gentamicin (Figure S4), indicating that reduced intracellular survival of IP2666 is not due to increased gentamicin internalization.
GAP activity of YopE is essential for reduced survival of Yersinia in macrophages
To investigate if the GAP activity of YopE is crucial for macrophages to restrict survival of intracellular Yersinia, experiments were carried out to compare survival of bacteria producing YopE or YopER144A. In YopER144A, a single substitution of arginine to alanine was introduced at amino acid 144 to yield a catalytically dead protein [14]. A plasmid vector encoding yopE or yopER144A was introduced into IP6 (Table 1). The production level of YopE or YopER144A from the vector in trans was similar to the native level in the wild-type strain as shown by SDS-PAGE and immunoblotting (Figure 3A, compare lanes 1, 3 and 4). The survival of IP6+pYopE and IP6+pYopER144A in macrophages was then compared. IP2666 or IP6 containing the empty vector (Table 1) were analyzed in parallel as controls. IP6+pYopE displayed a reduced intracellular survival phenotype, similar to IP2666+empty vector (Figure 3B). In contrast, IP6+pYopER144A showed increased intracellular survival, comparable to IP6+empty vector (Figure 3B). Unexpectedly, the empty vector (pMMB67HE) had a negative effect on Yersinia survival inside macrophages (Figure S5), possibly due to the metabolic burden introduced by the plasmid [28], [29]. Nevertheless, these results demonstrate that YopE GAP activity is indispensable for causing reduced survival of Yersinia in macrophages, and we hypothesize that YopE GAP activity is somehow recognized by macrophages, triggering increased killing of intracellular Yersinia.

Overexpression of YopT counteracts YopE-triggered killing of Yersinia in macrophages
Given the activity of YopT towards Rho GTPases and its crosstalk with YopE, the potential influence of YopT on survival of Yersinia inside macrophages was studied. IP2666 is a yopT mutant due to a naturally-occurring deletion in pYV in this strain [30]. Plasmids that overexpress YopT or catalytically-inactive YopTC139S were introduced into IP2666; control strains containing the empty vector or a plasmid producing native levels of YopT under its native promoter were also constructed (Table 1). Analysis of proteins secreted by the bacteria under low calcium conditions using SDS-PAGE and immunoblotting showed that YopT and YopTC139S were overproduced at equal levels, while the native level of YopT was undetectable (Figure 4A, compare lanes 2, 3 and 4). Interestingly, when these strains were used to infect macrophages, overexpression of YopT in IP2666 significantly increased Yersinia intracellular survival, giving the opposite effect of YopE (Figure 4B and Figure S6A). Yersinia survival in macrophages was moderately increased when YopTC139S was overexpressed in IP2666 (Figure 4B and Figure S6A), indicating that YopT catalytic activity is important for counteracting the YopE-triggered killing effect. Expression of YopT at native level in IP2666 also slightly improved Yersinia intracellular survival (Figure 4B and Figure S6A).

Using detergent extraction assay and immunoblotting, lysates of infected macrophage were analyzed to detect the amounts of YopE that were translocated by the different strains. Overexpression of YopT or YopTC139S in IP2666 diminished YopE translocation to 8% or 25% of wild-type level respectively (Figure S7AB). Native level of YopT in IP2666 slightly reduced YopE translocation (75% of wild-type level) (Figure S7AB).
Active and inactive YopT proteins were overexpressed in IP6 or IP37 to further examine the mechanism by which this effector counteracts killing of Yersinia in macrophages. Overexpression of YopT or YopTC139S in IP6 equally enhanced bacterial survival (Figure S6B), while overexpression of active or inactive YopT proteins in IP37 had no effect on Yersinia survival inside macrophages (Figure S6C).
Taken together, these results suggest that YopT has the ability to counteract YopE-triggered intracellular killing effect, which is partially dependent on YopT protease activity. YopT catalytic activity may counteract the YopE effect by competing with YopE for a Rho GTPase target or by reducing YopE translocation. Thus, inactivation of a Rho GTPase by a specific mechanism, i.e. GAP mechanism, appears to be sensed by macrophages, resulting in increased killing of intracellular Yersinia.
Membrane localization, stability and target specificity of YopE are important for killing of Yersinia in macrophages
Localization to membranes and stability of YopE are critical for functional GAP activity against Rho GTPases in host cells [15], [16], [19]. Since our results suggest that YopE GAP activity is sensed by macrophages, we hypothesize that membrane localization and stability of YopE will impact its ability to stimulate an intracellular killing response. To examine this possibility, plasmids encoding YopE variants that were defective for membrane localization (YopE3N) [17] or less stable (YopER62K) [19] were introduced into IP6. The resulting strains (Table 1) were used to infect macrophages and detergent extraction assays were used to compare the amounts of YopE, YopE3N and YopER62K that were translocated. The yopB mutant IP40, which is defective for Yop translocation, was used to infect macrophages as a negative control. The amount of YopE3N in the soluble fraction was comparable to wild-type YopE, indicating equal translocation of these proteins (Figure 5A, compare lanes 3 and 1). Some YopE proteins with reduced Rho GAP activity are translocated at higher levels as compared to the wild-type protein into epithelial cells infected with Y. pseudotuberculosis [31]. We did not observe hypertranslocation of YopE proteins with reduced Rho GAP activity in our experiments, possibly because YopE has a reduced ability to negatively regulate its own translocation into macrophages as compared to epithelial cells. The amount of YopER62K in the soluble fraction was lower compared to wild-type YopE, probably due to decreased stability as a result of increased ubiquitination (Figure 5A, compare lanes 5 and 1). The appearance of a slower migrating band for YopER62K was consistent with ubiquitination (Figure 5A, lane 5). IP6+YopE3N and IP6+YopER62K displayed improved survival in macrophages in comparison to IP6+YopE at 24 h post infection, as determined using immunofluorescence microscopy to detect intracellular Yersinia (Figure 5B). The results, quantified by the percentage of macrophages containing fluorescent intracellular Yersinia (Figure 5C), or CFU assay (Figure S8A), showed that IP6+YopE3N and IP6+YopER62K had increased bacterial survival compared to IP6+YopE at 24 h post infection. Since membrane localization and stability are important for YopE to efficiently inactivate Rho GTPases, these results provide additional evidence that macrophages sense the inactivation of one or more Rho GTPases, which results in killing of intracellular Yesinia.

YopE variants with altered Rho GTPase specificities [31] were compared to wild-type YopE for their capability to trigger intracellular killing response. YopEL109A has lower GAP activity towards RhoA (70% of wild-type level) and Rac2 (70% of wild-type level); YopE-SptP fusion protein, which contains the secretion and translocation domains of YopE and the GAP domain of SptP, has no GAP activity towards RhoA and decreased activity towards Rac1 (83% of wild-type level) and Rac2 (34% of wild-type level) [31]. The amounts of translocated YopEL109A and YopE-SptP were comparable to wild-type YopE in Yersinia-infected macrophages, and YopE-SptP displayed reduced mobility as expected due to its higher molecular weight (Figure 5A, compare lanes 1, 2 and 4). At 24 h post infection, IP6+YopEL109A and IP6+YopE-SptP showed improved survival inside macrophages compared to IP6+YopE, as demonstrated by immunofluorescence microscopy (Figure 6AB) and CFU assays (Figure S8B). These results indicate that the specificity of YopE GAP activity may impact its ability to trigger the intracellular killing. However, the results obtained with the YopEL109A and YopE-SptP variants did not reveal if inactivation of a specific Rho GTPase by YopE is important for intracellular killing. Since the activity of YopEL109A and YopE-SptP towards Rho GTPases other than RhoA, Rac1 and Rac2 is not clearly known, it is difficult to declare YopE interruption of which specific Rho GTPase is essential for macrophage recognition and intracellular killing.

Toxin B decreases Yersinia survival inside macrophages
To explore if this intracellular killing response applies to bacterial toxins targeting Rho GTPases, Clostridium difficile Toxin B was added to Yersinia infected macrophages. Toxin B has been well characterized to inactivate a wide range of Rho GTPases through glycosylation, including Rac1, RhoA/B/C, RhoG, TC10, and Cdc42 [32]–[34]. With Toxin B treatment, the survival of IP6, IP17 and IP40 was dramatically decreased as revealed by CFU assays (Figure 7A and B) and fluorescence microscopy in conjunction with mCherry induction (Figure 7C). Toxin B did not affect Yersinia growth in tissue culture media in the absence of macrophages; Toxin B did not cause significant cytotoxicity in macrophages in these experiments (data not shown). These results suggest that down-regulation of Rho GTPases by Toxin B is perceived by macrophages, inducing an intracellular killing response, mimicking the effect of YopE. Thus, the bactericidal effect triggered by Rho GTPase-inactivating toxins may be a general and conserved response to these bacterial toxins. In addition, the fact that Toxin B decreases IP40 survival inside macrophages implies that T3SS translocon is not essential for macrophage recognition of Rho GTPases-inactivating toxins to cause a bactericidal response.

Rac inhibition, but not Rho inhibition, decreases Yersinia survival inside macrophages
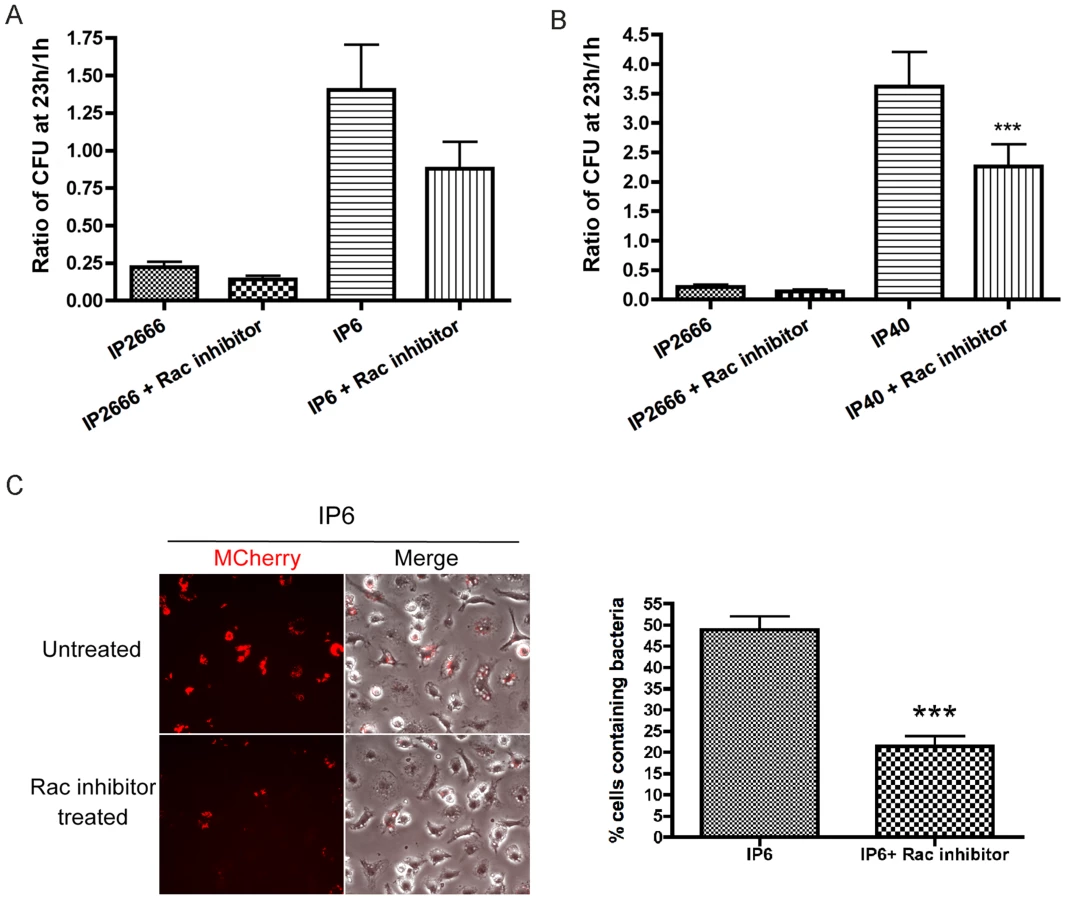
To identify the Rho GTPase target of YopE critical for causing intracellular killing, specific Rho GTPase inhibitors were studied for their capability to mimic the YopE effect. Treatment with Rac1 inhibitor NSC23766 negatively impacted IP6 and IP40 survival inside macrophages, as demonstrated by CFU assays (Figure 8AB) and fluorescence microscopy with mCherry induction (Figure 8C). The Rac1 inhibitor triggered a reduced bactericidal effect in comparison to Toxin B in the CFU assay (compare Figure 7AB and 8AB). In contrast to the Rac1 inhibitor, the RhoA inhibitor TAT-C3 did not significantly affect Yersinia survival inside macrophages (Figure S9BC). Dramatic morphological changes were observed in TAT-C3 treated macrophages as early as 4 h upon treatment, confirming the efficiency of TAT-C3 towards RhoA (Figure S9A). These results signify that macrophages restrict Yersinia intracellular survival in response to Rac1 inhibition, but not to Rho inhibition.
Presence of YopE induces higher levels of nitric oxide from infected macrophages
Several recent studies have shown that the activities of certain bacterial effectors can stimulate transcriptional changes in host cells, resulting in ETIRs [5]–[9]. For example, activation of Rac1 and Cdc42 by SopE from Salmonella enterica serovar Typhimurium is sensed through NOD1 receptor, eliciting NF-κB activation in the host cells as a protective response [9].
To study if YopE stimulates an altered host response that can occur at the transcriptional level, the production of nitric oxide (NO) from macrophages infected with IP2666, IP6, IP17 or IP40 was compared. Specifically, the concentration of nitrite (NO2−), an indicator of NO, was measured by Griess assay. At 23 h post infection, comparing to IP6-, IP17 - or IP40-infected macrophages, IP2666-infected macrophages produced significantly higher levels of NO (Figure 9). LPS - and IFN γ-treated macrophages were used as a positive control, while uninfected macrophages were used as a negative control (Figure 9).

To investigate whether YopE dependent-intracellular killing signals through NOD1 receptor, Nod1−/− BMDMs were compared to wild-type BMDMs for their ability to restrict intracellular survival of IP2666 or IP6 by CFU assay. No significant difference was observed for IP2666 survival inside wild-type or Nod1−/− BMDMs (Figure S10).
These results suggest that macrophages respond to wild-type Yersinia differently from yopE− mutant strains and produce higher levels of NO; however, YopE-triggered intracellular killing is not mediated by NOD1 receptor.
Discussion
The aims of this study were to determine T3SS-dependent factors that restrict Yersinia survival inside macrophages and characterize the mechanism of this “patterns of pathogenesis” triggered host response. Our findings reveal that primary naïve macrophages sense manipulation of Rho GTPases by Yersinia Yop effectors. Three known effector Yops directly inhibit host Rho GTPases: YopE, YpkA and YopT; a fourth effector, YopH, inhibits signals that activate these small GTPases. YopE is an important virulence factor for resistance of Yersinia to the innate immunity, as a Y. pseudotuberculosis yopE null mutant was defective for systemic spread following oral infection in the animal model [35]. However, on the other hand, here we show that YopE GAP activity towards Rho GTPases is recognized by macrophages, stimulating increased killing of intracellular Y. pseudotuberculosis (Figure 10). YopH cooperates with YopE to cause this killing effect, most likely by inhibiting a phosphotyrosine dependent signaling pathway that activates Rho GTPases in response to Yersinia infection (Figure 10). YpkA has very mild effect on Yersinia intracellular survival (data not shown), perhaps due to its low expression level in comparison to other Yops. Interestingly, we have observed that overexpression of YopT counteracts the YopE-triggered intracellular killing effect, which involves the protease activity of YopT. We speculate that an important biological function of YopT is to counteract sensing of YopE by the innate immune system, possibly by preventing YopE access to activated Rho GTPase targets or removing YopE-inactivated Rho GTPases from phagosome membranes (Figure 10). Zhang et al. studied a Y. pseudotuberculosis strain (32777), different from that used here (IP2666), and showed that a mutant of 32777 encoding catalytically inactive YopJ, YopT, YopE and YopH still triggered intracellular bacterial killing, to the same level as wild-type 32777 [25]. We speculate that 32777 has additional Rho GTPase-inactivating effector(s) causing bacterial killing, which remain to be identified.

We have obtained evidence that Toxin B decreases Yersinia survival in macrophages by inactivating several Rho GTPases (Figure 10). To date, at least 20 Rho GTPase proteins belonging to 8 subfamilies have been described in mammals [36], [37]. Interestingly, we have shown that the target preference of YopE impacts its ability to trigger bacterial killing. Thus, one intriguing question to ask is does the innate immune system monitor effector manipulation of a specific Rho GTPase? Our results showed that Rac inhibition, but not Rho inhibition, stimulates the macrophage killing response against intracellular Yersinia (Figure 10). However, the Rac inhibitor only partially promotes killing of Yersinia in macrophages in comparison to Toxin B or YopE, suggesting that disturbance of additional Rho GTPases contributes to the intracellular killing response. Other Rho GTPase candidates may include, but are not limited to, Rac2 and RhoG, which have been shown to be YopE targets and expressed in macrophages [31], [37]–[39]. Further investigation is required to reveal if additional Rho GTPases serve as surveillance points in response to pathogenic effector manipulation [37].
Rho GTPases act as molecular switches that regulate numerous cellular functions, like cytoskeletal dynamics, gene transcription, vesicular trafficking, cell growth and apoptosis [37], [40]. In order to ensure proper signaling responses, the activities of Rho GTPases are tightly regulated by multiple mechanisms, including the canonical regulators (GAPs, GEFs and GDIs) and direct post-translational modifications (like phosphorylation and ubiquitination) [40], [41]. The abnormal inactivation of Rho GTPases by YopE might interfere with multiple Rho GTPase-mediated signaling pathways and lead to many different consequences to trigger intracellular bacteria killing in macrophages. One possibility is that YopE may cause accumulation of inactivated GDP-bound Rho GTPases on phagosome (Figure 10), which could be modified by ubiquitination to stimulate signaling pathways. Activation of Rac1 by cytotoxic necrotizing factor 1 (CNF1) from Escherichia coli induces Rac1 poly - and mono-ubiquitination, the biological function of the latter remains unclear [42]. In line with this, GDP-bound RhoA is targeted by the ubiquitin E3 ligase Cullin-3 for poly-ubiquitination and degradation [43]. Thus, it is tempting to speculate that YopE-inactivated GDP-bound Rho GTPases could be mono-ubiquitinated and serve as signaling components; or they could be poly-ubiquitinated to mediate xenophagic degradation of bacteria-containing vesicles [44]. Alternatively, by modulating vesicular trafficking, YopE activity may interrupt formation of Y. pseudotuberculosis-containing autophagosomes, which have been shown to be impaired in acidification and support survival of the bacteria in macrophages [45]. On the other hand, given that the role of autophagy in Yersinia survival in macrophages is controversial [45], [46], it is possible that YopE activity promotes autophagy to eliminate intracellular bacteria. Deuretzbaher et al. showed that β1-integrin-mediated Y. enterocolitica internalization by macrophages was coupled to autophagy activation, which seemed to be deleterious for bacterial intracellular survival. Another possibility is that the disruption of the actin cytoskeleton by YopE is sensed by the innate immune system. It has been suggested that NOD receptors or inflammasome components associated with the actin cytoskeleton may act as surveillance mechanisms, becoming activated upon perturbations by pathogens [2]. Interesting, a recent study by Shao and colleagues showed that Rho-inactivating toxins such as Clostridium difficile Toxin B and Clostridium botulinum C3 trigger Pyrin inflammasome activation in BMDMs [47]. They further demonstrated that Burkholderia cenocepacia induced inactivation of Rho GTPase stimulates Pyrin inflammasome activation as an immune defense, which limits bacterial intra-macrophage growth and regulates lung inflammation in infected mice [47]. Whether YopE triggers Yersinia intracellular killing through the inflammasome pathway remains to be investigated.
The overall host cell innate immune response to a T3SS-containing bacterial pathogen is unique and multifactorial. MAMPs, the T3SS translocon channel, and the activities of bacterial effectors are likely recognized as combined pathogenic signals by the host cell. A two-signal model, requiring a MAMP and a pattern of pathogenesis, was proposed as an innate immune strategy to evaluate the virulence potential of a pathogen and adjust immune response appropriately to avoid self-damaging inflammation [48]. For example, a type IV secretion system allows Legionella pneumophila to deliver bacterial effectors into the host cell cytosol to inhibit host protein synthesis [6]. In this case, the effector-mediated interference of host protein synthesis, in concert with TLR signaling, results in prolonged activation of NF-κB as an ETIR [6]. Our data suggest that the YopBD translocon is not essential for Toxin B - or Rac inhibitor-triggered bacterial killing in macrophages. Further studies are needed to determine if TLRs or β1-integrins are possible receptors in MAMP-PRR pathways that facilitate the YopE-triggered killing effect. Alternatively, some studies in the literature support the idea that patterns of pathogenesis are sufficient to induce defense responses independently of classical MAMPs [5], [49], [50]. Boyer et al. demonstrated that Escherichia coli CNF1 elicited a vigorous ETIR in flies via activation of Rac2 and the IMD kinase pathway, which was observed even in the absence of PRR ligation [5]. Thus, it is possible that unbalanced disruption of Rho GTPases by YopE is adequate to stimulate a protective immune response, resulting in restriction of Yersinia survival in macrophages.
Roy and colleagues observed that during internalization of Salmonella enterica serovar Typhimurium or Y. pseudotuberculosis, the T3SSs of these pathogens stimulated SytVII-dependent phagolysosome fusion and bacterial killing in macrophages [26]. We have shown that YopE-triggered intracellular bacterial killing does not require SytVII, suggesting that there are at least two independent pathways by which killing of Yersinia internalized by macrophages can be stimulated. The YopE-dependent pathway senses inactivation of Rho GTPases, while the SytVII dependent pathway appears to recognize translocon insertion in the plasma membrane.
Various bacterial T3SS and T4SS effectors modulate Rho GTPase functions and interfere with corresponding host signaling pathways to benefit pathogenic infection [51]. Given that Rho GTPases play multiple roles in many signaling pathways critical for cellular functions, it is not surprising to envision surveillance mechanisms monitoring the status of Rho GTPases. Our work highlights that inactivation of Rac1, and possibly other GTPases, by YopE from Y. pseudotuberculosis is detected by macrophages as a danger signal, stimulating an ETIR that restricts intracellular bacterial survival. Detection of pathogens via Rho GTPase surveillance adds another layer of complexity to the mechanisms of innate immune recognition, improving our understanding of how the innate immune system responds to pathogenic infection.
Materials and Methods
Ethics statement
Use of mice for the preparation of BMDMs was carried out in accordance with a protocol that adhered to the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health (NIH) and was reviewed and approved (approval #206152) by the Institutional Animal Care and Use Committee at Stony Brook University, which operates under Assurance #A3011-01, approved by the NIH Office of Laboratory Animal Welfare.
Bacterial strains and plasmid constructions
The Y. pseudotuberculosis strains used in this study are shown in Table 1. These bacteria were grown on LB agar plates or in LB broth at 28°C supplemented with 100 µg/ml ampicillin, 25 µg/ml kanamycin or 30 µg/ml chloramphenicol as needed. The plasmids pMMB67HE [52], pYopE [14], pYopER144A [14], pPEYopE [30], pYopT [53], pPTYopT [30], pYopTC139S [18], p67GFP3.1 [23] and p207mCherry [54] have been previously described.
A new series of plasmids expressing yopE mutants were created as described below. Plasmids encoding yopEL109A, yopER62K and yopE3N were generated as follows. DNA fragments encoding yopEL109A, yopER62K or yopE3N were obtained by PCR using primers yopE-3 (5′-CGGATCCCATATGAAAATATCATCATTTATTTC-3′) and yopE-EcoRI (5′-CGCGGAATTCCCATATCACATCAATGACAGTAATTT-3′). Recombinant plasmid DNA (pBAD33/YopEL109A, a gift from Joan Mecsas, Tufts University), or Y. pseudotuberculosis virulence plasmid DNA (from 32777 yopER62K or 32777 yopE3N, Zhang et al. submitted) was used as template for the PCR to obtain yopEL109A, yopER62K and yopE3N, respectively. The resulting DNA fragments were inserted into pETBlue2 vector using blunt end ligation. To create a plasmid encoding the yopE-sptP fusion, a DNA fragment containing the first 100 codons of yopE (yopE1–100) was amplified from IP2666 virulence plasmid DNA with primers yopE-infusion-5 (5′-TAATAAATAGTCATATGAAAATATCATCATTTATTTCTACATCACTG-3′) and yopE-infusion-3 (5′-AGGTTGCTTACTTTCCGTAGGACTTGGCATTTGTGC-3′). A DNA fragment containing codons 166–293 of sptP (sptP166–293) was amplified with primers sptP-infusion-5 (5′-ATGCCAAGTCCTACGGAAAGTAAGCAACCTTTACTCAGTATCG-3′) and sptP-infusion-3 (5′-CAGCCAAGCTGAATTTTAGCCGGCTTCTATTTTCTCAAGTTC-3′) using chromosomal DNA from Salmonella enterica Typhimurium strain 14028 as template. A DNA fragment encoding the yopE1–100sptP166–263 fusion was made by overlapping PCR using the yopE1–100 and sptP166–293 fragments as templates and primers yopE-infusion-5 and sptP-infusion-3. The product was inserted into pETBlue2 by blunt end ligation. The sequences of the inserts in the plasmids described above were confirmed by DNA sequencing. DNA fragments encoding yopEL109A, yopER62K, yopE3N or yopE1–100sptP166–263 were obtained from the pETBlue2 vectors by digestion with NdeI and EcoRI, and ligated between the NdeI and EcoRI sites in pPEYopE, thereby replacing the wild-type yopE gene, and placing the mutant alleles under control of the native yopE promoter. The resulting plasmids pYopEL109A, pYopER62K, pYopE3N and pYopE-SptP were introduced into E. coli S17-1 λpir by electroporation and subsequently transferred into IP6 (Table 1) by conjugation as described previously [55].
Cell culture
Bone marrow-derived macrophages (BMDMs) were isolated and cultured from femurs of C57BL/6 wild-type mice (Jackson Laboratory) or SytVII−/− C57BL/6 mice (a generous gift from Dr. Norma Andrews, University of Maryland), or Nod1−/− C57BL/6 mice (a generous gift from Dr. Andreas Baumler, University of California-Davis) as previously described [56]. 24 h before infection, macrophages were seeded into 24-well tissue culture plate at a density of 1.5×105 cells/well in Dulbecco's modified Eagle medium supplemented with 10% fetal bovine serum (Hyclone), 15% L-cell conditioned medium, 1 mM sodium pyruvate and 2 mM glutamate.
Infection conditions
Y. pseudotuberculosis strains were grown at 28°C in LB broth with aeration overnight. The next day, overnight cultures were diluted 1∶40 into fresh LB broth containing 2.5 mM CaCl2 and sub-cultured at 37°C for 2 h to induce yop gene expression. Bacteria were washed once and resuspended in HBSS to obtain optical density at OD 600 nm. Next, bacteria were diluted into cell culture medium to infect macrophages at an MOI of 10, unless specified. After centrifugation for 5 min at 700 rpm to facilitate bacterial contact with macrophages, another 15 min incubation was performed at 37°C, giving the total infection time of 20 min. The end of 20 min incubation is considered as 0 h post infection. To eliminate extracellular bacteria, unless specified, the cells were then incubated in medium containing 8 µg/ml gentamicin for 1 h, and then maintained in fresh medium containing 4.5 µg/ml gentamicin until the end of incubation. When indicated, 40 ng/ml Toxin B (Calbiochem), 100 µM NSC23766 (Calbiochem), or 10 µg/ml TAT-C3 was added at 0 h post infection and maintained throughout the experiment. TAT-C3 was purified and kindly provided by Dr. Gloria Viboud, Stony Brook University [57].
CFU assay
At the time points indicated in the figures, the infected BMDMs were washed twice with HBSS, lysed and scraped with 500 µl 0.1% Triton X-100 in HBSS to release intracellular bacteria. After collecting the lysates, 500 µl HBSS was used to rinse the wells and collect any residual bacteria. The lysates and the wash were combined, serially diluted and spread on LB plates, and then incubated at 28°C for 2 days to enumerate output CFU.
Immunoblotting
The primary antibodies used were a cocktail of two monoclonal mouse anti-YopE antibodies designated 202 and 149 (unpublished data), the monoclonal mouse anti-YopH antibody designated 3D10 (a gift from Dr. Richard Siegel, NIH) diluted 1∶1000, a polyclonal rabbit anti-YopT antibody diluted 1∶500 [30], and a polyclonal rabbit anti-β-actin antibody (Cell signaling) diluted in 1∶1000. The secondary antibodies used were a goat anti-mouse antibody conjugated to IRD800 (Rockland) diluted 1∶5000 and a donkey anti-rabbit antibody conjugated to IRD800 (Rockland) diluted 1∶5000.
The protein samples were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, transferred to nitrocellulose membranes, and subjected to immunoblotting with specific primary and secondary antibodies. The membranes were then scanned and analyzed with the Odyssey system (Li-Cor Biosciences).
Fluorescence microscopy
BMDMs were prepared and infected as described above, except that they were seeded into wells with glass coverslips, which had been washed with acetone and heated at 180°C for 4 h to remove LPS. At indicated time points, infected BMDMs were washed three times with PBS and fixed with 2.5% PFA for 10 min. When needed, 0.5 mM Isopropyl-β-D-thiogalactopyranoside (IPTG) was added at 1 h before fixation to induce de novo GFP expression, or at 2 h before fixation to induce de novo mCherry expression. When indicated, immunofluorescence staining was performed as described previously [23]. Briefly, the fixed cells were permeabilized with 0.1% Triton X-100 in PBS for 1 min, followed by blocking with 3% bovine serum albumin in PBS for 10 min. The cells were then incubated with a polyclonal rabbit anti-Yersinia antibody SB349 diluted 1∶1000 for 30 min. After washing with PBS, the cells were incubated with FITC conjugated anti-rabbit antibody (Jackson Laboratories) diluted 1∶250 for 40 min. After washing, the coverslips were inverted onto 6 µl Prolong Gold anti-fade reagent (Invitrogen) on a microscope slide. The slides were examined by fluorescence microscopy using a Zeiss Axioplan2 microscope with a 32× objective. Three randomly selected fields of each slide were examined. In each field, about 50 BMDMs were examined from merged images of phase contrast and fluorescence, which were captured with a Spot camera (Diagnotic Instruments, Inc) and processed with Adobe Photoshop. Percentage of cells containing bacteria was quantified using three independent experiments.
Detergent extraction assay
Detergent extraction assays were performed as previously described in [58]. BMDMs were infected as described above, except that they were seeded in 6 well plates at a density of 8×105 cells/well and infected at an MOI of 30 for 2 h. The infected cells were washed twice with ice-cold HBSS and lysed with 50 µl 1% Triton X-100 in HBSS containing EDTA-free protease inhibitor cocktail (ROCHE). After 10 min on ice, the cells were scraped from the plate to collect the lysates. The soluble and insoluble fractions of the lysates were separated by centrifugation (14000 rpm, 10 min, 4°C) and subsequently analyzed using immunoblotting as described above.
Tail genotyping
Chromosome DNA was isolated from C57BL/6 wild-type or SytVII−/− mouse tails and used as templates for PCR amplification. Briefly, tail tips were digested in 500 µl lysis buffer (0.1M NaCl, 0.05M Tris-HCL pH 7.7, 1% SDS and 2.5 mM EDTA) with 40 µg/ml freshly added proteinase K (Sigma), and incubated at 55°C overnight. The resulting supernatant were collected and mixed with 500 µl isopropanol to precipitate chromosomal DNA. After centrifugation (14000 rpm, 10 min, RT), the pellets were washed twice with 70% ethanol, air-dried for 5 min, and dissolved in 100 µl TE buffer. Genotyping PCR were performed with the following primers: P1 (5′-CATCCTCCACTGGCCATGAATG-3′), P2 (5′-GCTTCACCTTGGTCTCCAG-3′), P3 (5′-CTTGGGTGGAGAGGCTATTC-3′) and P4 (5′-AGGTGAGATGACAGGAGATC-3′). PCR products were analyzed by agarose gel electrophoresis.
GRIESS assay
NO levels generated by infected macrophages were determined by measuring the accumulation of nitrite (NO2−) using the Griess assay as described previously [59]. Control macrophages were treated with E.coli LPS (100 µg/µl, Sigma) and IFN γ (0.1 units/µl, ROCHE) throughout the experiment. At 23 h post infection, conditioned medium were collected and centrifuged (14000 rpm, 10 min, RT). 100 µl of the supernatant was mixed with 100 µl Griess reagent (0.5% sulfanilamide and 0.05% N-(1-naphthyl)ethylenediamide in 2.5% acetic acid) and incubated for 10 min at room temperature. The samples were then measured at OD550 nm. The concentration of NO2− was calculated by using a standard curve prepared with sodium nitrite.
Accession numbers
The GenBank accession number for the YopE protein studied in this work is CAA68609.1.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. MedzhitovR (2007) Recognition of microorganisms and activation of the immune response. Nature 449 : 819–826.
2. VanceRE, IsbergRR, PortnoyDA (2009) Patterns of pathogenesis: discrimination of pathogenic and nonpathogenic microbes by the innate immune system. Cell Host Microbe 6 : 10–21.
3. StuartLM, PaquetteN, BoyerL (2013) Effector-triggered versus pattern-triggered immunity: how animals sense pathogens. Nat Rev Immunol 13 : 199–206.
4. BlanderJM, SanderLE (2012) Beyond pattern recognition: Five immune checkpoints for scaling the microbial threat. Nat Rev Immunol 12 : 215–225.
5. BoyerL, MagocL, DejardinS, CappillinoM, PaquetteN, et al. (2011) Pathogen-derived effectors trigger protective immunity via activation of the Rac2 enzyme and the IMD or Rip kinase signaling pathway. Immunity 35 : 536–549.
6. FontanaMF, BangaS, BarryKC, ShenX, TanY, et al. (2011) Secreted bacterial effectors that inhibit host protein synthesis are critical for induction of the innate immune response to virulent Legionella pneumophila. PLoS Pathog 7: e1001289.
7. FontanaMF, ShinS, VanceRE (2012) Activation of host mitogen-activated protein kinases by secreted Legionella pneumophila effectors that inhibit host protein translation. Infect Immun 80 : 3570–3575.
8. DunbarTL, YanZ, BallaKM, SmelkinsonMG, TroemelER (2012) C. elegans detects pathogen-induced translational inhibition to activate immune signaling. Cell Host Microbe 11 : 375–386.
9. KeestraAM, WinterMG, AuburgerJJ, FrassleSP, XavierMN, et al. (2013) Manipulation of small Rho GTPases is a pathogen-induced process detected by NOD1. Nature 496 : 233–237.
10. BliskaJB, WangX, ViboudGI, BrodskyIE (2013) Modulation of innate immune responses by Yersinia type III secretion system translocators and effectors. Cell Microbiol 15 : 1622–1631.
11. WrenBW (2003) The yersiniae–a model genus to study the rapid evolution of bacterial pathogens. Nat Rev Microbiol 1 : 55–64.
12. PerryRD, FetherstonJD (1997) Yersinia pestis–etiologic agent of plague. Clin Microbiol Rev 10 : 35–66.
13. ViboudGI, BliskaJB (2005) Yersinia outer proteins: role in modulation of host cell signaling responses and pathogenesis. Annu Rev Microbiol 59 : 69–89.
14. BlackDS, BliskaJB (2000) The RhoGAP activity of the Yersinia pseudotuberculosis cytotoxin YopE is required for antiphagocytic function and virulence. Mol Microbiol 37 : 515–527.
15. KrallR, ZhangY, BarbieriJT (2004) Intracellular membrane localization of pseudomonas ExoS and Yersinia YopE in mammalian cells. J Biol Chem 279 : 2747–2753.
16. IsakssonEL, AiliM, FahlgrenA, CarlssonSE, RosqvistR, et al. (2009) The membrane localization domain is required for intracellular localization and autoregulation of YopE in Yersinia pseudotuberculosis. Infect Immun 77 : 4740–4749.
17. ZhangY, BarbieriJT (2005) A leucine-rich motif targets Pseudomonas aeruginosa ExoS within mammalian cells. Infect Immun 73 : 7938–7945.
18. AuerbuchV, GolenbockDT, IsbergRR (2009) Innate immune recognition of Yersinia pseudotuberculosis type III secretion. PLoS Pathog 5: e1000686.
19. GausK, HentschkeM, CzymmeckN, NovikovaL, TrulzschK, et al. (2011) Destabilization of YopE by the ubiquitin-proteasome pathway fine-tunes Yop delivery into host cells and facilitates systemic spread of Yersinia enterocolitica in host lymphoid tissue. Infect Immun 79 : 1166–1175.
20. AndorA, TrulzschK, EsslerM, RoggenkampA, WiedemannA, et al. (2001) YopE of Yersinia, a GAP for Rho GTPases, selectively modulates Rac-dependent actin structures in endothelial cells. Cell Microbiol 3 : 301–310.
21. AepfelbacherM, TrasakC, WilharmG, WiedemannA, TrulzschK, et al. (2003) Characterization of YopT effects on Rho GTPases in Yersinia enterocolitica-infected cells. J Biol Chem 278 : 33217–33223.
22. WongKW, IsbergRR (2005) Yersinia pseudotuberculosis spatially controls activation and misregulation of host cell Rac1. PLoS Pathog 1: e16.
23. PujolC, BliskaJB (2003) The ability to replicate in macrophages is conserved between Yersinia pestis and Yersinia pseudotuberculosis. Infect Immun 71 : 5892–5899.
24. PujolC, BliskaJB (2005) Turning Yersinia pathogenesis outside in: subversion of macrophage function by intracellular yersiniae. Clin Immunol 114 : 216–226.
25. ZhangY, MurthaJ, RobertsMA, SiegelRM, BliskaJB (2008) Type III secretion decreases bacterial and host survival following phagocytosis of Yersinia pseudotuberculosis by macrophages. Infect Immun 76 : 4299–4310.
26. RoyD, ListonDR, IdoneVJ, DiA, NelsonDJ, et al. (2004) A process for controlling intracellular bacterial infections induced by membrane injury. Science 304 : 1515–1518.
27. BergsbakenT, FinkSL, den HartighAB, LoomisWP, CooksonBT (2011) Coordinated host responses during pyroptosis: caspase-1-dependent lysosome exocytosis and inflammatory cytokine maturation. J Immunol 187 : 2748–2754.
28. BentleyWE, MirjaliliN, AndersenDC, DavisRH, KompalaDS (1990) Plasmid-encoded protein: the principal factor in the “metabolic burden” associated with recombinant bacteria. Biotechnol Bioeng 35 : 668–681.
29. MeyerR (2009) Replication and conjugative mobilization of broad host-range IncQ plasmids. Plasmid 62 : 57–70.
30. ViboudGI, MejiaE, BliskaJB (2006) Comparison of YopE and YopT activities in counteracting host signalling responses to Yersinia pseudotuberculosis infection. Cell Microbiol 8 : 1504–1515.
31. SongsungthongW, HigginsMC, RolanHG, MurphyJL, MecsasJ (2010) ROS-inhibitory activity of YopE is required for full virulence of Yersinia in mice. Cell Microbiol 12 : 988–1001.
32. WilkinsTD, LyerlyDM (1996) Clostridium difficile toxins attack Rho. Trends Microbiol 4 : 49–51.
33. GenthH, DregerSC, HuelsenbeckJ, JustI (2008) Clostridium difficile toxins: more than mere inhibitors of Rho proteins. Int J Biochem Cell Biol 40 : 592–597.
34. BelyiY, AktoriesK (2010) Bacterial toxin and effector glycosyltransferases. Biochim Biophys Acta 1800 : 134–143.
35. LogsdonLK, MecsasJ (2003) Requirement of the Yersinia pseudotuberculosis effectors YopH and YopE in colonization and persistence in intestinal and lymph tissues. Infect Immun 71 : 4595–4607.
36. HeasmanSJ, RidleyAJ (2008) Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol 9 : 690–701.
37. WennerbergK, DerCJ (2004) Rho-family GTPases: it's not only Rac and Rho (and I like it). J Cell Sci 117 : 1301–1312.
38. MohammadiS, IsbergRR (2009) Yersinia pseudotuberculosis virulence determinants invasin, YopE, and YopT modulate RhoG activity and localization. Infect Immun 77 : 4771–4782.
39. RoppenserB, RoderA, HentschkeM, RuckdeschelK, AepfelbacherM (2009) Yersinia enterocolitica differentially modulates RhoG activity in host cells. J Cell Sci 122 : 696–705.
40. BusteloXR, SauzeauV, BerenjenoIM (2007) GTP-binding proteins of the Rho/Rac family: regulation, effectors and functions in vivo. Bioessays 29 : 356–370.
41. NetheM, HordijkPL (2010) The role of ubiquitylation and degradation in RhoGTPase signalling. J Cell Sci 123 : 4011–4018.
42. NetheM, AnthonyEC, Fernandez-BorjaM, DeeR, GeertsD, et al. (2010) Focal-adhesion targeting links caveolin-1 to a Rac1-degradation pathway. J Cell Sci 123 : 1948–1958.
43. ChenY, YangZ, MengM, ZhaoY, DongN, et al. (2009) Cullin mediates degradation of RhoA through evolutionarily conserved BTB adaptors to control actin cytoskeleton structure and cell movement. Mol Cell 35 : 841–855.
44. ManzanilloPS, AyresJS, WatsonRO, CollinsAC, SouzaG, et al. (2013) The ubiquitin ligase parkin mediates resistance to intracellular pathogens. Nature 501 : 512–516.
45. MoreauK, Lacas-GervaisS, FujitaN, SebbaneF, YoshimoriT, et al. (2010) Autophagosomes can support Yersinia pseudotuberculosis replication in macrophages. Cell Microbiol 12 : 1108–1123.
46. DeuretzbacherA, CzymmeckN, ReimerR, TrulzschK, GausK, et al. (2009) Beta1 integrin-dependent engulfment of Yersinia enterocolitica by macrophages is coupled to the activation of autophagy and suppressed by type III protein secretion. J Immunol 183 : 5847–5860.
47. XuH, YangJ, GaoW, LiL, LiP, et al. (2014) Innate immune sensing of bacterial modifications of Rho GTPases by the Pyrin inflammasome. Nature doi:10.1038/nature13449
48. FontanaMF, VanceRE (2011) Two signal models in innate immunity. Immunol Rev 243 : 26–39.
49. ShinS, CaseCL, ArcherKA, NogueiraCV, KobayashiKS, et al. (2008) Type IV secretion-dependent activation of host MAP kinases induces an increased proinflammatory cytokine response to Legionella pneumophila. PLoS Pathog 4: e1000220.
50. BrunoVM, HannemannS, Lara-TejeroM, FlavellRA, KleinsteinSH, et al. (2009) Salmonella Typhimurium type III secretion effectors stimulate innate immune responses in cultured epithelial cells. PLoS Pathog 5: e1000538.
51. AktoriesK (2011) Bacterial protein toxins that modify host regulatory GTPases. Nat Rev Microbiol 9 : 487–498.
52. FursteJP, PansegrauW, FrankR, BlockerH, ScholzP, et al. (1986) Molecular cloning of the plasmid RP4 primase region in a multi-host-range tacP expression vector. Gene 48 : 119–131.
53. ViboudGI, BliskaJB (2001) A bacterial type III secretion system inhibits actin polymerization to prevent pore formation in host cell membranes. EMBO J 20 : 5373–5382.
54. PujolC, KleinKA, RomanovGA, PalmerLE, CirotaC, et al. (2009) Yersinia pestis can reside in autophagosomes and avoid xenophagy in murine macrophages by preventing vacuole acidification. Infect Immun 77 : 2251–2261.
55. BliskaJB, BlackDS (1995) Inhibition of the Fc receptor-mediated oxidative burst in macrophages by the Yersinia pseudotuberculosis tyrosine phosphatase. Infect Immun 63 : 681–685.
56. CeladaA, GrayPW, RinderknechtE, SchreiberRD (1984) Evidence for a gamma-interferon receptor that regulates macrophage tumoricidal activity. J Exp Med 160 : 55–74.
57. MejiaE, BliskaJB, ViboudGI (2008) Yersinia controls type III effector delivery into host cells by modulating Rho activity. PLoS Pathog 4: e3.
58. RyndakMB, ChungH, LondonE, BliskaJB (2005) Role of predicted transmembrane domains for type III translocation, pore formation, and signaling by the Yersinia pseudotuberculosis YopB protein. Infect Immun 73 : 2433–2443.
59. PujolC, GrabensteinJP, PerryRD, BliskaJB (2005) Replication of Yersinia pestis in interferon gamma-activated macrophages requires ripA, a gene encoded in the pigmentation locus. Proc Natl Acad Sci U S A 102 : 12909–12914.
60. SimonetM, FalkowS (1992) Invasin expression in Yersinia pseudotuberculosis. Infect Immun 60 : 4414–4417.
61. BlackDS, BliskaJB (1997) Identification of p130Cas as a substrate of Yersinia YopH (Yop51), a bacterial protein tyrosine phosphatase that translocates into mammalian cells and targets focal adhesions. EMBO J 16 : 2730–2744.
62. PalmerLE, PancettiAR, GreenbergS, BliskaJB (1999) YopJ of Yersinia spp. is sufficient to cause downregulation of multiple mitogen-activated protein kinases in eukaryotic cells. Infect Immun 67 : 708–716.
63. LiloS, ZhengY, BliskaJB (2008) Caspase-1 activation in macrophages infected with Yersinia pestis KIM requires the type III secretion system effector YopJ. Infect Immun 76 : 3911–3923.
64. ZhangY, RomanovG, BliskaJB (2011) Type III secretion system-dependent translocation of ectopically expressed Yop effectors into macrophages by intracellular Yersinia pseudotuberculosis. Infect Immun 79 : 4322–4331.
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2014 Číslo 8
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Disruption of Fas-Fas Ligand Signaling, Apoptosis, and Innate Immunity by Bacterial Pathogens
- Ly6C Monocyte Recruitment Is Responsible for Th2 Associated Host-Protective Macrophage Accumulation in Liver Inflammation due to Schistosomiasis
- Host Responses to Group A Streptococcus: Cell Death and Inflammation
- Pathogenicity and Epithelial Immunity
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy