
The C-Terminal Domain of the Arabinosyltransferase EmbC Is a Lectin-Like Carbohydrate Binding Module
The d-arabinan-containing polymers arabinogalactan (AG) and lipoarabinomannan (LAM) are essential components of the unique cell envelope of the pathogen Mycobacterium tuberculosis. Biosynthesis of AG and LAM involves a series of membrane-embedded arabinofuranosyl (Araf) transferases whose structures are largely uncharacterised, despite the fact that several of them are pharmacological targets of ethambutol, a frontline drug in tuberculosis therapy. Herein, we present the crystal structure of the C-terminal hydrophilic domain of the ethambutol-sensitive Araf transferase M. tuberculosis EmbC, which is essential for LAM synthesis. The structure of the C-terminal domain of EmbC (EmbCCT) encompasses two sub-domains of different folds, of which subdomain II shows distinct similarity to lectin-like carbohydrate-binding modules (CBM). Co-crystallisation with a cell wall-derived di-arabinoside acceptor analogue and structural comparison with ligand-bound CBMs suggest that EmbCCT contains two separate carbohydrate binding sites, associated with subdomains I and II, respectively. Single-residue substitution of conserved tryptophan residues (Trp868, Trp985) at these respective sites inhibited EmbC-catalysed extension of LAM. The same substitutions differentially abrogated binding of di - and penta-arabinofuranoside acceptor analogues to EmbCCT, linking the loss of activity to compromised acceptor substrate binding, indicating the presence of two separate carbohydrate binding sites, and demonstrating that subdomain II indeed functions as a carbohydrate-binding module. This work provides the first step towards unravelling the structure and function of a GT-C-type glycosyltransferase that is essential in M. tuberculosis.
Published in the journal:
. PLoS Pathog 7(2): e32767. doi:10.1371/journal.ppat.1001299
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1001299
Summary
The d-arabinan-containing polymers arabinogalactan (AG) and lipoarabinomannan (LAM) are essential components of the unique cell envelope of the pathogen Mycobacterium tuberculosis. Biosynthesis of AG and LAM involves a series of membrane-embedded arabinofuranosyl (Araf) transferases whose structures are largely uncharacterised, despite the fact that several of them are pharmacological targets of ethambutol, a frontline drug in tuberculosis therapy. Herein, we present the crystal structure of the C-terminal hydrophilic domain of the ethambutol-sensitive Araf transferase M. tuberculosis EmbC, which is essential for LAM synthesis. The structure of the C-terminal domain of EmbC (EmbCCT) encompasses two sub-domains of different folds, of which subdomain II shows distinct similarity to lectin-like carbohydrate-binding modules (CBM). Co-crystallisation with a cell wall-derived di-arabinoside acceptor analogue and structural comparison with ligand-bound CBMs suggest that EmbCCT contains two separate carbohydrate binding sites, associated with subdomains I and II, respectively. Single-residue substitution of conserved tryptophan residues (Trp868, Trp985) at these respective sites inhibited EmbC-catalysed extension of LAM. The same substitutions differentially abrogated binding of di - and penta-arabinofuranoside acceptor analogues to EmbCCT, linking the loss of activity to compromised acceptor substrate binding, indicating the presence of two separate carbohydrate binding sites, and demonstrating that subdomain II indeed functions as a carbohydrate-binding module. This work provides the first step towards unravelling the structure and function of a GT-C-type glycosyltransferase that is essential in M. tuberculosis.
Introduction
Tuberculosis (TB) affects large parts of the world's population, particularly in developing countries [1]. The antibiotics isoniazid (INH) and ethambutol (EMB) [2] have been used for decades as frontline drugs to treat Mycobacterium tuberculosis infections, the causative agent of TB, but the rise of multi-drug resistant (MDR) and extensively drug resistant (XDR) strains poses a serious threat to present treatment options [3]. Both, INH and EMB inhibit the synthesis of essential components of the mycobacterial cell wall. This unique and highly impermeable barrier surrounds a single phospholipid bilayer membrane and is composed of an outer segment of solvent-extractable lipids, glycans and proteins, and a covalently linked inner segment, known as the mycolyl-arabinogalactan-peptidoglycan (mAGP) core [4]. Perturbations to the mAGP core tend to undermine viability of M. tuberculosis, a major reason why mAGP biosynthesis constitutes an attractive target for drug design efforts. The mycobacterial cell wall also encompasses various membrane-anchored lipoglycans, a group that includes lipoarabinomannan (LAM), which plays a key role in modulating the host immune response [5]. The arabinogalactan (AG) segment of the mAGP core and LAM both contain d-arabinan polymer, composed of α(1→5), α(1→3) and β(1→2)-linked arabinofuranosyl (Araf) residues that are assembled in distinct structural motifs (Fig. 1A) [4], [5].

In recent years, substantial progress has been made in defining the enzymatic processes resulting in the complete synthesis of AG and LAM [6]–[14]. Probing susceptibility to EMB, initial studies established that this inhibitor acted on a set of closely related arabinofuranosyl (Araf) transferases, EmbC (Rv3793), EmbA (Rv3794) and EmbB (Rv3795) [6], [7], collectively referred to as the Emb enzymes. These three proteins belong to the glycosyltransferase superfamily C (GT-C), which encompasses a diverse set of membrane-embedded glycosyltransferases that utilise lipid-linked as opposed to nucleotide-linked sugars as donor substrates (Fig. 1A) [15]. The Emb enzymes of M. tuberculosis display a common architecture of 13 transmembrane helices in conjunction with a hydrophilic C-terminal domain [10], [14] (Fig. 1B), and share the same polyprenyl donor-substrate, β-D-arabinofuranosyl-1-monophosphoryldecaprenol (DPA) [16], [17].
Owing to their hydrophobic nature, generating recombinant Emb proteins in soluble form has proved difficult, hampering in vitro characterisation. As a result, the function of the Emb enzymes has been delineated by genetics, phenotypic analysis of the cell envelope and cell-free assays. Single gene deletions of embC, embB in M. tuberculosis are lethal [18], [19], but corresponding knock-outs in Mycobacterium smegmatis or Corynebacterium glutamicum yield viable, albeit slow growing mutants, whose cell wall defects can be analysed [8], [9]. Following attachment of the initial Araf residue to the linear galactan polymer [Galf-β(1→5)Galf-β(1→6)]n, catalysed by the Araf-transferase AftA [12], EmbA and EmbB extend the arabinan chain in AG synthesis, transferring Araf residues from DPA to polysaccharide acceptors [8], [9]. Highly similar in amino acid sequence (∼40% identity, see also Supporting Fig. S1), EmbA,B and EmbC have differential roles: the ΔembA,B deletions inhibit AG synthesis, but leave LAM synthesis intact, whereas the ΔembC deletion only affects LAM synthesis. Chimaeric forms of the Emb enzymes, where the hydrophilic C-terminal domain of EmbC was swapped for that of EmbB led to a hybrid-LAM, bearing an AG-specific, branched Araf6 group instead of the characteristic LAM-specific linear Araf4 [9]. These data indicated that the hydrophilic C-terminal domain makes a critical contribution to determining the structure of the resulting AG or LAM segments.
To date, the Emb enzymes have remained poorly characterised in structural terms, despite their central significance as targets of the TB antibiotic EMB and their link to drug resistance [20]. Herein, we present the crystal structure of the C-terminal hydrophilic domain of M. tuberculosis EmbC (residues 719–1094, henceforth EmbCCT), as a first step towards the elucidation of the 3D structure of the full-length enzyme.
Results
Structure determination and domain architecture
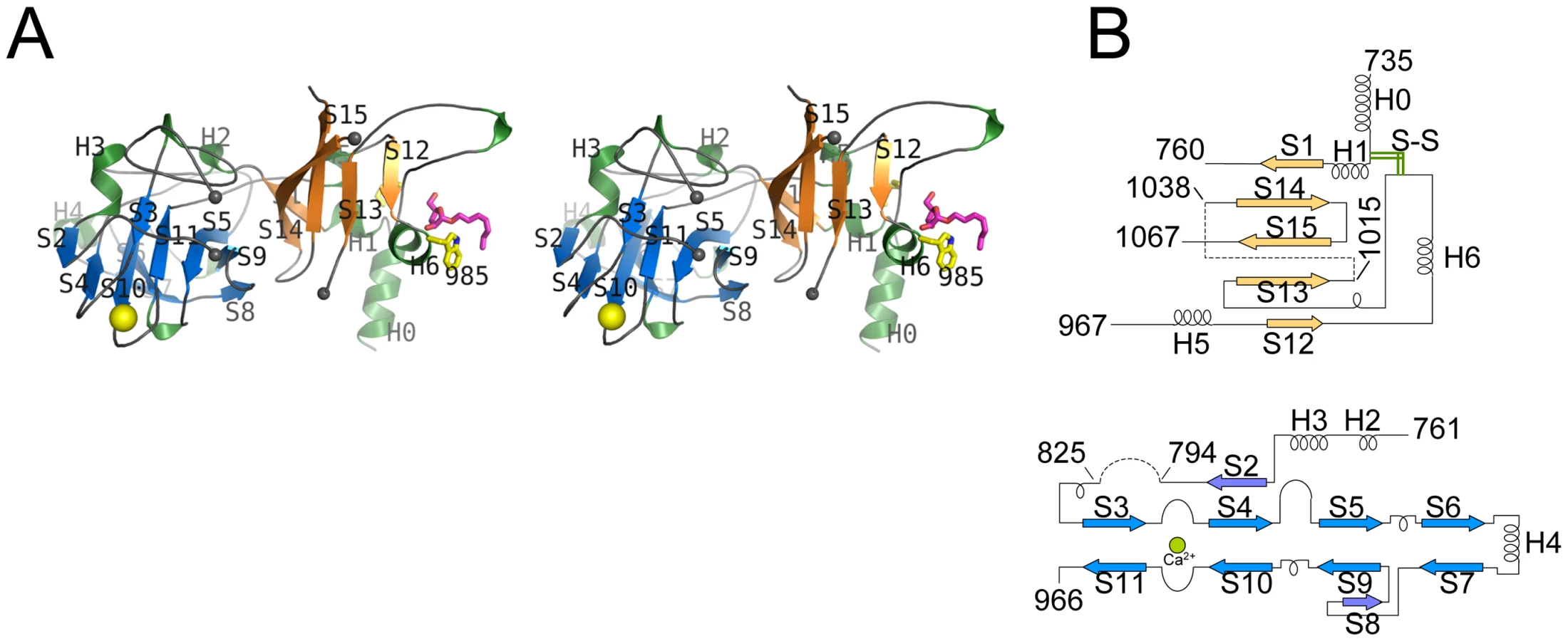
EmbCCT crystallised in space group P6522 over a diverse range of reservoir conditions, with one molecule in the crystallographic asymmetric unit. Crystals were generated with or without an Araf acceptor analogue (see below) present in the crystallisation droplet. The experimental density, phased by multi-wavelength anomalous dispersion (2.7 Å, Table 1), was of very good quality (Fig. S2A), defining the structure for residues 735–1067, except for two disordered loops (795–824 and 1016–1037, Fig. 2A). EmbCCT is composed of two distinct subdomains, separated by a deep crevice marked by the disordered loops (residues 795–824 and 1016–1037). Subdomain I, which encompasses residues 746–760 and 967–1067, displays a mixed α/β structure, with a 5-stranded β-sheet forming a semi-barrel (Fig. 2A). The long H6-S13 loop, which forms a minor crystal packing interface, protrudes from the core of subdomain I with a helical half-turn at its tip (Fig. 2A). Subdomain II (residues 761–966) forms an anti-parallel β-sandwich structure, of which the ‘outer’ sheet (S2, S4, S10, S6, S7) faces solvent while the ‘inner’ sheet (S3, S11, S5, S9, S8) packs against the core of the domain (Fig. 2A). The β-sandwich of subdomain II assumes a jellyroll fold (Fig. 2B), a fold typical for polysaccharide binding units in plant lectins and carbohydrate active enzymes [21]. Although not part of the formal jellyroll description, strands S2 and S8 extend the ‘outer’ and ‘inner’ sheet, respectively, while helix H4 forms a boundary to the ‘outer’ sheet. A high-density peak (14σ, anomalous density difference map, Fig. 3A) is embedded between loops S3–S4 and S10–S11. Quasi-octahedral coordination geometry and the distribution of peak-ligand distances from 2.40 to 2.63 Å (Fig. 3A) suggest a bound Ca2+ ion [22]. The metal ion appears shielded from solvent, although including 10 mM EDTA in the cryoprotectant buffer significantly diminished the height of the density peak (Fig. S2B). Substitution of Asp949 by serine in EmbCCT, the only side chain in direct contact with the Ca2+ ion (2.6 Å, bidentate, Fig. 3A), resulted in very poor recombinant expression compared to wild-type and other point mutants probed in this study (see below). Together these observations suggest that the Ca2+ ion is important for the structural integrity of EmbCCT.


Structural neighbours
The fold of subdomain II is consistent with the proposed role of EmbCCT as an acceptor saccharide recognition module. The comparison with structural homologues, identified via distance matrix alignment using the DALI program (http://ekhidna.biocenter.helsinki.fi/dali_server/, [23]) reinforces this notion. The vast majority of PDB entries retrieved by DALI (over 300 entries above the default significance threshold of Z = 2) match the β-sandwich fold of subdomain II and represent ‘carbohydrate binding modules’ (CBM), structural domains that confer carbohydrate-binding specificity, but that lack intrinsic catalytic activity [21]. CBMs occur frequently as a part of glycoside hydrolase enzymes and fall into (to date) 61 distinct CBM families (http://www.cazy.org/). While none of the structural homologues is particularly close to subdomain II (Z-scores≤6.9, root mean square deviation (RMSD)≥3.0 Å), the top 10 hits include the calcium-containing CBM families 6 and 36 (Fig. S3A–C). Interestingly, in the DALI-generated superposition of EmbCCT with Paenibacillus polymyxa endo-1,4-β-xylanase (PDB entry 1UX7, CBM 36), the Ca2+ sites match to within 0.9 Å, and in the latter, the Ca2+ ion makes direct contact with the bound xylobiose ligand (Fig. S3A). In contrast, only three hits were obtained for subdomain I of which only the best (PDB entry 2ZAG, Z = 3.0, RMSD 3.4 Å for 66 Cα pairs) showed weak similarity in terms of secondary structure topology in a limited region of overlap (Fig. S4). This PDB entry describes the hydrophilic C-terminal domain of oligosaccharyltransferase STT3 from Pyrococcus furiosus [24], a membrane-embedded glycosyltransferase of the GT-C superfamily that catalyses transfer of glycosyl groups from a lipid donor to Asn-glycosylation sites of the acceptor protein.
Self-assembly in solution
Crystal packing contacts, analysed using the PISA server (http://www.ebi.ac.uk/msd-srv/prot_int/pistart.html), highlighted three prominent interaction surfaces burying 390 Å2, 670 Å2 and 1100 Å2 of solvent accessible surface (SAS) per monomer, respectively (Fig. S5). We probed self-assembly of EmbCCT by sedimentation velocity at three different protein concentrations (Fig. 4A). The distribution C(S) of the sedimentation coefficient S indicates a dynamic equilibrium between three different molecular species at 3.1S, 4.6S and 7S, which correspond to apparent molecular weights of 46.5 kDa, 75.8 kDa and 138.0 kDa, respectively, compared to the calculated monomer mass of 39.9 kDa. Bearing in mind that under - or overestimates of apparent masses can occur as a result of fitting a single frictional coefficient for an ensemble of species with different frictional ratios, the dominant peak at 4.6S most likely represents a dimer. The higher molecular weight peak at 7.6S, could be a trimer or tetramer, but strongly suggests that more than one of the crystal packing interfaces is able to mediate oligomerisation of EmbCCT in vitro.
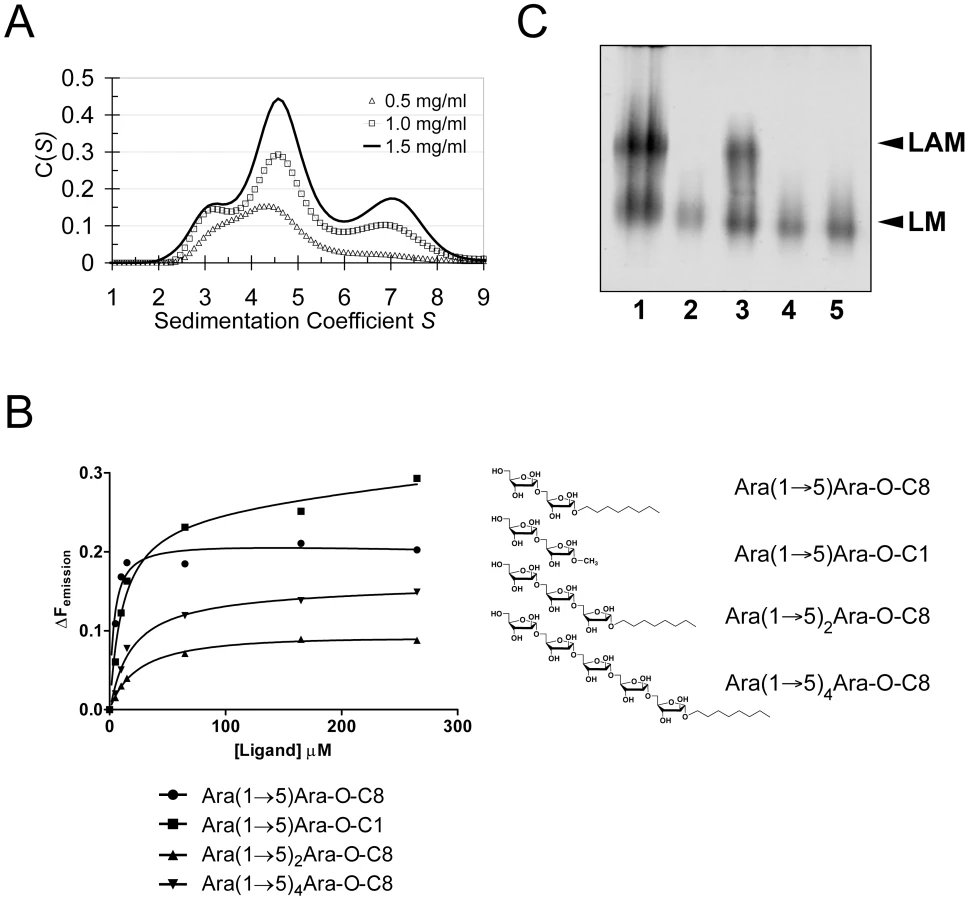
Carbohydrate binding
Previous studies had attributed to the C-terminal domain of the Emb proteins a critical role in arabinan chain extension [9], [11]. Therefore, we asked whether the isolated domain is able to bind synthetic acceptor analogues. As the physiological substrate is chemically complex and diverse, using synthetic acceptor analogues offered the best chance to obtain an experimental acceptor-bound complex structure. In previous work, our laboratory had chemically synthesised neo-glycolipid acceptors that were modelled on motifs found in mycobacterial AG and LAM. When incubated with [14C]-labelled Araf-donor substrate DPA and isolated mycobacterial membranes in a cell-free Araf transferase, these molecules acted as potent acceptor mimics [25]. One of these acceptors was the di-arabinoside α-D-Araf-(1→5)-α-D-Araf-O-(CH2)7CH3 (for short: Ara(1→5)Ara-O-C8, Fig. 4B). The O-linked octyl tail allowed extraction of the reaction products for qualitative characterisation in vitro. Importantly, the closely related di-arabinoside α-D-Araf-(1→5)-α-D-Araf-O-CH3 (Ara(1→5)Ara-O-C1) exhibited similar levels of acceptor activity, demonstrating the O-linked octyl was dispensable for activity [25]. By way of intrinsic tryptophan fluorescence, we probed binding of Ara(1→5)Ara-O-C8 to EmbCCT, as well as that of analogous tri - and penta-arabinofuranosides, [α-D-Araf-(1→5)]2-α-D-Araf-O-(CH2)7CH3 (Ara-α(1→5)2-Ara-O-C8) and [α-D-Araf-(1→5)]4-α-D-Araf-O-(CH2)7CH3 (Ara-α(1→5)4-Ara-O-C8, Fig. 4B). Fitting the binding curves to a single-site saturation model, yielded an equilibrium dissociation constant Kd of 3.6 µM for the di-arabinofuranoside Ara(1→5)Ara-O-C8 (Table 2), while the disaccharide lacking the octyl chain, Ara(1→5)Ara-O-C1, resulted in a Kd of 11.0 µM. These data confirmed that in the solution state the octyl chain is not essential for binding, although it may enhance affinity. Soaking EmbCCT crystals in cryoprotectant solution containing 27 mM Ara(1→5)Ara-O-C8 (∼3-fold excess of ligand relative to protein concentration in the crystal) reproducibly resulted in defined ligand density (Fig. 3B), allowing us to unequivocally build one Araf unit and the octyl chain of Ara(1→5)Ara-O-C8, while the second Araf ring remained invisible, even when contouring the map at near-noise level. Soaking experiments using the other acceptor analogues, for which solution binding was examined, failed to reveal electron density for the ligand. The soaked di-arabinofuranoside ligand is positioned between two symmetry-related copies of EmbCCT, forming non-covalent contacts only with residues in subdomain I, but not with the CBM-like subdomain II, in contrast to our expectation. The Araf moiety packs against helix H6 and the H6-S13 loop (Fig. 2), forming three direct H-bond contacts with protein: O2 binds to carbonyl O of Trp985 (2.53 Å), O1 to Nε1 of Trp985 (2.99 Å), and O3 to Nδ2 of Asn740′ (primed residues indicating the symmetry mate). In contrast, the octyl chain binds between helix H0 and the S13–S14 loop of the symmetry mate (Fig. 3B). Ligand binding promotes ordering of the N-terminus of helix H0, where 3 additional residues become visible compared to apo, and induces a conformational shift of aspartate residues 1051 and 1052 in the S13–S14 loop (Fig. S6). While this crystallographic complex structure did not reveal binding to the CBM-like subdomain II, it is possible that crystal lattice formation of EmbCCT interferes with binding at a site on subdomain II. We, therefore, asked whether the structural superimposition with saccharide-bound CBM domains could be exploited to predict potential additional binding sites. We note that ligand binding modes and substrate specificity of CBM domains can differ even within the same CBM family [21], [26]. Thus, structural alignments of the protein scaffolds are unlikely to accurately predict the precise modes of binding and potential specificity-determining interactions. Nevertheless, superimposing carbohydrate-bound structures of CBM domains with the 10-highest DALI Z-scores (with respect to the non-redundant PDB90 subset) shows two clusters of putative ligand binding sites in subdomain II (Fig. 3C): (1) near the Ca2+ site and the S3–S4 loop, and (2) on the open surface of the ‘outer’ β-sheet (strands S2, S4, S10, S6, S7). Virtually all ligands in the first cluster sterically clash with the loops that coordinate the Ca2+ site. Without invoking a conformational change that exposes the Ca2+ to solvent, this site appears unable to accommodate a ligand. In contrast, in the second cluster, only minor steric hindrance occurs between EmbCCT and the superimposed ligands, and thus this site appeared more plausible as a carbohydrate-binding site.

Mutagenesis and activity in full-length EmbC
The crystallographic complex of EmbCCT bound to Ara(1→5)Ara-O-C8 and the structural superposition with carbohydrate-bound homologues had indicated two distinct regions in EmbCCT as potential sites for carbohydrate binding (Fig. S7A). In order to probe the relevance of these two sites, we asked whether replacement of endogenous EmbC with recombinant EmbC carrying appropriate point mutations would alter the cell wall composition of M. smegmatis. Aromatic residues frequently mediate binding of carbohydrate ligands to CBMs [21]. Given the H-bond contacts between Trp985 and Ara(1→5)Ara-O-C8 in subdomain I, and the central position of Trp868 of the ‘outer’ (solvent-exposed) β-sheet of subdomain II (Fig. 3C and Fig. S7A), we probed these two residues in the first instance.
Using a phage-mediated transduction method for allelic exchange [27], we generated an EmbC-deficient strain of M. smegmatis (M. smegmatis ΔembC), which was complemented with plasmids encoding either wild-type (full length) M. tuberculosis EmbC or mutant forms thereof. In accordance with previously reported data [9], our M. smegmatis ΔembC strain retains lipomannan (LM) synthesis, but is deficient in LAM (Fig. 4C – lane 2). The abrogation of LAM biosynthesis can be directly attributed to the loss of EmbC, which is involved in the early synthesis of α(1→5)-Araf arabinan elongation of LM, the immediate LAM precursor (Fig. 1A) [9]. We utilised this phenotype by analysing LM/LAM resulting from complementation of M. smegmatis ΔembC with plasmid pVV16-Mt-embC, encoding full-length M. tuberculosis EmbC, and plasmids pVV16-Mt-embCW868A or pVV16-Mt-embCW985A, which encode point mutants W868A and W985A of full-length M. tuberculosis EmbC, respectively. Complementation with wild type EmbC largely restored the normal phenotype (Fig. 4C – lane 3), whereas complementation with the point mutants failed to re-establish LAM synthesis (Fig. 4C – lanes 4, 5). We verified by Western blot that loss of LAM synthesis was not due to failure of the plasmid-encoded protein to incorporate into the membrane of M. smegmatis ΔembC (Supporting Fig. S7B). These results suggest that the structural perturbations caused by the individual single-site mutations are sufficient to disrupt the function of EmbC.
Differential acceptor binding of EmbCCT mutants
In order to establish whether loss of activity was linked to compromised acceptor binding, we introduced the single-residue mutations W868A or W985A into expression plasmids encoding EmbCCT. In addition, we prepared analogous expression plasmid constructs bearing mutations on Asn740 (to Ala, binding site subdomain I), Gln899 (to Ser) and His911 (to Ala, binding site subdomain II) and Asp949 (to Ser, Ca2+ binding site, see Supporting Fig. S7A). Two constructs (Q899S, D949S) did not express well enough to yield protein suitable for in vitro assays. For those proteins that were produced successfully, proper folding was verified by far-UV circular dichroism spectroscopy (Supporting Fig. S7C). When comparing binding of the di - and penta-arabinoside acceptor analogues (Fig. 4B and Fig. 5) that both carry the O-linked octyl tail, it was striking that the substitutions W868A and W985A affected binding of these ligands in a differential fashion. While the W985A mutation virtually abrogated binding of the disaccharide Ara(1→5)Ara-O-C8, the W868A substitution preserved binding of this particular ligand, with only a modestly higher Kd (Table 2, Fig. 5A). In contrast, binding of the penta-arabinoside Ara(1→5)4Ara-O-C8 was insensitive to the W985A mutation, but completely inhibited in response to the W868A mutation. Likewise, mutating Asn740 to Ala weakened binding of the disaccharide (Table 2), consistent with its position within H-bond distance of the ordered Araf in subdomain I, whereas the distant H911A mutation in subdomain II had no effect on this ligand. Thus, the differential effect of mutations in the putative binding sites in subdomain I and II on binding of acceptor analogues that differ only in length, strongly suggests that these bind preferentially to distinct sites on EmbCCT.

Discussion
Polyprenyl-dependent glycosyltransferases of superfamily GT-C are still awaiting the determination of a structure of an intact, full-length enzyme, but structures of individual hydrophilic domains have begun to emerge [24] (see also PDB entry 3BYW). As a first step towards the complete structural characterisation of the Emb Araf-transferases in M. tuberculosis, we have determined the crystal structure of the hydrophilic C-terminal domain of EmbC, the enzyme responsible for arabinan chain elongation in LAM synthesis and a target for the front line antibiotic EMB [5]. We found that the architecture of this domain comprises two subdomains, one of which folds as a lectin - or CBM-like domain, the other one shows weak similarity to the C-terminal hydrophilic domain of an unrelated GT-C glycosyltransferase, oligosaccharyl transferase STT3 [24]. The match between subdomain I and the so-called CC region of STT3 is poor (Fig. S4), and is limited to core secondary structure elements. Nevertheless, the DALI-derived superposition aligns the second Trp in STT3's highly conserved WWDYG motif with EmbC's Trp985, a side chain we showed is critical for enzymatic activity. Thus the alignment lends additional support to the notion of Trp985 sitting at a critical junction of the C-terminal domain of EmbC.
Sequence comparison of the Emb C-terminal domains (Fig. S1) strongly suggests that the disulfide bond Cys749-Cys993 is a conserved structural feature. Forming a topologically intuitive demarcation of this domain, this covalent link presumably enhances the stability of the C-terminal domain at physiological conditions in the host. The disordered loops (residues 794–825, 1016–1037) encompass regions of high sequence diversity as opposed to otherwise remarkably conserved regions of the structure. Given the latter, one could speculate that these disordered regions are linked to acceptor discrimination, and/or that ordering might be induced by contacts with adjacent structural elements in the context of the full-length enzyme.
It has previously been proposed that the Emb enzymes may function as dimers, possibly in the combination EmbA/EmbB and EmbC/EmbC [11], [28]. Our sedimentation velocity data now provide supporting evidence for self-assembly of EmbC, although we cannot rule out that the observed oligomerisation occurs solely as a result of separating EmbCCT from the rest of the protein. However, the presence of dimers and trimers (or tetramers) (Fig. 4A) in solution demonstrated that at least two of the observed crystal packing interfaces were able to mediate self-assembly of EmbCCT. While thile the most-extended packing interface (SAS buried 1100 Å2) is mediated by structural elements (helices H0 and H6) that are close the truncation site, the second-largest interface (SAS buried 670 Å2) is mediated by strand S2, and distant to the truncation site. Indeed, the latter self-assembly interface generates a continuous β-sheet that extends across the monomer-monomer boundary (Fig. S5C), hinting that it could be preserved in the full-length enzyme.
The presence of a CBM-like subdomain in EmbCCT is consistent the proposed role of the C-terminal domain in acceptor substrate recognition [10], [11]. Among these structurally diverse carbohydrate binding modules, the β-sandwich fold seen in EmbCCT is most common [21]. The differential response of the ligands of different length to the Trp mutations in subdomains I and II provides compelling evidence for the presence of two separate ligand binding sites in EmbCCT. This response also links the loss of Araf transferase activity in the Trp mutants to compromised acceptor binding. Although we were not successful in crystallising a complex structure that directly demonstrates binding of an acceptor analogue to the CBM-like subdomain II, the dramatic loss of binding affinity of the penta-arabinoside acceptor for the mutant EmbCCT(W868A) (Fig. 5B, Table 2) and the corresponding loss of LAM synthesis, are strong indications that subdomain II indeed functions as a carbohydrate binding module. We note that the W868A mutation has also a modest effect on binding of Ara(1→5)Ara-O-C8 (∼2.5-fold increase in Kd, Table 2), despite the obvious preference of this ligand for binding to subdomain I, as shown by the structure and the response to the W985A mutation. This observation could indicate that Ara(1→5)Ara-O-C8 also associate with the CBM-like subdomain II, albeit with considerably lower affinity. The converse may be true for the penta-saccharide as well, although the affinities we measured show no corresponding signature. Comparison of the affinities for binding of the tri - and pentasaccharide to wild type EmbCCT clearly indicates that binding to subdomain II is tighter for longer polysaccharides, as these can be expected to make additional contacts. However, the apparent switch in binding preference from the site in subdomain I to that in subdomain II on going from two to five Araf units is less straightforward to explain. If, as the structure suggests, only the octyl tail and the first Araf unit were the major determinants of binding to subdomain I, one would expect to see evidence for binding of Ara(1→5)4Ara-O-C8 to subdomain I, that is, a significant change in affinity when mutating Trp985. Thus, while the octyl tail clearly influences binding of the di-saccharide, this appears to be less the case for the tri - and penta-saccharides. This observation is in line with the dispensable nature of the octyl chain when the above ligands are used as acceptor mimics in cell-free Araf transferase assays [25].
Overall, a string of genetic and biochemical evidence consistently indicated that enzymatic activity of the Emb Araf-transferases is associated with loops displayed on the extra-cellular face of the membrane. For instance, the most frequent point mutation present in EMB-resistant clinical isolates of M. tuberculosis concerns residue Met306 in EmbB ( = Met300 in EmbC, see Fig. 1) [20], only a few residues downstream of the GT-C-specific, strictly conserved DDX motif in the E2 loop [15]. Berg et al. showed that loop E6 carries a functionally relevant, conserved proline-containing sequence motif [10], consistent with findings in the Emb protein of C. glutamicum [14]. Moreover, a crystal structure of the first extracellular loop of the Emb Araf-transferase of the related organism Corynebacterium diphtheriae has become available very recently (PDB entry 3BYW; Tan K., Hatzos C., Abdullah J., Joachimiak A., unpublished). The domain of the E1 loop displays a β-sandwich fold with similarity to the fold of galectin [29], but is not superimposable on that of subdomain II of EmbCCT. The galectin-like fold again hints to a potential function in carbohydrate binding – perhaps the sugar moiety of the Araf-donor DPA. In conclusion, the present structure of the C-terminal domain of M. tuberculosis EmbC provides a first corner stone towards assembling the structure of the full-length enzyme, and allows us to begin probing this essential enzyme in a rational and targeted fashion.
Methods
Reagents
Plasmids were propagated during cloning in E. coli Top10 cells (Invitrogen). All restriction enzymes, T4 DNA ligase and Phusion DNA polymerase enzymes were sourced from New England Biolabs. Oligonucleotides were from MWG Biotech Ltd and PCR fragments were purified using the QIAquick gel extraction kit (Qiagen). Plasmid DNA was purified using the QIAprep purification kit (Qiagen).
Recombinant protein
A 1125-bp region coding for the C-terminal domain (residues 719–1094) of EmbC was cloned from genomic DNA of M. tuberculosis H37Rv using PCR primers (restriction sites underlined) GATCGATCCATATGGAGGTGGTATCGCTGACCCAG (forward) and GATCGATCCTCGAGCTAGCCTCTGCGCAACGGC (reverse). The PCR product was ligated into plasmid pET23b (NdeI, XhoI restriction sites), yielding the His6-tagged pET23b-EmbCCT construct, whose sequence was verified (School of Biosciences Genomics Facility, University of Birmingham). For expression, E. coli C41(DE3) cells were transformed with pET23b-EmbCCT using the rubidium chloride method. Overnight cultures (5 ml LB medium, 100 µg/ml ampicillin) were used to inoculate bulk cultures (4×1 litre LB, 100 µg/ml ampicillin, 37°C, 200 rpm). Seleno-methionine derivatised EmbCCT was produced using the same expression plasmid and host, but following the feedback inhibition protocol described in [30]. Cultures were induced at OD600 = 0.5 using 1 mM IPTG (12 h, 16°C). Cells were harvested (6000×g, 15 min), washed with 20 ml phosphate buffered saline, and frozen. Pellets were re-suspended in 50 mM KH2PO4 (pH 7.9), 300 mM NaCl, 1 mM PMSF, 15 µg/ml benzamidine, DNAse and RNAse (50 µg/ml), and sonicated (30 sec ON/OFF cycles, total of 8 cycles). The lysate was cleared (30 min, 28000×g, 4°C) and passed over a HiTRAP Ni2+-NTA column (GE Healthcare), equilibrated in 50 mM KH2PO4 (pH 7.9), 300 mM NaCl, and eluted using a step-gradient of 50–500 mM imidazole. The purification was monitored by 12% SDS-PAGE. Fractions containing EmbCCT (250, 500 mM imidazole) were pooled and dialysed against 50 mM KH2PO4 (pH 7.9), 300 mM NaCl, and concentrated by ultrafiltration to ∼15 mg/ml.
Structure determination
Hanging drop vapour diffusion was used to grow crystals of EmbCCT over a reservoir of 0.1 M sodium acetate pH 4.4, 80 mM ammonium phosphate, mixing 1 µl of protein with 1 µl of reservoir solution. Crystals were cryoprotected in reservoir solution, adding up to 12% ethylene glycol and 12% glycerol, and flash frozen in liquid nitrogen. Native and 3-wavelength SeMet MAD data were recorded on beamline ID23-1 (ESRF, Grenoble, France). Diffraction images were processed using XDS and XSCALE [31] (Table 1). Selenium sites and phases were obtained using standard procedures (SHELXD [32], SHARP v2.2 [33] SOLOMON [34]) leading to a readily interpretable electron density map (Fig. S2A). The ARP/wARP-built [35] initial model was rebuilt in COOT [36], with intermittent refinement against native data (REFMAC5 [37], PHENIX.REFINE [38]). Temperature factor modelling included TLS refinement [39]. The final model has good stereochemistry and comprises EmbC residues 735–794, 825–1015 and 1038–1067, 113 water molecules, one molecule of Ara(1→5)Ara-O-C8, one Ca2+ and one phosphate ion (Table 1).
Solution binding assay by intrinsic tryptophan fluorescence
Intrinsic tryptophan fluorescence (ITF) experiments were carried out using a PTI QuantaMaster 40 spectrofluorimeter, recording data with the FeliX32 software package (PTI, Birmingham, New Jersey, USA). The excitation wavelength was set to 294 nm and the fluorescence emission (Femission) was recorded between 300–400 nm for each ligand aliquot added to a 200 µl solution containing 20 µM EmbCCT in 50 mM KH2PO4 (pH 7.9), 300 mM NaCl. For EmbCCT, the emission maximum (Femissionmax) was at λ = 338 nm, providing a basal Femission coordinate for the collection of subsequent ITF data. The change in fluorescence emission (ΔFemission) was calculated by subtracting Femission (recorded 2 min after each ligand addition) from Femissionmax, and the data was then plotted against ligand concentration, [L] (3 independent experiments). A plot of ΔFemission at λ = 338 nm vs. [L] was fitted to the saturation binding equation using GraphPad Prism software:
Circular dichroism spectroscopy
Far-UV circular dichroism (CD) spectra were recorded at 25°C using a Jasco J-715 spectropolarimeter and a cell of 0.01 cm path length. Proteins EmbCCT, EmbCCT(N740A), EmbCCT(W868A), EmbCCT(H911A) and EmbCCT(W985A) were dialysed into 50 mM KH2PO4 (pH 7.9), 50 mM NaF to a final concentration of 0.5 mg/ml each. Spectra were recorded of 250 µl aliquots of each protein by measuring ellipticity from 195–260 nm, using a bandwidth of 2 nm and a scan speed of 100 nm/min. Spectra were normalised by subtracting the spectrum of buffer alone (baseline).
Analytical ultracentrifugation
Sedimentation velocity experiments were performed using a Beckman Proteome XL-I analytical ultracentrifuge equipped with absorbance optics. EmbCCT was dialysed into 50 mM KH2PO4 (pH 7.9), 300 mM NaCl, and loaded into cells with two channel Epon centre pieces and quartz windows. A total of 100 absorbance scans (280 nm) were recorded (40,000 rpm, 4°C) for each sample, representing the full extent of sedimentation of the sample. Data analysis was performed using the SEDFIT software, fitting a single friction coefficient [40].
Generation of embC-deficient M. smegmatis and complementation plasmids
Approximately 1 kb of upstream and downstream flanking sequences of the embC gene (MSMEG2785) were PCR amplified from M. smegmatis mc2155 genomic DNA using the primer pairs MSEMBCLL, MSEMBCLR, MSEMBCRL and MSEMBCRR, respectively (sequences listed in Supporting Information Table S1). Following restriction digestion of the primer incorporated AlwNI sites, the PCR fragments were cloned into AlwNI-digested p0004S to yield the knockout plasmid pΔMSMEGEMBC which was then packaged into the temperature sensitive mycobacteriophage phAE159 as described previously [27] to yield phasmid DNA of the knockout phage phΔMSMEGEMBC. Generation of high titre phage particles and specialized transduction were performed as described earlier [27], [41]. Deletion of MSMEGEMBC in one hygromycin-resistant transductant was confirmed by Southern blot. For complementation, M. tuberculosis embC was cloned using primer pairs Mt-embC-forward and Mt-embC-reverse (sequences listed in Supporting Information Table S1) and blunt-end ligated into SmaI digested pUC18. For QuikChange mutagenesis (Stratagene) of pUC18-Mt-embC W868A and W985A codons, primer pairs W868A-sense/-antisense and W985A-sense/-antisense (sequences in Supporting Information Table S1, each with 5′-phosphate modifications) were used. The 3301 bp product was extracted from plasmids (pUC18-Mt-embC, pUC18-Mt-embCW868A and pUC18-Mt-embCW985A) digested with NdeI and HindIII, and sub-cloned into the similarly digested mycobacterial shuttle vector pVV16 to yield pVV16-Mt-embC, pVV16-Mt-embCW868A and pVV16-Mt-embCW985A. These plasmids were then used to transform M. smegmatisΔembC to yield clones resistant to both hygromycin and kanamycin.
Point mutations in recombinant EmbCCT
QuikChange mutagenesis (Stratagene) was carried out using pET23b-Mt-embCCT (generated as described above). Primer pairs used for the codon alterations N740A, W868A, Q899S, H911A and W985A are listed in the Supporting Information Table S1. Mutant plasmids were subsequently transformed individually into E. coli C41 (DE3). Mutant proteins were expressed and purified as described above.
Analysis of lipoglycans
Lipoglycans form M. smegmatis strains were extracted as described previously [42]. Dried cells were resuspended in de-ionized water and disrupted by sonication (MSE Soniprep 150, 12 µm amplitude, 60 s on, 90 s off for 10 cycles, at 4°C). An equal volume of ethanol was added to the cell suspension and the mixture was refluxed at 68°C, for 12 h intervals, followed by centrifugation and recovery of the supernatant. The C2H5OH/H2O extraction process was repeated five times and the combined supernatants dried. The dried supernatant was then subjected to hot-phenol treatment by addition of phenol/H2O (80%, w/w) at 70°C for 1 h, followed by centrifugation and the aqueous phase was dialyzed using a 1500 MWCO membrane (Spectrapore) against de-ionized water. The retentate was dried, resuspended in water and sequentially digested with α-amylase, DNase, RNase, chymotrypsin and trypsin. The retentate was further dialyzed using a 1500 MWCO membrane (Spectrapore) against deionized water. The eluates were collected, extensively dialysed against deionized water, concentrated and analyzed by 15% SDS-PAGE using a Pro-Q emerald glycoprotein stain (Invitrogen).
Accession numbers
The accession number for the coordinates and structure factors of the C-terminal domain of EmbC in the Protein Data Bank (http://www.rcsb.org) is 3PTY.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. World Health Organisation 2009 Global Tuberculosis Control: a short update to the 2009 report (http://www.who.int/entity/tb/publications/2009/factsheet_tb_2009update_dec09.pdf)
2. HarriesAD
DyeC
2006 Tuberculosis. Ann Trop Med Parasitol 100 415 431
3. JainA
MondalR
2008 Extensively drug-resistant tuberculosis: current challenges and threats. FEMS Immunol Med Microbiol 53 145 150
4. CrickDC
MahapatraS
BrennanPJ
2001 Biosynthesis of the arabinogalactan-peptidoglycan complex of Mycobacterium tuberculosis. Glycobiology 11 107R 118R
5. BrikenV
PorcelliSA
BesraGS
KremerL
2004 Mycobacterial lipoarabinomannan and related lipoglycans: from biogenesis to modulation of the immune response. Mol Microbiol 53 391 403
6. BelangerAE
BesraGS
FordME
MikusovaK
BelisleJT
1996 The embAB genes of Mycobacterium avium encode an arabinosyl transferase involved in cell wall arabinan biosynthesis that is the target for the antimycobacterial drug ethambutol. Proc Natl Acad Sci U S A 93 11919 11924
7. TelentiA
PhilippWJ
SreevatsanS
BernasconiC
StockbauerKE
1997 The emb operon, a gene cluster of Mycobacterium tuberculosis involved in resistance to ethambutol. Nat Med 3 567 570
8. EscuyerVE
LetyMA
TorrellesJB
KhooKH
TangJB
2001 The role of the embA and embB gene products in the biosynthesis of the terminal hexaarabinofuranosyl motif of Mycobacterium smegmatis arabinogalactan. J Biol Chem 276 48854 48862
9. ZhangN
TorrellesJB
McNeilMR
EscuyerVE
KhooKH
2003 The Emb proteins of mycobacteria direct arabinosylation of lipoarabinomannan and arabinogalactan via an N-terminal recognition region and a C-terminal synthetic region. Mol Microbiol 50 69 76
10. BergS
StarbuckJ
TorrellesJB
VissaVD
CrickDC
2005 Roles of conserved proline and glycosyltransferase motifs of EmbC in biosynthesis of lipoarabinomannan. J Biol Chem 280 5651 5663
11. ShiL
BergS
LeeA
SpencerJS
ZhangJ
2006 The carboxy terminus of EmbC from Mycobacterium smegmatis mediates chain length extension of the Arabinan in lipoarabinomannan. J Biol Chem 281 19512 19526
12. AlderwickLJ
SeidelM
SahmH
BesraGS
EggelingL
2006 Identification of a novel arabinofuranosyltransferase (AftA) involved in cell wall arabinan biosynthesis in Mycobacterium tuberculosis. J Biol Chem 281 15653 15661
13. SeidelM
AlderwickLJ
BirchHL
SahmH
EggelingL
2007 Identification of a novel arabinofuranosyltransferase AftB involved in a terminal step of cell wall arabinan biosynthesis in Corynebacterianeae, such as Corynebacterium glutamicum and Mycobacterium tuberculosis. J Biol Chem 282 14729 14740
14. SeidelM
AlderwickLJ
SahmH
BesraGS
EggelingL
2007 Topology and mutational analysis of the single Emb arabinofuranosyltransferase of Corynebacterium glutamicum as a model of Emb proteins of Mycobacterium tuberculosis. Glycobiology 17 210 219
15. LiuJ
MushegianA
2003 Three monophyletic superfamilies account for the majority of the known glycosyltransferases. Protein Sci 12 1418 1431
16. LeeRE
MikusovaK
BrennanPJ
BesraGS
1995 Synthesis of the mycobacterial arabinose donor beta-D-arabinofuranosyl-1-monophosphoryldecaprenol, development of a basic arabinosyl-transferase assay, and identification of ethambutol as an arabinosyl transferase inhibitor. J Am Chem Soc 117 11829 11832
17. WoluckaBA
McNeilMR
de HoffmannE
ChojnackiT
BrennanPJ
1994 Recognition of the lipid intermediate for arabinogalactan/arabinomannan biosynthesis and its relation to the mode of action of ethambutol on mycobacteria. J Biol Chem 269 23328 23335
18. AminAG
GoudeR
ShiL
ZhangJ
ChatterjeeD
2008 EmbA is an essential arabinosyltransferase in Mycobacterium tuberculosis. Microbiology 154 240 248
19. GoudeR
AminAG
ChatterjeeD
ParishT
2008 The critical role of embC in Mycobacterium tuberculosis. J Bacteriol 190 4335 4341
20. RamaswamySV
AminAG
GokselS
StagerCE
DouSJ
2000 Molecular genetic analysis of nucleotide polymorphisms associated with ethambutol resistance in human isolates of Mycobacterium tuberculosis. Antimicrob Agents Chemother 44 326 336
21. BorastonAB
BolamDN
GilbertHJ
DaviesGJ
2004 Carbohydrate-binding modules: fine-tuning polysaccharide recognition. Biochem J 382 769 781
22. HardingMM
2001 Geometry of metal-ligand interactions in proteins. Acta Crystallogr D Biol Crystallogr 57 401 411
23. HolmL
SanderC
1996 Mapping the protein universe. Science 273 595 603
24. IguraM
MaitaN
KamishikiryoJ
YamadaM
ObitaT
2008 Structure-guided identification of a new catalytic motif of oligosaccharyltransferase. EMBO J 27 234 243
25. LeeRE
BrennanPJ
BesraGS
1997 Mycobacterial arabinan biosynthesis: the use of synthetic arabinoside acceptors in the development of an arabinosyl transfer assay. Glycobiology 7 1121 1128
26. PiresVM
HenshawJL
PratesJA
BolamDN
FerreiraLM
2004 The crystal structure of the family 6 carbohydrate binding module from Cellvibrio mixtus endoglucanase 5a in complex with oligosaccharides reveals two distinct binding sites with different ligand specificities. J Biol Chem 279 21560 21568
27. BardarovS
BardarovSJJr
PavelkaMSJJr
SambandamurthyV
LarsenM
2002 Specialized transduction: an efficient method for generating marked and unmarked targeted gene disruptions in Mycobacterium tuberculosis, M. bovis BCG and M. smegmatis. Microbiology 148 3007 3017
28. BhamidiS
SchermanMS
McNeilMR
2009 Mycobacterial cell wall arabinogalactan: a detailed perspective on structure, biosynthesis, functions and drug tageting.
UllrichM
Bacterial polysaccharides Norfolk, UK Caister Academic Press 39 65
29. WalserPJ
HaebelPW
KunzlerM
SargentD
KuesU
2004 Structure and functional analysis of the fungal galectin CGL2. Structure 12 689 702
30. Van DuyneGD
StandaertRF
KarplusPA
SchreiberSL
ClardyJ
1993 Atomic structures of the human immunophilin FKBP-12 complexes with FK506 and rapamycin. J Mol Biol 229 105 124
31. KabschW
1993 Automatic processing of rotation diffraction data from crystals of initially unknown cell constants and symmetry. J Appl Crystallogr 26 795 800
32. SheldrickGM
2008 A short history of SHELX. Acta Crystallogr A 64 112 122
33. de la FortelleE
BricogneG
1997 Maximum-likelihood heavy-atom parameter refinement for multiple isomorphous replacement and multi-wavelength anomalous diffraction methods. Methods Enzymol 276 472 494
34. AbrahamsJP
LeslieAG
1996 Methods used in the structure determination of bovine mitochondrial F1 ATPase. Acta Crystallogr D Biol Crystallogr 52 30 42
35. MorrisRJ
PerrakisA
LamzinVS
2002 ARP/wARP's model-building algorithms. I. The main chain. Acta Crystallogr D Biol Crystallogr 58 968 975
36. EmsleyP
CowtanK
2004 Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60 2126 2132
37. MurshudovGN
VaginAA
DodsonEJ
1997 Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr 53 240 255
38. AdamsPD
Grosse-KunstleveRW
HungLW
IoergerTR
McCoyAJ
2002 PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr 58 1948 1954
39. WinnMD
IsupovMN
MurshudovGN
2001 Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr D Biol Crystallogr 57 122 133
40. SchuckP
2000 Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys J 78 1606 1619
41. StoverCK
de la CruzVF
FuerstTR
BurleinJE
BensonLA
1991 New use of BCG for recombinant vaccines. Nature 351 456 460
42. NigouJ
GilleronM
CahuzacB
BouneryJD
HeroldM
1997 The phosphatidyl-myo-inositol anchor of the lipoarabinomannans from Mycobacterium bovis bacillus Calmette Guerin. Heterogeneity, structure, and role in the regulation of cytokine secretion. J Biol Chem 272 23094 23103
43. GouetP
CourcelleE
StuartDI
MetozF
1999 ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics 15 305 308
44. Jamal-TalabaniS
BorastonAB
TurkenburgJP
TarbouriechN
DucrosVM
2004 Ab initio structure determination and functional characterization of CBM36; a new family of calcium-dependent carbohydrate binding modules. Structure 12 1177 1187
45. van BuerenAL
MorlandC
GilbertHJ
BorastonAB
2005 Family 6 carbohydrate binding modules recognize the non-reducing end of beta-1,3-linked glucans by presenting a unique ligand binding surface. J Biol Chem 280 530 537
46. HenshawJ
Horne-BitschyA
van BuerenAL
MoneyVA
BolamDN
2006 Family 6 carbohydrate binding modules in beta-agarases display exquisite selectivity for the non-reducing termini of agarose chains. J Biol Chem 281 17099 17107
47. DavisIW
Leaver-FayA
ChenVB
BlockJN
KapralGJ
2007 MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res 35 W375 83
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2011 Číslo 2
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Genetic Mapping Identifies Novel Highly Protective Antigens for an Apicomplexan Parasite
- Type I Interferon Signaling Regulates Ly6C Monocytes and Neutrophils during Acute Viral Pneumonia in Mice
- Infections in Cells: Transcriptomic Characterization of a Novel Host-Symbiont Interaction
- The ESCRT-0 Component HRS is Required for HIV-1 Vpu-Mediated BST-2/Tetherin Down-Regulation
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy