
NS2 Protein of Hepatitis C Virus Interacts with Structural and Non-Structural Proteins towards Virus Assembly
Growing experimental evidence indicates that, in addition to the physical virion components, the non-structural proteins of hepatitis C virus (HCV) are intimately involved in orchestrating morphogenesis. Since it is dispensable for HCV RNA replication, the non-structural viral protein NS2 is suggested to play a central role in HCV particle assembly. However, despite genetic evidences, we have almost no understanding about NS2 protein-protein interactions and their role in the production of infectious particles. Here, we used co-immunoprecipitation and/or fluorescence resonance energy transfer with fluorescence lifetime imaging microscopy analyses to study the interactions between NS2 and the viroporin p7 and the HCV glycoprotein E2. In addition, we used alanine scanning insertion mutagenesis as well as other mutations in the context of an infectious virus to investigate the functional role of NS2 in HCV assembly. Finally, the subcellular localization of NS2 and several mutants was analyzed by confocal microscopy. Our data demonstrate molecular interactions between NS2 and p7 and E2. Furthermore, we show that, in the context of an infectious virus, NS2 accumulates over time in endoplasmic reticulum-derived dotted structures and colocalizes with both the envelope glycoproteins and components of the replication complex in close proximity to the HCV core protein and lipid droplets, a location that has been shown to be essential for virus assembly. We show that NS2 transmembrane region is crucial for both E2 interaction and subcellular localization. Moreover, specific mutations in core, envelope proteins, p7 and NS5A reported to abolish viral assembly changed the subcellular localization of NS2 protein. Together, these observations indicate that NS2 protein attracts the envelope proteins at the assembly site and it crosstalks with non-structural proteins for virus assembly.
Published in the journal:
. PLoS Pathog 7(2): e32767. doi:10.1371/journal.ppat.1001278
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1001278
Summary
Growing experimental evidence indicates that, in addition to the physical virion components, the non-structural proteins of hepatitis C virus (HCV) are intimately involved in orchestrating morphogenesis. Since it is dispensable for HCV RNA replication, the non-structural viral protein NS2 is suggested to play a central role in HCV particle assembly. However, despite genetic evidences, we have almost no understanding about NS2 protein-protein interactions and their role in the production of infectious particles. Here, we used co-immunoprecipitation and/or fluorescence resonance energy transfer with fluorescence lifetime imaging microscopy analyses to study the interactions between NS2 and the viroporin p7 and the HCV glycoprotein E2. In addition, we used alanine scanning insertion mutagenesis as well as other mutations in the context of an infectious virus to investigate the functional role of NS2 in HCV assembly. Finally, the subcellular localization of NS2 and several mutants was analyzed by confocal microscopy. Our data demonstrate molecular interactions between NS2 and p7 and E2. Furthermore, we show that, in the context of an infectious virus, NS2 accumulates over time in endoplasmic reticulum-derived dotted structures and colocalizes with both the envelope glycoproteins and components of the replication complex in close proximity to the HCV core protein and lipid droplets, a location that has been shown to be essential for virus assembly. We show that NS2 transmembrane region is crucial for both E2 interaction and subcellular localization. Moreover, specific mutations in core, envelope proteins, p7 and NS5A reported to abolish viral assembly changed the subcellular localization of NS2 protein. Together, these observations indicate that NS2 protein attracts the envelope proteins at the assembly site and it crosstalks with non-structural proteins for virus assembly.
Introduction
The hepatitis C virus (HCV) has a high propensity to establish a persistent infection in the human liver. Approximately 170 million people suffer from chronic hepatitis C and are at risk to develop cirrhosis and hepatocellular carcinoma [1]. Current antiviral therapy is based on the use of polyethylene glycol conjugated interferon alpha in combination with ribavirin. However, this treatment is expensive, relatively toxic and effective in only approximately half of the treated patients [2]. A better understanding of the HCV life cycle is therefore essential for the development of more efficacious and better tolerated anti-HCV treatments.
HCV is an enveloped virus that belongs to the Hepacivirus genus in the Flaviviridae family [3]. HCV has a positive strand RNA genome encoding a single polyprotein that is cleaved by cellular and viral proteases into 10 different proteins: core, E1, E2, p7, NS2, NS3, NS4A, NS4B, NS5A and NS5B [3]. The non-structural proteins NS3 to NS5B are involved in the replication of the viral genome, whereas the structural proteins (core, E1 and E2) are the components of the viral particle (reviewed in [4]). The remaining proteins, p7 and NS2, are dispensable for RNA replication and there is no evidence that they are part of the viral particle [5], [6].
For reasons still unknown, HCV clinical isolates do not propagate in cell culture. However, with the development of a cell culture system that enables a relatively efficient amplification of HCV (HCVcc) [7], [8], [9], all the steps of the HCV life cycle can be investigated. Due to the accumulation of HCV core protein around lipid droplets (LDs) (reviewed in [10]), a role of these lipid bodies in HCV assembly has been suspected for a long time. Moreover, it has recently been shown that viral non-structural proteins like NS5A and NS3 and double stranded viral RNA are also present around LDs [11], [12]. The association between core and LDs seems to play a role in the recruitment of the other viral proteins and for virus production [12], [13]. Furthermore, NS5A plays a double role in both replication and assembly processes as a potential switch between these two steps [14], [15], [16], [17].
Since it is dispensable for HCV RNA replication and it does not seem to be incorporated into viral particles [6], NS2 has been suspected to be involved in the assembly process of HCV particle. Recently, experimental evidence supporting this hypothesis has been obtained [18], [19], [20]. Although the role of NS2 in the assembly process remains elusive, some data suggest that NS2 might interact with viral partners involved in virion morphogenesis. Indeed, construction of chimeric viruses between different genotypes identified the C-terminus of the first transmembrane segment of NS2 as the optimum crossover point [21]. Thus, a genetic interaction was implied between the N-terminus of NS2 and the upstream structural proteins. In the context of a chimeric virus containing genotype 1a and 2a sequences, adaptive mutations in E1, p7, NS2 and NS3 were identified, also suggesting genetic interactions between these proteins [22]. Moreover, a detailed rescue mutant analysis recently showed genetic interactions between NS2, E1E2 and NS3-4A [23]. However, despite these genetic analyses we have almost no understanding about NS2 interactions with other viral proteins and the role of these interactions in the production of infectious particles. Here, we report molecular interactions between NS2 and p7 and E2 proteins. Using a functional HCVcc virus with a reporter epitope at the N-terminus of NS2, we found that NS2 accumulates in dotted structures derived from the endoplasmic reticulum (ER) and colocalizes with E1, E2, NS3 and NS5A in close proximity to the core protein and LDs. Mutations and deletions in the p7-NS2 region affecting the subcellular localization of NS2 and its physical protein-protein interactions abolished viral assembly. Moreover, mutations in other viral proteins reported to inhibit the assembly process induced consistent changes in NS2 subcellular localization. Together, these data suggest that p7, NS2 and E2 form a functional unit which drives the proteins in the proximity of the LDs where NS2 crosstalks with other viral proteins during the virion assembly process.
Results
Mutation rationale in NS2
NS2 is a polytopic transmembrane protein containing 3 putative transmembrane segments [19](Figure 1). The p7 polypeptide and E1E2 heterodimer, which are putative partners of NS2, are also membrane proteins that contain transmembrane segments [24], [25]. Due to their respective topologies (Figure 1A), it is expected that interactions between these three proteins would involve helix-helix contacts in their transmembrane segments. Furthermore, helix-helix interactions between transmembrane segments of NS2 are also likely to take place.

To analyze the role of the transmembrane domain of NS2 in potential protein-protein interactions, we used a previously reported deletion mutant of NS2 (ΔTM12) which lacks the first two transmembrane segments [19]. For a more refined analysis, we also used alanine scanning insertion mutagenesis, a technique which has been shown to disrupt helix-helix interactions in a membrane environment [26], [27], [28], [29]. As illustrated in Figure 1C for the first transmembrane segment of NS2, this approach is based on the fact that insertion of a single amino acid into a transmembrane helix displaces the residues on the N-terminal side of the insertion by 110° relative to those on the C-terminal side of the insertion. The subsequent perturbation of the residue side-chain distribution could disrupt a potential helix-helix packing interface involving residues on both sides of the insertion. Such mutations are therefore expected to disrupt helix-helix interactions between the transmembrane segments of NS2 and/or between NS2 and other putative partners like p7 or E1E2.
Based on the NMR structure of the transmembrane segments of NS2 [19](Jirasko et al., 16th International Symposium on HCV and Related Viruses, Nice, October 3–7, 2009), we designed insertion mutations in the transmembrane domain of NS2. Three different mutants were designed by inserting alanine residues close to the middle of the transmembrane segments of NS2 (Figure 1, B–C). The positions for alanine insertions were carefully chosen within the putative helical segments to preserve the overall fold of the corresponding helices. According to previous reports [26], [27], [28], [29], alanine insertions near the center of putative transmembrane helices were expected to be the most efficient to disrupt inter-helices potential interactions.
NS2 transmembrane domain mediates E2-NS2 interaction
Firstly, we wanted to evaluate the impact of our mutations on the viral life cycle. As negative controls for virus assembly, we used assembly-deficient viruses JFH-ΔE1E2-HA and JFH-Δp7-HA, containing a deletion in the regions encoding HCV envelope glycoproteins and the p7 polypeptide, respectively (Figure S1). As template for all our constructs, we used a full-length JFH1 plasmid containing adaptive mutations [30] in which the N-terminal sequence of E1 has been modified to reconstruct the A4 monoclonal antibody (Mab) epitope of the H77 isolate in order to facilitate the immunodetection of this envelope protein [31]. Moreover, we introduced an HA epitope in the N-terminus of NS2 to be used for protein detection (JFH-HA). Huh-7 cells were electroporated with different mutated viral genomes and the production of infectious virus was assessed at 72h by supernatant titration (Figure 2A). The insertion of the HA epitope did not affect the virus production as compared to the wild-type virus (JFH). As previously reported, deletion of the first two transmembrane segments prevented the production of infectious particles similar to the negative controls [19] (Figure 2A and S1). Furthermore, alanine insertions also drastically affected the viral production. While mutants A16 and A41 presented residual infectivity, A82 was not able to produce infectious particles (Figure 2A).

To identify the stage where the virus production was affected, we started with the evaluation of the replication capacity of our viruses. To this aim, we used Renilla luciferase reporter viruses (Figure S1A). As a negative control for replication, we used the replication-deficient construct JFHGND-Luc which contains a mutation in NS5B that prevents viral genome replication [7]. As the RNA input after electroporation is potentially variable, we evaluated the capacity of our viruses to replicate by determining the ratio between the luciferase activity at 72h and 4h post-electroporation when only the luciferase activity of the input RNA is present as previously shown [32]. As shown in Figure S1B, all the mutant viruses presented a similar replication capacity, which was comparable to the control viruses, whereas replication was abolished in the JFHGND-Luc mutant (Figure S1B-upper panel).
Since the replication was not affected, the effect of alanine insertion on NS2 protein stability was further investigated. As shown in Figure S1C, A82 insertion was detrimental for the protein integrity. For A16 mutant, the level of expression of NS2 was slightly lower as compared to the JFH-HA control. However, the level of expression of HCV proteins was higher for the JFH-HA control in this particular experiment. We therefore measured the NS2/E2 ratios which were similar for A16, A41 and JFH-HA, indicating that A16 and A41 insertions did not affect NS2 stability. Therefore, we focused our analysis on A16 and A41 mutants.
To test whether the lack of infectivity could be due to a defect in virus secretion or the release of non-infectious particles, we determined the level of core protein in the supernatants. The quantity of core protein in supernatants decreased drastically as compared to wild-type, paralleling the decrease in infectivity (Figure 2B). It has to be noted that for the mutants showing a residual infectivity (A16, A41 and RR/QQ), the release of core was at the same level as the dead mutants (ΔTM12, A82 and Δp7). This likely reflects a difference of sensitivity between the two assays. Then, we measured the viral RNA in the supernatants by qRT-PCR as previously described [17]. For all the alanine insertions, the release of viral RNA was close to the background level observed for the assembly-deficient control viruses (Figure S1B – lower panel), indicating a defect in particle secretion. These data suggest that either the process of assembly is affected at an early stage or the secretion of assembled infectious particles is impaired. To answer this question, we compared the intra and extracellular level of the core protein (Figure 2B). The ratio between intra and extracellular core was similar to JFH-Δp7-HA which was shown to be defective in early assembly steps. We also measured the intracellular infectivity of the alanine mutants in the Rluc reporter viruses context as previously reported [20]. As shown in Figure S1B (middle panel), there was no accumulation of intracellular infectivity for any of the mutants. These results suggest that mutations in the transmembrane domain of NS2 prevent the assembly process rather than the secretion of particles.
Considering the rationale of our mutagenesis, we investigated protein-protein interactions in E1E2-p7-NS2 region. Indeed, due to their position within the HCV polyprotein, it is reasonable to think that these proteins might potentially interact. Genetic and co-immunoprecipitation data suggest that NS2 interacts with E2 [23], [32], [33], [34]. Interaction assays between NS2 and E2 were performed in the context of the JFH-HA virus, which allows to immunoprecipitate NS2 with the HA tag (Figure 2C, JFH-HA). As shown in Figure 2C (JFH-HA), E2 and NS2 proteins migrated at the expected molecular mass, indicating that the polyprotein was correctly processed in these viruses. It has to be noted that in the absence of p7, E2 migrated slightly faster. This is likely due to the absence of E2-p7, which has a slightly slower migration profile in the band corresponding to E2.
The E2-NS2 interaction was then tested by co-immunoprecipitation with an anti-HA antibody followed by the detection of E2 by Western blotting. As shown in Figure 2C (JFH-HA), E2 co-precipitated with NS2, confirming that these proteins interact together in the context of the virus. In contrast, deletion of the first two transmembrane segments prevented the interaction between NS2 and E2 (Figure 2C, ΔTM12), whereas alanine insertions in the transmembrane region had different effects. While A16 (within TM1) did not affect the E2-NS2 interaction, A41 (within TM2) induced a consistent decrease in the amount of E2 co-precipitated by NS2 (Figure 2C, A16 and A41).
We further investigated the effect of p7 on the E2-NS2 interaction. To this aim, we used two mutants of p7, a deletion mutant (JFH-Δp7-HA) and a mutant having the two arginine residues in the cytosolic loop of p7 replaced by glutamine residues (JFH-RR/QQ-HA) (Figure 2C). This RR/QQ mutation is believed to abolish the ion channel activity of p7, and it induces a drastic decrease in virus production as previously reported [32], [35] and confirmed by us as shown in Figure 2A. As shown in Figure 2C (JFH-Δp7-HA and JFH-RR/QQ-HA), E2 and NS2 proteins migrated at the expected molecular mass, indicating that the polyprotein was correctly processed in these viruses. However, the two mutations had no significant effect on E2-NS2 interaction (Figure 2C JFH-Δp7-HA and JFH-RR/QQ-HA), suggesting that p7 does not modulate E2-NS2 interaction.
NS2 accumulates in dotted structures
To obtain more insight into the assembly defects of our viral mutants, we decided to determine their potential effect on the subcellular localization of NS2. To this aim, we first characterized the subcellular localization of the wild-type NS2 in the context of an infectious virus, which has never been reported before. NS2 presented an ER-like reticulated pattern, and in some cells, accumulated in dotted structures (Figure 3A and data not shown). The number of these NS2-positive dot-like structures increased over time, suggesting a transition from an initial reticulate pattern to these dotted structures. At 72 hours post-electroporation, 34±15% of infected cells from 8 different electroporations displayed NS2 dots. The size of these structures was 0.84±0.38 µm (mean±SD, n = 402).

To further characterize these NS2 structures, we performed co-localization analyses with different cellular markers. As expected, the reticulate and perinuclear pattern of NS2 overlapped with calreticulin staining as well as other ER markers like calnexin and PDI (data not shown). Interestingly, NS2 dots also colocalized with ER markers (Figure 3B), but they did not colocalize with other organelle markers of the secretory pathway (data not shown). Importantly, NS2 dots were also found in close proximity of LDs (Figure 3B), suggesting that they might play a role in HCV assembly.
To better understand the potential role of NS2 dots in virus assembly, we analyzed the subcellular localization of NS2 in relationship with the other viral proteins. NS2 dots overlapped with HCV envelope glycoproteins E2 (Figure 3B) and E1 (Figure S2). NS5A and NS3 were also shown to colocalize with NS2 in its dotted pattern (Figures 3B and S2). Importantly, the structures containing both NS2 and NS5A were observed in close proximity to LDs (Figure 4A). This is illustrated by the presence of magenta dots (red NS2 and blue NS5A) in the proximity of LD (green). Finally, as observed with the LDs, NS2 dots were also found in close proximity to core protein (Figure S2), and as expected this association was observed in close proximity to LDs (Figure 4B). As shown in Figure 4B, core (blue) is tightly associated to LD (green) as the LD becomes cyan due to colocalization, NS2 (red) localizes in regions juxtaposed to the cyan LD (Figure 4B). Relying on the spatial proximity of NS2 dots, core, E1E2, NS3, NS5A proteins and LDs, we speculated that NS2 present in these structures is involved in the assembly process. Thus, we established some criteria of functionality for NS2 positive structures. They have to localize in the proximity of LDs and core protein and more importantly to colocalize with NS5A protein, which we used as a criteria for quantification purposes.

It has to be pointed out that a low number of cells contained NS2-positive structures with a different pattern of subcellular localization (Figure S3). This different pattern was indeed observed in approximately 1 to 3% of the cells at 72h post-infection or post-electroporation. These structures colocalized less with ER markers and they overlapped with ERGIC-53, a marker of the ER-to-Golgi intermediate compartment [36] (Figure S3). Furthermore, these NS2 dots were detected in close proximity to the ER exit sites, which were identified by markers of the COP II coatomer, Sec31 and p125 [37], [38] (Figure S3 and data not shown). However, cells containing these NS2 positive structures showed dramatic alterations of the secretory compartments as observed by immunofluorescence analysis of ER-to-Golgi intermediate compartment and Golgi morphology using ERGIC-53 and GM130 as markers (Figure S3). It is worth noting that NS2 did not colocalize with NS3 or NS5A and it was not found in the proximity of core and LDs in these cells (Figure S3). Due to these alterations, it is unlikely that these cells are involved in the production of infectious virus.
We asked further the relevance of NS2 dots for the production of infectious particles. After electroporation, we determined the titer of virus production at different time points (Figure 4C, upper panel). In parallel, we counted the number of cells which presented NS2/NS5A positive dots for reasons detailed above (Figure 4C, lower panel). The kinetics of virion production and the percentage of cells presenting NS2/NS5A positive dots showed a high correlation with a correlation coefficient of 0.9. To exclude the possibility that the NS2 phenotype depends on the cell culture adaptive mutations, we performed a similar experiment with a virus that does not contain the mutations. As for the adaptive mutant, virus production in the absence of mutation paralleled the NS2/NS5A dots formation with a significant correlation coefficient (data not shown). These data reinforce the idea that NS2/NS5A positive dots are involved in the virus production process.
To further investigate the NS2 localization, we performed immuno-electron microscopy with an anti-HA antibody on Huh7 cells which were electroporated with JFH-HA RNA and prepared by cryosubstitution. As shown in Figure 4D, we could identify clusters of gold particles in the proximity of LDs. Moreover, two of the clusters are lying on preserved ER bilayers. For one of the clusters of gold particles, a connection between the ER bilayer and the LDs could be observed (Figure 4D). Thus, the immuno-electron microscopy confirms the juxtaposition between NS2 dots and LDs in a 0.2µm range, which is consistent with the observations in confocal microscopy. It has to be noted that gold particles were detected on both sides of the membrane even if the HA epitope is supposed to be located in the ER lumen. This is compatible with the length of two antibodies (primary+secondary) since the gold particles were never further away than 30 nm from the membrane. However, we cannot exclude a double topology for NS2 as recently suggested [39].
NS2 transmembrane region is crucial for its subcellular localization
The next obvious step was to determine the subcellular localization of NS2 for the different mutants defective in assembly. In the case of ΔTM12, NS2 localized in confined structures which did not colocalize with NS5A and they were not associated with the core protein or LD (Figure 5, panels A and C and data not shown). Moreover, there was no colocalization between the truncated NS2 and the E2 glycoprotein (Figure 5B), which correlates with the lack of interaction between the two proteins (Figure 2C). Then, we analyzed the subcellular localization of NS2 for the alanine insertion mutants. While A16 mutant presented NS2 dotted structures as wild-type, A41 mutation induced a drastic decrease in the percentage of cells with NS2 dots (Figure 5, panels A and C). These data suggest that NS2 transmembrane region is an important localization determinant.

Core, envelope and p7 proteins influence NS2 subcellular localization
A peculiarity of the Flaviviridae family is the involvement of both structural and non-structural proteins in the assembly process (reviewed in [40]). Thus, we wanted to investigate the NS2 subcellular localization in the context of assembly deficient viruses having mutations in different viral proteins.
Recruitment of core protein to LDs was reported to be essential for a productive assembly process [12], [13]. The proline residues 138 and 143 in domain D2 of the core protein are crucial for virus production and core recruitment to LDs [13], [41]. Furthermore, the mutation of these proline residues has been previously shown to prevent the core induced recruitment of NS5A to the LDs [12]. Therefore, we introduced these mutations in the context of JFH-HA virus (Figure 6, JFH-HA-PP). As previously reported [13], the mutation prevented the production of infectious virions (Figure 9A), and the core protein was not redistributed to LDs, which in turn remained spread in the cytoplasm rather than the perinuclear localization induced by a functional core protein (Figure S4). As shown in Figure 6, in the context of this mutation, NS2 protein maintained the capacity to accumulate in dotted structures that colocalized with NS5A. In contrast to the wild-type, NS2 dotted structures were not found in the vicinity of LDs in the context of the PP mutation, suggesting that NS2 does not have the signals to localize by itself around the LDs (Figure S4). Importantly, in this context, the number of cells presenting NS2 dotted structures increased tremendously in comparison to the wild-type (Figure 6B). These observations suggest that the PP mutation induces a block in the assembly process, which favors the accumulation of NS2 protein in the dotted structures.
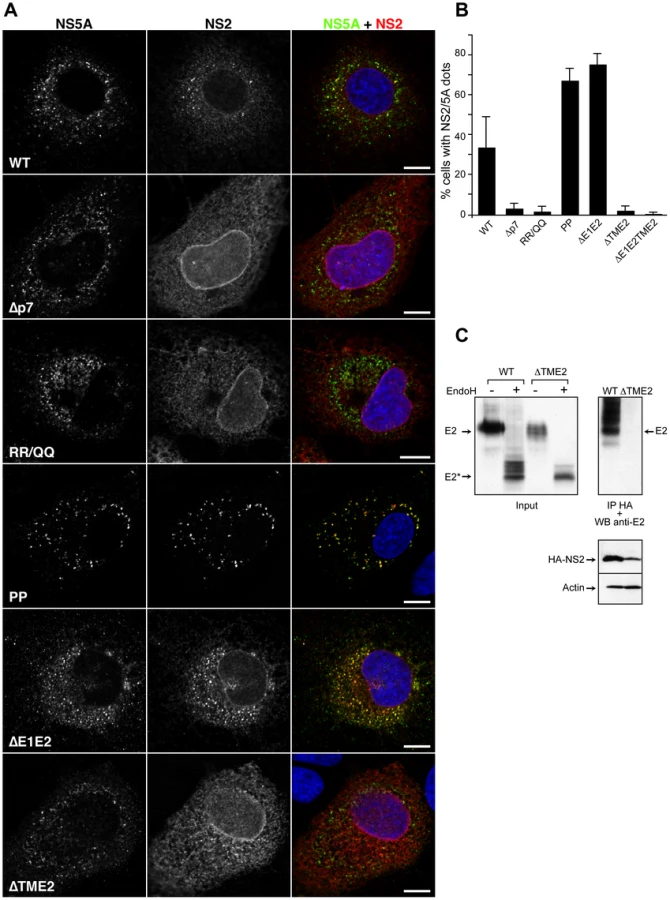
As structural proteins, the envelope glycoproteins are involved in the assembly process [42]. The envelope proteins play a crucial role in the assembly of enveloped viruses, which can be due for some viruses to the capacity of the envelope proteins to establish lateral interactions and to generate a pushing force necessary for the budding process [43]. A deletion in the envelope region would therefore block the assembly process as shown by an in-frame deletion of 351 amino acids in the envelope proteins region [7]. In the context of our JFH-HA virus, this deletion mutant is also fully replicative, and it does not produce infectious particles (data not shown). To further understand the interplay between NS2 and HCV envelope glycoproteins, we analyzed the subcellular localization of NS2 protein in the context of the E1E2 deletion. In this context, NS2 localized in NS5A positive structures juxtaposed to the LDs and core protein (Figure 6 and S4). Interestingly, as for the JFH-HA-PP virus, the number of cells presenting NS2 dotted structures also increased in the case of JFH-ΔE1E2-HA, correlating also with a block in the assembly process favoring an accumulation of NS2 dotted structures (Figure 6B).
The deletion introduced in the envelope region is predicted to generate a chimeric protein comprising the N-terminus of E1 and the C-terminus of E2 protein. Since we introduced the A4 epitope in the N-terminus of E1, we were able to detect the subcellular localization of this small chimeric protein. It is worth mentioning that we also detected this truncated chimeric protein in NS2/NS5A dotted structures, suggesting that the E2 transmembrane domain is sufficient for the recruitment of HCV envelope proteins to NS2 dotted structures (Figure S4).
The p7 protein has been shown to be crucial for the assembly process [32], [35]. Moreover, the ion channel activity of p7 correlates with the virus assembly process since mutations predicted to abolish the ion channel activity have a strong inhibitory effect on the virus production [32], [35]. We therefore also analyzed the subcellular localization of NS2 in the context of p7 mutants corresponding to a complete deletion (JFH-Δp7-HA) or an amino acid substitution (JFH-RR/QQ-HA) previously reported to affect the assembly and release of the virus [32], [35]. The two constructs behaved as expected. While JFH-Δp7-HA produced no infectious particles, JFH-RR/QQ-HA presented a 2 log10 decrease in virus titers at 72h post-electroporation (Figure 2A). Importantly, the two mutants induced a drastic decrease in the number of cells presenting NS2/NS5A dotted structures (Figure 6B). Together, these data indicate that NS2 needs a functional p7 polypeptide to colocalize with NS5A in dotted structures.
Other partners than p7 are likely necessary for NS2 accumulation in dotted structures. Indeed, as shown in Figure 5, A41 and ΔTM12 mutants, which fail to interact with E2, also present a drastically reduced number of NS2 dotted structures. This suggests that NS2-E2 interaction might be crucial for NS2 subcellular localization. Thus, one additional determinant could be represented by the transmembrane domain of E2, which is most likely the interacting region with NS2 due to topological constraints. In order to check this hypothesis, we used both JFH-HA and JFH-ΔE1E2-HA constructs in which we replaced the transmembrane region of E2 with the autoprotease 2A from foot and mouth disease virus (FMDV) (JFH-ΔTME2-HA and JFH-ΔE1E2TME2-HA)(Figure S1). We first verified that a proper processing of the polyprotein mediated by FMDV 2A protease has occurred in these constructs. To check our constructs, we analyzed the molecular mass of E2 following deglycosylation with EndoH or PNGase endoglycosidases. As shown in Figure 6C, E2 from JFH-HA and JFH-ΔTME2-HA presented a similar molecular mass after deglycosylation with EndoH, suggesting that the FMDV 2A protease is functional since FMDV 2A and the transmembrane domain of E2 have similar sizes. Indeed, if FMDV 2A protease had not been functional, we would have observed a difference of 7kD corresponding to the molecular mass of unprocessed p7 (Figure 6C and data not shown). Interestingly, in contrast to the wild-type envelope protein, the truncated E2 did not interact with NS2 (Figure 6D). Furthermore, in contrast to what was observed for JFH-HA and JFH-ΔE1E2-HA (Figure 6A), NS2 protein of the ΔTME2 mutant presented an ER like pattern and the formation of NS2 dotted structures was prevented (Figure 6A and B). Similar data were also obtained with JFH-ΔE1E2TME2-HA construct (Figure 6B). Thus, it seems that p7-NS2 and the transmembrane domain of E2 form a functional unit that targets these proteins to NS5A positive structures.
NS2 interacts with p7 in a co-immunoprecipitation assay
The above data suggest a possible interaction between p7 and NS2. We therefore explored this putative interaction in a biochemical assay, by analyzing p7-NS2 association in a co-immunoprecipitation assay. Due to the difficulties in analyzing p7-NS2 interactions in the context of an infectious virus, we analyzed these interactions by co-transfecting cells with plasmids expressing these two proteins only. In this approach, the p7 polypeptide and NS2 were tagged with a Flag or a HA epitope, respectively (Figure 7A, p7-Flag and HA-NS2).

The p7-NS2 interaction was tested after co-expression of the tagged proteins in 293T cells. Co-immunoprecipitation experiments were performed with an anti-Flag antibody linked to agarose beads. The immunoprecipitates were separated by SDS-PAGE and probed with anti-HA antibodies by Western blotting. As shown in Figure 7B, NS2 of different genotypes coprecipitated with p7. Since only p7 of genotype 1a was used in these experiments, it suggests that p7 interacts with NS2 in a genotype independent manner. However, we cannot exclude that the system is not sensitive enough to discriminate between slight changes in affinities.
Further, we constructed two chimeric proteins, NS2 tagged with a green fluorescent protein (NS2-GFP) and NS2-GTM (Figure 7A). In NS2-GFP, the cytosolic domain of NS2 was replaced by GFP protein, whereas for NS2-GTM, we replaced the transmembrane domain of NS2 by the transmembrane domain of glycoprotein G of VSV. As shown in Figure 7B, the NS2-GFP could be precipitated by p7-FLAG, while NS2-GTM could not. This clearly shows that the transmembrane region is the main determinant of p7-NS2 interaction.
NS2 interacts with p7 in a fluorescence resonance energy transfer with fluorescence lifetime imaging microscopy (FRET-FLIM) assay
To confirm the p7-NS2 interaction with another approach, we used the FRET-FLIM technique. FRET-FLIM requires the presence of two fluorophores (a donor and an acceptor) fused in frame to the studied proteins. If the two proteins interact, an energy transfer occurs between the two fluorophores and the fluorescence life time of the donor (a parameter of the energy emitted by the donor) will decrease. To measure p7-NS2 interactions by FRET-FLIM, Cerulean fluorescent protein (CFP) and Venus yellow fluorescent protein (YFP) were fused to the N-terminus of p7 and NS2, respectively (Figure 8A).

As previously reported, CFP-p7 and YFP-NS2 showed a reticulate perinuclear distribution (Figure 8B), which is characteristic of ER proteins [34], [44]. Western blotting analyses indicated that CFP-p7 and YFP-NS2 migrate at the expected molecular mass with some degradation products of lower molecular weight (Figure 8C). The energy transfer in FRET-FLIM assay needs the integrity of the fluorophores and the correct positioning of the interacting partners. The degradation products could fall into two categories: either soluble fluorophores or membrane bound truncated chimeras. In either case the energy transfer is unlikely to occur with the cleavage products. Thus, the presence of degradation byproducts is unlikely to influence the accuracy of the FRET-FLIM acquisitions.
After biphoton laser excitation and data analysis, fluorescence life time maps were built. Interestingly, the regions showing interactions were located in distinct spots throughout the cells as illustrated in a fluorescence life time color-coded map (Figure 8D). A summary of FRET-FLIM analysis is presented in Table 1. The mean life time of fluorescence decreased from 2.69±0.12 ns (n = 10) in cells transfected with the donor only (CFP-p7) to 2.34±0.09 ns (n = 10) for double transfections (CFP-p7+YFP-NS2). The variation of the mean donor life time is characteristic for energy transfer between two fluorescent proteins in FRET-FLIM analyses as previously observed for other protein-protein interactions [45], [46]. As a positive control, we used the transmembrane domains of HCV glycoproteins E1 and E2 which are known to interact and to have the same subcellular localization as p7 and NS2 [25]. The positive control couple presented a comparable decrease in the mean lifetime of the donor to CFP-p7/YFP-NS2 couple. Indeed, the mean life time decreased from 2.56±0.03 ns (n = 14) in cells transfected with the donor only (CFP-E2) to 2.35±0.08 ns (n = 14) for double transfections (CFP-E2+YFP-E1). As a negative control, we used CFP fused to the transmembrane domain of yellow fever virus E protein (CFP-EYF), a donor protein with the same topology and localization as p7 [28], [47]. As shown in Table 1, the mean life time of the donor in monotransfection did not change in double transfections confirming the lack of interaction between CFP-EYF and YFP-NS2 (n = 11). The biphoton pictures for the positive and the negative control are shown in Figure S5. Thus, these data strongly suggest that p7 and NS2 proteins interact intracellularly.

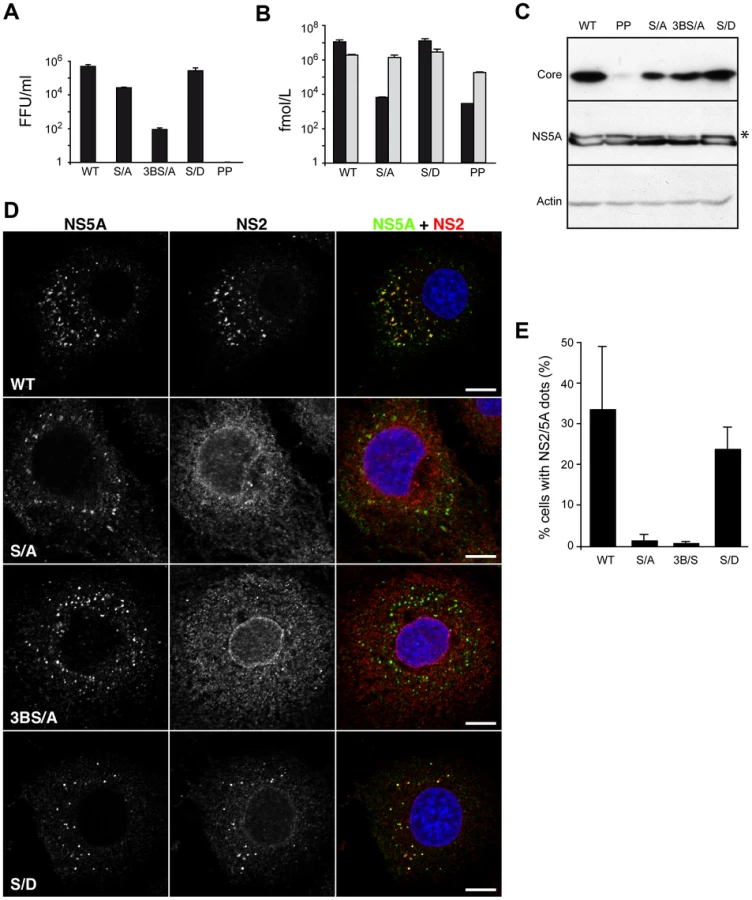
NS5A stabilizes NS2 dotted structures
Among the non-structural proteins, NS5A is the most characterized in terms of its role in the assembly process. NS5A is recruited through direct interaction by the core protein around LDs where its domain III is involved in the assembly process potentially by its phosphorylation [14], [16], [17]. By deletion mutagenesis, Tellinghuisen et al. identified a cluster of serine residues at positions 452, 454 and 457, which are crucial for virus production [17]. Furthermore, by alanine scanning mutagenesis, Masaki et al. reported that the same serine cluster is involved in the direct interaction between NS5A and core protein [16]. While Tellinghuisen et al. reported that serine 457 alone is essential for virus production, Masaki et al. showed that only double mutants had a significant impact on virus production [16], [17]. The apparent contradiction might be explained by the different viruses and time points for virus production assessment. While Tellinghuisen et al. used a chimeric virus consisting of the structural proteins of J6 strain up to NS2 protein, Masaki et al. used the wild type JFH strain [16], [17]. Thus, we constructed the two mutants in the context of our JFH-HA virus – JFH-S/A-HA and JFH-3BS/A-HA, respectively. We analyzed the phenotype of the mutants as well as the polyprotein processing. The results fitted the literature with JFH-S/A-HA virus infectivity moderately reduced at 72h and JFH-3BS/A-HA profoundly impaired in infectious virus production, while the replication and protein integrity were unaltered (Figure 9A, B, C). As reported, we showed that JFH-S/A-HA and JFH-3BS/A-HA present less hyperphosphorylated NS5A (Figure 9C). Surprisingly, for both mutants, NS2 localized mainly in an ER-like pattern and the number of cells with NS2/NS5A dots decreased dramatically (Figure 9D, E). The serine 457 may be replaced by an aspartate residue, which mimics a phosphoserine [17]. We therefore introduced the same mutation (JFH-S/D-HA) and as reported the virus titers were restored at wild-type levels (Figure 9A). Interestingly, this mutant partially recovered the NS2 subcellular localization both qualitatively and quantitatively (Figure 9D, E). Together, these results suggest that NS5A phosphorylation might stabilize the NS2 dotted structures in the assembly process.
Discussion
Our understanding of the HCV morphogenesis process is still in its infancy. Different viral components were identified as players in the morphogenesis process. As expected, the structural proteins are essential in the virus makeup [7]. The scenario gets more complicated with the involvement of non-structural proteins in the assembly process. The p7 polypeptide, NS2, NS3, NS4B, NS5A were reported to be involved in viral assembly [14], [17], [20], [22], [32], [48]. However, the mechanism of the complex interplay between the structural and non-structural proteins towards the virion production is not understood. In this paper, we provide evidence for molecular interactions between NS2, p7 and E2, respectively. Furthermore, we show that NS2 accumulates over time in ER-derived dotted structures and colocalizes with the envelope glycoproteins and components of the replication complex in close proximity to the core protein and LDs. Characterized assembly deficient mutants in both structural and non-structural proteins present qualitative and quantitative modifications in NS2 subcellular localization. Indeed, specific mutations within NS2, p7 or E2 modify the subcellular localization of NS2 and impair virus production. Mutations in core, envelope proteins or NS5A affect the NS2 subcellular localization along with the virus titers. Altogether, these observations indicate that NS2 protein crosstalks with both structural and non-structural proteins during virus assembly.
Our data demonstrate a physical interaction between NS2 and p7. This interaction correlates with the previously reported genetic interactions present in the C-NS2 region [21], [22]. The HCV p7 polypeptide is a viroporin involved in viral assembly [20], [32], [49]. Viroporins represent a class of viral proteins that are involved in the viral morphogenesis process in different and largely unknown manners. Alphavirus 6K interacts with E1 and p62 envelope glycoproteins and is involved in optimal assembly and release of the virion by an unknown mechanism [50], [51]. The E protein of coronaviruses interacts with the M protein and is crucial for the assembly of virus-like particles and virions [52], [53], [54]. To our knowledge, the HCV p7 polypeptide is the first viroporin which interacts with a non-structural protein (NS2) and this might be a peculiarity of the members of the Flaviviridae family where the non-structural proteins are involved in particle assembly [40].
Topologically, the transmembrane domain of NS2 (NS2TM) would be the main region available for interactions with the upstream transmembrane proteins p7 and E1E2. Based on this assumption, we wanted to characterize NS2 interactions with p7 and E2 by deletion and chimeric mutants. Both NS2-E2 interaction and NS2-p7 interaction were mapped in the transmembrane region of NS2 as expected. However, since the lack of p7 does not affect the E2-NS2 interaction, we could imagine that NS2 uses separate domains to interact with p7 and the envelope proteins. Interestingly, when we deleted the transmembrane region of E2, the NS2-E2 interaction was impaired and the NS2 subcellular localization changed. Altogether, these observations suggest that p7, the transmembrane domain of NS2 and the transmembrane domain of E2 contain signals which act synergistically to direct the NS2 protein towards the NS5A positive membranes in the LD proximity.
The drastic effects of alanine insertion mutagenesis on HCV infectivity reflect more intrinsic effects on NS2 function in virus assembly. As shown for lactose permease, single alanine residue insertions into transmembrane helices of a polytopic membrane protein can be highly disruptive to protein structure and function due to their effects on intramolecular helix-helix interactions [26]. Furthermore, within the same protein, different transmembrane helices can have differential sensitivities to single residue insertions [26]. In the case of NS2, our data indicate that an insertion in the third putative transmembrane helix strongly reduces the stability of the protein, suggesting a drastic alteration of NS2 structure by this mutation. The decrease in infectivity for the mutations in the first two transmembrane helices also indicates a drastic alteration in NS2 function that can be linked to local alteration of its structure, as suggested by the change in subcellular localization of NS2 mutant A41. Finally, the drastic decrease in infectivity of mutant A16 in spite of NS2 localization in dotted structures indicates that the subcellular localization of NS2 in these structures is not sufficient by itself for infectivity. Rather, it likely needs to play additional function(s) at the site of virus assembly and such function(s) would be disrupted by the A16 mutation. One explanation could be that a weak E2-NS2 interaction as seen for A41 is not able to direct the p7-NS2 unit to the LDs and a potential strong interaction as for A16 does not allow the p7-NS2 unit to release the envelope proteins heterodimer to the assembly site and the subsequent recycling/dissociation of the unit.
Over time, a substantial part of NS2 accumulates in dotted structures localized in the ER in close proximity to the core protein which is associated to LDs. Since the LD/ER interface is considered as the potential particle assembly site [12], NS2 localization close to the LDs is expected to correlate with its function in a late step of the viral life cycle. Interestingly, NS2 accumulation in dotted structures parallels the colocalization of NS2 with NS5A, NS3 proteins and most likely the replication complex. Moreover, the NS2 dotted structures are juxtaposed to the core protein and colocalize with the envelope proteins E1 and E2. Thus the NS2 dots contain all the assembly players and are located in the microenvironment of the LDs, the proposed virus assembly site. It is worth mentioning that the formation of NS2 dotted structures is not due to a non-specific effect of the viral genome replication since some of our mutants showing the same replication rate had very different subcellular localizations of NS2 (e.g. A16 vs. A41). It seems rather that the formation of NS2 related dots represents a transition rather than an end state in a productive assembly process. Indeed, mutations in core (JFH-HA-PP) or in the envelope proteins (JFH-ΔE1E2-HA) induced an obvious increase in the number of cells presenting NS2 dots. This could mean that the p7-NS2-E1E2 complexes pre-exist in the NS5A positive subcompartment. In addition, the core-mediated redistribution of LDs could induce the recruitment of the assembly components to LDs followed by the envelope protein incorporation into the virion during the budding process. Finally, after budding, NS2 might relocate to another subcellular compartment or be degraded. This scenario is supported by the fact that NS2 dots accumulate around the LDs and core protein when the envelope proteins are deleted, which is expected to inhibit the lateral interactions between the envelope proteins [42], preventing the budding process to occur.
For the moment, the connection between NS2 and the replication complex (RC) is just inferred from genetic data and colocalization in immunofluorescence experiments [23], [34](this report). We show here that mutations in NS5A, which are reported to affect the phosphorylation status of the protein, abolish the accumulation of NS2 in dotted structures. Moreover, we could partially restore the phenotype by an aspartate mutant, which mimics phosphoserine. Thus, NS2 colocalization with NS5A is favored by the phosphorylation state of the latter. It is possible that NS5A charged state enhances a potential NS2-RC interaction, which translates in NS2 dot formation. Removing the charges would shift the interaction equilibrium and would affect the virus production kinetics in different extents depending on the number of charges and virus strain. Hence, if serine 457 is replaced, the virus titers are moderately reduced at 72h. In contrast, removing serine residues 452, 454, 457 determines a profound defect in infectious virus production. However, there is no NS2 accumulation for either of the two mutants. Thus, the NS2 dots may represent transition states in the assembly process. Indeed, some mutations may prevent the arrival of dots components to NS5A structures (JFH-ΔTM12-HA, JFH-A41-HA, JFH-Δp7-HA and JFH-ΔTME2-HA). Alternatively, other mutations may block the assembly and stabilize them (JFH-HA-PP, JFH-ΔE1E2-HA). If we combine a mutation from the former category (JFH-ΔTME2-HA) with one from the latter (JFH-ΔE1E2-HA) in the mutant JFH-ΔE1E2TME2-HA, we prevent the NS2 dots formation. This suggests that the formation of NS2 complexes and their arrival to the NS5A structures precede the accumulation of NS2 around the LDs during the assembly process. Furthermore, changes in the phosphorylation state of NS5A could regulate the stability of NS2 dots and virion production efficacy.
In our current view, the assembly process would involve several steps. Upon viral genome translation and polyprotein processing, formation of different complexes occurs: the E1E2 native heterodimer, the p7NS2 unit and the RC. The core protein and other viral proteins (e.g. NS4B) create the LD-ER microenvironment by redistribution of the LDs and intracellular membranes. The LDs surrounded by core protein are recruited to the RC. E1E2 complex interacts with p7NS2 unit and E1E2p7NS2 arrives to NS5A positive membranes in the proximity of LDs due to a combination of signals in p7, NS2 and E2 proteins. NS5A switches from the replication to assembly mode by phosphorylation, which stabilizes the presence of NS2 in dotted structures favoring the assembly process (Figure 10). Finally, our data indicate a crucial role played by NS2 in the assembly process and highlight the complexity of the mechanism of its action. In conclusion, NS2 emerges as an essential mediator between the structural and non-structural proteins in HCV assembly process.

Materials and Methods
Cell culture
293T human embryo kidney cells (HEK293T), U2OS human osteosarcoma cells (American Type Culture Collection) and Huh-7 human hepatoma cells [55] were grown in Dulbecco's modified essential medium (Invitrogen) supplemented with 10% fetal bovine serum.
Antibodies
Anti-HCV Mabs A4 (anti-E1) [56] and 3/11 (anti-E2; kindly provided by J.A. McKeating, University of Birmingham, UK) [57] were produced in vitro by using a MiniPerm apparatus (Heraeus) as recommended by the manufacturer. Anti E2 Mab AP33 was kindly provided by A. H. Patel, University of Glasgow, UK. Anti-capsid ACAP27 [58] and anti-NS3 (486D39) Mabs were kindly provided by JF Delagneau (Bio-Rad, France). The anti-NS5A Mab 9E10 [8] and polyclonal antibody were kindly provided by CM Rice (Rockefeller University, NY, USA) and M Harris (University of Leeds, UK), respectively. The anti-NS2 Mab 6H6 [18] and polyclonal antibody were kindly provided by CM Rice (Rockefeller University, NY, USA) and R Bartenschlager (University of Heidelberg, Germany), respectively. The anti-Sec31 antibody [38] was kindly provided by F Gorelick (Yale University School of Medicine, CT, USA). The anti-p125 Mab [37] was kindly provided by K Tani (University of Tokyo, Japan). The following antibodies were purchased: the anti-ERGIC-53 Mab (Alexis), the anti-actin (Santa Cruz Biotechnology), anti-calnexin polyclonal (Stressgen), anti-calreticulin polyclonal (Stressgen), anti-PDI (Stressgen), anti-GFP (Roche) and the anti-hemagglutinin (HA) Mab 3F10 (Roche) and Mab HA11 (Covance). Alexa 488, Alexa 555 and Alexa 633 conjugated goat anti–rabbit, anti-rat and anti-mouse immunoglobulin G (IgG) were purchased from Invitrogen.
Plasmids
All plasmids were constructed by standard molecular biology methods and the constructs were confirmed by sequencing. The following plasmids were assembled for FRET-FLIM analyses in the background of pCMV plasmid (Addgene): pCMV/YFP-E1TM, pCMV/CFP-E2TM, pCMV/CFP-p7, pCMV/CFP-EYF, pCMV/YFP-NS2. For these constructs, the plasmids encoding YFP and CFP were obtained from DW Piston (Vanderbilt University, USA) and A Miyawaki (Riken Institute, Japan), respectively. YFP-E1TM and CFP-E2TM are YFP and CFP in fusion with the transmembrane domain of E1 and E2 (H strain), respectively. In these two constructs, a two amino acid linker (serine and glycine) was inserted between the fluorescent protein and the transmembrane domains. CFP-p7 and CFP-EYF are CFP proteins fused to the N-terminus of p7 and the transmembrane domain of yellow fever virus envelope protein E (EYF), respectively. YFP-NS2 is a YFP protein fused to the N-terminus of NS2. In all the constructs, the calreticulin signal sequence was fused to the N-terminus of CFP and YFP for translocation in the ER lumen, allowing the study of the recombinant proteins in their native topology.
For p7-NS2 co-immunoprecipitation experiments, the following plasmids were constructed in the background of pTriex (Novagen) or pCI plasmids (Promega): pTriex/p7-Flag, pTriex/EYF-Flag, pCI/HA-NS2, pCI/HA-NS2-GFP and pCI/HA-NS2-GTM. p7-Flag and EYF-Flag are HCV p7 of genotype 1a (H strain) and YFE in fusion with the Flag epitope (DYKDDDDK). HA-NS2 has a HA epitope fused to the N-terminus of NS2. In HA-NS2-GFP, the cytosolic domain of NS2 was replaced by GFP protein, whereas for HA-NS2-GTM, we replaced the transmembrane domain of NS2 by the transmembrane domain of VSV-G protein.
In this work, we used a modified version of the plasmid encoding JFH1 genome (genotype 2a; GenBank access number AB237837), kindly provided by T. Wakita (National Institute of Infectious Diseases, Tokyo, Japan) [7]. Mutations were introduced in a JFH1 plasmid containing a Renilla Luciferase reporter gene [59] and mutations leading to amino acids changes F172C and P173S which have been shown to increase the viral titers [30]. Furthermore, the E1 sequence encoding residues 196TSSSYMVTNDC has been modified to reconstitute the A4 epitope (SSGLYHVTNDC) [56] as described [31]. Overlapping PCR was used to construct all the JFH1 mutants. The JFHGND-Luc construct was obtained by inserting the previously described GND mutation [7] in JFH-Luc plasmid. The JFH-HA construct was obtained by inserting the sequence of the HA epitope (YPYDVPDYA) followed by a GGG linker at the N-terminus of NS2. The JFH-Δp7-HA keeps the first 2 amino acids of p7 followed by the HA tag sequence and the SGG linker at the N-terminus of NS2. The JFH-ΔE1E2-Luc plasmid has been described previously [31]. It contains an in-frame deletion of amino acids 217–567. JFH-ΔTM12-HA has the first two transmembrane segments of NS2 deleted as described [19], in the context of our JFH-HA virus. JFH-HA-PP has proline residues 138 and 143 in domain D2 of the core protein replaced by alanine residues as described [13], [41]. JFH-S/A-HA has serine 457 of NS5A replaced by an alanine as described [17]. JFH-3BS/A-HA has serine residues at positions 452, 454 and 457 of NS5A replaced by alanine residues as described [16]. The JFH-S/D-HA has serine 457 of NS5A replaced by an aspartate residue as described [17]. JFH-ΔTME2-HA and JFH-ΔE1E2TME2-HA have the transmembrane region of E2 glycoprotein replaced by FMDV 2A autoprotease. JFH-ΔTME2-HA contains an in-frame deletion of amino acids 720–750, corresponding to the transmembrane domain of E2 of JFH1, whereas JFH-ΔE1E2TME2-HA contains in-frame deletions of amino acids 217–567 and 720–750. To construct the JFH-ΔTME2-HA, we replaced the C-terminal region of E2 (aa 720–750 of JFH1) by QLLNFDLLKLAGDVESNPGP FMDV 2A autoprotease peptide preceded by a GGG linker sequence. A similar strategy was used for the construction of JFH-ΔE1E2TME2-HA using as a backbone plasmid the JFH-ΔE1E2-HA. JFH-RR/QQ-HA has the arginines 33 and 35 of p7 replaced by glutamine residues. The primers and enzymes used for the constructs are presented in Table S1. Schematic representation of the constructs used in this study is presented in Figure S1.
DNA transfections
Twenty-four hours before transfection, 293T cells were seeded in 100 mm tissue culture plates to reach a 70–80% confluency the next day. Cells were tranfected with 6 µg of DNA/plate at a ratio of 1∶4 with PEI transfection reagent (Eurogentec). In cotransfection experiments, equal quantities of each plasmid were used. At 24h post-transfection, cells were processed for co-immunoprecipitation analyses.
Twenty-four hours before transfection, U2OS cells were seeded in 6 well plates on 32mm slides to reach a confluency of 70–80% the next day. Cells were transfected with 1µg of CFP-expressing plasmid (donor) and 125 ng of YFP-expressing plasmid (acceptor) mixed with Fugene reagent (Roche) following the instructions of the manufacturer. For CFP-E1 and YFP-E2 co-transfection experiments, we used 300 ng of donor and 600 ng of acceptor plasmids, respectively.
Two-photon fluorescence lifetime microscopy and data analysis (FRET-FLIM)
Twenty-four hours after transfection, U2OS cells were selected for FRET-FLIM acquisition. We analyzed cells with similar expression levels of donor and acceptor fusion proteins. We also chose cells with normal reticulate ER morphology avoiding those where the overexpression of recombinant proteins was present.
In order to detect the FRET events, the Time Correlated Single Photon Counting FLIM system (TCSPC) was used [60], [61], [62]. The analyses were performed with a Leica SP5.X confocal Microscope (Leica Microsystem) with an internal FLIM detector. A dedicated photo-counting and timing electronic card (SPC 830 TCSPC card, Becker and Hickl) was coupled to the Leica internal detector and used to classify the photon emission in time to determine the lifetime of the donor protein. To excite the samples, Chameleon Ultra2 (Chorent Inc) biphoton was used at 830 nm at an average power of 0.13 mW/µm2 [60], [61], [62].
The fluorescence events of the donor protein result in a photon decay curve generated by the FLIM method. The decay curve was directly used to determine the donor's lifetime. The least square fitting method was used to describe the non linear responses commonly observed in FLIM analysis [60], [61], [62]. We used TITAN (“in the house” designed) and SPCImage (Becker and Hickl) software for advanced FLIM data analysis and curve fittings. In order to reduce the impact of background and improve the Signal to Noise Ratio (SNR), we excluded from our analysis the pixels located in the nuclear region or from ER-like regions where the donor protein was overexpressed. After setting these thresholds, we made a summation of all the pixels of interest to achieve a fitting statistically significant for the TITAN software. To compensate the possible large scattering of points in the curve, we used a Newton trust region algorithm [63] and an extraction of mean lifetimes was performed in order to determine the FRET events from the multi-exponential model [64].
Immunoprecipitation
Cell pellets were lysed in phosphate-buffered saline (PBS) lysis buffer (1% Triton 100-X, 20mM NEM, 2mM EDTA, protease inhibitors cocktail Roche) and they were precleared with 20 µl Prot G for 2h at 4C. The precleared lysates were incubated with anti-HA antibodies (HA11) or Sepharose beads covalently bound to HA11 antibody (Covance) overnight at 4h. The immunocomplexes were pulled down with 50 µl of Protein G and washed three times with lysis buffer.
For p7-NS2 interaction, cell lysates were incubated with 20 µl of agarose-anti-Flag beads over night at 4°C. The immunocomplexes were treated the same as above and the Western blots were revealed by an anti-HA antibody.
PNGase and EndoH digestion
The endoglycosidase digestions were performed following the manufacture's instructions (NEB). Briefly, cell lysates containing 20 µg of protein were denatured in EndoH (PNGase) denaturing buffer (0.5% SDS, 1% 2-mercaptoethanol) for 10 min at 100°C. Then, the lysates were incubated or not with 1 µl of EndoH (PNgase) for 20h at 37°C.
Western blotting
After separation by SDS-PAGE, proteins were transferred to nitrocellulose membranes (Hybond-ECL, Amersham) by using a Trans-Blot apparatus (Biorad) and revealed with specific antibodies followed by secondary immunoglobulin conjugated to peroxidase. The proteins of interest were revealed by enhanced chemiluminescence detection (ECL, Amersham) as recommended by the manufacturer.
Replication and infectivity assays
Plasmids encoding wild-type (WT) and mutated genomes were linearized at the 3′ end of the HCV cDNA with the restriction enzyme XbaI and treated with the Mung Bean Nuclease (New England Biolabs). In vitro transcripts were generated using the Megascript kit according to the manufacturer's protocol (Ambion). The in vitro reaction was set up and incubated at 37°C for 4 h and transcripts were precipitated by the addition of LiCl. Ten micrograms of RNA were delivered into Huh-7 cells by electroporation as described previously [30]. Replication was assessed at 72 h by measuring Renilla Luciferase activities in electroporated cells as indicated by the manufacturer (Promega). Supernatants containing HCVcc were harvested 72 h after electroporation and filtered through 0.45 µm pore-sized membrane for infectivity measurements. HCVcc were incubated for 3 h with Huh-7 cells seeded the day before in 24-well plates. At 72 h post-infection, Luciferase assays were performed on infected cells as indicated by the manufacturer (Promega). For supernatants titration, Huh7 electroporated cells were seeded in 6-well plates. 72h post-electroporation, naïve Huh-7 cells were inoculated with serial dilutions of the supernatant. 48h post-inoculation, the infected cells were fixed in ice-cold methanol, they were immunostained with anti-E1 antibody and the focus forming units (FFUs) were counted.
HCV Core quantification
HCV Core was quantified by a fully automated chemiluminescent microparticle immunoassay according to manufacturer's instructions (Architect HCVAg, Abbott, Germany) [65], [66]. For the determination of intracellular core quantity, the electroporated cells were lysed in PBS lysis buffer (1% Triton 100-X, 20mM NEM, 2mM EDTA, protease inhibitors cocktail Roche) and the lysates were cleared for 20 min at 14,000g.
HCV RNA quantification
Mutated HCV genomes were delivered into Huh-7 cells. At day 3 post-electroporation, cells were trypsinized, washed once with fresh medium and reseeded into cell culture dishes. At day 5 post-electroporation, total RNA in cell lysates and HCV RNA in supernatants were extracted using the Qiagen RNeasy kit and Qiagen QiaAmp viral RNA mini kit, respectively. cDNA was synthesized using High Capacity cDNA Reverse Transcription kit as described by the manufacturer (Applied BioSystems) and titrated by quantitative real-time RT-PCR assay (RT-qPCR) using TaqMan and minor groove binding (MGB) probe detection. The primer pair and the probe were located in the 5′ HCV non-coding region [67].
Immunofluorescence
Huh-7 cells transfected with HCV RNA were grown on 12-mm glass coverslips. At the indicated time points, cells were fixed with 3% paraformaldehyde and then permeabilized with 0.1% Triton X-100 in PBS. Both primary - and secondary-antibody incubations were carried out for 30 min at room temperature with PBS containing 10% goat serum. LDs were stained for 10 minutes in 300 ng/ml BODIPY 493/503 (Invitrogen). Nuclei were stained with 4,6′-diamidino-2-phenylindole (DAPI). The coverslips were mounted on slides by using Mowiol 4–88 (Calbiochem) containing mounting medium. Confocal microscopy was performed with an LSM710 laser-scanning confocal microscope (Zeiss) using a 63×/1.4 numerical aperture oil immersion objective. Signals were sequentially collected by using single fluorescence excitation and acquisition settings to avoid crossover. Images were processed using Adobe Photoshop software. Cells showing NS2/NS5A-positive dot-like structures were counted on images collected with a 40× oil immersion objective.
Immunoelectron microscopy
Huh-7 cells transfected with HCV RNA were grown on 75 cm2 flasks. At 72h post-electroporation, cells were fixed by incubation in a solution containing 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.2) for 20 h. The cells were collected by centrifugation and the cell pellet was then dehydrated in a graded series of ethanol solutions at −20°C, using an automatic freezing substitution system (AFS, Leica), and embedded in London Resin Gold (LR Gold, Electron Microscopy Science). The resin was allowed to polymerize at −25°C, under UV light, for 72 h. Ultrathin sections were cut and blocked by incubation with 3% fraction V bovine serum albumin (BSA, Sigma) in PBS. They were then incubated with anti-HA Mab (Covance) diluted 1∶50 in PBS supplemented with 1% BSA, washed and incubated with goat anti-mouse antibodies conjugated to 15 nm gold particles (British Biocell International, Cardiff, UK) diluted 1∶40 in PBS supplemented with 1% BSA. Ultrathin sections were cut, stained with 5% uranyl acetate, 5% lead citrate, and placed on electron microscopy grids coated with collodion. The sections were then observed with a Jeol 1230 transmission electron microscope (Tokyo, Japan) connected to a Gatan digital camera driven by Digital Micrograph software (Gatan, Pleasanton, CA) for image acquisition.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. WasleyA
AlterMJ
2000
Epidemiology of hepatitis C: geographic differences and temporal trends.
Semin Liver Dis
20
1
16
2. FeldJJ
HoofnagleJH
2005
Mechanism of action of interferon and ribavirin in treatment of hepatitis C.
Nature
436
967
972
3. LindenbachBD
ThielHJ
RiceCM
2007
Flaviviridae: the viruses and their replication.
KnipeDM
HowleyPM
Fields Virology. 5th ed
Philadelphia, Pa
Lippincott Williams & Wilkins
1101
1152
4. MoradpourD
PeninF
RiceCM
2007
Replication of hepatitis C virus.
Nat Rev Microbiol
5
453
463
5. BlightKJ
McKeatingJA
RiceCM
2002
Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication.
J Virol
76
13001
13014
6. LohmannV
KörnerF
KochJ-O
HerianU
TheilmannL
1999
Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line.
Science
285
110
113
7. WakitaT
PietschmannT
KatoT
DateT
MiyamotoM
2005
Production of infectious hepatitis C virus in tissue culture from a cloned viral genome.
Nat Med
11
791
796
8. LindenbachBD
EvansMJ
SyderAJ
WolkB
TellinghuisenTL
2005
Complete replication of hepatitis C virus in cell culture.
Science
309
623
626
9. ZhongJ
GastaminzaP
ChengG
KapadiaS
KatoT
2005
Robust hepatitis C virus infection in vitro.
Proc Natl Acad Sci U S A
102
9294
9299
10. McLauchlanJ
2009
Lipid droplets and hepatitis C virus infection.
Biochim Biophys Acta
1791
552
559
11. RouilléY
HelleF
DelgrangeD
RoingeardP
VoissetC
2006
Subcellular localization of hepatitis C virus structural proteins in a cell culture system that efficiently replicates the virus.
J Virol
80
2832
2841
12. MiyanariY
AtsuzawaK
UsudaN
WatashiK
HishikiT
2007
The lipid droplet is an important organelle for hepatitis C virus production.
Nat Cell Biol
9
1089
1097
13. BoulantS
Targett-AdamsP
McLauchlanJ
2007
Disrupting the association of hepatitis C virus core protein with lipid droplets correlates with a loss in production of infectious virus.
J Gen Virol
88
2204
2213
14. AppelN
ZayasM
MillerS
Krijnse-LockerJ
SchallerT
2008
Essential role of domain III of nonstructural protein 5A for hepatitis C virus infectious particle assembly.
PLoS Pathog
4
e1000035
15. EvansMJ
RiceCM
GoffSP
2004
Phosphorylation of hepatitis C virus nonstructural protein 5A modulates its protein interactions and viral RNA replication.
Proc Natl Acad Sci U S A
101
13038
13043
16. MasakiT
SuzukiR
MurakamiK
AizakiH
IshiiK
2008
Interaction of hepatitis C virus nonstructural protein 5A with core protein is critical for the production of infectious virus particles.
J Virol
82
7964
7976
17. TellinghuisenTL
FossKL
TreadawayJ
2008
Regulation of hepatitis C virion production via phosphorylation of the NS5A protein.
PLoS Pathog
4
e1000032
18. DentzerTG
LorenzIC
EvansMJ
RiceCM
2009
Determinants of the hepatitis C virus nonstructural protein 2 protease domain required for production of infectious virus.
J Virol
83
12702
12713
19. JiraskoV
MontserretR
AppelN
JanvierA
EustachiL
2008
Structural and functional characterization of non-structural protein 2 for its role in hepatitis C virus assembly.
J Biol Chem
283
28546
28562
20. JonesCT
MurrayCL
EastmanDK
TasselloJ
RiceCM
2007
Hepatitis C virus p7 and NS2 proteins are essential for production of infectious virus.
J Virol
81
8374
8383
21. PietschmannT
KaulA
KoutsoudakisG
ShavinskayaA
KallisS
2006
Construction and characterization of infectious intragenotypic and intergenotypic hepatitis C virus chimeras.
Proc Natl Acad Sci U S A
103
7408
7413
22. YiM
MaY
YatesJ
LemonSM
2007
Compensatory mutations in E1, p7, NS2, and NS3 enhance yields of cell culture-infectious intergenotypic chimeric hepatitis C virus.
J Virol
81
629
638
23. PhanT
BeranRK
PetersC
LorenzIC
LindenbachBD
2009
Hepatitis C virus NS2 protein contributes to virus particle assembly via opposing epistatic interactions with the E1-E2 glycoprotein and NS3-NS4A enzyme complexes.
J Virol
83
8379
8395
24. Carrère-KremerS
Montpellier-PalaC
CocquerelL
WychowskiC
PeninF
2002
Subcellular localization and topology of the p7 polypeptide of hepatitis C virus.
J Virol
76
3720
3730
25. CocquerelL
Op de BeeckA
LambotM
RousselJ
DelgrangeD
2002
Topologic changes in the transmembrane domains of hepatitis C virus envelope glycoproteins.
Embo J
21
2893
2902
26. BraunP
PerssonB
KabackR
von HeijneG
1997
Alanine insertion scanning mutagenesis of lactose permease transmembrane helices.
J Biol Chem
272
29566
29571
27. MingarroI
WhitleyP
LemmonM
von HeijneG
1996
Ala-insertion scanning mutagenesis of the glycophorin A transmembrane helix : A rapid way to map helix-helix ineractions in integral membrane protein.
Protein Sci
5
1339
1341
28. Op De BeeckA
MolenkampR
CaronM
Ben YounesA
BredenbeekP
2003
Role of the transmembrane domains of prM and E proteins in the formation of yellow fever virus envelope.
J Virol
77
813
820
29. Op De BeeckA
MontserretR
DuvetS
CocquerelL
CacanR
2000
Role of the transmembrane domains of hepatitis C virus envelope proteins E1 and E2 in the assembly of the noncovalent E1E2 heterodimer.
J Biol Chem
275
31428
31437
30. DelgrangeD
PillezA
CastelainS
CocquerelL
RouilléY
2007
Robust production of infectious viral particles in Huh-7 cells by introducing mutations in hepatitis C virus structural proteins.
J Gen Virol
88
2495
2503
31. GoueslainL
AlsalehK
HorellouP
RoingeardP
DescampsV
2010
Identification of GBF1 as a cellular factor required for hepatitis C virus RNA replication.
J Virol
84
773
787
32. SteinmannE
PeninF
KallisS
PatelAH
BartenschlagerR
2007
Hepatitis C virus p7 protein is crucial for assembly and release of infectious virions.
PLoS Pathog
3
e103
33. SelbyMJ
GlazerE
MasiarzF
HoughtonM
1994
Complex processing and protein∶protein interactions in the E2∶NS2 region of HCV.
Virology
204
114
122
34. YiM
MaY
YatesJ
LemonSM
2009
Trans-complementation of an NS2 defect in a late step in hepatitis C virus (HCV) particle assembly and maturation.
PLoS Pathog
5
e1000403
35. GriffinSD
HarveyR
ClarkeDS
BarclayWS
HarrisM
2004
A conserved basic loop in hepatitis C virus p7 protein is required for amantadine-sensitive ion channel activity in mammalian cells but is dispensable for localization to mitochondria.
J Gen Virol
85
451
461
36. SchweizerA
FransenJAM
BächiT
GinselL
HauriH-P
1988
Identification, by a monoclonal antibody, of a 53-kD protein associated with tubulo-vesicular compartment at the cis-side of the Golgi apparatus.
J Cell Biol
107
1643
1653
37. ShimoiW
EzawaI
NakamotoK
UesakiS
GabreskiG
2005
p125 is localized in endoplasmic reticulum exit sites and involved in their organization.
J Biol Chem
280
10141
10148
38. ShugrueCA
KolenER
PetersH
CzernikA
KaiserC
1999
Identification of the putative mammalian orthologue of Sec31P, a component of the COPII coat.
J Cell Sci
112
4547
4556
39. MaY
AnantpadmaM
TimpeJM
ShanmugamS
SinghSM
2011
Hepatitis C virus NS2 protein serves as a scaffold for virus assembly by interacting with both structural and nonstructural proteins.
J Virol
85
86
97
40. MurrayCL
JonesCT
RiceCM
2008
Architects of assembly: roles of Flaviviridae non-structural proteins in virion morphogenesis.
Nat Rev Microbiol
6
699
708
41. BoulantS
MontserretR
HopeG
RatinierM
Targett-AdamsP
2006
Structural determinants that target the hepatitis C virus core protein to lipid droplets.
J Biol Chem
281
22236
22247
42. VieyresG
ThomasX
DescampsV
DuverlieG
PatelAH
2010
Characterization of the envelope glycoproteins associated with infectious hepatitis C virus.
J Virol
84
10159
10168
43. VillanuevaR
RouilléY
DubuissonJ
2005
Interactions between virus proteins and host cell membranes.
Int Rev Cytol
245
171
244
44. HaqshenasG
MackenzieJM
DongX
GowansEJ
2007
Hepatitis C virus p7 protein is localized in the endoplasmic reticulum when it is encoded by a replication-competent genome.
J Gen Virol
88
134
142
45. SprietC
TrinelD
WaharteF
DesleeD
VandenbunderB
2007
Correlated fluorescence lifetime and spectral measurements in living cells.
Microsc Res Tech
70
85
94
46. YamadaH
Padilla-ParraS
ParkSJ
ItohT
ChaineauM
2009
Dynamic interaction of amphiphysin with N-WASP regulates actin assembly.
J Biol Chem
284
34244
34256
47. ZhangW
ChipmanPR
CorverJ
JohnsonPR
ZhangY
2003
Visualization of membrane protein domains by cryo-electron microscopy of dengue virus.
Nat Struct Biol
10
907
912
48. JonesDM
PatelAH
Targett-AdamsP
McLauchlanJ
2009
The hepatitis C virus NS4B protein can trans-complement viral RNA replication and modulates production of infectious virus.
J Virol
83
2163
2177
49. PavlovicD
NevilleDC
ArgaudO
BlumbergB
DwekRA
2003
The hepatitis C virus p7 protein forms an ion channel that is inhibited by long-alkyl-chain iminosugar derivatives.
Proc Natl Acad Sci U S A
100
6104
6108
50. LiljestromP
LusaS
HuylebroeckD
GaroffH
1991
In vitro mutagenesis of a full-length cDNA clone of Semliki Forest virus: the small 6,000-molecular-weight membrane protein modulates virus release.
J Virol
65
4107
4113
51. LoewyA
SmythJ
von BonsdorffCH
LiljestromP
SchlesingerMJ
1995
The 6-kilodalton membrane protein of Semliki Forest virus is involved in the budding process.
J Virol
69
469
475
52. CorseE
MachamerCE
2003
The cytoplasmic tails of infectious bronchitis virus E and M proteins mediate their interaction.
Virology
312
25
34
53. FischerF
StegenCF
MastersPS
SamsonoffWA
1998
Analysis of constructed E gene mutants of mouse hepatitis virus confirms a pivotal role for E protein in coronavirus assembly.
J Virol
72
7885
7894
54. VennemaH
GodekeGJ
RossenJW
VoorhoutWF
HorzinekMC
1996
Nucleocapsid-independent assembly of coronavirus-like particles by co - expression of viral envelope protein genes.
Embo J
15
2020
2028
55. NakabayashiH
TaketaK
MiyanoK
YamaneT
SatoJ
1982
Growth of human hepatoma cells lines with differentiated functions in chemically defined medium.
Cancer Res
42
3858
3863
56. DubuissonJ
HsuHH
CheungRC
GreenbergHB
RussellDG
1994
Formation and intracellular localization of hepatitis C virus envelope glycoprotein complexes expressed by recombinant vaccinia and Sindbis viruses.
J Virol
68
6147
6160
57. FlintM
MaidensC
Loomis-PriceLD
ShottonC
DubuissonJ
1999
Characterization of hepatitis C virus E2 glycoprotein interaction with a putative cellular receptor, CD81.
J Virol
73
6235
6244
58. MaillardP
KrawczynskiK
NitkiewiczJ
BronnertC
SidorkiewiczM
2001
Nonenveloped nucleocapsids of hepatitis C virus in the serum of infected patients.
J Virol
75
8240
8250
59. Rocha-PeruginiV
MontpellierC
DelgrangeD
WychowskiC
HelleF
2008
The CD81 partner EWI-2wint inhibits hepatitis C virus entry.
PLoS ONE
3
e1866
60. O'ConnorDV
Phillips
1984
Time-correlated single photon counting
London
Academic Press
61. WaharteF
SprietC
HeliotL
2006
Setup and characterization of a multiphoton FLIM instrument for protein-protein interaction measurements in living cells.
Cytometry A
69
299
306
62. DraperNR
SmithH
1998
Applied regression analysis
New York
Wiley-Interscience
63. ColemanTF
LiY
1994
On the convergence of reflective Newton methods for large-scale nonlinear minimization subject to bounds.
Mathematical Programming
67
189
224
64. LakowiczJR
1999
Principles of fluorescence Spectroscopy
New York
Kluwer Academic / Plenum Publishers
65. MederackeI
WedemeyerH
CiesekS
SteinmannE
RaupachR
2009
Performance and clinical utility of a novel fully automated quantitative HCV-core antigen assay.
J Clin Virol
46
210
215
66. MorotaK
FujinamiR
KinukawaH
MachidaT
OhnoK
2009
A new sensitive and automated chemiluminescent microparticle immunoassay for quantitative determination of hepatitis C virus core antigen.
J Virol Methods
157
8
14
67. CastelainS
DescampsV
ThibaultV
FrancoisC
BonteD
2004
TaqMan amplification system with an internal positive control for HCV RNA quantitation.
J Clin Virol
31
227
234
68. ShapiroSS
WilkMB
1965
An analysis of variance test for normality.
Biometrika
52
591
611
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2011 Číslo 2
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Genetic Mapping Identifies Novel Highly Protective Antigens for an Apicomplexan Parasite
- Type I Interferon Signaling Regulates Ly6C Monocytes and Neutrophils during Acute Viral Pneumonia in Mice
- Infections in Cells: Transcriptomic Characterization of a Novel Host-Symbiont Interaction
- The ESCRT-0 Component HRS is Required for HIV-1 Vpu-Mediated BST-2/Tetherin Down-Regulation
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy