
Late Repression of NF-κB Activity by Invasive but Not Non-Invasive Meningococcal Isolates Is Required to Display Apoptosis of Epithelial Cells
Meningococcal invasive isolates of the ST-11 clonal complex are most frequently associated with disease and rarely found in carriers. Unlike carriage isolates, invasive isolates induce apoptosis in epithelial cells through the TNF-α signaling pathway. While invasive and non-invasive isolates are both able to trigger the TLR4/MyD88 pathway in lipooligosaccharide (LOS)-dependant manner, we show that only non-invasive isolates were able to induce sustained NF-κB activity in infected epithelial cells. ST-11 invasive isolates initially triggered a strong NF-κB activity in infected epithelial cells that was abolished after 9h of infection and was associated with sustained activation of JNK, increased levels of membrane TNFR1, and induction of apoptosis. In contrast, infection with carriage isolates lead to prolonged activation of NF-κB that was associated with a transient activation of JNK increased TACE/ADAM17-mediated shedding of TNFR1 and protection against apoptosis. Our data provide insights to understand the meningococcal duality between invasiveness and asymptomatic carriage.
Published in the journal:
. PLoS Pathog 7(12): e32767. doi:10.1371/journal.ppat.1002403
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1002403
Summary
Meningococcal invasive isolates of the ST-11 clonal complex are most frequently associated with disease and rarely found in carriers. Unlike carriage isolates, invasive isolates induce apoptosis in epithelial cells through the TNF-α signaling pathway. While invasive and non-invasive isolates are both able to trigger the TLR4/MyD88 pathway in lipooligosaccharide (LOS)-dependant manner, we show that only non-invasive isolates were able to induce sustained NF-κB activity in infected epithelial cells. ST-11 invasive isolates initially triggered a strong NF-κB activity in infected epithelial cells that was abolished after 9h of infection and was associated with sustained activation of JNK, increased levels of membrane TNFR1, and induction of apoptosis. In contrast, infection with carriage isolates lead to prolonged activation of NF-κB that was associated with a transient activation of JNK increased TACE/ADAM17-mediated shedding of TNFR1 and protection against apoptosis. Our data provide insights to understand the meningococcal duality between invasiveness and asymptomatic carriage.
Introduction
The exclusive human bacterium Neisseria meningitidis (the meningococcus) is a major cause of infectious diseases worldwide, including meningitis and fulminant sepsis that are associated with significant morbidity and case fatality rates ranging from 10 to 50% in patients with severe septicaemia [1], [2] and up to 20% of survivors sustain neurological sequelae [3]. Despite this notoriety, N. meningitidis is a frequent inhabitant of the nasopharyngeal mucosa being asymptomatically carried by 10–35% of the adult population [4], [5]. A combination of bacterial factors and host susceptibility (including age, prior viral infection, and genetic polymorphisms [6]–[8]), may ultimately lead to meningococcal disease. Multilocus sequence typing (MLST) has been used to characterize the genotypes of meningococcal isolates determined by the sequence types (STs) and grouping these genotypes into distinct phylogenetic lineages referred to as “clonal complexes” [9]. Comparisons of the genotypes of meningococcal isolates have shown that asymptomatic carriage isolates are genetically and antigenically highly diverse, whereas most meningococcal disease is caused by a limited number of clonal complexes known as the “hyper-invasive clonal complexes” [10]–[13]. Genomic analysis failed to identify which bacterial features are responsible for the different epidemiologies [14]. Moreover, bacterial virulence factors such as pili and capsule, although important in the establishment of the disease, are widely distributed among carriage and invasive isolates. Therefore, a better understanding of the pathogenesis of this disease, notably the interaction with host cells, is central in developing new anti-meningococcal strategies.
There is increasing evidence that invasive meningococcal infections lead to cytopathic effects [15]–[20]. These observations are consistent with the extensive cell injury and tissue damage seen in autopsy material from cases of human disease [21]. We have recently shown a strong association of cytopathic effect to epithelial cells with isolates belonging to the hyper-invasive clonal complex ST-11. Infected cells presented features of apoptosis. The apoptotic pathway induced by these isolates is mediated in part by lipooligosaccharides (LOS), the major bacterial endotoxin, and involved tumor necrosis factor alpha (TNF-α) signaling through its cognate receptor TNFR1. In contrast, carriage isolates interfered with TNF-α-dependent apoptotic signaling by increasing extracellular shedding of TNFR1 leading to attenuation of the biological activity of TNF-α [22]. Several signaling pathways are known to regulate apoptosis, but the transcription factor NF-κB lies at the nexus of both anti-apoptotic and proinflammatory cascades (reviewed in references [23], [24]). In resting cells, NF-κB is sequestered in the cytosol through interactions with its inhibitor, IκB. Proinflammatory stimuli, such as lipopolysaccharide (LPS) and TNF-α, activate a signaling pathway that results in phosphorylation and subsequent degradation of IκB by the proteasome machinery. The liberated NF-κB translocates then to the nuclear compartment, where it activates the transcription of both proinflammatory and anti-apoptotic genes [25]. Given the role of NF-κB in both inflammation and apoptosis, it is not surprising that certain pathogens have also evolved mechanisms to modulate NF-κB activity during infection [26]. In this work we aimed to explore the differential ability of meningococcal invasive and non-invasive isolates to modulate the NF-κB activity.
Results
TLR4 contribute to apoptosis induced by the invasive ST-11 isolates
We have shown that apoptosis of epithelial cells promoted by the ST-11 isolates is partially dependent on the meningococcal lipooligosaccharide LOS [22]. Indeed, both invasive ST-11 isolates and the non-invasive carriage isolates were able to induce the expression of TLR4 at the surface of Hec-1B epithelial cells (Supporting Figure S1). However, only invasive ST-11 isolates but not non-invasive carriage isolates or LOS-devoid mutants were able to induce apoptosis in Hec-1B epithelial cells (Figure 1C and Table 1). Anti-TLR4 neutralizing mAb but not an isotype-matching IgG control Ab was able to inhibit apoptosis in epithelial cells infected by the ST-11 invasive isolates (Table 1). Furthermore, the induction of apoptosis was abolished when TLR4 was knocked-down by siRNA-mediated TLR4 silencing strategy using specific TLR4 silencing duplex oligonucleotides siTLR4-1 or siTLR4-2 but not a non-specific control oligonucleotide (siCTRL) (Supporting Table S1 and Figures 1A and 1B). The induction of apoptosis by the invasive isolate LNP19995 was correlated with a significant high level of caspases-3 activity that was significantly reduced in the isogenic LOS-devoid mutant Z0305 or upon siRNA-mediated TLR4 silencing (Figure 1D). Caspase-3 activity in cells infected with the carriage isolate or its isogenic LOS mutant AD1001 were comparable to the background level. Altogether, these data demonstrate that TLR4 is required to promote apoptosis by the ST-11 meningococcal isolates. The following experiments were performed using LNP19995 and LNP21019 as a representative candidate of each, the invasive ST-11 isolates and the non-invasive carriage isolates respectively, unless otherwise specified.


ST-11 isolates but not carriage isolates promote apoptosis of epithelial cells in MyD88-dependent manner
TLR4 links to both MyD88 and TRIF to transduce signals to downstream effectors [27]. MyD88 has an NH2-terminal death domain which links it to downstream effectors in the TLR signaling pathways and a COOH-terminal TIR domain which interacts with the cytoplasmic portion of various TLRs. Each domain expressed alone functions as a dominant negative form [28]. The TIR domain of TRIF has similar behaviour [29], [30]. To further explore the extent of LOS signaling pathway downstream TLR4 in the apoptosis promoted by the invasive ST-11 isolates, Hec-1B cells were knocked-down for functional MyD88 or TRIF by transfecting either TIR domain and apoptosis was analyzed after 9 h of infection in comparison with cells transfected with the empty vector control. Expression of AU1-tagged dnMyD88 or Myc-tagged dnTRIF was confirmed in the transfected cells by immunoblotting using specific Abs directed against each tag (Figure 2A). As expected, the apoptotic level promoted by the wild-type (WT) ST-11 isolate LNP19995 in empty vector-transfected cells was dramatically decreased after infection with the LOS-deficient isogenic mutant Z0305. Comparably to TLR4 knock-down, expression of the dnMyD88 considerably impeded apoptosis promoted by the WT ST-11 isolate LNP19995, while expression of the dnTRIF did not improve the survival of LNP19995-infected cells (Figure 2B, 11.70±5.03% of dnMyD88-transfected cells underwent apoptosis while 37.01±2.28% of pcDNA3 or 31.58±2.98% of dnTRIF cells were already apoptotic). Both transfected constructs (dnMyD88 or dnTRIF) resulted in apoptotic level in cells infected with the non invasive carriage isolate LNP21019 comparable to uninfected cells (Figure 2B). Results similar to dnMyD88 were obtained in cells transfected with a dnIRAK1, an effector protein downstream MyD88 (data not shown). Taken together, our results indicate that LOS-mediated apoptotic signaling elicited by the ST-11 isolates through TLR4 involved a MyD88 - but not TRIF-dependent pathway.

Invasive ST-11 isolates and non-invasive carriage isolates differentially modulate NF-κB activity
The subsequent steps in TLR4/MyD88 signalling lead to the activation of NF-κB which translocates into the nucleus to activate pro-inflammatory and pro-survival gene expression including TNF-α [31]. We therefore sought to determine the role of NF-κB activity in apoptosis induced by the meningococcal ST-11 isolates. We first followed the kinetic of NF-κBtrans-activation in response to meningococcal infection. For that purpose, Hec-1B cells were transiently transfected with the plasmid (Igκ)3conaLuc, where expression of the luciferase reporter gene is driven by an NF-κB-dependent promoter. Luciferase activity normalized to the constitutive β-galactosidase activity control was assayed in a time course infection. As depicted in Figure 3A, infection of Hec-1B cells with either isolates induced luciferase activity which peaked at 4h post-infection to nearly 20 fold relative to uninfected cells. This early activation required LOS and occurred in TLR4-dependent manner as lack of LOS from both, the invasive or the carriage isolates (Z0305 or AD1001 mutants, respectively) or silencing of TLR4 considerably reduced transactivation of NF-κB (Figure 3B). Surprisingly, NF-κB-dependent luciferase activity decreased beyond 6 h of challenge with the invasive ST-11 isolates while persisted in response to infection with the carriage isolates (Figure 3A). These data corroborate with the EMSA assays. Indeed, the DNA-binding activity of NF-κB to a specific radio-labeled probe peaked transiently at 4 h of infection then decreased beyond 6 h of infection with the ST-11 invasive isolate LNP19995, while persisted longer in cells infected with the carriage isolate LNP21019 (Supporting Figure S2). Interestingly the decrease of NF-κB-dependent luciferase and DNA-binding activities in cells infected with the ST-11 isolates was concomitant to induction of apoptosis by these isolates. Collectively, these data show a differential modulation of NF-κB-dependent transcriptional activity between the invasive ST-11 and the non-invasive carriage isolates.

LOS purified from the invasive LNP19995 or the carriage LNP21019 isolates were able to trigger a persistent transactivation of NF-κB similarly to the carriage isolates, as judged by EMSA and luciferase reporter assays. No alteration of NF-κB activity was observed at least up to 9 h of treatment with the purified LOS of both isolates (Supporting Figure S3). In absence of serum, the lack of PorB expression in the mutant NM0401 resulted in slight decrease of the early activation of NF-κB comparing to the parental ST-11 strain LNP19995. In contrast to the PorB mutant, the LOS-devoid mutant Z0305 or the mutant lacking both LOS and PorB were strongly affected (Figure 3C). Interestingly, activation of NF-κB was decreased in the later time points in cells infected with the PorB mutant NM0401 similarly to cells infected with the wild type parental strain LNP19995 (Figure 3C). Overall, our data suggest that LOS is a potent activator of NF-κB comparing to PorB. However, the late reduction of NF-κB activity provoked by the invasive ST-11 isolates seems to be independent of the expression of both PorB and LOS.
It is worth to note that expression of NF-κBp65 and p50 subunits was not affected over the time of infection with each isolates, excluding the modulation of NF-κB transcriptional activity due to the alteration of NF-κB expression (Supporting Figure S4).
Non-invasive isolates promote sustained NF-κB activity to protect epithelial cells from apoptosis
To further explore the role of NF-κB in the apoptosis induced by the meningococcal ST-11 isolates, early activation of NF-κB was blocked 1 h prior to infection, using MG-132 inhibitor. MG-132 is a peptide-aldehyde protease inhibitor that blocks NF-κB activation via inhibition of the proteasome function [32], [33]. Hec-1B cells were then infected for 9 h in presence of a non-toxic concentration of MG-132 or the carrier solvent DMSO (0.1%). Neither MG-132 nor DMSO alone resulted in apoptosis above background levels in uninfected cells (data not shown). Unexpectedly, pre-treatment of cells with MG-132 effectively protected cells from apoptosis brought about infection with the invasive isolate comparing to cells infected in presence of DMSO (Figure 4A). Similar results were obtained using the specific NF-κB inhibitor IKK-Nemo Binding Domain (NBD) peptide (data not shown). Early activation of NF-κB seems then to be a pre-requisite to promote apoptosis of epithelial cells with the invasive isolates. This requirement contrasts the cytoprotective role of NF-κB reported by several groups [34]–[38]. Nevertheless, the kinetic of NF-κB transactivation reported in Figure 3A showed that both invasive and non-invasive isolates are able to promote NF-κB activity after 4h of infection. However, only invasive ST-11 isolates are able to induce apoptosis after 9h of infection that was correlated with reduced NF-κB activity. Non-invasive isolates promoted sustained NF-κB activity that correlated with protection against apoptosis (Figure 3). We therefore explored the extent of the late down-regulation of NF-κB activity on the apoptotic cell death induced by the ST-11 invasive isolates. Hec-1B cells were transfected with a FLAG tagged-constitutively active form of IKK2 (CA-IKK2), leading to a constitutive phosphorylation and degradation of IκBα and subsequent increase of NF-κB activity, [39] or pCMV2 empty vector control (Figure 4B, immunoblot insert). Expression of CA-IKK2 was able to abrogate LNP19995-induced apoptosis by four fold comparing to empty vector-transfected cells (Figure 4B). These results strongly suggest that the late down regulation of NF-κB activity is required for invasive ST-11 isolates to promote LOS-mediated apoptosis of epithelial cells.

One possible explanation for this dual effect of NF-κB activation is that early activation of NF-κB is required to promote expression of TNF-α, which acts lately to promote apoptosis of epithelial cells following the decrease of NF-κB activity. Indeed, MG-132 dramatically reduced the level of secreted TNF-α with respect to DMSO-treated cells (Supporting Figure S5). To test this hypothesis, MG-132 was incorporated after 4 h of bacterial challenge (the period of which NF-κB transactivation was peaked). In these conditions, apoptosis was strongly induced irrespective to infection with the invasive or the carriage isolates (Figure 4C). The presence of anti-TNF-α neutralizing antibody significantly abrogated this effect (Figure 4C). To further analyse the role of TNF-α in meningococcal induced apoptosis, TLR4-mediated NF-κB activation was blocked by transfecting cells with the dnMyD88-expressing vector. Indeed, expression of the dnMyD88 as MG-132 pre-treatment strongly reduced the levels of secreted TNF-α in cells infected with both the invasive or the carriage isolates when compared to cells transfected with the empty vector control (Supporting Figure S5). The apoptotic level promoted by the invasive isolate LNP19995 also decreased significantly in cells transfected with the dnMyD88 (Figure 4A). Addition of TNF-α rescued apoptosis regarding cells transfected with the dnMyD88 and infected in absence of TNF-α. In all tested conditions, the carriage isolate induced low apoptosis similar to the background level (Figure 4A). These results underline the key role of TNF-α in promoting apoptosis by the invasive, but not the carriage isolates. In contrast to LOS, TNF-α also activates NF-κB but in MyD88-independent manner [40] and Figure 4D. After 9 h of incubation, TNF-α-induced NF-κB activity was also deceased upon infection with the invasive isolate LNP19995 but not with the carriage isolate LNP21019. Collectively, these data suggest that TNF-α secreted early upon infection with the ST-11 isolates is required to sensitize cells to apoptosis in synergy with the late down-regulation of NF-κB transcriptional activity.
Increased shedding of TNFR1 in cells infected with non-invasive isolates occurs in TACE/ADAM17-dependent manner
We have previously shown that carriage non-invasive isolates inhibit TNF-α-dependent apoptotic pathway through increasing the shedding of soluble TNFR1 (sTNFR1) which interfere with the biological activity of TNF-α. Increased shedding of sTNFR1 occurred concomitantly to the decreased level of the membrane-associated TNFR1 (mTNFR1) [22]. Shedding of TNFR1 is mediated by TNF-α converting enzyme (TACE also known as ADAM17), a metalloproteinase localized to the cytoplasmic membrane [41], [42]. One possible explanation for the increased shedding of sTNFR1 in cells infected with the carriage isolates could be the increased activity of TACE/ADAM17. We therefore analyzed the ability of the specific TACE/ADAM17 inhibitor, TNF-α protease inhibitor–1 (TAPI-1), to block TNFR1 release in response to infection with the carriage isolates. At the concentration employed (1 µM), TAPI-1 did not compromise cell viability as judged by PI staining (data not shown). As depicted in Figure 5, cells infected for 9 h with the carriage isolates in presence of TAPI-1 failed to induce shedding of sTNFR1 comparing to DMSO-treated cells (Figure 5, left panel; 34.56±7.37 pg/ml for TAPI-treated cells vs. 433.3±58.75 pg/ml for DMSO-treated cells, P<0.001) and concomitantly displayed significant surface levels of membrane bound-TNFR1 (Figure 5, right panel; MFI 81.12±15.49 in TAPI-treated cells vs. 24.94±4.15 in DMSO-treated cells, P<0.001). TAPI-1 treatment further increased mTNFR1 level in cells infected with the ST-11 invasive isolates comparing to cells infected in presence of DMSO (Figure 5, right panel; MFI 146.4±35.79 vs. 73.88±5.48, P<0.001). These data were also confirmed by immunofluorescence microscopy examination (Supporting Figure S6). These results suggest that increased shedding of sTNFR1 in cells infected with the carriage non-invasive isolates involved TACE/ADAM17 activity.
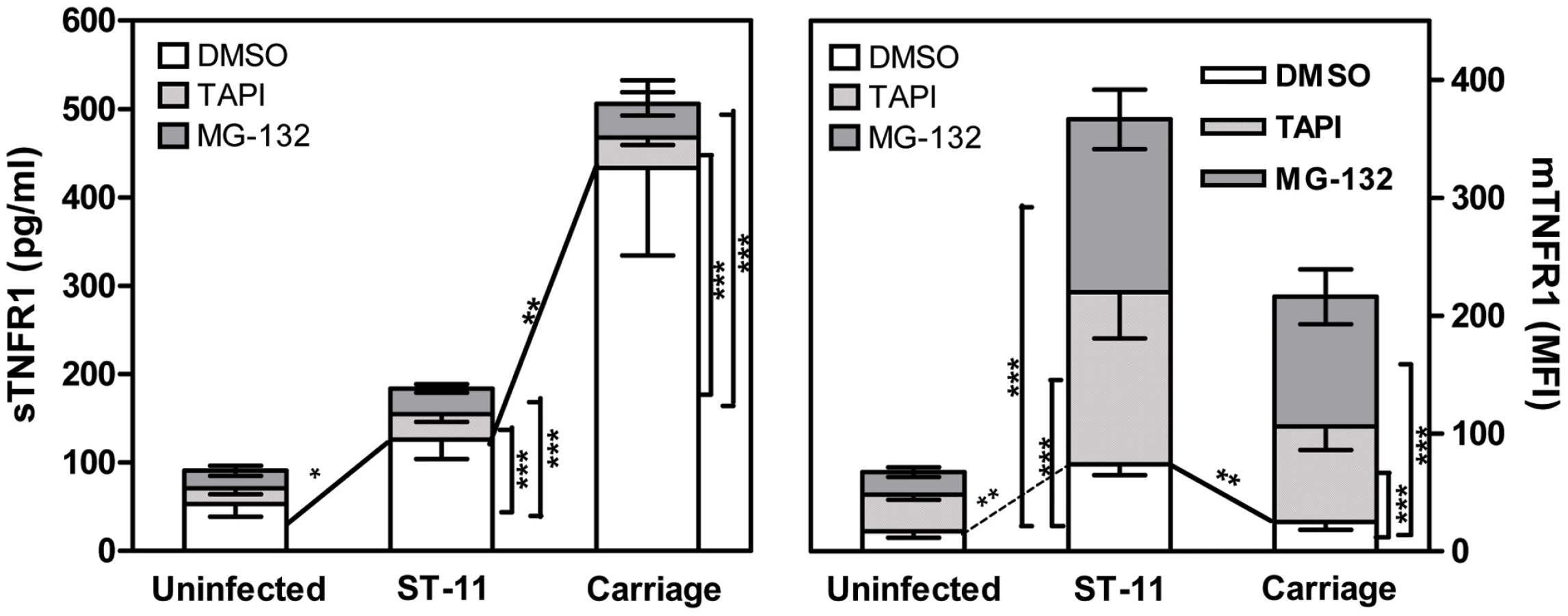
Increased TACE/ADAM17-mediated shedding of TNFR1 depends on sustained NF-κB activation
Given that carriage and invasive ST-11 isolates differentially modulate the late activity of NF-κB, we sought to determine whether the modulation of NF-κB transcriptional activity may impact the surface display and extracellular shedding of TNFR1. To address this issue, MG-132 was added after 4 h of bacterial challenge to not compromise the early expression of TNF-α. Comparing to DMSO vehicle-treated cells, MG-132-treatment led to significant decrease of sTNFR1 shedding and increase of mTNFR1 level in cells infected with the carriage isolates (Figure 5). To test whether persistent transcriptional activity of NF-κB would inverse the surface display of TNFR1 due to infection with the ST-11 isolates, Hec-1B cells were transiently transfected with the plasmid pCA-IKK2 that was modified by insertion of EGFP marker to visualize transfected cells. As control, cells were transfected with pcDNA3 harbouring the same marker (empty vector). As shown in Figure 6A, 39-47% and 35-40% of cells were transfected with empty vector control and pCA-IKK2, respectively (left panels). After 9 h of infection, surface expression of TNFR1 (mTNFR1) was examined in this sub-population of transfected cells (GFP+ events gated in region R1, Figure 6A, left panels). As expected, among empty vector-transfected cells, infection with the ST-11 isolate LNP19995 resulted in higher level of mTNFR1 compared to cells infected with the carriage isolate LNP21019 (MFI 67.13 vs. MFI 11, respectively). Interestingly, among pCA-IKK2-transfected cells, infection with the ST-11 isolate LNP19995 led to almost 50% decrease of TNFR1 surface level compared to empty vector-transfected cells (Figure 6A, right panels). Consistent with these results, the level of sTNFR1 significantly increased in CA-IKK2 transfected-, LNP19995-infected cells compared to empty vector-transfected, LNP19995 infected cells or cells infected with the carriage isolate LNP21019 (Figure 6B). Taken together, these results suggest that the differential modulation of NF-κB activity between the invasive ST-11 and the non invasive carriage isolates resulted in differential surface display of TNFR1 in TACE/ADAM17-dependent way.

Apoptosis of epithelial cells promoted by ST-11 isolates involves a sustained JNK activation
MAP kinase (MAPK) signaling pathways are activated in inflammatory reactions and have been shown to play important role in cell growth and death [43]. In general, p38 and c-Jun N-terminal kinase (JNK) are involved in cell death mechanisms, whereas Extracellular signal-regulated kinase (Erk1/2) is critical for cell survival [44]. We therefore investigated the potential role of MAP kinases in apoptosis of epithelial cells induced by the pathogenic meningococcal isolates. First, the effect of meningococci on the activation of the above mentioned kinases was examined in time course infection using phospho-specific antibodies. We monitored for total levels of each kinase to ensure that any change in phosphorylated protein levels was an actual measure of activation. Phosphorylation of Erk1/2 increased after 2h of cell exposure to each meningococcal isolate and then decreased after 6 h (Figure 7A, upper panel). Phosphorylation of p38 MAP kinase increased beyond 6 h of infection and persisted thereafter (Figure 7A, middle panel). Interestingly, phosphorylation of JNK increased gradually up to 6 h post infection, and then decreased afterwards to almost reach the basal level in cells infected with the carriage non invasive isolate. At the opposite, JNK phosphorylation was sustained in cells infected with the ST-11 isolate LNP19995 (Figure 7A, lower panel). As the carriage isolate, JNK was transiently activated upon treatment of Hec-1B cells with the LOS purified from the invasive or the carriage isolates (Supporting Figure S3). These data corroborate with the sustained activation of NF-κB. In all tested conditions, the total expression of each kinase was not altered (Figure 7A). To examine more thoroughly the role of the prolonged JNK activation in the cell death promoted by the invasive ST-11 isolates, we determined the effects of a dose range of the specific JNK inhibitor SP600125 [45] on apoptotic cell death induced by the invasive isolate LNP19995. Pre-treatment of cells with SP600125 markedly subdued the extent of the invasive isolate LNP19995-mediated cell death in dose-dependent manner with a maximum effective dose of 140 nM (Figure 7B, lower panel). This dose had no significant effect on viability of uninfected cells. In contrast to JNK, inhibition of p38 MAPK and Erk1/2 phosphorylation with their respective specific inhibitors had no beneficial effect on LNP19995-induced epithelial cell death and no effect on the viability of uninfected cells (Figure 7B, middle and upper panels, respectively). These data pointed out a selective differential ability of isolates to modulate the activation of JNK and suggest that infection with ST-11 invasive isolates induced death signals involving JNK, while attenuating survival signals in epithelial cells.

Sustained JNK activation promoted by ST-11 isolates is associated with reduction of NF-κB activity and occurs in TNFR1-dependent manner
Previous observations reported that sustained JNK activation associated with inhibition of NF-κB activation, contributes to TNF-α-induced apoptosis [46], [47]. To determine the extent of the late reduction of NF-κB activity mediated by the ST-11 isolates on the sustained JNK activation, Hec-1B cells were transiently transfected with the CA-IKK2 and the activation of JNK was analyzed after 9 h of infection with the invasive isolate LNP19995. Comparing to the control empty vector-transfected cells, maintenance of NF-κB activation in CA-IKK2-transfected cells resulted in dramatic impairment of JNK phosphorylation (Figure 8) that was associated with improvement of cell survival (Figure 4C). Our data establish a direct cause-effect of interference of ST-11 invasive isolates with NF-κB activity to allow a sustained JNK activation that is necessary to promote apoptosis of epithelial cells.

Discussion
Isolates of several bacterial species such as N. meningitidis may exist as symbiote in their host but may also invade internal compartments of the host with important local and systemic inflammatory responses. The consequences of the interaction with epithelial cells at the nasopharynx (the portal of entry of N. meningitidis) are therefore crucial in the pathophysiology of meningococcal infections. Induction of cytokines and particularly TNF-α has been implicated in local disruption of epithelial barrier functions [48], [49] and inducing epithelial cell apoptosis [50]. This hallmark may pave the way for further meningococcal invasion and dissemination to deeper sites (septicaemia and meningitis) [19]. Indeed, it has been reported that patients with meningococcal sepsis or meningitis often describe signs of pharyngitis before the onset of invasive disease while carriage isolates persist in the pharynx asymptomatically [51]. We have previously reported the impressive ability of the invasive ST-11 meningococcal isolates to induce apoptosis of epithelial cells. This overwhelming process, driven in part by the major bacterial endotoxin LOS, is dependent on an autocrin mechanism of TNF-α signaling through its receptor TNFR1 [22]. In contrast, infection with the non-invasive carriage isolates is associated with protection of epithelial cells mediated by the shedding of sTNFR1 resulting in alteration of the biological activity of TNF-α through soluble receptor-ligand complex formation and abrogation of apoptosis [22]. In this work, we further identified the actors in the signalling pathways leading to this different behaviour of invasive and non invasive isolates.
Our data with siTLR4 approach confirm the role of TLR4 as a potential transducer of the LOS induction of apoptotic signalling as in other Gram-negative pathogens such as Yersinia and Salmonella [52], [53]. Our data further show that TLR4 signaling induced by N. meningitidis through occurs in MyD88-dependent manner [54], [55], but not through the TRIF adaptor molecule (MyD88-independent pathway) [54], [56]. Indeed, these results are in agreement with the previous reports showing that MyD88 and IRAK1 are involved in apoptotic signaling upon stimulation with bacteria or bacterial components [57], [58]. TRIF-dependent pathway (MyD88-independent pathway) is more involved in generating a type I IFN-dependent response that is essential to host defence against viral infection [54], [56]. Nevertheless, many TLRs signal through the adaptor protein MyD88. In this regard, our data cannot exclude the role of other TLRs (which also signal through MyD88 such as TLR2) in the regulation of apoptosis upon infection with the pathogenic meningococcal isolates. Expression of dn-MyD88 as TLR4 knock-down, considerably altered the apoptosis induced by the ST-11 isolates and dramatically reduced expression of TNF-α. Moreover, our data suggest that the initial TLR4-dependent activation of NF-κB is required to establish apoptosis most likely through induction of TNF-αexpression. Treatment with MG-132 after 4 h of infection promoted apoptosis in cells infected with the carriage isolates and further increased apoptosis induced by the ST-11 isolates. However, sustained NF - κB activity seems to be protective against apoptosis as constitutive NF-κB activation mediated by the CA-IKK2, rescued the viability of the ST-11-infected cells. Interference with the NF-κB transcriptional activity to promote apoptosis of host cells has been described for other pathogens. Uropathogenic E. coli (UPEC) was able to abrogate urothelial responses by blocking NF-κB translocation to the nucleus and by inhibiting NF-κB-dependent transcription in response to either LPS or TNF-α stimulation [59]. Yersinia induces apoptotic cell death in macrophages by type III secretion system-mediated suppression of NF-κB activation [60]. In our hands, expression level of NF-κB p65 and p50 subunits were not affected during meningococcal infection, excluding the possibility that invasive ST-11 isolates modulate the activity of NF-κB through alteration of its expression. It has been shown that PorB activates NF-κB in TLR2-dependent manner [61]. The late reduction of NF-κB activity seems to be independent on PorB expression although the early activation slightly decreased comparing to the wild type strain. These results comfort our previous results showing that ST-11 isolates induce apoptosis in two independent pathways an extrinsic pathway promoted by LOS and an intrinsic pathway promoted by PorB [22]. How ST-11 meningococcal isolates attenuate the activity of NF-κB to promote apoptosis of epithelial cells is currently under investigation. Bacterial factor(s) may be responsible for this difference in the fate of NF - κB activity upon meningococcal infection.
As a biological consequence of the differential modulation of NF-κB activity between ST-11 and carriage isolates, cells exhibited: i) a differential display of TNFR1 expression at the surface of infected cells dependent on TACE/ADAM17 activity (higher mTNFR1 and low sTNFR1 in cells infected with the ST-11 isolates versus low mTNFR1 and higher sTNFR1 in cells infected with the carriage isolates) and ii) a differential profile of JNK activation (sustained activation in cells infected with the ST-11 invasive isolates and transient activation in cells infected with the carriage non invasive isolates). Indeed, we demonstrated that the inducible shedding of TNFR1 from cells infected with carriage isolates can be blocked by TAPI-1, an inhibitor of TACE/ADAM17. Furthermore NF-κB inhibitor suppressed the shedding of TNFR1 in carriage isolates infected cells while constitutive activation of NF-κB resulted in increased TNFR1 shedding from ST-11-infected cells. Our data are in agreement that sustained NF-κB activity is associated with up-regulation, maturation and increased activity of TACE/ADAM17/ADAM17 [62]. Nevertheless, the precise mechanism by which the meningococcal isolates modulate TACE/ADAM17 activity has yet to be identified. Recently, the involvement of the MAPK pathway in response to infection by N. meningitidis has been reported. It has been demonstrated that N. meningitidis can induce a sustained activation of p38 MAPK and JNK in vitro [63]. However, the role of these MAPKs in meningococcal-induced cell death has not been documented. It has been suggested that p38 and JNK are in general involved in cell death mechanisms, whereas Erk1/2 is critical for cell survival [64]. Our findings provide evidence for the participation of sustained JNK phosphorylation in the regulation of epithelial cell apoptosis in response to infection by ST-11 meningococcal invasive isolates. The JNK activity was only transiently detected with non invasive isolates. Using microarray analysis, we have recently reported that TACE/ADAM17 expression is reduced in blood of infected mice with N. meningitidis ST-11 ( - 2.4 fold change and Z score = -1.7) [65]. On the other hand, it has been shown that TACE/ADAM17 activity increases upon loss of c-Jun, a downstream target of JNK [66]. Sustained JNK activation promoted by ST-11 isolates may therefore be involved in the increase of TNFR1 surface expression through activation of c-Jun.
Based on these results, our data lean toward a biphasic model to promote apoptosis by ST-11 isolates (Figure 9): First, LOS mediates early activation of NF-κB during meningococcal infection in TLR4/MyD88/IRAK1-dependent manner leading to induction of expression and early secretion of the pro-inflammatory cytokine TNF-α and its receptor TNFR1 (Figure 9A). The sustained NF - κB activity may then promote TACE/ADAM17 activity that allows shedding of TNFR1 and prevents a sustained activation of the apoptotic factor JNK. This may then protect epithelial cells against apoptosis when they encounter non invasive carriage isolates. At the opposite, invasive isolates may produce factor(s) that inhibit the NF - κB activity leading to the accumulation of membrane bound TNFR1 and a sustained activation of the apoptotic factor JNK. (Figure 9B).

N. meningitidis-host cell interaction seems to involve a complex process in which bacterial heterogeneity impact differentially on the modulation of host cell signaling. Knowledge of the mechanism of alteration of NF-κB activity related to the detrimental effect of ST-11 invasive isolates may therefore provide better understanding and rational approaches for the control of invasive meningococcal infection.
Materials and Methods
Reagents and antibodies
RPMI 1640, HBSS and trypsin-EDTA were obtained from Invitrogen (France). Cocktail of protease inhibitors and phenylmethylsulfonyl fluoride (PMSF) were from Boehringer Mannheim (France). Human TNF-α (hTNF-α) was from Clinisciences (France). MG-132, SP600125, SB203580 and PD98059 were obtained from Sigma-Aldrich. Firefly D-luciferin was purchased from Caliper (France). Mouse anti-AU1, anti-Myc, anti-FLAG, anti-HA, anti-hTNFR1 mAbs and anti-p65 and anti-p50 rabbit polyclonal antibodies were purchased from abcam (France). human anti-TLR4 and anti-actin mAbs were from sigma-aldrich. Neutralizing anti-TLR4 mAb (clone HTA125) was from eBioscience (Hatfield, UK). Rabbit antibodies directed against JNK, p38 MAPK, Erk1/2, and their phosphorylated forms were obtained from Cell signaling. Horseradish peroxidase (HRP)-conjugated IgG antibodies were obtained from Jackson ImmunoResearch. FITC - and Texas Red-conjugated secondary antibodies were obtained from Invitrogen.
Plasmid constructs and siRNA oligonucleiotides
The AU1-MyD88152-296 (dnMyD88) and MycHis-IRAK11-215 (dnIRAK1) constructs were a generous gift from Dr. Muzio Marta (San Raffaele Scientific Institute, Division of Molecular Oncology, Milano, Italy). The pSH241 plasmid [67] carrying the TIR domain of TRIF (pcDNA-TRIF-TIR397-530, dnTRIF) was kindly provided by Dr. Wilbert A. Derbigny (Department of Microbiology and Immunology, Indiana University, School of Medicine). The (Igκ)3conaLuc plasmid harbouring NF-κB-dependent luciferase reporter construct was kindly provided from the Dr. Alain Israel's laboratory (Institut Pasteur, Unité de signalisation moléculaire et Activation cellulaire). The β-galactosidase expression plasmid pCMVβ was a generous gift from Dr. Laurence Arbibe, (Institut Pasteur, Unité de Pathogénie Microbienne Moléculaire). The plasmid IKK-2 S177E S181E carrying the FLAG-tagged constitutively active form IKK-2 (pCA-IKK2) (due to two point mutations in the S177E and S181E [39]) was purchased from Addgen (Cambridge, MA). To insert the EGFP into pCA-IKK2, the EGFP cassette under the control of the CMV promoter was obtained by digesting the plasmid pEGFP-C1 (Clontech) using AseI and XhoI restriction enzymes. The gel-purified cassette was then blunt-ended using the Klenow fragment of DNA polymerase (Fermentas) and was inserted into the blunt-ended AgeI restriction site of pCA-IKK2 plasmid. This recombinant plasmid was named pCA-IKK2-GFP.. pcDNA3 and pCMV2 empty vectors were respectively from Invitrogen and Addgen. All plasmid DNAs were prepared with a Endofree Maxi prep kit (Qiagen). siRNA oligonucléiotides used in this study are listed in the Table S1 and were purchased from Sigma proligo (France).
Bacterial strains and growth conditions
Escherichia coli DH5α strain [68] was used for plasmid propagations. DH5α was grown in Luria - Bertani (LB) medium supplemented with 100 µg/ml ampicilin. Meningococcal clinical isolates in France are sent to the National Reference Centre for Meningococci (NRCM) for full determination and typing. Bacteria were grown in GCB medium with Kellogg supplements [69]. Phenotypes (serogroup: serotype: serosubtype) and MLST genotypes were determined as previously described [9]. Sequence types (ST) and clonal complexes were assigned using the Neisseria MLST database (http://pubmlst.org/neisseria). All N. meningitidis strains used in this study were previously characterized [22] and their characteristics are listed in Supporting Table S2. The mutant strains NM0401, Z0305 and NM0701 were described previously [22]. The strain AD1001 inactivated in lpxA gene involved in LOS biosynthesis was generated by transforming the genomic DNA of Z0305 strain (lpxA::aph3') in the parental strain LNP21019. Positive clones were selected on GCB agar plates supplemented with 100 µg/ml kanamycin. Knock-out mutant was further confirmed by PCR and Southern blot.
Cell culture, transient transfection, short interfering RNA silencing (siRNA) and infection
Human endometrial epithelial cell line (Hec-1B) has been widely used as a model for meningococcal infections [70], [71]. This cell line express a surface CD14 and TNFR1 and secrete TNF-α upon infection [22], [72]. These features make it therefore suitable for this study. Hec-1B cells were maintained in RPMI-1640 (Invitrogen, France) supplemented with 5% foetal bovine serum (Sigma), 50 U/ml penicillin and 50 µg/ml streptomycin. Hec-1B cells were washed extensively and harvested by Trypsin-EDTA. Depending to experiments, cells were plated in 10 cm-culture dish, or seeded in 12 - or 96-well culture plates (Costar) at a density of ∼ 5×105 per cm2. Transfection experiments were carried-out prior to infection with 3 µg of plasmid DNA or 25 nM siRNA (Sigma) using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. All transfection experiments were performed in FBS - and antibiotics-free medium. Forty eight hours post-transfection, cells were washed and infected in absence of antibiotics and in presence of serum (unless otherwise indicated) with bacteria at multiplicity of infection (MOI) 20∶1. In all infection experiments, bacteria were centrifuged for 3 min, 3000 rpm at 25°C to synchronize the adhesion of bacteria to cells. Where indicated, TNF-α was added at a final concentration of 5 ng/ml. MG-132 was added either 1 h prior to or 4 h after bacterial challenge at a final concentration of 10 µM and maintained during the whole period of infection. SP600125, SB203580 or PD98059 were added 30 min before infection and maintained during the period of infection. After infection, samples were washed extensively before processing. As apoptotic level was more pronounced after 9h of infection [22] with the ST-11 isolates, this time point was therefore undertaken for most experiments, unless otherwise indicated.
Nuclear extraction and electrophoresis mobility gel shift assay (EMSA)
Nuclear extracts were prepared from uninfected or infected Hec-1B (at different time points), as described previously [73] and bandshift assay was performed by combining 5 µg of nuclear extract with a 32P-labeled oligonucleotide probe corresponding to the NF-κB consensus binding site (Santa Cruz Biotechnology, France) and then run on 5% (wt/vol) polyacrylamide gel in Tris-borate-EDTA buffer [74]. Native gel was dried under vacuum, and scanned in a Molecular Imager Faros FX plus (Bio-Rad, France).
Luciferase reporter assay
Cells co-transfected with (Igκ)3conaluc and pCMVβ plasmids were harvested in 200 µl of lysis buffer (50 mM HEPES (pH 7.6), 150 mM NaCl, 1 mM DTT, 1 mM EDTA, 0.5% NP-40, 10% glycerol, and protease inhibitors cocktail) and were incubated for 20 min at 4°C. The supernatants were collected from cell lysates after centrifugation at 12 000 x g for 15 min. The luciferase activity was monitored with 60 µl of the cell lysate and 20 µl of luciferase assay buffer (85 mM DTT, 0.85 mM K2HPO4 [pH 7.8], 50 mM ATP and D-luciferin substrate). Luciferase activity was measured using a MicroLumat Plus luminometer (EGG BERTHOLD Technologies, Toiry, France). In parallel, samples were assayed for β-galactosidase activity to normalize for transfection efficiency using the ortho-nitrophenyl β-D-galactopyranoside (ONPG)-based assay as described previously [75]. The results are reported as fold induction of relative luciferase units (RLU) over the control cells after normalizing for β-galactosidase activity and protein concentration.
Quantitative measurement of apoptosis and caspase-3 activity assay
Cells were carefully harvested using cold PBS/0.02% EDTA and washed in PBS. Double staining with FITC-Annexin V (specific for apoptotic cells) and Propidium iodide (PI, specific for necrotic cells) was carried out using the FITC-Annexin V kit (Sigma-Aldrich) following the manufacturer's recommendations. Samples were then analysed by flow cytometry. For some experiments, cells infected in 96-well plates in presence of anti-human TLR4 neutralizing mAb (clone HTA125, Abcam) or irrelevant isotype matched IgG control were washed and stained for apoptotic cells using the colorimetric detection and measurement APOPercentage apoptosis assay kit following manufacturer instructions (Biocolor Ltd, N. Ireland). Levels of caspase-3 activity were determined using the colorimetric caspase-3 assay kit (Sigma, France) as described previously [22].
TNFR1 surface staining and flow cytometry
Cells were labelled with anti-TNFR1, anti-TLR4 mAbs or irrelevant isotype-matched IgG controls as described elsewhere [22]. Stained samples were subjected to Fluorescence Activated Cell Sorting (FACS) analysis using a FACSCalibur flow cytometer (BD Biosciences, France). Fluorescence was recorded from a total of 10,000 events per sample. The acquired fluorescence data were subsequently analysed using WinMDI 2.8 software.
Fluorescence microscopy
Cells were allowed to adhere for 24 h on coverslips in 24-well plates. After infection with the previously generated DsRed-expressing meningococcal strains (Supporting Table S2 and [22]), coverslips were fixed in 4% paraformaldehyde in PBS and blocked with 1% normal goat serum in PBS. Anti-TNFR1 mAb was used at 1∶50 and FITC-conjugated goat anti-mouse IgG was used at a dilution of 1∶1000. Coverslips were mounted on microscope slides in DAPI-incorporated ProLong Gold antifade reagent (Invitrogen) to both minimize photobleaching and stain nuclei. Slides were then examined by digital confocal microscopy using Zeiss Axio Imager. D1 fluorescent microscope coupled to AxioCam MRm vers.3 (Carl Zeiss, Germany). Digital images were acquired using appropriate filters and combined using the Axiovision Rel. 4.6 software (Carl Zeiss).
TNF-α and soluble TNFR1 (sTNFR1) specific enzyme linked Immuno-sorbant assay (ELISA)
Quantitation of TNF-α or sTNFR1 in the culture supernatants was performed by specific ELISA as described elsewhere [22].
Immunoblotting
Cell lysates fractionated by SDS-PAGE, were transferred to polyvinylidene difluoride (PVDF) membrane which was then probed with primary antibody. The immunoreactive band was visualized using appropriate secondary HRP-conjugated secondary IgG antibody and ECL detection reagents (Amersham Pharmacia Biotech, France). The membranes were visualized using ChemiDoc XRS imager and QuantityOne 4.6 software (Bio-Rad).
Statistical analysis
The t test was used to compare two groups, and P values<0.05 were considered statistically significant.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. PeltolaH 1983 Meningococcal disease: still with us. Rev Infect Dis 5 71 91
2. SchuchatARobinsonKWengerJDHarrisonLHFarleyM 1997 Bacterial meningitis in the United States in 1995. Active Surveillance Team. N Engl J Med 337 970 976
3. van DeurenMBrandtzaegPvan der MeerJW 2000 Update on meningococcal disease with emphasis on pathogenesis and clinical management. Clin Microbiol Rev 13 144 166 table of contents
4. CartwrightKAStuartJMJonesDMNoahND 1987 The Stonehouse survey: nasopharyngeal carriage of meningococci and Neisseria lactamica. Epidemiol Infect 99 591 601
5. OrrHJGraySJMacdonaldMStuartJM 2003 Saliva and meningococcal transmission. Emerg Infect Dis 9 1314 1315
6. GoldschneiderIGotschlichECArtensteinMS 1969 Human immunity to the meningococcus. II. Development of natural immunity. J Exp Med 129 1327 1348
7. DensenP 1989 Interaction of complement with Neisseria meningitidis and Neisseria gonorrhoeae. Clin Microbiol Rev 2 Suppl S11 17
8. EmontsMHazelzetJAde GrootRHermansPW 2003 Host genetic determinants of Neisseria meningitidis infections. Lancet Infect Dis 3 565 577
9. MaidenMCBygravesJAFeilEMorelliGRussellJE 1998 Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc Natl Acad Sci U S A 95 3140 3145
10. CaugantDA 1998 Population genetics and molecular epidemiology of Neisseria meningitidis. Apmis 106 505 525
11. YazdankhahSPKrizPTzanakakiGKremastinouJKalmusovaJ 2004 Distribution of serogroups and genotypes among disease-associated and carried isolates of Neisseria meningitidis from the Czech Republic, Greece, and Norway. J Clin Microbiol 42 5146 5153
12. JolleyKAKalmusovaJFeilEJGuptaSMusilekM 2000 Carried meningococci in the Czech Republic: a diverse recombining population. J Clin Microbiol 38 4492 4498
13. ZarantonelliMLLancellottiMDeghmaneAEGiorginiDHongE 2008 Hyperinvasive genotypes of Neisseria meningitidis in France. Clin Microbiol Infect 14 467 472
14. SchoenCBlomJClausHSchramm-GluckABrandtP 2008 Whole-genome comparison of disease and carriage strains provides insights into virulence evolution in Neisseria meningitidis. Proc Natl Acad Sci U S A 105 3473 3478
15. KleinNJIsonCAPeakmanMLevinMHammerschmidtS 1996 The influence of capsulation and lipooligosaccharide structure on neutrophil adhesion molecule expression and endothelial injury by Neisseria meningitidis. J Infect Dis 173 172 179
16. VirjiMKayhtyHFergusonDJAlexandrescuCHeckelsJE 1991 The role of pili in the interactions of pathogenic Neisseria with cultured human endothelial cells. Mol Microbiol 5 1831 1841
17. MercierJCBeaufilsFHartmannJFAzemaD 1988 Hemodynamic patterns of meningococcal shock in children. Crit Care Med 16 27 33
18. BirknessKASwisherBLWhiteEHLongEGEwingEPJr 1995 A tissue culture bilayer model to study the passage of Neisseria meningitidis. Infect Immun 63 402 409
19. StephensDSFarleyMM 1991 Pathogenic events during infection of the human nasopharynx with Neisseria meningitidis and Haemophilus influenzae. Rev Infect Dis 13 22 33
20. ReadRCFoxAMillerKGrayTJonesN 1995 Experimental infection of human nasal mucosal explants with Neisseria meningitidis. J Med Microbiol 42 353 361
21. WarrenHSJrGonzalezRGTianD 2003 Case records of the Massachusetts General Hospital. Weekly clinicopathological exercises. Case 38-2003. A 12-year-old girl with fever and coma. N Engl J Med 349 2341 2349
22. DeghmaneAEVeckerleCGiorginiDHongERucklyC 2009 Differential modulation of TNF-alpha-induced apoptosis by Neisseria meningitidis. PLoS Pathog 5 e1000405
23. BarnesPJKarinM 1997 Nuclear factor-kappaB: a pivotal transcription factor in chronic inflammatory diseases. N Engl J Med 336 1066 1071
24. RothwarfDMKarinM The NF-kappa B activation pathway: a paradigm in information transfer from membrane to nucleus. Sci STKE 1999: RE1
25. WangCYMayoMWKornelukRGGoeddelDVBaldwinASJr 1998 NF-kappaB antiapoptosis: induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science 281 1680 1683
26. RhenMErikssonSPetterssonS 2000 Bacterial adaptation to host innate immunity responses. Curr Opin Microbiol 3 60 64
27. KawaiTAkiraS 2006 TLR signaling. Cell Death Differ 13 816 825
28. MedzhitovRPreston-HurlburtPKoppEStadlenAChenC 1998 MyD88 is an adaptor protein in the hToll/IL-1 receptor family signaling pathways. Mol Cell 2 253 258
29. YamamotoMSatoSMoriKHoshinoKTakeuchiO 2002 Cutting edge: a novel Toll/IL-1 receptor domain-containing adapter that preferentially activates the IFN-beta promoter in the Toll-like receptor signaling. J Immunol 169 6668 6672
30. YamamotoMSatoSHemmiHSanjoHUematsuS 2002 Essential role for TIRAP in activation of the signalling cascade shared by TLR2 and TLR4. Nature 420 324 329
31. AderemAUlevitchRJ 2000 Toll-like receptors in the induction of the innate immune response. Nature 406 782 787
32. JensenTJLooMAPindSWilliamsDBGoldbergAL 1995 Multiple proteolytic systems, including the proteasome, contribute to CFTR processing. Cell 83 129 135
33. RockKLGrammCRothsteinLClarkKSteinR 1994 Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell 78 761 771
34. BegAABaltimoreD 1996 An essential role for NF-kappaB in preventing TNF-alpha-induced cell death. Science 274 782 784
35. LiuZGHsuHGoeddelDVKarinM 1996 Dissection of TNF receptor 1 effector functions: JNK activation is not linked to apoptosis while NF-kappaB activation prevents cell death. Cell 87 565 576
36. Van AntwerpDJMartinSJKafriTGreenDRVermaIM 1996 Suppression of TNF-alpha-induced apoptosis by NF-kappaB. Science 274 787 789
37. WangCYMayoMWBaldwinASJr 1996 TNF - and cancer therapy-induced apoptosis: potentiation by inhibition of NF-kappaB. Science 274 784 787
38. WuMLeeHBellasRESchauerSLArsuraM 1996 Inhibition of NF-kappaB/Rel induces apoptosis of murine B cells. Embo J 15 4682 4690
39. MercurioFZhuHMurrayBWShevchenkoABennettBL 1997 IKK-1 and IKK-2: cytokine-activated IkappaB kinases essential for NF-kappaB activation. Science 278 860 866
40. PalombellaVJRandoOJGoldbergALManiatisT 1994 The ubiquitin-proteasome pathway is required for processing the NF-kappa B1 precursor protein and the activation of NF-kappa B. Cell 78 773 785
41. BlackRARauchCTKozloskyCJPeschonJJSlackJL 1997 A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature 385 729 733
42. MossMLJinSLBechererJDBickettDMBurkhartW 1997 Structural features and biochemical properties of TNF-alpha converting enzyme (TACE). J Neuroimmunol 72 127 129
43. GalloisCHabibATaoJMoulinSMacloufJ 1998 Role of NF-kappaB in the antiproliferative effect of endothelin-1 and tumor necrosis factor-alpha in human hepatic stellate cells. Involvement of cyclooxygenase-2. J Biol Chem 273 23183 23190
44. JohnsonSEDormanCMBolanowskiSA 2002 Inhibition of myogenin expression by activated Raf is not responsible for the block to avian myogenesis. J Biol Chem 277 28742 28748
45. BennettBLSasakiDTMurrayBWO'LearyECSakataST 2001 SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc Natl Acad Sci U S A 98 13681 13686
46. TangGMinemotoYDiblingBPurcellNHLiZ 2001 Inhibition of JNK activation through NF-kappaB target genes. Nature 414 313 317
47. De SmaeleEZazzeroniFPapaSNguyenDUJinR 2001 Induction of gadd45beta by NF-kappaB downregulates pro-apoptotic JNK signalling. Nature 414 308 313
48. CoyneCBVanhookMKGamblingTMCarsonJLBoucherRC 2002 Regulation of airway tight junctions by proinflammatory cytokines. Mol Biol Cell 13 3218 3234
49. SchmitzHFrommMBentzelCJScholzPDetjenK 1999 Tumor necrosis factor-alpha (TNFalpha) regulates the epithelial barrier in the human intestinal cell line HT-29/B6. J Cell Sci 112 (Pt 1) 137 146
50. GitterAHBendfeldtKSchulzkeJDFrommM 2000 Leaks in the epithelial barrier caused by spontaneous and TNF-alpha-induced single-cell apoptosis. Faseb J 14 1749 1753
51. MattilaPSCarlsonP 1998 Pharyngolaryngitis caused by Neisseria meningitidis. Scand J Infect Dis 30 198 200
52. HsuLCParkJMZhangKLuoJLMaedaS 2004 The protein kinase PKR is required for macrophage apoptosis after activation of Toll-like receptor 4. Nature 428 341 345
53. ZhangYBliskaJB 2003 Role of Toll-like receptor signaling in the apoptotic response of macrophages to Yersinia infection. Infect Immun 71 1513 1519
54. VogelSNFentonM 2003 Toll-like receptor 4 signalling: new perspectives on a complex signal-transduction problem. Biochem Soc Trans 31 664 668
55. TakedaKAkiraS 2004 TLR signaling pathways. Semin Immunol 16 3 9
56. FitzgeraldKARoweDCBarnesBJCaffreyDRVisintinA 2003 LPS-TLR4 signaling to IRF-3/7 and NF-kappaB involves the toll adapters TRAM and TRIF. J Exp Med 198 1043 1055
57. ChoiKBWongFHarlanJMChaudharyPMHoodL 1998 Lipopolysaccharide mediates endothelial apoptosis by a FADD-dependent pathway. J Biol Chem 273 20185 20188
58. RuckdeschelKMannelOSchrottnerP 2002 Divergence of apoptosis-inducing and preventing signals in bacteria-faced macrophages through myeloid differentiation factor 88 and IL-1 receptor-associated kinase members. J Immunol 168 4601 4611
59. KlumppDJWeiserACSenguptaSForrestalSGBatlerRA 2001 Uropathogenic Escherichia coli potentiates type 1 pilus-induced apoptosis by suppressing NF-kappaB. Infect Immun 69 6689 6695
60. MonackDMMecsasJBouleyDFalkowS 1998 Yersinia-induced apoptosis in vivo aids in the establishment of a systemic infection of mice. J Exp Med 188 2127 2137
61. SingletonTEMassariPWetzlerLM 2005 Neisserial porin-induced dendritic cell activation is MyD88 and TLR2 dependent. J Immunol 174 3545 3550
62. TakamuneYIkebeTNaganoOShinoharaM 2008 Involvement of NF-kappaB-mediated maturation of ADAM-17 in the invasion of oral squamous cell carcinoma. Biochem Biophys Res Commun 365 393 398
63. SokolovaOHeppelNJagerhuberRKimKSFroschM 2004 Interaction of Neisseria meningitidis with human brain microvascular endothelial cells: role of MAP - and tyrosine kinases in invasion and inflammatory cytokine release. Cell Microbiol 6 1153 1166
64. JohnsonGLLapadatR 2002 Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 298 1911 1912
65. SzatanikMHongERucklyCLedroitMGiorginiD 2011 Experimental meningococcal sepsis in congenic transgenic mice expressing human transferrin. PLoS ONE 6 e22210
66. Guinea-ViniegraJZenzRScheuchHHniszDHolcmannM 2009 TNFalpha shedding and epidermal inflammation are controlled by Jun proteins. Genes Dev 23 2663 2674
67. DerbignyWAHongSCKerrMSTemkitMJohnsonRM 2007 Chlamydia muridarum infection elicits a beta interferon response in murine oviduct epithelial cells dependent on interferon regulatory factor 3 and TRIF. Infect Immun 75 1280 1290
68. HanahanD 1983 Studies on transformation of Escherichia coli with plasmids. J Mol Biol 166 557 580
69. KelloggDSJrPeacockWLJrDeaconWEBrownLPirkleDI 1963 Neisseria Gonorrhoeae. I. Virulence Genetically Linked to Clonal Variation. J Bacteriol 85 1274 1279
70. DeghmaneAEGiorginiDLarribeMAlonsoJMTahaMK 2002 Down-regulation of pili and capsule of Neisseria meningitidis upon contact with epithelial cells is mediated by CrgA regulatory protein. Mol Microbiol 43 1555 1564
71. TahaMKMorandPCPereiraYEugeneEGiorginiD 1998 Pilus-mediated adhesion of Neisseria meningitidis: the essential role of cell contact-dependent transcriptional upregulation of the PilC1 protein. Mol Microbiol 28 1153 1163
72. JarvisGALiJSwansonKV 1999 Invasion of human mucosal epithelial cells by Neisseria gonorrhoeae upregulates expression of intercellular adhesion molecule 1 (ICAM-1). Infect Immun 67 1149 1156
73. PhilpottDJBelaidDTroubadourPThibergeJMTankovicJ 2002 Reduced activation of inflammatory responses in host cells by mouse-adapted Helicobacter pylory isolates. Cell Microbiol 4 285 296
74. Schmidt-UllrichRMemetSLilienbaumAFeuillardJRaphaelM 1996 NF-kappaB activity in transgenic mice: developmental regulation and tissue specificity. Development 122 2117 2128
75. DeghmaneAEPetitSTopilkoAPereiraYGiorginiD 2000 Intimate adhesion of Neisseria meningitidis to human epithelial cells is under the control of the crgA gene, a novel LysR-type transcriptional regulator. Embo J 19 1068 1078
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2011 Číslo 12
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Controlling Viral Immuno-Inflammatory Lesions by Modulating Aryl Hydrocarbon Receptor Signaling
- Fungal Virulence and Development Is Regulated by Alternative Pre-mRNA 3′End Processing in
- Cryo Electron Tomography of Herpes Simplex Virus during Axonal Transport and Secondary Envelopment in Primary Neurons
- Epstein-Barr Virus Nuclear Antigen 3C Stabilizes Gemin3 to Block p53-mediated Apoptosis
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy