
versus the Host: Remodeling of the Bacterial Outer Membrane Is Required for Survival in the Gastric Mucosa
Modification of bacterial surface structures, such as the lipid A portion of lipopolysaccharide (LPS), is used by many pathogenic bacteria to help evade the host innate immune response. Helicobacter pylori, a gram-negative bacterium capable of chronic colonization of the human stomach, modifies its lipid A by removal of phosphate groups from the 1 - and 4′-positions of the lipid A backbone. In this study, we identify the enzyme responsible for dephosphorylation of the lipid A 4′-phosphate group in H. pylori, Jhp1487 (LpxF). To ascertain the role these modifications play in the pathogenesis of H. pylori, we created mutants in lpxE (1-phosphatase), lpxF (4′-phosphatase) and a double lpxE/F mutant. Analysis of lipid A isolated from lpxE and lpxF mutants revealed lipid A species with a 1 or 4′-phosphate group, respectively while the double lpxE/F mutant revealed a bis-phosphorylated lipid A. Mutants lacking lpxE, lpxF, or lpxE/F show a 16, 360 and 1020 fold increase in sensitivity to the cationic antimicrobial peptide polymyxin B, respectively. Moreover, a similar loss of resistance is seen against a variety of CAMPs found in the human body including LL37, β-defensin 2, and P-113. Using a fluorescent derivative of polymyxin we demonstrate that, unlike wild type bacteria, polymyxin readily associates with the lpxE/F mutant. Presumably, the increase in the negative charge of H. pylori LPS allows for binding of the peptide to the bacterial surface. Interestingly, the action of LpxE and LpxF was shown to decrease recognition of Helicobacter LPS by the innate immune receptor, Toll-like Receptor 4. Furthermore, lpxE/F mutants were unable to colonize the gastric mucosa of C57BL/6J and C57BL/6J tlr4 -/ - mice when compared to wild type H. pylori. Our results demonstrate that dephosphorylation of the lipid A domain of H. pylori LPS by LpxE and LpxF is key to its ability to colonize a mammalian host.
Published in the journal:
. PLoS Pathog 7(12): e32767. doi:10.1371/journal.ppat.1002454
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1002454
Summary
Modification of bacterial surface structures, such as the lipid A portion of lipopolysaccharide (LPS), is used by many pathogenic bacteria to help evade the host innate immune response. Helicobacter pylori, a gram-negative bacterium capable of chronic colonization of the human stomach, modifies its lipid A by removal of phosphate groups from the 1 - and 4′-positions of the lipid A backbone. In this study, we identify the enzyme responsible for dephosphorylation of the lipid A 4′-phosphate group in H. pylori, Jhp1487 (LpxF). To ascertain the role these modifications play in the pathogenesis of H. pylori, we created mutants in lpxE (1-phosphatase), lpxF (4′-phosphatase) and a double lpxE/F mutant. Analysis of lipid A isolated from lpxE and lpxF mutants revealed lipid A species with a 1 or 4′-phosphate group, respectively while the double lpxE/F mutant revealed a bis-phosphorylated lipid A. Mutants lacking lpxE, lpxF, or lpxE/F show a 16, 360 and 1020 fold increase in sensitivity to the cationic antimicrobial peptide polymyxin B, respectively. Moreover, a similar loss of resistance is seen against a variety of CAMPs found in the human body including LL37, β-defensin 2, and P-113. Using a fluorescent derivative of polymyxin we demonstrate that, unlike wild type bacteria, polymyxin readily associates with the lpxE/F mutant. Presumably, the increase in the negative charge of H. pylori LPS allows for binding of the peptide to the bacterial surface. Interestingly, the action of LpxE and LpxF was shown to decrease recognition of Helicobacter LPS by the innate immune receptor, Toll-like Receptor 4. Furthermore, lpxE/F mutants were unable to colonize the gastric mucosa of C57BL/6J and C57BL/6J tlr4 -/ - mice when compared to wild type H. pylori. Our results demonstrate that dephosphorylation of the lipid A domain of H. pylori LPS by LpxE and LpxF is key to its ability to colonize a mammalian host.
Introduction
Helicobacter pylori, a gram-negative bacterium with only one well-defined niche, the human stomach, can persist for several years without manifestation of symptoms. However, over time serious complications may appear including peptic ulcer disease and gastric cancer, allowing for classification of H. pylori as a class I carcinogen by the World Health Organization [1], [2]. Similar to most gram-negative bacteria the outer membrane of H. pylori is composed of lipopolysaccharide (LPS), a surface exposed glycolipid.
LPS is anchored to the bacterial membrane by its hydrophobic lipid A domain. Extended from lipid A is the core oligosaccharide followed by the O-antigen creating a uniform hydrophilic surface layer that interphases with the surrounding environment. The first sugar of the core, Kdo (3-deoxy-D-manno-octulosonic acid), serves as a bridge to link the lipid anchor to the carbohydrate domains of LPS. The core polysaccharide is well conserved within a bacterial species; however, this is not the case with O-antigen. H. pylori shows great diversity in expression of its O-antigen [3], achieving a form of molecular mimicry by assembling surface polysaccharides resembling human blood group antigens, contributing to its ability to evade immune detection [4].
The biosynthetic pathway for assembly of Kdo2-lipid A (Figure 1), is well conserved throughout gram-negative bacteria and proceeds via the nine-step enzymatic “Raetz pathway”; however, great variety is seen in Kdo2-lipid A structures when comparing gram-negative bacterial species [5]. One of the best examples of Kdo-lipid A diversity is the structure produced on the surface of H. pylori (Figure 1). Variation in the Kdo-lipid A domain of LPS is generated in part through the action of modification enzymes [6]. The structural diversity among human pathogens might have arisen through evolutionary pressures applied to the bacterium by the host innate immune system.

To evade the host innate immune system by modification of its lipid A, pathogenic bacteria employ several approaches. One approach involves masking of negatively charged phosphate groups present on the lipid A disaccharide backbone by adding positively charged substituents, such as phosphoethanolamine or L-4-aminoarabinose, while a second approach involves the complete removal of phosphate groups from the backbone [7]. Both approaches result in a net loss of negative surface charges, resulting in a bacterial membrane more resistant to cationic antimicrobial peptides (CAMPs). CAMPs are positively charged peptides (e.g. defensins) that bind to negatively charged surface structures (e.g. lipid A) present on the bacterial cell surface, presumably creating pores in the membrane, resulting in cell lysis and eventual death [8], [9]. These structurally diverse CAMPs are found in macrophages, neutrophils and at the mucosal surface, making them an important component of the host innate immune response [8]. It has also been reported that changes in lipid A acylation or removal of Kdo (3-deoxy-D-manno-octulosonic acid) core sugars, through the activity of modification machinery, play a role in resistance to CAMPs [10], [11].
The lipid A domain is responsible for the endotoxic properties associated with LPS due to recognition and activation of the human Toll-like receptor 4-myeloid differentiation factor 2 (hTLR4-MD2) complex [12]. The human Toll-like receptor 2 (hTLR2), known to recognize several conserved bacterial structures including lipoteichoic acid and lipoproteins, has also been shown to recognize atypical forms of LPS [13], [14]. During infection by gram-negative organisms, dissociated LPS is recognized by the hTLR4-MD2 complex present on many cell types [15]. In preventing dissemination of infection, LPS serves as a molecular signal helping to clear the invading microbe; however, over-stimulation of inflammatory mediators (e.g. TNF-α), can result in the syndrome of septic shock [16]. LPS from enteric bacteria such as Escherichia coli produce the typical highly stimulatory lipid A (Figure 1), easily recognized by hTLR4-MD2; however, many pathogenic bacteria produce altered lipid A structures resulting in attenuated activation of hTLR4-MD2, allowing for immune evasion [6]. Several tactics are employed to produce hTLR4-MD2 attenuated lipid A structures, including the incorporation of longer acyl chains (16 or 18 carbons) instead of the standard (12 or 14 carbons) or by decreasing the overall number of acyl-chains from hexa-acylated to penta - or tetra-acylated lipid A forms [16]. In some organisms, the masking or removal of phosphate groups on the disaccharide backbone serves a dual role in CAMP resistance and attenuation of hTLR4-MD2 activation [17].
H. pylori produce a minimal Kdo-lipid A structure that is tetra-acylated with a phosphoethanolamine residue attached at the 1 position of the disaccharide (Figure 1), presumably contributing to high CAMP resistance and attenuated hTLR4-MD2 activation reported for this pathogen [18]. Since no other reservoirs exist for H. pylori, a unique balance must be established during infection in order to permit long-term survival of both the bacterium and its human host. Unlike a number of bacterial pathogens [6], modification of H. pylori lipid A appears to be constitutive and is not regulated by specific environmental cues [19]. Furthermore, the H. pylori lipid A modification pathway is highly ordered and efficient giving rise to a single lipid A species. This is in contrast to the lipid A variation seen on the surface of other pathogens [19]. Given that only one known reservoir exists for H. pylori, the human stomach, adaptation to changing environmental conditions is presumably unnecessary and explains this lack of regulation and single form of lipid A.
De novo lipid A synthesized by H. pylori is modified from a bis-phosphorylated hexa-acylated Kdo2-lipid A structure (Figure 1) by a five-step enzymatic pathway (Figure 2) and a single distinct lipid A species is produced [19], [20], [21], [22]. Previous studies from our laboratory have characterized the enzymes responsible for four of these steps; however, the identity of the lipid A 4′-phosphatase in H. pylori has yet to be identified or characterized. Thus, what role the 4′-phosphatase plays in CAMP resistance or in modulation of hTLR4-MD2 activation has not been addressed. Furthermore, although the lipid A 1-phosphatase (LpxE) of H. pylori was shown to be important for CAMP resistance, its role in hTLR4-MD2 activation remains unclear [17]. Here we report the identification of the lipid A 4′-phosphatase in H. pylori and demonstrate that the 1 and 4′-phosphatases act synergistically to produce a bacterial surface that is highly resistant to CAMP attack. We also show that dephosphorylation of H. pylori lipid A at the 1 and/or 4′ position results in LPS with attenuated hTLR4-MD2 activation. Most exciting, our results suggest that modification of H. pylori lipid A phosphate groups are important for colonization of a host.

Results
The H. pylori Lipid A 4′-Phosphatase Is Encoded by jhp1487
The published structure for wild type H. pylori is Kdo-lipid A that is tetra-acylated with a phosphoethanolamine attached at the 1-position (Figure 1) [18]. Previous work in our laboratory identified and characterized the Kdo2-lipid A modification machinery in H. pylori, including LpxE (1-phosphatase), EptA (phosphoethanolamine transferase), KdoH1/2 (Kdo Hydrolase) and LpxR (3′-O-deacylase), which all act in an ordered efficient manner to produce a single lipid A species found on the bacterial surface (Figure 2) [19], [20], [22]. To date, all enzymes have been identified and characterized except for the enzyme responsible for removal of the lipid A 4′-phosphate group, also known as LpxF (4′-phosphatase).
Previous attempts to identify H. pylori lpxF by our laboratory have proven unsuccessful, as heterologous expression of possible homologs in E. coli have failed to demonstrate LpxF activity in whole cells [17]. Searching the H. pylori genome for proteins homologous to LpxE (Hp0021) using the Blastp algorithm [23] revealed three possibilities, identified as belonging to a family of phosphatidic acid type 2 phosphatases (PAP2), Jhp0324, Jhp0787 and Jhp1487. Along with LpxE these proteins are members of COG0671, a cluster of putative orthologs of E. coli PgpB. PgpB is a membrane-bound phosphatidylglycerol phosphatase involved in phospholipid biosynthesis. A ClustalW2 alignment of Jhp0324, Jhp0787 and Jhp1487 against LpxE revealed a score of 8%, 7% and 20%, respectively. Score is defined as percent identities divided by number of residues compared. Given that Jhp1487 showed the highest score (20%) when aligned with LpxE, it became the likely target. A knockout in jhp1487 was created by interruption of the coding sequence with an antibiotic resistance cassette in H. pylori strain J99 and named J99 lpxF. Complementation of J99 lpxF was achieved by re-introduction of lpxF into the chromosome and named J99 lpxF, lpxF+.
To screen for LpxF activity in membrane fractions, in vitro assays were performed using radiolabelled Kdo2-[4′32P]lipid A as the substrate (Figure 3). Following incubation of the enzyme source with the radioactive substrate, the reaction products were separated via thin-layer chromatography (TLC) such that the more hydrophobic reaction products migrated faster. As expected, wild type H. pylori J99 membranes show robust LpxF activity (lane 2) as indicated by the appearance of free inorganic phosphate (32Pi), while membranes isolated from J99 lpxF were completely free of 4′-phosphatase activity (lane 3). LpxF activity was restored in membranes isolated from the complemented strain J99 lpxF, lpxF+ (lane 4), indicating that the H. pylori lipid A 4′-phosphatase is encoded by jhp1487. Reaction products from the previously characterized 1-phosphatase (LpxE) [20] and Kdo hydrolase (KdoH1/H2) [19], [21] were also detected. Loss of jhp1487 had no effect on 1-phosphatase or Kdo hydrolase activity. It was demonstrated previously [17] that membranes lacking a functional LpxE were unable to catalyze removal of the 1-phosphate group of lipid A substrates. Thus, ruling out H. pylori LpxF as a lipid A 1-phosphatase.

Characterization of Lipid A from Select H. pylori Mutants
To clearly ascertain the role LpxE and LpxF play in the pathogenesis of H. pylori, a series of mutants were generated, including a single lpxE, lpxF and a double lpxE/F mutant. Single lpxE and double lpxE/F mutants were created by introduction of a previously described lpxE mutation [17] into wild type J99 and J99 lpxF background by natural transformation, creating J99 lpxE and J99 lpxE/F, respectively. Complementation of J99 lpxE was achieved by re-introduction of lpxE into the chromosome and named J99 lpxE, lpxE+. Additionally, all mutations were moved into the mouse adapted H. pylori strains, B128 and X47 [24], [25], [26].
To thoroughly characterize this series of mutants, the lipid A species produced by each strain was determined. To begin, all strains were grown in the presence of 32Pi, the lipid A purified, separated by TLC and visualized by phosphorimaging. H. pylori wild type strains J99, B128 and X47 all revealed a single identical species of lipid A indicating no variation between backgrounds (Figure 4A). In contrast, lipid A from the single lpxE, lpxF and double lpxE/F mutants showed differences in migration compared to that of wild type. Chromosomal complementation of either the lpxE or lpxF mutations resulted in production of wild type lipid A (Figure 4B).

To confirm changes in lipid A and determine the structure, lipid A from all strains was subjected to analysis by MALDI-TOF (matrix-assisted laser desorption/ionization-time of flight) mass spectrometry in the negative ion mode. The wild type J99 spectrum showed a predominant peak at m/z 1546.9 consistent with the [M–H]- ion of the wild type structure of H. pylori lipid A (predicted [M–H]- at m/z 1547.1), which is tetra-acylated without a phosphate group at the 4′-position and a phosphoethanolamine residue at the 1-position (Figure 5). The lpxF mutant spectrum showed a predominant peak at m/z 2091.0 consistent with the [M–H]- ion of a lipid A species that is hexa-acylated with a phosphate group at the 4′-position and a phosphoethanolamine residue at the 1-position (predicted [M–H]- at m/z 2091.5), confirmation that the H. pylori LpxF is encoded by jhp1487. The presence of hexa-acylated lipid A species in the lpxF mutant suggest an ordered modification system in which H. pylori LpxR (3′-O-deacylase) activity is dependent on removal of the lipid A 4′-phosphate group by LpxF. The lpxE mutant spectrum (Figure 5) showed a predominant peak at m/z 1504.8 consistent with the [M–H]- ion of a lipid A species that is tetra-acylated bearing a single phosphate group (predicted [M–H]- at m/z 1505.1), in agreement with the published lipid A structure of lpxE deficient H. pylori strains [17]. The double lpxE/F mutant spectrum showed a predominant peak at m/z 2048.2 consistent with the [M–H]- ion of a lipid A species that is hexa-acylated with a phosphate group at the 1 - and 4′-positions (predicted [M–H]- at m/z 2048.5) (Figure 5). Complemented lpxE and lpxF strains showed a predominate peak consistent with that of wild type H. pylori (Figure 5). Moreover, MALDI-TOF mass spectrometry analysis of lipid A purified from all mutants in B128 (Figure S1 in Text S1) and X47 (Figure S2 in Text S1) backgrounds, agreed with corresponding J99 stains. The proposed structure for the lipid A species found in each strain is illustrated in Figure 5.

Dephosphorylation of Lipid A by LpxE and LpxF Results in Resistance to CAMPs
Resistance to CAMPs is of great importance to all mucosal pathogens. This is especially true for H. pylori whose only known reservoir is the human gastric mucosal surface. H. pylori is highly resistant to the CAMP polymyxin B (PMB) with the minimal inhibitory concentration (MIC) of wild type strains ranging from 250–1000 µg/ml peptide [19]. PMB is an experimental substitute for CAMPs and commonly used to demonstrate CAMP resistance in laboratory settings because of a similar mechanism of action [27]. MICs were determined for all strains using PMB Etest strips. H. pylori wild type strain J99 and all complemented strains exhibited a PMB MIC (469.3±73.9 µg/ml) comparable to published determinations [17], [19]. The lpxE, lpxF and double lpxE/F mutants in background J99 showed a 16, 360 and 1020 fold decrease in resistance to PMB, with MICs of 29.3±4.6, 1.3±0.3 and 0.46±0.03 µg/ml, respectively (Table 1). Thus, although LpxE and LpxF work in concert to produce a highly resistance bacterial membrane, removal of the 4′-phosphate group has the most influence on CAMP resistance in H. pylori.

To rule out changes in acylation in the single lpxF and double lpxE/F mutant as playing a role in CAMP resistance, the MIC of a previously characterized lpxR mutant was also determined and found to be comparable (512±0.0 µg/ml) to that shown by wild type H. pylori (Table 1). The lipid A of the lpxR deficient strain is hexa-acylated without a phosphate group at the 4′-position and a phosphoethanolamine residue at the 1-position [22] suggesting that, in H. pylori, deacylation plays no role in resistance to CAMPs. MICs were also determined for mutants in the mouse adapted backgrounds, B128 and X47, and were found to be comparable to J99 stains (Table 1).
The lifecycle and transmission of H. pylori is not well understood. Most literature agrees that oral familial passage of the bacterium is the most likely route of transmission, suggesting that H. pylori must not only resist CAMPs located at the gastric mucosal surface, but also those found in the oral cavity [28]. Moreover, the bacterium must resist CAMPs secreted by a variety of leukocytes, constantly surveying for invading pathogens [8]. To confirm our PMB MIC findings, we repeated MIC determinations using CAMPs possibly encountered by H. pylori during its lifecycle. We chose a number of peptides relevant to human infection for this study including: (i) the human cathelicidin LL-37 produced by both leukocytes and epithelial cells, (ii) human β-defensin 2 (HBD-2) found throughout the gastrointestinal tract, (iii) P-113, a fragment of histatin 5 found within the oral cavity, (iv) and HNP-2, an α–defensin produced by neutrophils [9], [29].
To determine the MICs, a standard microtiter broth dilution method was utilized. J99 wild type was highly resistant to all CAMPs analyzed (Table 2). The lpxE, lpxF and double lpxE/F mutants all showed a decrease in resistance against LL37, P-113, and HBD-2, similar to what is seen against PMB (Table 2). Once again, LpxF activity seemed to have the largest influence on resistant CAMPs. No change in resistance was seen for the α–defensin HNP2 at the indicated concentrations (Table 2). The lpxR deficient strain was identical to that of wild type, once again confirming that deacylation of LPS plays no role in resistance to CAMPs in H. pylori (Table 2). Together, these results indicate that dephosphorylation of lipid A in H. pylori, by LpxE and LpxF, is essential for resistance to CAMPs.

Interestingly, H. pylori produces an antibacterial peptide referred to as Hp (2.20), related to the insect cecropins that bind to the phosphate groups of lipid A [30]. H. pylori is naturally resistant to Hp (2.20), and it has been suggested that release of this peptide into the stomach may contribute to clearance of other gastrointestinal pathogens providing some benefit to H. pylori infected individuals [30]. Again, the double lpxE/F mutant shows a large decrease (≥ 60 fold) in resistance against Hp (2.20) (Table 2). The level of Hp (2.20) production by Helicobacter during colonization of the gastric mucosa is unclear; however, it is likely that modification of the lipid A structure provides protection against this Helicobacter derived antimicrobial compound.
Dephosphorylation of Lipid A by LpxE and LpxF Produce a Surface More Resistant to CAMP Binding
CAMPs primary mode of action against bacteria is to bind negatively charged moieties present on the bacterial surface (e.g. phosphate groups) [8]. Unlike most gram-negative bacteria, wild type strains of H. pylori completely lack unsubstituted phosphate moieties on its LPS [18]. However, LpxE/F deficient H. pylori strains produce an LPS anchored to the membrane by bis-phosphorylated lipid A (Figure 5) providing a negatively charged target for positively charged CAMPs.
To visually assess this, we devised a PMB binding assay by use of a biologically active fluorescent Oregon Green 514 derivative of polymyxin B (PMB-OG). Briefly, H. pylori wild type and double lpxE/F mutant in strain J99 were incubated for 10 minutes in the presence of 0, 1, 25, or 250 µg/ml PMB-OG and visualized by phase contrast and fluorescence microscopy (Figure 6). Overlay images clearly illustrate that unlike wild type, the double lpxE/F mutant fluoresced when incubated with 1 µg/ml PMB-OG, indicating increased surface-bound or entry of PMB-OG. Even at 25 µg/ml of peptide, essentially no fluorescence was observed for wild type bacteria. Both strains fluoresced when incubated with 250 µg/ml PMB-OG, although wild type was lower in intensity. These images visually demonstrate the protective effect lipid A dephosphorylation by LpxE and LpxF has against CAMP attack of the bacterial surface of H. pylori.
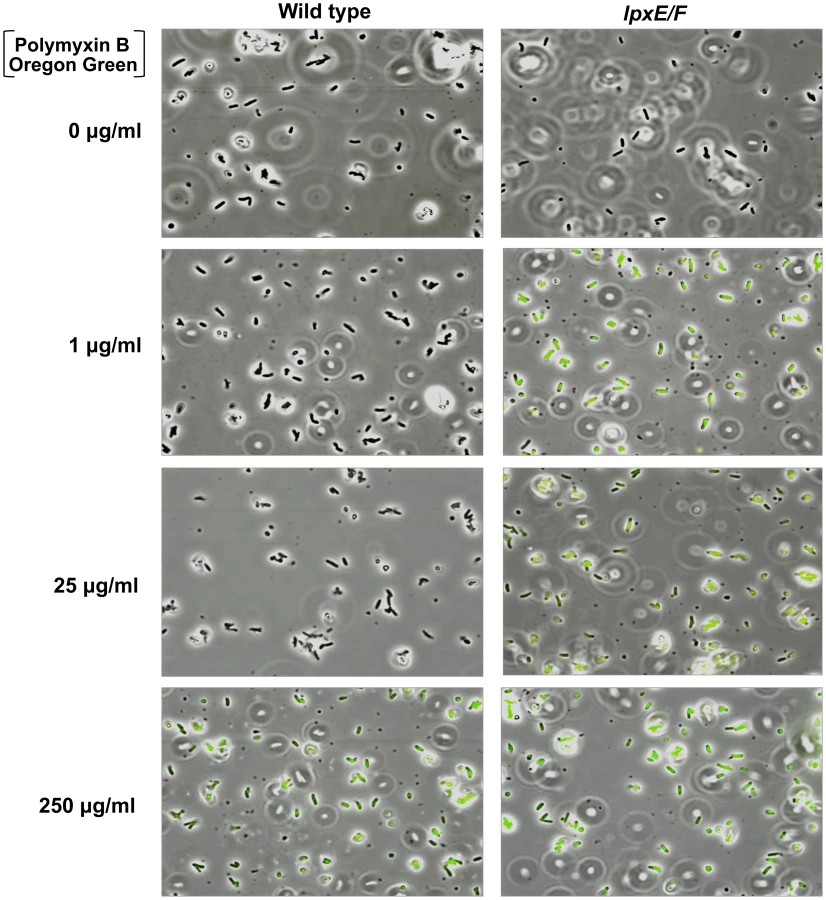
To quantify the binding and/or entry of PMB-OG to H. pylori strains, stained cells were re-suspended in PBS and fluorescent intensity measured (Figure S3 in Text S1). The double lpxE/F mutant showed significantly increased fluorescence when compared to wild type confirming our visual interpretation of the fluorescence microscopy. Furthermore, quantitative fluorescent analysis of PMB-OG binding to the lpxE, lpxF, lpxR and complemented mutants corresponded to the loss of CAMP resistance seen in our MIC experiments (Table 2) solidifying these findings.
Dephosphorylation of Lipid A by LpxE and LpxF Results in Attenuated TLR4-MD2 Activation and Has No Effect on TLR2 Activation
H. pylori LPS is characterized by strikingly low endotoxicity which may contribute to the long-term carriage state of the organism [18]. LPS from F. tularensis and P. gingivalis also exhibit very low endotoxicity and both organisms harbor lipid A phosphatases [31], [32]. LPS purified from LpxF deficient F. tularensis mutants which produce a penta-acylated form of lipid A failed to show increases in hTLR4-MD2 activation, indicating no role in attenuation [31]. However, given that hTLR4-MD2 is unable to recognize LPS anchored to the membrane by a penta-acylated form of lipid A, this result is not surprising [33]. Alternatively, LPS purified from LpxE and/or LpxF deficient P. gingivalis, triggered an increased hTLR4-MD2 response, indicating lipid A dephosphorylation plays a role in its low endotoxicity [32]. The ability of LPS from a number of bacteria to elicit an immune response has been examined. However, much of the literature describes experiments using LPS isolated from bacteria (e.g. P. gingivalis) producing a mixture of lipid A species, complicating the findings [34]. Further confusing the literature are reports that heavily modified forms of lipid A, like that found in H. pylori, P. gingivalis, and Leptospira interrogans elicit an immune response through hTLR2 [14], [35], [36], [37], [38]. Fortuitously, H. pylori and the mutants generated herein produce an abundant single species of lipid A (Figures 4 & 5), giving us the ability to investigate the contribution of single lipid A modification in hTLR4-MD2 and hTLR2 activation.
The Toll-activation profiles of intact LPS were examined using samples purified using by the Hirschfield method [39] that allows for removal of potential contaminating lipoproteins. Activation of TLRs was monitored using HEK-Blue 293 cells stably transfected with TLR machinery and a secreted alkaline phosphatase (SEAP) reporter gene placed under the control of an NF-κB inducible promoter allowing for easy detection of TLR activation using a colorimetric assay. Changes in hTLR4-MD2 activation in the H. pylori strains are clearly illustrated in Figure 7A. LPS from the H. pylori single lpxE, lpxF and double lpxE/F mutant show 2, 6 and 10 fold significant (P ≤0.001) increases in activation of hTLR4-MD2 at 10000 ng/ml when compared to wild type, indicating that LpxF imparts greater influence on attenuated hTLR4-MD2 when compared to LpxE. This agrees with the published crystal structure of the hTLR4-MD2-LPS complex showing that the 4′ phosphate group is involved for ligand recognition [40]. Similar results were seen using 1000 ng/ml of LPS (Figure 7A). LPS isolated from the lpxR deficient mutant activated hTLR4-MD2 similar to wild type LPS indicating that lipid A dephosphorylation plays a larger role in lowering endotoxicity compared to deacylation of Helicobacter lipid A (Figure 7A). Although these findings are of interest, it is important to note that in comparison to E. coli LPS, higher concentrations of Helicobacter LPS are required for activation of TLR4. For each experiment LPS from R. sphaeroides (1000 ng/ml), a known TLR4 antagonist [41], and E. coli (10 and 1000 ng/ml) were used as a negative and positive controls for activation of hTLR4-MD2, respectively (Figure 7B).

LPS from the H. pylori single lpxE, lpxF and double lpxE/F mutants showed no significant activation of hTLR2 (p>0.05), even at LPS concentrations as high as 10,000 ng/ml when compared to wild type LPS (Figure 8A). LPS from E. coli (1000 ng/ml) and Pam3CSK4 (10 and 1000 ng/ml), a synthetic lipopeptide, were used as a negative and positive control for activation of hTLR2, respectively (Figure 8B). H. pylori LPS has been reported to act as both a hTLR4-MD2 and hTLR2 agonist [42]; however, the ability of H. pylori LPS to activate hTLR2 is more controversial. Our data suggest that H. pylori LPS does not activate hTLR2. Some of these discrepancies may arise because of contaminating lipoproteins in LPS preparations. Recent literature documenting activation of hTLR2 by H. pylori LPS used concentrations as high as 10,000 ng/ml [42], increasing the possibility that activation was not the result of LPS but rather contamination. All TLR activation studies were performed using LPS purified from strains in background J99.

Previous research has demonstrated that hTLR4-MD2 and murine TLR4-MD2 (mTLR4-MD2) display differential recognition of LPS [33]. In anticipation of a future colonization study using a murine host, we felt it necessary to address this by examining the ability of H. pylori LPS to activate mTLR4-MD2. Activation of TLRs was monitored using HEK-Blue 293 cells stably transfected with murine TLR machinery using the same reporter system described above. Changes in mTLR4-MD2 activation in the H. pylori strains are clearly illustrated in Figure 7C. LPS from the H. pylori single lpxE, lpxF, lpxR and double lpxE/F mutant show 10, 8, 9 and 5 fold significant (p ≤0.001) increases in activation of mTLR4-MD2 at 10000 ng/ml when compared to wild type (Figure 7B), suggesting that LpxE, LpxF and LpxR all play similar roles in mTLR4-MD2 ligand recognition and confirming species-specific differential recognition of LPS. Differential recognition by mTLR4-MD2 of LPS purified from an lpxR deficient background is not surprising considering the promiscuity of this receptor for lipid A isoforms (i.e. tetra - and penta-acylated lipid A) [33]. For each experiment endotoxin free water and E. coli (10 and 1000 ng/ml) were used as negative and positive controls for activation of mTLR4-MD2, respectively (Figure 7D). LPS from the H. pylori single lpxE, lpxF, lpxR and double lpxE/F mutants showed no significant activation of mTLR2 (p>0.05), even at LPS concentrations as high as 10,000 ng/ml when compared to wild type LPS (data not shown).
Dephosphorylation of Lipid A by LpxF Is Required for Host Colonization in H. pylori
To date, nothing is known regarding participation of H. pylori LPS modifications in host colonization. Presumably, dephosphorylation by LpxE and LpxF provides an advantage through resistance to CAMPs at the gastric mucosal surface and attenuated hTLR-MD2 activation. Francisella mutants lacking lpxF were greatly attenuated in animal studies and considered avirulent [31]. However, F. tularemia does not establish a long-term colonization of its human host but rather, results in quick progression to severe illness and/or death [43]. Considering our current findings, we felt it was important to proceed with an in vivo animal study to emphasize the role played by LpxE and LpxF in host colonization.
For these experiments, all mutations were transferred into the mouse-adapted strains B128 and X47 [24]. Two bacterial strains were used to rule out the influence of variation between strains and for confirmation of findings. Recent publications in Campylobacter jejuni and H. pylori have demonstrated that lipid A modification enzymes can modulate motility and O-antigen expression, respectively [19], [44]. Both of these have been shown to be important for host colonization. Given these findings, we felt it important to examine both phenotypes prior to colonization studies. The LPS profiles (O-antigen polysaccharide and core oligosaccharide composition) of all mutants were indistinguishable from that of wild type during SDS-PAGE (Figure S4 in Text S1) and all strains showed normal motility in a soft agar assay (Figure S5 in Text S1), suggesting LpxE and LpxF do not influence these phenotypes. Mutants in H. pylori background J99 were used to illustrate these findings. However, identical results were found in both mouse-adapted strains, B128 and X47 (data not shown).
Two mice populations, C57BL/6J and C57BL/6J tlr4-/-, were colonized. The TLR4 deficient mice were used in conjunction with wild type mice to help determine which plays a larger role in host colonization: CAMP resistance or attenuated hTLR4-MD2 activation. Mice were infected orogastrically with 2×108 bacteria per mouse. Colonization rates were determined by enumeration of colony forming units per gram (CFU/g) of stomach. Two independent colonization experiments were performed. The first was a 15-day time point using both B128 and X47 strains in C57BL/6J. The second was a time course colonization experiment at days 3, 15, and 45 using only strain B128 in both C57BL/6J and C57BL/6J tlr4-/- mice.
Results from the first experiments revealed that in wild type C57BL/6J mice the single lpxE, lpxF and double lpxE/F mutants, in background B128 and X47, showed a statistically significant colonization defect (p<0.0001) (Figure 9A and 9B). In B128, complementation of lpxE deficient strains failed to restore the colonization defect. However, complementation of the lpxF deficient strain successfully restored colonization to wild type levels (p>0.05). In X47, complementation of lpxE and lpxF deficient strains resulted in partial recovery of the colonization defect but still significantly different from wild type (p<0.05). Data from both cohorts of mice at day 15 were combined to increase significance and robustness of our analysis. The double lpxE/F mutant in strain X47 was not included because the addition of a chloramphenicol cassette shows a strain specific decrease in the fitness of the bacteria in colonization experiments (unpublished observation). Overall, lpxF deficient strains in both B128 and X47 background showed the largest defect in colonization, suggesting LpxF activity is essential for host colonization.

A second colonization experiment was performed using B128 strains in C57BL/6J and C57BL/6J tlr4-/- mice. A 3, 15 and 45 day time course was included in this study to determine if H. pylori strains were cleared soon after infection or if it was a gradual loss of colonization. Wild type strain B128 colonized C57BL/6J and C57BL/6J tlr4-/- mice equally well (p>0.05) throughout the time course (Figure 10A). Single LpxF and double LpxE/F deficient strains were mostly unable to colonize C57BL/6J or C57BL/6J tlr4-/- mice mice, demonstrating a colonization defect by day 3 (Figure 10C and 10D) and no significant differences were seen when comparing C57BL/6J and C57BL/6J tlr4-/- mice (p>0.05). However, the LpxE deficient strain colonized well until day 45 (Figure 10B) and a significant difference was seen when comparing C57BL/6J and C57BL/6J tlr4-/- mice (p<0.01). This suggests that resistance to CAMPs, resulting from the dephosphorylation of lipid A by LpxF, is important for survival within a host. The role LpxF dependent attenuated TLR4-MD2 activation plays in the colonization of a host cannot be determined because they are so quickly cleared from the mouse stomach, presumably due to CAMP sensitivity. However, LpxE deficient strains retain partial resistance to CAMPs (Table 2), giving us the ability to compare mTLR4-MD2 dependent colonization. Interestingly, a colonization defect seen at day 45 using wild type mice disappears in tlr4-/- mice. Given that the LPS from lpxE mutants shows a 10-fold increase in activation of mTLR4-MD2 (Figure 7C), one can speculate that dephosporylation of lipid A by LpxE results in attenuated TLR4-MD2 activation required for long term colonization of a host. Taken together, these findings indicate that dephosphorylation of lipid A by LpxE and LpxF in H. pylori plays a role in effective colonization and survival within a host.

Discussion
The primary surface component of nearly all gram-negative bacteria is the glycolipid LPS. LPS is localized to the outer leaflet of the outer membrane, providing an additional layer of protection from the external environment and interfacing with the surrounding environment. Modification of the bioactive Kdo-lipid A domain of LPS is a common theme among gram-negative pathogens, most likely providing an advantage to the invading pathogen [6]. H. pylori is a great example, producing a highly modified form of lipid A that aids in camouflaging the bacterium from the innate immune response of its host allowing it to set up a life long chronic infection of the gastric mucosa.
The identification and characterization of LpxF in this work completes the five-step enzymatic pathway by which H. pylori lipid A is modified (Figure 2), resulting in an outer surface that is highly resistant to CAMPs and LPS that shows attenuated TLR4-MD2 activation [19]. Given that deletion of some enzymes in this five-step pathway (e.g. LpxE, KdoH1/2, and LpxF), result in the complete or partial loss of “downstream” modifications, the order for modification of H. pylori lipid A can now be determined (Figure 2). A recent publication demonstrated that efficient ligation of O-antigen to the core-lipid A molecule is modulated by KdoH1/2 activity in H. pylori, suggesting that surface LPS glycosylation is also an ordered process linked to lipid A modification [19]. Thus, it appears that the enzymatic machinery responsible for the maturation of LPS in H. pylori evolved a highly ordered pathway, ensuring a single and very specific surface-exposed LPS that is essentially invisible to its host innate immune system. Moreover, the only known variable region of the H. pylori LPS molecule is the O-antigen, most likely allowing for adaptation to antigenic variation in host cell surfaces [4]. One could speculate that lack of multiple hosts for H. pylori was the key evolutionary pressure leading to an ordered pathway; hence, any genetic drift resulting in changes to the lipid A structure were quickly selected against by the host innate immune system.
CAMP resistance is important for any pathogen, especially those that establish long-term colonization of a host and H. pylori is an excellent example. Our research identifies the dephosphorylation of lipid A in H. pylori by LpxE and LpxF as the most important determinant of CAMP resistance identified to date in this organism. Furthermore, our data suggest that although both LpxE and LpxF act synergistically to provide resistance to CAMPs, LpxF imparts the most influence. The removal of a Kdo core sugar by KdoH1/2 was shown to influence CAMP resistance in previous studies, with KdoH1/2 deficient strains having a polymyxin B MIC of between 0.5 and 1.0 µg/ml, similar to what is seen in LpxF deficient strains [19]. This can be attributed to the inefficient removal of the 4′-phosphate group by LpxF in KdoH1/2 deficient strains of H. pylori.
It is well documented that removal or “masking” of phosphate groups is the primary feature involved in CAMP resistance in several organisms [6]. However, the exact mechanism for resistance is more speculative and thought to occur by reducing the binding efficiency of these peptides. By use of a biologically active fluorescent PMB, our data demonstrates that membranes composed of fully phosphorylated lipid A show an increased ability to bind CAMPs. Moreover, the use of biologically relevant CAMPs found at multiple locations within a human host provides evidence that resistance to CAMPs is not only important for survival at the site of colonization, but also during transmission of H. pylori through the oral cavity.
LPS serves as one of the most powerful activators of the innate immune system. Presumably, H. pylori must avoid activation of hTLR4-MD2 to set up long-term colonization of the gastric mucosa. Producing an LPS characterized by strikingly low endotoxicity likely contributes to this immune deception. But which lipid A modifications in H. pylori have the greatest impact on attenuated hTLR4-MD2 activation? Our data suggest that dephosphorylation of lipid A by LpxE and LpxF contributes to the low endotoxicity of H. pylori LPS, but is not the only factor. It should be noted that LPS prepared from the double LpxE/F mutant, synthesizing lipid A that is bis-phosphorylated and hexa-acylated, still shows attenuated hTLR4-MD2 activity when compared to bis-phosphorylated hexa-acylated lipid A of LPS prepared from E. coli K-12 stains (Figure 7), requiring high concentrations of LPS (100 ng/ml) for in vitro activation of hTLR4-MD2. It is likely these differences arise from the increased length of acyl chains (16 and 18 carbons) found on H. pylori lipid A. Additionally, our data suggest that H. pylori LPS does not activate hTLR2 even at high concentrations of LPS, helping to clarify some conflicting reports in the literature [42].
Previous to this work, nothing was known regarding participation of H. pylori lipid A in host colonization. This work clearly demonstrates that dephosphoryation of H. pylori lipid A by LpxE and LpxF is essential for efficient host colonization, with LpxF activity being the largest determinant of bacterial survival within a host. LpxF deficient strains are unable to colonize host and are quickly cleared within 3 days of inoculation (Figure 10A and 10B), presumably due to loss of CAMP resistance (Table 2). However, LpxE deficient strains retain partial resistance to CAMPs (Table 2) and show longer-term survival within a murine model but are cleared in a mTLR4-MD2 dependent manner by day 45 (Figure 10B). Given that LPS isolated from LpxE deficient strains show a 10-fold increase in mTLR4-MD2 activation (Figure 7B), mTLR4-MD2 dependent clearance from the murine stomach seems likely. Furthermore, considering the 10-fold increase in hTLR4-MD2 seen with LPS purified from LpxF deficient strains, an even greater role in immune evasion within the human stomach is suggested and highlights the importance of lipid A modification in the pathogenesis of H. pylori.
To date, all LpxF enzymes identified only catalyze the removal of the 4′-phosphate group from penta-acylated forms of lipid A [31], [45]. H. pylori LpxF differs in that it can dephosphorylate hexa-acylated forms of lipid A (Figure 3). The use of H. pylori LpxF for the synthesis of 4′-monophosphoryl lipid A variants could prove useful in the development of vaccine adjuvants. Lipid A derivatives that lack the 1-phosphate group can be prepared by chemical synthesis, acid hydrolysis of LPS, or LpxE phosphatase treatment and are referred to as 1-monophosphoryl lipid A [46], [47], [48], [49]. More recently, some 4′-monophosphoryl lipid A variants have been approved as adjuvants for use in human vaccines. These lipid A variants retain full adjuvant activity while demonstrating low endotoxicity [50], [51].
In this research, we identify the enzyme responsible for lipid A 4′-phosphatase activity as Jhp1487 (LpxF) in H. pylori. We also demonstrate that dephosphorylation of lipid A by LpxE and LpxF in H. pylori provides resistance to CAMPs and results in attenuated hTLR4-MD2 activation, with LpxF having the largest impact in both cases. Removal of phosphate groups from the lipid A backbone is essential for colonization of H. pylori in a mammalian host. This research highlights the importance of membrane lipid composition in the pathogenesis of gram negative bacteria and provides clear evidence that lipid A is an important virulence factor.
Materials and Methods
Ethics Statement
This study was carried out in strict accordance with the European Union Directive 2010/63/EU (and its revision 86/609/EEC) on the protection of animals used for scientific purposes. Our laboratory has the administrative authorization for animal experimentation (Permit Number 75–1250) and the protocol was approved by the Institut Pasteur Review Board that is part of in the Regional Committee of Ethics of Animal Experiments of Paris region (Permit Number: 99–174).
Bacterial Strains and Growth Conditions
The bacterial strains and plasmids used in the study are summarized in Table 3. Cultures of H. pylori were started from methyl-cellulose stocks routinely stored at −80°C, plated onto 5% sheep blood agar medium (BAP) and incubated at 37°C for 36 to 60 h in a microaerobic atmosphere (5% O2, 10% CO2, 85% N2). The methyl-cellulose storage media is composed of cellulose-methyl ether 4000 CPS grade (1.0% w/v) and MgSO4 (0.1 M) and the BAP is composed of Tryptic soy agar (acumedia Cat. 7100A) and defibrinated sheep blood (Remel Cat. R54020). Liquid cultures of H. pylori were started from overnight culture on BAP and inoculated into brucella broth supplemented with 7% fetal bovine serum (Hyclone) and vancomycin (10 µg/ml). Liquid cultures of H. pylori were grown to an A600 of ∼1.0 at 37°C under microaerobic conditions for 24 to 36 h with shaking. E. coli was routinely grown at 37°C in LB broth with appropriate antibiotics using standard conditions.

Recombinant DNA Techniques
Plasmids were isolated using a QIAprep Spin Miniprep Kit (Qiagen). Custom primers (Table S1 in Text S1) were obtained from Integrated DNA Technologies. PCR reagents were purchased from Stratagene. PCR clean up was performed using a Qiaquick PCR Purification Kit (Qiagen). DNA fragments were isolated from agarose gels using a Qiaquick Gel Extraction Kit (Qiagen). Restriction endonucleases, T4 DNA ligase, and antarctic phosphatase were purchased from New England Biolabs. All modifying enzymes were used according to the manufacturer's instructions.
Construction of H. pylori Mutants
To construct the lipid A 4′-phosphatase mutant, plasmid pILL570 containing hp1580 (lpxF), from H. pylori 26695 was obtained from a previously published library [52]. This plasmid was reverse amplified using primers 5 and 6 (Table S1 in Text S1), incorporating BamH1 and Kpn1 restriction sites and leaving 300 bps at the 5′ and 3′ end of the gene. A non-polar kanamycin cassette was digested out of plasmid pUC18-Km2 [53] using BamH1 and Kpn1 and ligated into the reverse PCR product in the same orientation as hp1580 using standard cloning techniques. The generated plasmid, pILLhp1580:kan, was used to create 4′-phosphatase defective mutants in H. pylori J99, B128 and X47 by natural transformation [54]. Resistant colonies were selected on blood agar plates containing 8 µg/ml of kanamycin, patched onto kanamycin-containing plates, and verified by PCR of genomic DNA. Gene hp1580 corresponds to jhp1487 in H. pylori J99. Mutation of jhp0019 (lpxE) and jhp0634 (lpxR) in H. pylori B128 and X47 was achieved by introduction of a chloramphenicol or kanamycin cassette into the coding sequence of each gene resulting in the interruption and removal of a large portion of its sequence, using previously described suicide vectors [17], [22]. The lpxE/F double mutant was created by introduction of the lpxE mutation into a lpxF defective mutant background using natural transformation as described above.
Chromosomal Complementation of H. pylori lpxE and lpxF Defective Mutants
The interruption of hp0954 (rdxA) renders H. pylori resistant to metronidazole making it an ideal candidate for chromosomal complementation [55]. Using methods previously described [22], lpxE and lpxF defective mutants were complemented by insertion and consequent disruption of rdxA. Briefly, the lpxE and lpxF genes plus 500 bps upstream were amplified by PCR (primers 3,4 and primers 1,2, respectively), Table S1 in Text S1, from H. pylori J99 genomic DNA using Pfu Turbo (Stratagene) according to the manufacturer's instructions. The lpxE and lpxF amplicons were digested with BamH1 and EcoR1, gel purified, and subsequently ligated into the previously described H. pylori rdxA complementation plasmid pET634comp [22]. Previous to ligation, plasmid pET634comp was digested with BamH1 and EcoR1, gel purified, and treated with antarctic phosphatase. The resulting plasmids pETjhp1487comp and pETjhp0019comp were transformed into H. pylori J99 lpxE and lpxF deficient mutants by natural transformation [54]. The same method was used for complementation in X47 and B128 backgrounds. Resistant colonies were selected on blood agar plates containing 12 µg/ml metronidazole, patched onto metronidazole-containing plates, and verified by PCR of genomic DNA.
Isolation and Analysis of Lipid A Species from 32Pi-Labelled Cells
H. pylori (25 ml) and E. coli (5 ml) cultures were grown in the presence of 5.0 µCi/ml 32Pi (PerkinElmer) to an A600 of ∼1.0 using standard liquid growing conditions described above and the cells harvested by centrifugation. 32P-labelled lipid A and phospholipids were isolated using published protocols [20] and spotted onto a Silica Gel 60 TLC plate (EMD) at 2500 cpm/lane. Lipid species were separated using the solvent chloroform, pyridine, 88% formic acid and water (50∶50∶16∶5, v/v). The TLC plates were exposed overnight to a phosphorimager screen and visualized using a Bio-Rad Molecular Imager phosphorimager equipped with Quantity One software.
Isolation of Lipid A for Mass Spectrometry Analysis
H. pylori cultures (25 ml) were grown to an A600 of ∼1.0 using standard liquid growing conditions described above and the cells harvested by centrifugation. Lipid A was purified from bacteria as described previously [20] and stored frozen at −20 °C. The lipid A species were analyzed using a MALDI-TOF (ABI Voyager-DE PRO) mass spectrometer equipped with a N2 laser (337 nm) using a 20 Hz firing rate. The spectra were acquired in negative ion reflectron mode and each spectrum represented the average of a minimum of 4000 shots. The matrix used was a saturated solution of 6-aza-2-thiothymine in 50% acetonitrile and 10% tribasic ammonium citrate (9∶1, v/v). The samples were dissolved in chloroform-methanol (4∶1, v/v) and deposited on the sample plate, followed by an equal portion of matrix solution (0.3 µl).
Preparation of Cell-free Extracts, Double-spun Cytosol, and Washed Membrane
H. pylori cultures (200 ml) were harvested at A600 of ∼1.0 and the cells harvested by centrifugation. Cell free extracts, membrane-free cytosol, and washed membranes were prepared as previously described [19] and were stored in aliquots at −20°C. Protein concentration was determined by the bicinchoninic acid method [56].
Assay of the 4′-phosphatase Activity
The 4′-phosphatase activity of Jhp1487 (LpxF) was assayed under optimized conditions in a 10 - µl reaction mixture containing 50 mM MES, pH 6.0, 0.2% Triton X-100, and Kdo2-[4′-32P]lipid A (at ∼3000–5000 cpm/nmol) as the substrate. Kdo2-[4′-32P]lipid A was prepared as previously described [21]. Washed membranes (1.0 mg/ml) were employed as the enzyme source. The dephosphorylation reaction was allowed to proceed at 30 °C for 3.0 h. Enzymatic reactions were terminated by spotting 4.5 µl of the mixtures on Silica Gel 60 TLC plates and the plate was dried under a cool air stream for 20 min. Reaction products were separated using the solvent chloroform, pyridine, 88% formic acid, water (30∶70∶16∶10, v/v). Reaction products were detected and analyzed using a Bio-Rad Molecular Imager PhosphorImager equipped with Quantity One Software.
Fluorescent Polymyxin B Binding Assay
Liquid cultures of H. pylori were grown to an A600 of ∼0.8 to 1.0 at 37°C under microaerobic conditions for 24 to 36 h. The cells were washed with PBS (3X) followed by dilution to A600 of 0.05 in PBS. Polymyxin B Oregon Green 514 conjugate (Invitrogen) was added to 50 µl diluted cells at select concentrations and incubated at 37°C for 10 minutes. Following the incubation cells were washed with PBS (3X), resuspended in 50 µl of PBS, 15 µl placed onto poly-L-lysine coated slides (Electron Microscopy Sciences), and coverslips added. Bacteria were viewed at a magnification of 1000X with a Nikon Eclipse 80i microscope equipped with a 100x 1.4NA PLAN APO lens, a GFP band pass emission filter set with a 480±15 nm excitation range, a 535±20 nm emission range and a Photometrics Cool SNAP HQ2 camera. NIS-Elements AR 3.0 software was used to capture and create the merged images. To quantify PMB-OG binding, strains were cultured and stained as described above but in a volume of 200 µl. After washing, the cells were resuspended in PBS and placed into 96-well plates for analysis. Fluorescence (480 nm excitation and 535 nm emission) and A600 of each well was determined using a Synergy Mx multi-mode microplate reader (BioTek). Each experiment was repeated in triplicate and data reported as a ratio of Fluorescence intensity to A600. The results of three experiments were pooled and a one tailed Mann–Whitney test was used to determine statistical significance of observed differences (GraphPad Prism v5.0; GraphPad Software, CA).
LPS Preparation
LPS from H. pylori was isolated from a 500 ml liquid culture grown under standard conditions to an A600 of ∼1.0 using the hot water-phenol method [57]. Isolated LPS was further purified to remove contaminating immuno-stimulatory proteins (e.g. lipoproteins) that could falsely alter TLR assays using the previously described Hirschfeld method [39]. Purified LPS was quantified using a Mettler Toledo XS105 Dual Range analytical balance (sensitivity≥0.1 ng) and resuspended in HyPure Cell Culture Grade Endotoxin Free Water (HyClone) to a concentration of 5 mg/ml.
Determination of Minimum Inhibitory Concentrations (MIC)
MIC determination using Polymyxin B Etest strips (Biomérieux) was determined as previously described [19]. For MIC determination in liquid medium, the Hancock Laboratory Microtiter Broth Dilution Method was used and modified as needed [58]. H. pylori strains were grown overnight in brucella broth supplemented with 7% fetal bovine serum. The overnight cultures were inoculated into a 96-well microtiter plates at a starting A600 of 0.05, in a total volume of 100 µl standard H. pylori growing medium, supplemented with select concentration of CAMPs. The 96-well plates were incubated at 37°C in a microaerobic atmosphere (5% O2, 10% 1 CO2, and 85% N2) with constant shaking and A600 of each well determined using a Synergy-Mx monochromator based multi-mode microplate reader at 24 and 48 hours. Each experiment was repeated in triplicate. A positive growth control containing no CAMP and a negative control containing no bacteria was performed with every replicate. The MIC was taken as the lowest concentration of CAMP that reduced growth (A600) by more than 50% when compared to the positive growth control. The following CAMPs were purchased and used in the MIC experiment: polymyxin B (Sigma), human β defensin-2 (Phoenix Pharm), human cathelicidin LL-37 (Anaspec), histatin-5 P-113 (Sigma), human neutrophil peptide-2 (Bachem), and H. pylori peptide HP2-20 (Invitrogen). All peptides were stored according to manufacturers' instructions. Because of cost, MICs were only determined for mutants in the J99 background. Complemented strains were also excluded.
TLR Signaling Assay
For TLR4 and TLR2, human epithelial kidney (HEK) 293 cells stably co-transfected with m or hTLR4, m or hMD2, and m or hCD14 (denoted as HEK-m/hTLR4) or m/hTLR2 and m/hCD14 (denoted as HEK-m/hTLR2) were purchased from InvivoGen (CAT. hkb-htlr4 and hkb-htlr2 or hkb-mtlr4 and hkb-mtlr2, respectively). All cell lines stably express secreted embryonic alkaline phosphatase (SEAP) under the control of a promoter inducible by NF-κB and activator protein 1 (AP-1). Thus, stimulation of m/hTLR4-MD2 or m/hTLR2 will result in an amount of extracellular SEAP in the supernatant that is proportional to the level of NF-κB induction. The cell lines were maintained in standard Dulbecco's modified Eagle's medium (DMEM) with 10% heat-inactivated fetal bovine serum (FBS) (Gibco) supplemented with 4.5 g/L glucose, 2 mM L-glutamine, 50 U/mL penicillin, 50 ug/ml streptomycin and 1X HEK-Blue selection (InvivoGen) in a 5% saturated CO2 atmosphere at 37°C.
The induction of TLR signaling in HEK-m/hTLR4 and HEK-m/hTLR2 cell lines was assessed by measuring SEAP activity using QUANTI-BlueTM colorimetric assay (InvivoGen). The assay was performed according to manufacturer's protocols. Briefly, cells were seeded in a 96-well plate in triplicate (2.5×104 cells/well for HEK-m/hTLR4 and 5×104 cells/well for HEK-m/hTLR2) in the presence or absence of 10-fold dilutions of purified LPS. Controls included Rhodobacter sphaeroides LPS (TLR4 antagonist) (InvivoGen), E. coli W3110 LPS (TLR4 agonist) and the synthetic triacylated lipoprotein Pam3CSK4 (TLR2 agonist) (InvivoGen). After 20–24 h incubation, supernatants (20 ul) were transferred to a 96-well plate and incubated at 37°C with QUANTI-Blue (180 ul) for 1-2. SEAP activity was measured by reading optical density at 655 nm with a Synergy Mx multi-mode microplate reader (BioTek). The results of several (at least three) experiments were pooled and a one tailed Mann–Whitney test was used to determine statistical significance of observed differences (GraphPad Prism v5.0; GraphPad Software, CA).
Mouse Colonization Assays
C57BL/6J female mice purchased from Charles River Laboratories and C57BL/6J tlr4 -/ - female mice from Pasteur Institute (kindly provided by Shizuo Akira) aged 4 to 5 weeks were infected orogastrically with feeding needles with either X47 or B128 strains (2×108 bacteria per mouse). Colonization rates were determined after 3, 15 or 45 days by enumeration of colony forming units (CFU) per gram of stomach. Mice were euthanized with CO2 and the stomachs were ground and homogenized in peptone broth. The samples were then diluted and spread on blood agar plates supplemented with 200 µg/ml of bacitracin, and 10 µg/ml of nalidixic acid, to inhibit the growth of resident bacteria from the mouse forestomach. The CFUs were enumerated after 8 days of incubation under microaerobic conditions. The results of several (at least two) colonization experiments were pooled and a one tailed Mann–Whitney test was used to determine statistical significance of observed differences (GraphPad Prism v5.0; GraphPad Software, CA).
Supporting Information
Zdroje
1. ParsonnetJFriedmanGDVandersteenDPChangYVogelmanJH 1991 Helicobacter pylori infection and the risk of gastric carcinoma. N Engl J Med 325 1127 1131
2. BlaserMJ 1996 The bacteria behind ulcers. Sci Am 274 104 107
3. AppelmelkBJShiberuBTrinksCTapsiNZhengPY 1998 Phase variation in Helicobacter pylori lipopolysaccharide. Infect Immun 66 70 76
4. WangGGeZRaskoDATaylorDE 2000 Lewis antigens in Helicobacter pylori: biosynthesis and phase variation. Mol Microbiol 36 1187 1196
5. RaetzCRWhitfieldC 2002 Lipopolysaccharide endotoxins. Annu Rev Biochem 71 635 700
6. RaetzCRReynoldsCMTrentMSBishopRE 2007 Lipid A modification systems in gram-negative bacteria. Annu Rev Biochem 76 295 329
7. GunnJSLimKBKruegerJKimKGuoL 1998 PmrA-PmrB-regulated genes necessary for 4-aminoarabinose lipid A modification and polymyxin resistance. Mol Microbiol 27 1171 1182
8. DiamondGBeckloffNWeinbergAKisichKO 2009 The roles of antimicrobial peptides in innate host defense. Cur Pharma Design 15 2377 2392
9. PazgierMHooverDMYangDLuWLubkowskiJ 2006 Human beta-defensins. Cell Mol Life Sci 63 1294 1313
10. MatsonJSYooHJHakanssonKDiritaVJ 2010 Polymyxin B resistance in El Tor Vibrio cholerae requires lipid acylation catalyzed by MsbB. J Bacteriol 192 2044 2052
11. ClementsATullDJenneyAWFarnJLKimSH 2007 Secondary acylation of Klebsiella pneumoniae lipopolysaccharide contributes to sensitivity to antibacterial peptides. J Biol Chem 282 15569 15577
12. KimHMParkBSKimJIKimSELeeJ 2007 Crystal structure of the TLR4-MD-2 complex with bound endotoxin antagonist Eritoran. Cell 130 906 917
13. ErridgeC 2010 Endogenous ligands of TLR2 and TLR4: agonists or assistants? J Leuk Biol 87 989 999
14. WertsCTappingRIMathisonJCChuangTHKravchenkoV 2001 Leptospiral lipopolysaccharide activates cells through a TLR2-dependent mechanism. Nat Immunol 2 346 352
15. TobiasPS 1999 Lipopolysaccharide-Binding Protein. BradeHOpalSMVogelSNMorrisionDC Endotoxin in Health and Disease New York Marcel Dekker, Inc 359 367
16. TrentMSSteadCMTranAXHankinsJV 2006 Diversity of endotoxin and its impact on pathogenesis. J Endotoxin Res 12 205 223
17. TranAXWhittimoreJDWyrickPBMcGrathSCCotterRJ 2006 The lipid A 1-phosphatase of Helicobacter pylori is required for resistance to the antimicrobial peptide polymyxin. J Bacteriol 188 4531 4541
18. TranAXSteadCMTrentMS 2005 Remodeling of Helicobacter pylori lipopolysaccharide. J Endotoxin Res 11 161 166
19. SteadCMZhaoJRaetzCRTrentMS 2010 Removal of the outer Kdo from Helicobacter pylori lipopolysaccharide and its impact on the bacterial surface. Mol Microbiol 78 837 52
20. TranAXKarbarzMJWangXRaetzCRMcGrathSC 2004 Periplasmic cleavage and modification of the 1-phosphate group of Helicobacter pylori lipid A J Biol Chem 279 55780 55791
21. SteadCTranAFergusonDJrMcGrathSCotterR 2005 A novel 3-deoxy-D-manno-octulosonic acid (Kdo) hydrolase that removes the outer Kdo sugar of Helicobacter pylori lipopolysaccharide. J Bacteriol 187 3374 3383
22. SteadCMBeasleyACotterRJTrentMS 2008 Deciphering the unusual acylation pattern of Helicobacter pylori lipid A J Bacteriol 190 7012 7021
23. SchafferAAAravindLMaddenTLShavirinSSpougeJL 2001 Improving the accuracy of PSI-BLAST protein database searches with composition-based statistics and other refinements. Nucleic Acids Res 29 2994 3005
24. PeekRM 2008 Helicobacter pylori infection and disease: from humans to animal models. Dis Models Mech 1 50 55
25. IsraelDASalamaNArnoldCNMossSFAndoT 2001 Helicobacter pylori strain-specific differences in genetic content, identified by microarray, influence host inflammatory responses. J Clin Invest 107 611 620
26. ErmakTHGiannascaPJNicholsRMyersGANedrudJ 1998 Immunization of mice with urease vaccine affords protection against Helicobacter pylori infection in the absence of antibodies and is mediated by MHC class II-restricted responses. J Exp Med 188 2277 2288
27. GutsmannTHaggeSODavidARoesSBohlingA 2005 Lipid-mediated resistance of Gram-negative bacteria against various pore-forming antimicrobial peptides. J Endotoxin Res 11 167 173
28. BassoDPlebaniMKustersJG 2010 Pathogenesis of Helicobacter pylori infection. Helicobacter 15 Suppl 1 14 20
29. SalzmanNH 2008 Defensins versus bacteria: not just antibiotics anymore. Gastroenterol 134 2174 2177
30. PutsepKBrandenCIBomanHGNormarkS 1999 Antibacterial peptide from H. pylori. Nature 398 671 672
31. WangXRibeiroAAGuanZAbrahamSNRaetzCR 2007 Attenuated virulence of a Francisella mutant lacking the lipid A 4′-phosphatase. Proc Natl Acad Sci USA 104 4136 4141
32. CoatsSRJonesJWDoCTBrahamPHBainbridgeBW 2009 Human Toll-like receptor 4 responses to P. gingivalis are regulated by lipid A 1 - and 4′-phosphatase activities. Cell Microbiol 11 1587 1599
33. HajjarAMErnstRKTsaiJHWilsonCBMillerSI 2002 Human Toll-like receptor 4 recognizes host-specific LPS modifications. Nature Immunol 3 354 359
34. ChenYYPengBYangQGlewMDVeithPD 2011 The outer membrane protein LptO is essential for the O-deacylation of LPS and the co-ordinated secretion and attachment of A-LPS and CTD proteins in Porphyromonas gingivalis. Mol Microbiol 79 1380 1401
35. Que-GewirthNLRibeiroAAKalbSRCotterRJBulachDM 2004 A methylated phosphate group and four amide-linked acyl chains in leptospira interrogans lipid A. The membrane anchor of an unusual lipopolysaccharide that activates TLR2. J Biol Chem 279 25420 25429
36. DarveauRPPhamTTLemleyKReifeRABainbridgeBW 2004 Porphyromonas gingivalis lipopolysaccharide contains multiple lipid A species that functionally interact with both toll-like receptors 2 and 4. Infect Immun 72 5041 5051
37. SmithMFJrMitchellALiGDingSFitzmauriceAM 2003 Toll-like receptor (TLR) 2 and TLR5, but not TLR4, are required for Helicobacter pylori-induced NF-kappa B activation and chemokine expression by epithelial cells. J Biol Chem 278 32552 32560
38. YokotaSOhnishiTMuroiMTanamotoKFujiiN 2007 Highly-purified Helicobacter pylori LPS preparations induce weak inflammatory reactions and utilize Toll-like receptor 2 complex but not Toll-like receptor 4 complex. FEMS Immunol Med Mic 51 140 148
39. HirschfeldMMaYWeisJHVogelSNWeisJJ 2000 Cutting edge: repurification of lipopolysaccharide eliminates signaling through both human and murine toll-like receptor 2. J Immunol 165 618 622
40. ParkBSSongDHKimHMChoiBSLeeH 2009 The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature 458 1191 1195
41. QureshiNTakayamaKKurtzR 1991 Diphosphoryl lipid A obtained from the nontoxic lipopolysaccharide of Rhodopseudomonas sphaeroides is an endotoxin antagonist in mice. Infect immun 59 441 444
42. SmithSMMoranAPDugganSPAhmedSEMohamedAS 2011 Tribbles 3: a novel regulator of TLR2-mediated signaling in response to Helicobacter pylori lipopolysaccharide. J Immunol 186 2462 2471
43. SanticMAl-KhodorSAbu KwaikY 2010 Cell biology and molecular ecology of Francisella tularensis. Cell Microbiol 12 129 139
44. CullenTWTrentMS 2010 A link between the assembly of flagella and lipooligosaccharide of the Gram-negative bacterium Campylobacter jejuni. Proc Natl Acad Sci U S A 107 5160 5165
45. IngramBOMasoudiARaetzCR 2010 Escherichia coli mutants that synthesize dephosphorylated lipid A molecules. Biochem 49 8325 8337
46. LoppnowHBradeHDurrbaumIDinarelloCAKusumotoS 1989 IL-1 induction-capacity of defined lipopolysaccharide partial structures. J Immunol 142 3229 3238
47. QureshiNMascagniPRibiETakayamaK 1985 Monophosphoryl lipid A obtained from lipopolysaccharides of Salmonella minnesota R595. Purification of the dimethyl derivative by high performance liquid chromatography and complete structural determination. J Biol Chem 260 5271 5278
48. QureshiNTakayamaKRibiE 1982 Purification and structural determination of nontoxic lipid A obtained from the lipopolysaccharide of Salmonella typhimurium. J Biol Chem 257 11808 11815
49. KarbarzMJSixDARaetzCR 2009 Purification and characterization of the lipid A 1-phosphatase LpxE of Rhizobium leguminosarum. J Biol Chem 284 414 425
50. KundiM 2007 New hepatitis B vaccine formulated with an improved adjuvant system. Expert Rev Vaccines 6 133 140
51. BaldridgeJRMcGowanPEvansJTCluffCMossmanS 2004 Taking a Toll on human disease: Toll-like receptor 4 agonists as vaccine adjuvants and monotherapeutic agents. Expert Opin Biol Ther 4 1129 1138
52. JenksPJChevalierCEcobichonCLabigneA 2001 Identification of nonessential Helicobacter pylori genes using random mutagenesis and loop amplification. Res Microbiol 152 725 734
53. SkouloubrisSLabigneADe ReuseH 1997 Identification and characterization of an aliphatic amidase in Helicobacter pylori. Mol Microbiol 25 989 998
54. HaasRMeyerTFvan PuttenJP 1993 Aflagellated mutants of Helicobacter pylori generated by genetic transformation of naturally competent strains using transposon shuttle mutagenesis. Mol Microbiol 8 753 760
55. SmeetsLCBijlsmaJJBoomkensSYVandenbroucke-GraulsCMKustersJG 2000 comH, a novel gene essential for natural transformation of Helicobacter pylori. J Bacteriol 182 3948 3954
56. SmithPKKrohnRIHermansonGTMalliaAKGartnerFH 1985 Measurement of protein using bicinchoninic acid. Anal Biochem 150 76 85
57. WestphalOJannK 1965 Bacterial lipopolysaccharides. Extraction with phenol-water and further applications of the procedure. WhistlerRL Methods in carbohydrate chemistry New York Academic Press, Inc 83 91
58. HancockREW Hancock Laboratory Methods: Modified MIC Method for Cationic Antimicrobial Peptides Vancouvor, Canada Department of Microbiology and Immunology, University of British Columbia http://www.cmdr.ubc.ca/bobh/methods.htm. June 2009 ed
59. MoranAPLindnerBWalshEJ 1997 Structural characterization of the lipid A component of Helicobacter pylori rough - and smooth-form lipopolysaccharides. J Bacteriol 179 6453 6463
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2011 Číslo 12
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Controlling Viral Immuno-Inflammatory Lesions by Modulating Aryl Hydrocarbon Receptor Signaling
- Fungal Virulence and Development Is Regulated by Alternative Pre-mRNA 3′End Processing in
- Cryo Electron Tomography of Herpes Simplex Virus during Axonal Transport and Secondary Envelopment in Primary Neurons
- Epstein-Barr Virus Nuclear Antigen 3C Stabilizes Gemin3 to Block p53-mediated Apoptosis
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy