
Deficiency of a Niemann-Pick, Type C1-related Protein in Is Associated with Multiple Lipidoses and Increased Pathogenicity
Several proteins that play key roles in cholesterol synthesis, regulation, trafficking and signaling are united by sharing the phylogenetically conserved ‘sterol-sensing domain’ (SSD). The intracellular parasite Toxoplasma possesses at least one gene coding for a protein containing the canonical SSD. We investigated the role of this protein to provide information on lipid regulatory mechanisms in the parasite. The protein sequence predicts an uncharacterized Niemann-Pick, type C1-related protein (NPC1) with significant identity to human NPC1, and it contains many residues implicated in human NPC disease. We named this NPC1-related protein, TgNCR1. Mammalian NPC1 localizes to endo-lysosomes and promotes the movement of sterols and sphingolipids across the membranes of these organelles. Miscoding patient mutations in NPC1 cause overloading of these lipids in endo-lysosomes. TgNCR1, however, lacks endosomal targeting signals, and localizes to flattened vesicles beneath the plasma membrane of Toxoplasma. When expressed in mammalian NPC1 mutant cells and properly addressed to endo-lysosomes, TgNCR1 restores cholesterol and GM1 clearance from these organelles. To clarify the role of TgNCR1 in the parasite, we genetically disrupted NCR1; mutant parasites were viable. Quantitative lipidomic analyses on the ΔNCR1 strain reveal normal cholesterol levels but an overaccumulation of several species of cholesteryl esters, sphingomyelins and ceramides. ΔNCR1 parasites are also characterized by abundant storage lipid bodies and long membranous tubules derived from their parasitophorous vacuoles. Interestingly, these mutants can generate multiple daughters per single mother cell at high frequencies, allowing fast replication in vitro, and they are slightly more virulent in mice than the parental strain. These data suggest that the ΔNCR1 strain has lost the ability to control the intracellular levels of several lipids, which subsequently results in the stimulation of lipid storage, membrane biosynthesis and parasite division. Based on these observations, we ascribe a role for TgNCR1 in lipid homeostasis in Toxoplasma.
Published in the journal:
. PLoS Pathog 7(12): e32767. doi:10.1371/journal.ppat.1002410
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1002410
Summary
Several proteins that play key roles in cholesterol synthesis, regulation, trafficking and signaling are united by sharing the phylogenetically conserved ‘sterol-sensing domain’ (SSD). The intracellular parasite Toxoplasma possesses at least one gene coding for a protein containing the canonical SSD. We investigated the role of this protein to provide information on lipid regulatory mechanisms in the parasite. The protein sequence predicts an uncharacterized Niemann-Pick, type C1-related protein (NPC1) with significant identity to human NPC1, and it contains many residues implicated in human NPC disease. We named this NPC1-related protein, TgNCR1. Mammalian NPC1 localizes to endo-lysosomes and promotes the movement of sterols and sphingolipids across the membranes of these organelles. Miscoding patient mutations in NPC1 cause overloading of these lipids in endo-lysosomes. TgNCR1, however, lacks endosomal targeting signals, and localizes to flattened vesicles beneath the plasma membrane of Toxoplasma. When expressed in mammalian NPC1 mutant cells and properly addressed to endo-lysosomes, TgNCR1 restores cholesterol and GM1 clearance from these organelles. To clarify the role of TgNCR1 in the parasite, we genetically disrupted NCR1; mutant parasites were viable. Quantitative lipidomic analyses on the ΔNCR1 strain reveal normal cholesterol levels but an overaccumulation of several species of cholesteryl esters, sphingomyelins and ceramides. ΔNCR1 parasites are also characterized by abundant storage lipid bodies and long membranous tubules derived from their parasitophorous vacuoles. Interestingly, these mutants can generate multiple daughters per single mother cell at high frequencies, allowing fast replication in vitro, and they are slightly more virulent in mice than the parental strain. These data suggest that the ΔNCR1 strain has lost the ability to control the intracellular levels of several lipids, which subsequently results in the stimulation of lipid storage, membrane biosynthesis and parasite division. Based on these observations, we ascribe a role for TgNCR1 in lipid homeostasis in Toxoplasma.
Introduction
Toxoplasma gondii is an obligate intracellular parasite that resides in a unique vacuole formed in the cytoplasm of mammalian cells during invasion. The parasitophorous vacuole (PV) of Toxoplasma protects the parasite from hostile cytosolic innate immune-surveillance pathways and potent inflammatory signaling cascades. Although separated from the nutrient-rich cytosol by the PV membrane, the parasite has nevertheless evolved efficient strategies to co-opt multiple host cellular pathways and host organelles, to acquire essential nutrients and fuel its growth. The parasite expresses many surface transporters that mediate the internalization of host molecules [1]. T. gondii contains large amounts of cholesterol that it scavenges from plasma low-density lipoproteins (LDL) after processing in host endocytic compartments [2]. Interference with LDL endocytosis or cholesterol egress from host lysosomes arrests parasite development. We demonstrated that Toxoplasma can sequester nutrient-filled host lysosomes within invaginations of the PV membrane, which allows access to cholesterol supplied by the host endocytic network [3]. Cholesterol incorporation into the parasite is abolished after treatment with various proteases [4], and we have recently identified a transport system of cholesterol to the parasite involving parasite ABCG proteins [5]. Although much is known about host cholesterol delivery to Toxoplasma, very little is known about the regulatory mechanisms and trafficking routes of cholesterol (and other lipids) within the parasite. We have reported a role for a D-bifunctional protein containing two sterol-carrier protein-2 domains in promoting the circulation of phospholipids, cholesterol and fatty acids between parasite organelles and the plasma membrane in T. gondii [6].
In higher organisms, intracellular cholesterol transport is a fundamental process required for cell division, growth and differentiation. The distribution of cholesterol in different subcellular compartments is maintained by a combination of vesicle-mediated interorganelle transport and protein-mediated monomeric transfer through the aqueous cytosol [7], [8]. Among the cholesterol-binding proteins, the Niemann-Pick C (NPC) proteins, NPC1 and NPC2, are required for cholesterol export from endocytic organelles [9]. Loss of function of either of these proteins causes the sequestration of LDL-derived cholesterol and other lipids in these organelles and leads to a progressive neurodegenerative disorder: the NPC disease [10]. NPC1 is a multispanning transmembrane protein residing predominantly in the limiting membrane of late endosomes/lysosomes while NPC2 is a soluble lysosomal protein [11], [12]. Both proteins operate in the trafficking of cholesterol in the endo-lysosomal vesicle system; cholesterol liberated from LDL is first bound to NPC2 that then hands off the lipid to NPC1 that expels cholesterol out of the lysosomal compartment [13], [14]. NPC2 has a hydrophobic interior containing small cavities that can accommodate the steroid core while NPC1 has a high affinity binding domain to cholesterol at the N-terminus, called the sterol-binding domain (SBD). In addition, NPC1 has five to six transmembrane regions constituting the putative ‘sterol-sensing domain’ (SSD) that is common to many proteins that have key roles in cholesterol homeostasis or cholesterol-linked signaling [15]. However, while several potential interaction surfaces of the NPC proteins have been identified, the molecular mechanism of NPC interaction to transfer cholesterol remains still elusive.
NPC1 and NPC2 are conserved throughout much of eukaryotic evolution [16]. An authentic NPC1 ortholog appears in fungi, worms, insects, slime molds, plants and all mammals. Within the genome of most multicellular organisms, the NPC1 gene is represented at least twice. The retention of NPC1 genes throughout eukaryotic evolution permits the identification of conserved sequence motifs that include the SSD [15]. In addition to NPC1, 3-hydroxy-3-methylglutaryl CoA reductase (HMGCoA reductase, the key enzyme of the mevalonate pathway producing sterols), SCAP [the sterol regulatory element-binding protein (SREBP)-cleavage activating protein] (a regulator of the sterol-dependent transcription of cholesterol biosynthetic genes), 7-dehydrocholesterol reductase (an enzyme involved in cholesterol biosynthesis), Patched (a tumor suppressor involved in the signal transduction cascade mediated by the cholesterol-modified morphogen Hedgehog), Dispatched (a protein that facilitates the secretion of Hedgehog) and the Patched-related protein (a protein closely related in sequence and predicted topology to Patched) share a SSD. Mutations in the SSD of these proteins in mammalian cells render insensitivity not only to sterols but also to other lipids such as oxysterols, fatty acids, sphingolipids and phospholipids, suggesting that the SSD is a motif for membrane anchorage that can sense and respond to various agents that perturb membranes [17]–[20]. In yeast, a dominant mutation in the putative SSD of NCR1, a homolog of NPC1, confers resistance to inhibitors of inositophosphorylceramide and accumulation of complex sphingolipids, but no defect in sterol metabolism, pointing to a primary role of yeast NCR1 in sphingolipid recycling instead of sterol homeostasis [21].
To gain more insight into the key cellular pathways that govern lipids, e.g., cholesterol movement within T. gondii, we have characterized a parasite protein harboring a canonical SSD. This protein is a close relative of human NPC1, and we named it TgNCR1. When targeted to mammalian endo-lysosomes, TgNCR1 restores lipid trafficking from these organelles in mammalian NPC1 mutant cells. We generated a parasite cell line lacking NPC1 that is viable. The mutant parasites exhibit perturbations in their content in lipids, e.g. cholesteryl esters and sphingolipids, but not in free cholesterol. These alterations are reflected by the amassing of large lipid bodies in the parasite cytoplasm, and the stimulation of membrane biosynthesis and parasite replication. This suggests the involvement of TgNCR1 in monitoring the status of various lipids in Toxoplasma, a regulatory function likely important for proper parasite growth.
Results
Cloning and molecular characterization of a Toxoplasma protein that harbors an archetypal ‘sterol sensing-like domain’ and shares identity with human NPC1
To identify parasite proteins involved in cholesterol homeostasis in T. gondii, we searched for sequence homology to the sterol-sensing domain (SSD) in the Toxoplasma genome database (ToxoDB.org). A gene coding for a transmembrane polypeptide containing a sterol-sensing-like domain, annotated ‘Patched transmembrane domain-containing protein’ was retrieved (TGME49_090870; location on chromosome IX). Another gene with the same annotation was also present but its expression level was predicted to be very low (TGME49_120500; location on chromosome IV). As expected, no genes with homologous sequences to sterol biosynthetic enzymes, HMGCoA reductase and 7-dehydrocholesterol reductase, could be identified from the genomic database of T. gondii. The regulatory SREBP-SCAP machinery and the Patched/Hedgehog system are also missing from the parasite genome. The ORF of TGME49_090870 was amplified by PCR from a sporozoite cDNA library; it is 3,534 nucleotides long, consists of 6 exons and encodes a polypeptide of 1,178 amino acids, predicting a protein of 130.6-kDa (Figure S1). A parallel search of the Eukaryotic Pathogens Database Resources (EuPathDB.org) for sequence homology to TGME49_090870 using Jalview 2.6.1 [22] revealed the presence of one or two versions of NPC1-related protein in several apicomplexan parasites including Neospora caninum, Cryptosporidium sp. and Plasmodium sp. but no NPC1 homolog in any species of Theileria or Babesia (Figure S2).
Analysis of the deduced amino acid sequence of the sterol sensing-like domain of TGME49_090870 reveals high similarity to the SSD of the members of the cholesterol-sensing protein family (Figure 1A). The parasite sterol sensing-like domain encompasses 180 amino acids and is organized into five membrane spanning α-helices (Figure S3). This domain shares 31% identity and 51% similarity with the SSD of human NPC1. Interestingly, the sterol sensing-like domain in the Toxoplasma protein contains many residues implicated in the human NPC disease [23]–[26]. Of the miscoding mutations identified in the SSD of human NPC1 that are responsible for severe biochemical defects in patients, seven amino acids (70%) are identical between the parasite protein and human NPC1 (Y-634, G-660, G-673, P-691, D-700, D-786 and R-789). Figure 1A highlights the SSD containing a number of conserved amino acids that have been implicated in the NPC disease and that are present in the Toxoplasma protein.
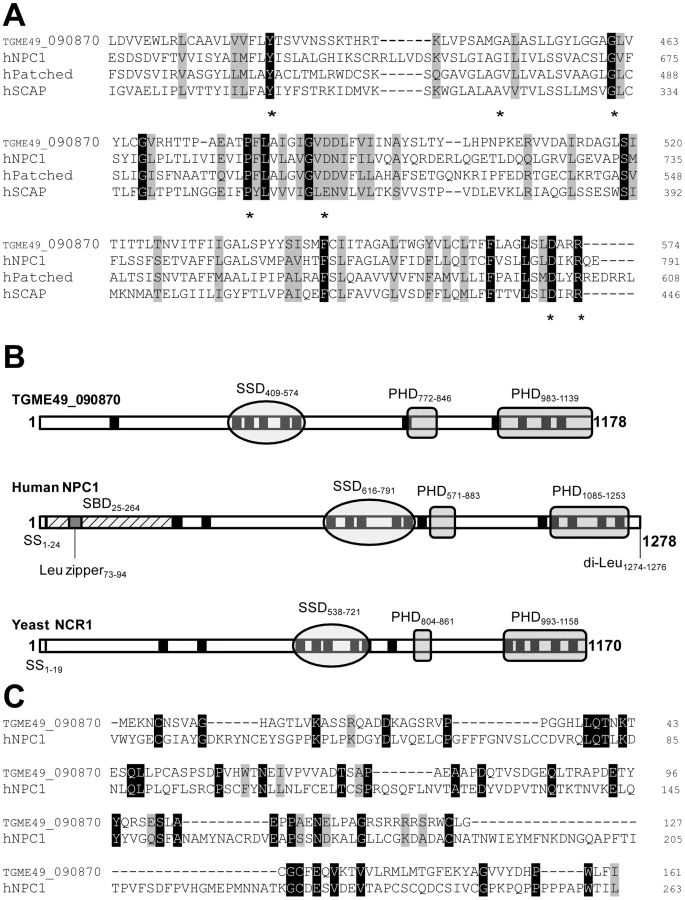
In addition to a putative SSD, TGME49_090870 has multiple membrane spanning domains and two Patched Homology Domains (PHD) that typify the NPC family (Figure 1B). The parasite sequence does not possess endosomal targeting signals such as a C-terminal di-Leu motif [27], probably due to the lack of endocytic compartments in the parasite. The N-terminal sequence of human NPC1 harbors the sterol-binding domain (SBD) on luminal loop-1 that is known to interact with cholesterol and oxysterols with high affinity [28], [29]. Sequence alignment of the N-terminal ends of TGME49_090870 and human NPC1 encompassing the SBD reveals the presence of 35 identical residues (22%), which suggests that the N-terminus of the parasite protein may have potential sterol binding activity (Figure 1C). Moreover, several conserved Pro and Gly residues present in the parasite amino-terminal sequence are critical residues whose substitutions correspond to naturally occurring mutations in patients with NPC disease. At the protein level, the parasite full-length sequence has 37% identity and 61% similarity with human NPC1; 61 of the functionally important residues (59%) for the sterol transfer function are present, and 47 of those are identical to human NPC1. Based on these observations, we therefore renamed the protein, TgNCR1 (for NPC-related gene 1). A schematic representation of the topology of TgNCR1 and human NPC1 shown in Figure 2A highlights their structural similarities.

Expression of TgNCR1 corrects the cholesterol-trafficking defect in NPC1 mutant cells
To test the functional equivalence of TgNCR1 and mammalian NPC1, we planned to express TgNCR1 in a CHO cell line lacking functional NPC1 and examine the potential ability of the parasite protein to restore NPC1 activity in the mutant cells, namely the abatement of lipid accumulation in endo-lysosomes. However, as Toxoplasma does not possess an endocytic membranous system involved in the internalization of extracellular cholesterol, TgNCR1 does not contain any targeting information for endocytic organelles as found on human NPC1. We first analyzed the intracellular localization of TgNCR1 when ectopically expressed in mammalian cells. CHO cells were transfected with pHA-TgNCR1 and immunolabeled with antibodies against HA (Figure 2B). A fluorescence pattern corresponding to cortical and perinuclear ER was discernible. Expectedly, no staining was observed on endo-lysosomal compartments. We therefore decided to engineer a chimeric protein to correctly address TgNCR1 to endo-lysosomes in mammalian cells. Mammalian NPC1 localization to endo-lysosomes is mediated by several COOH-terminal motifs [26]. We constructed a hybrid protein in which the last 64 residues of human NPC1 that encompass most of the functional endosomal targeting motifs (hNPC11214-1278) were fused to TgNCR1 lacking its last 78 amino acids (TgNCR1Δ1101-1178) as schematized in Figure 2A. A myc tag was introduced at the N-terminus of the chimeric protein designated myc-TgNCR1-hNPC1. The expression construct was transfected into CHO cells for double immunofluorescence assays (IFA) using antibodies against myc and various markers of endosomes or lysosomes (Figure 2C). Results show that the TgNCR1-hNPC1 fusion protein largely localized to structures positively labeled for EEA1, LAMP1 or LysoTracker, indicating delivery of the exogenous protein to endocytic organelles.
We used the somatic 2-2 mutant of CHO cells as a model of NPC1 mutant cells. These cells are characterized by the excessive accumulation of lipids including cholesterol in endocytic compartments [30]. We confirmed this phenotype by staining the 2-2 mutant cells with filipin, a fluorescent compound that binds to the β-hydroxyl group of sterols, and data revealed enlarged, filipin-positive structures corresponding to perinuclear lysosomes (Figure S4A). Our ultrastructural observations of sterol-overloaded lysosomes in filipin-treated 2-2 mutant cells illustrated the corrugated aspect of membranes as a result of the formation of sterol-filipin complexes (Figure S4B). The 2-2 mutant cells were transfected with pmyc-TgNCR1-hNPC1 to assess sterol transport by filipin staining and fluorescence microscopy. We first confirmed the localization of the TgNCR1-hNPC1 fusion protein to perinuclear structures in 2-2 mutant cells (Figure 3A). Expression of the TgNCR1-hNPC1 fusion protein largely restored cholesterol clearance from endo-lysosomes (Figure 3B) as quantified by a ∼4-fold reduction in filipin intensity levels in endo-lysosomes of the transfected cells with TgNCR1-hNPC1 compared to mock-transfected cells (Figure 3C). Fluorescence levels in mutant cells expressing TgNCR1-hNPC1 were not significantly different from those associated with endo-lysosomes in wild-type cells (Figure 3C). Thin layer chromatography (TLC) analysis of a neutral lipid fraction of cellular extracts of TgNCR1-hNPC1-expressing mutant cells confirmed that the levels of free cholesterol have returned to normal (Figure 3D). No change has been observed in the levels of esters of cholesterol between wild-type cells, NPC1 mutant cells and mutant cells expressing TgNCR1-hNPC1 (Figure S5). The 2-2 mutant cells expressing TgNCR1 did not show any decrease in endo-lysosomal filipin staining, in accordance to the mislocalization of TgNCR1 in the ER. Negative controls for these experiments included mutant cells transfected with a truncated construct containing a short sequence of the C-terminus of TgNCR1 (from residues 978 to 1100) combined with the C-terminal end of hNPC1 containing the endosomal targeting motifs (Tr.TgNCR1-hNPC1). This chimeric construct localized both to endocytic compartments and the ER, and the filipin fluorescence levels in these cells were similar to those in TgNCR1-expressing cells.

The subcellular accumulation of sphingolipids, particularly gangliosides (e.g., GM1), is another characteristic of the NPC1 syndrome [31]. TLC analysis of an acidic lipid fraction from cellular extracts confirmed that these 2-2 mutant cells contained high levels of GM1, whereas in CHO wild-type cells the levels of this lipid were undetectable (Figure 3D). TgNCR1-hNPC1 expression in mutant cells caused an obvious decrease in the levels of GM1 compared to NPC1 mutant cells, and quantitative measurement indicated a reversion of the GM1 accumulation by ∼6-fold.
Altogether, these results suggest that, when provided with the correct localization motif for endocytic compartments, the TgNCR1constructs functions in a similar way as human NPC1 to promote LDL-cholesterol and GM1 release from endocytic organelles, which indicates a conserved activity of the two proteins.
TgNCR1 localizes to the inner membrane complex in the parasite
Our data showed that in mammalian cells, the parasite and human NPC1 are interchangeable with respect to LDL-cholesterol transport from endocytic compartments. However, Toxoplasma never internalizes host cholesterol into endo-lysosomes, which predicts a function for TgNCR1 unrelated to exogenous sterol transport. This raises an intriguing issue about the biochemical function of TgNCR1 in T. gondii. As the first step towards understanding the molecular function of TgNCR1 in the parasite, we decided to examine its intracellular localization. For this purpose, a recombinant peptide corresponding to residues 162 to 501 of TgNCR1 was produced in E. coli to generate polyclonal antibodies in rabbits (Figure S6). Using anti-TgNCR1162-501 antibodies, immunofluorescence of intracellular wild-type parasites showed a diffuse peripheral staining along the parasite body, which seemed to be excluded from the apical and basal extremities of the parasite (Figure S7). Despite several attempts to purify these antibodies, a high level of fluorescence background remained both on the parasites and in host cells, which precluded the use of these antibodies for in-depth morphological studies.
To circumvent this issue and have a better resolution of the TgNCR1 localization, we created a stable line of the parasite expressing, under the tubulin promoter, TgNCR1 with a HA tag at the N-terminus. Lysates of transgenic parasites were resolved in SDS-PAGE and immunoblot analysis using anti-HA antibodies, confirming the expression of exogenous HA-TgNCR1, which migrated as a single band at ∼140-kDa (Figure 4A). Immunofluorescence of intracellular HA-TgNCR1-expressing T. gondii showed a peripheral staining along the parasite body as observed on wild-type parasites labeled with anti-TgNCR1162-501 antibodies (Figure 4B). The apical and basal extremities of the parasite transfectants clearly excluded the staining, and this pattern is reminiscent of resident proteins of the inner membrane complex (IMC). EM observations reveal that the IMC is a continuous patchwork of flattened vesicular cisternae located beneath the plasma membrane and overlying the cytoskeletal network [32], [33]. The IMC arises from vesicles derived from the secretory pathway which flatten during parasite maturation to form large membranous sheets that envelop the parasite, leaving only a small gap at both parasite ends. TgNCR1 localization was further confirmed by immunoEM staining showing gold particles on the IMC (Figure 4C). Toxoplasma divides by endodyogeny, a mode of replication in which two daughter cells are produced within an intact mother parasite [34], [35]. A critical step in building daughter cells is the construction of their IMC, which forms a bud into which replicated organelles are packaged. Figure 4D reveals TgNCR1 labeling on the cone-shaped cistern forming nascent IMC, indicating the association of the protein with the daughter parasites at the onset of their formation. Immunogold staining confirmed an association of TgNCR1 with the IMC of nascent parasites (Figure 4E). Interestingly, the presence of gold particles was also visible on vesicles adjacent to the IMC (Figure 4F). Quantitative distribution of gold particles on various parasite compartments indicated that more than 80% of the gold staining was associated with both the IMC and vesicles, while the ER counted for 8% of the gold labeling density (Figure 4G). Finally to corroborate this IMC localization, we engineered two other constructs: TgNCR1, under the NTPase promoter, tagged at its N-terminus with the HA epitope and TgNCR1, under the tubulin promoter, tagged at its C-terminus with the HA epitope. Parasites were transiently transfected with each construct, and the original localization of TgNCR1 on the IMC was confirmed in these transgenic parasites expressing HA-TgNCR and TgNCR1-HA (Figure S8).

Parasites expressing dominant negative mutants of TgNCR1 have no striking phenotype
As the second step towards clarifying the role of TgNCR1 in the parasite, we expressed in the wild-type T. gondii four different dominant negative mutants derived from HA epitope-tagged TgNCR1: HA-TgNCR1D571N, HA-TgNCR1P913A, HA-TgNCR1I957T and HA-TgNCR1L1100V. These mutants are predicted to be dominant negative based on identified mutations in NPC1 patients [24]–[26]. When transiently transfected in the parasite for 16 h to 24 h, these constructs produced a signal that localized to the IMC/ER. The four dominant negative mutants were viable and gave no unusual phenotype, such as the abnormal accumulation of lipids based on TLC analysis, which precludes further functional studies (data not shown). It is possible that dominant negative mutants have lower expression levels than TgNCR1 wild-type protein, which renders the dominant-negative effect silent. Another possibility could be that the time-course for lipid accumulation takes longer than 24 h before giving a clear phenotype. While the expression level of the mutant proteins or the timing of the lipid accumulation phenotype may effect the lack of phenotype, it is also plausible that the amino acids mutated in the TgNCR1 sequence are not critical for its function in the parasite, which may be viewed as evidence that TgNCR1 plays distinct roles from human NPC1.
Depletion of NCR1 does not affect parasite replication
As the third approach to study the function of TgNCR1 in T. gondii, we focused on genetically disrupting NCR1 using a fusion PCR-based method to replace NCR1 in the parasite genome with a selectable HXGPRT marker. Clones were tested by PCR and Southern blotting for the absence of NCR1 and the presence of a single copy of the HXGPRT cassette (Figure 5 and Figure S9). Clones lacking NCR1 were obtained, demonstrating that TgNCR1 was not necessary for the in vitro propagation of T. gondii. In fibroblasts, the ΔNCR1 strain formed large vacuoles that associated with host organelles, e.g., mitochondrion, as observed for wild-type parasites (Figure S10). The knockout parasites were organized in rosettes. During normal endodyogeny, the cytoplasm of the mother parasite is equally distributed between the daughter cells; a cleavage furrow is initiated at the anterior pole and extends between the daughters throughout the mother cell, leaving only a residual body at the posterior end connecting the two daughters. The residual body contains mother cell components that are not incorporated into the daughter parasites, and this structure usually disappears after the completion of parasite division. Interestingly, we observed that the progeny of TgNCR1-deficient parasites staid mostly connected by their posterior ends to a common and enlarged residual body. Similar prominent residual bodies were also apparent in light microscope images. Disruption of TgNCR1 did not result in any obvious defect in parasite invasion or egress (data not shown).

The growth rate of the ΔNCR1 strain in fibroblasts was quantified by uracil incorporation assays. Compared to the parental strain the (ΔKu80ΔHXGPRT or ΔK80), the ΔNCR1 strain incorporated significantly higher amounts of uracil (1.4-fold increase) at 16 h, 24 h and 48 h p.i. (Figure 6A). To verify the specificity of the mutant phenotype, we genetically complemented the ΔNCR1 strain with TgNCR1. TgNCR1-complemented clones were verified by PCR analyses (Figure S11). Values of uracil incorporation were comparable in the complemented and parental strains, corresponding to 2,392±504 cpm and 2,693±134 cpm, respectively, which indicates that the uracil incorporation defect of the ΔNCR1 strain was due to the deletion of NCR1.

TgNCR1-deficient parasites are insensitive to excess cholesterol in the medium and contain normal amount of free cholesterol
We previously reported that Toxoplasma growth is directly dependent upon exogenously supplied cholesterol [2]. Mammalian NPC1 has a primordial function in exogenous sterol distribution throughout the cell [36]. TgNCR1-deficient parasites were then exposed to excess amounts of LDL-cholesterol to determine whether loss of NCR1 has an effect on parasite viability as a result of the potential accumulation or mislocalization of cholesterol within mutant parasites. Results show that the addition of excess LDL in the medium, i.e. 10-times more than normal medium (10% FBS), did not affect the intravacuolar development of TgNCR1-deficient parasites (Figure 6B). A ∼150% increase in replication rate was observed for the ΔNCR1 strain compared to the parental strain grown in excess LDL, which parallels the increased replication rate of the ΔNCR1 strain in normal medium. We showed previously that exposure of T. gondii to a medium depleted in LDL results in a slowdown of parasite development as smaller vacuoles were observed compared to parasites developing in complete medium [2]. When TgNCR1-deficient parasites were incubated in the absence of LDL, they grew slower than in medium containing 10% FBS, but still showed a growth advantage over the parental strain grown in LDL-free medium (Figure 6C). Thus, the ΔNCR1 strain displays no significant change in sensitivity to extracellular cholesterol compared to parental parasites. This may predict a primary role for TgNCR1 that is unrelated to cholesterol transport.
The NPC disease is characterized by lysosomal sequestration of endocytosed LDL-cholesterol, abnormal enrichment of unesterified cholesterol in trans-Golgi cisternae and anomalies in intracellular sterol trafficking [9]. We then monitored the cholesterol status in TgNCR1-deficient parasites compared to control parasites. In a first set of experiments, parasites were stained with filipin (Figure 7A). A fluorescence pattern was associated with the parasite's pellicle and apical rhoptries in both the ΔNCR1 and parental strains when cultivated in 10% FBS. When excess LDL was added to the medium, TgNCR1-deficient parasites did not show any major sites of cholesterol accumulation as is the case in NPC1-deficient mammalian cells. To validate our microscopic observations, we measured the levels of free cholesterol molecules, at monomeric and oligomeric states by mass spectrometry (Figure 7B). Our results established that the overall concentrations of monomeric, dimeric and trimeric cholesterol in ΔNCR1 were similar to the parental strain.

TgNCR1-deficient parasites contain numerous lipid bodies in their cytoplasm and accumulate several species of cholesteryl esters
To examine whether the lack of NCR1 might impact the metabolism of lipids other than cholesterol, TgNCR1-deficient and control parasites were stained with Nile Red. This dye fluoresces when trapped inside lipid bodies allowing the examination of the cytosolic stores of neutral lipids in cells. Data show the presence of abundant lipid bodies in the ΔNCR1 strain, which were greater in size and number than those found in the parental strain (Figure 8A). The ΔNCR1 parasites could produce up to nine lipid bodies, with an average of five lipid bodies per cell whereas control parasites contained two lipid bodies per cell on average (Figure 8B). We previously described that Toxoplasma can store both esters of cholesterol [37] and triglycerides [38] in lipid bodies. The nature of neutral lipids accumulated in TgNCR1-deficient parasites was then investigated by mass spectrometry. Quantitative analysis of the cholesteryl ester species present in wild-type Toxoplasma was performed in detail (Table 1). As predicted by the detection of acyl-CoA:cholesterol acyltransferase (ACAT) activities in the parasite, cholesteryl esters were abundant in the parasite. Of the 21 molecular species detected in Toxoplasma, cholesteryl oleate C18 : 1 (42%) and palmitate C16 : 0 (26%) were the main esters as is the case in mammalian cells but the parasite also had uniquely large amounts of cholesteryl eicosanoate C20 : 1 (7%). In addition, the parasite contained to a lesser extent, cholesterol palmitoleate C16 : 1, stearate C18 : 0, linoleate C18 : 2, arachidonate C20 : 4 and some polyunsaturated C22 fatty acids. The other cholesteryl ester fatty acid species shown in Table 1 represented 3.5% of the total species. Interestingly, TgNCR1-deficient parasites showed an overall increase in the cholesteryl ester content, with the greatest accumulation of cholesteryl linoleate, arachidonate, C20 : 3 and some very-long-chain polyunsaturated fatty acids (Figure 9).



In parallel, we examined the triglyceride content of the parasites (Figure 10). Toxoplasma contained pools of triglycerides with either palmitate:oleate:stearate or palmitate:oleate:linoleate, and these lipid species were also detected in TgNCR1-deficient parasites at similar levels. These results suggest that lipid bodies in the ΔNCR1 parasites are particularly enriched in cholesterol esters, as a result of an overproduction of these lipids and/or a dysregulation of the cholesterol cycle.

TgNCR1-deficient parasites accumulate several species of very-long-chain fatty acid sphingolipids
The NPC mutation has been also shown to impact the levels of sphingolipids [21], [39]. We then determined the status of sphingolipids in ΔNCR1 parasites. A previous quantitative study reported that sphingomyelins and ceramides represent 1.5 and 4.7% of total polar lipids respectively, in T. gondii [40]. We used quantitative mass spectrometry analyses to identify 10 different species of sphingomyelins (Table 2) and 11 species of ceramides (Table 3) in Toxoplasma controls. The main molecular species of sphingomyelins contained residues of arachidic acid C20 : 0 (44%), erucic acid C22 : 1 (28%) and stearic acid C18 : 0 (13%). Sphingomyelin species with eicosenoic acid C20 : 1 and behenic acid C22 : 0 were detected though in lesser abundance. Other species represented about 2% of the total sphingomyelins, with palmitic acid corresponding to 1%. The main species of ceramides in the parasite was palmitic acid (83%). Ceramides with stearic acid totaled 8% while the other ceramide species represented about 10%, with oleic acid less than 1%. Overall, the profiles of sphingomyelins and ceramides in Toxoplasma were quite different from those in mammalian cells in which oleic acid and palmitic acid are prominently represented. The sphingolipid profiles of TgNCR1-deficient parasites showed significant increases in both sphingomyelin and ceramide species containing lignoceric acid C24 : 0 and selacholeic acid C24 : 1 (Figures 11 and 12). In TgNCR1-deficient parasites, the ceramide C22 : 0 and the sphingomyelin containing palmitic acid and palmitoleic acid were also more abundant compared to the parental strain. Finally, to provide a complete overview of the lipid profiles in the ΔNCR1 strain, the status of phospholipids other than sphingomyelins was analyzed by quantitative mass spectrometry. It has been previously observed that the parasite phospholipid class distribution is significantly different from that of mammalian cells [40], [41]. Phosphatidylcholine is the most prevalent lipid, accounting for 61–75% of total phospholipids. The next most abundant lipids are phosphatidylethanolamine (10–16%), phosphatidylinositol (3.6–7.5%) and phosphatidylserine (5.6–6%). Data showed no difference in the levels of phosphatidylcholine and phosphatidylinositol between the ΔNCR1 and parental strains (Table S1). Only very few species of phosphatidylethanolamine and phosphatidylserine were significantly higher (up to a 3-fold increase) in TgNCR1-deficient parasites as compared to controls. Altogether, these data suggest that the main role of TgNCR1 in the parasite is in regulating the homeostasis of many different lipids.




TgNCR1-deficient parasites shift to the endopolygeny replication mode
We next examined the morphology of the TgNCR1-deficient parasites at the ultrastructural level. Astonishingly, nearly all EM sections demonstrated profiles of dividing parasites, indicating that these knockout parasites relentlessly underwent endodyogeny (Figure S10). While images capturing parasite endodyogeny can also be observed in wild-type parasites, it is at a much lower frequency. A representative PV containing many dividing TgNCR1-deficient parasites is shown in panel A of Figure 13, in which nascent daughter cells were visible within the cytoplasm of each mother cell. The sequential steps in parasite endodyogeny were then analyzed in detail (Figure S12). At the onset of daughter cell formation, the cytoplasm of the mother cell contained one or two thin, and either elongated (Figure S12, panel a) or horseshoe-shaped structures (Figure S12, panel b) that had the same electron-density as the IMC beneath the mother plasma membrane (Figure S12, panel a). These tubules likely represent the initial pieces of the new IMC that will assemble to provide the cytoskeletal scaffolding of developing daughter cells. As endodyogeny progresses, the nascent IMC extended in size and enveloped the dividing organelles, e.g., nucleus (Figure S12, panel c), Golgi and apicoplast (Figure S12, panel d), and the organelles produced de novo, e.g., rhoptries (Figure S12, panel Bd-e) and micronemes (Figure S12, panel e). The plasmalemma then invaginated around the daughters. Concomitant to the emergence of daughter parasites with an intact pellicle, the mother mitochondrial network moved to the posterior end of nascent parasites, bifurcated and entered the daughter scaffolds (Figure S12, panels f and g). These EM observations did not reveal any obvious defects in organellar partioning in daughters lacking NCR1, based on studies tracking fluorescent organelles in wild-type parasites by time-lapse microscopy. This suggests that the process of division of the ΔNCR1 strain occurs in a similar fashion as found for wild-type parasites.

The frequency of multiple daughter formation is usually rare in wild-type parasites and is typically in the range of 0.5 to 3% [42], [43]. Interestingly, we observed a significant high number of cases in which more than two daughters were formed within a single mother, as illustrated by immunofluorescence microscopy using the protein marker IMC1 to observe the IMC of nascent daughters within the mother cells (Figure 13B). We could observe the different steps of the construction of the multiple daughters per mother cell (Figure 13C). The budding seemed synchronized, and the time for multiple daughter formation did not reveal and delay compared to parasites undergoing normal binary division in the same vacuole. Transmission EM analysis also confirmed the presence of several individualized nuclei encircled by the IMC as a sign of assembly of new parasites (Figure 13D). To quantify this growth defect, we stained the ΔNCR1 and parental strain with DAPI and IMC1 and counted the PV containing parasites producing more than 2 daughters per mother (Figure 13E). Data show a dramatic increase in the number of endopolygeny events in the mutants with some mother parasites assembling as many as five daughters at the same time.
TgNCR1-deficient parasites have an unusual cell cycle with a long S phase and short G1 phase
Toxoplasma tachyzoite replication differs from the classic animal cell cycle as they divide using a three-phase cycle (G2 may be short or missing) with the G1 interphase period comprising 40–60% of the parasite's doubling time [reviewed in 35]. S phase distributions in T. gondii are also peculiar, with late S phase parasites (1.8 N) being more numerous than early S phase parasites. Internal daughter budding appears to initiate in late S phase. We performed flow cytometry analysis on the ΔNCR1 strain to examine the cell cycle profiles of these mutants, in comparison with control parasites and VERO cells from which these parasites have been isolated (Figure 14). Data show that the percentages of parasites in G2/M phase were comparable between the mutant and parental strains and represented consistently a fraction between ∼10–12% of the total population. However, the average percentage of TgNCR1-deficient-parasites in S phase was significantly higher than for control parasites (about 61% vs. 41%). Consequently, the population of parasite mutants during the G1 phase corresponded to ∼24% only.

The phenotype of TgNCR1-deficient parasites is associated with long PV membranous extensions
We next wanted to focus our attention to the PV of the ΔNCR1 strain to detect any peculiar morphological changes compared to the parental strain. A feature commonly shared by many intravacuolar pathogens is the formation of membranous tubules or fibers that extend away from their vacuole to facilitate their replication [44]–[46]. These filamentous structures contain many pathogen-derived proteins. Toxoplasma also forms long tubules extending from the PV membrane containing proteins released from dense granules, e.g., GRA7, GRA10, GRA14 proteins [3], [45] (Figure S13). We then examined the morphology of the PV membrane, with special emphasis on the tubules emanating from this membrane by fluorescence microscopy.
Fibroblasts were infected with the ΔNCR1 or parental strain for 36 h for double IFA to detect GRA3 and GRA7 on the PV membrane (Figure 15A). Both strains form long extensions originating from the PV. In multi-infected cells, some of these tubules were connecting two PV. The fluorescent signal for GRA3 and GRA7 appeared much brighter and homogenous for the extensions of the PV containing TgNCR1-deficient parasites than those generated by the parental strain, suggestive of more robust extensions. Morphometric quantification of the diameter and length of PV membranous extensions confirmed that they were significantly thicker and longer for TgNCR1-deficient Toxoplasma (Figure 15B).

Interestingly, we observed that several PV of TgNCR1-deficient parasites, although located to different host cells, were connected together by these PV membranous extensions (Figure 16A). In some occasions, two PV of the parental strain residing in adjacent cells were also seen joined together by PV tubules, suggesting that this phenotype plays a physiological role during parasite development. Close observations of the PV of TgNCR1-deficient parasites revealed that the closer two PV were to each other, the thicker the connecting tubules were (Figure 16B, panels a to c). A plausible scenario would be that the tubular structures bridging two PV may be generated consequently to the split of a PV into two vacuoles that stay connected by their PV membrane which elongates as the two PV separate from each other. Finally, we observed that these membranous connections between PV were maintained after host cell division, and dramatically increased in length as the two PV move apart (Figure 16B, panels d to e). The tubules originated from the PV membrane are likely made of lipids. The abundant and thick membranous extensions formed by TgNCR1-deficient parasites may suggest that these mutants may have an unusual membrane lipid biogenesis.

Disruption of NCR1 results in increase in virulence in mice
It still remains unclear why TgNCR1-deficient parasites can generate more than two daughters from one mother cell. Nevertheless, if mutant daughter cells are viable and infective, the increase in their production may contribute to the more rapid expansion of the knockout population in vivo.The fast development of TgNCR1-deficient parasites in vitro prompted us to examine whether the ΔNCR1 strain is particularly virulent in mice. To examine the role of TgNCR1 in Toxoplasma infectivity, mice were infected with either the ΔNCR1 or parental strain after intraperitoneal injection, and mice survival was monitored daily. All mice infected with the parental strain died by day 10 (Figure 17). By comparison, mice parasitized with TgNCR1-deficient T. gondii showed higher susceptibility to infection as all mice systematically died one day earlier, as monitored in three independent trials. This suggests that TgNCR1-deficient parasites are slightly more virulent than control parasites.

Discussion
As a starting point for studies on lipid transport and regulatory processes within the parasite, the aim of this work was to investigate the role of a unique SSD-containing protein, TgNCR1, expressed by T. gondii. The sequence of TgNCR1 predicts an uncharacterized Niemann-Pick, type C1-related protein with significant identity (37%) to human NPC1. The TgNCR1 sequence predicts a 1178-residue protein (131-kDa) with four potential N-glycosylation sites, and regions homologous to the Patched Homology Domain (PHD), the SSD and the sterol-binding domain (SBD) [5]. Hydropathy plots and topology predictions of TgNCR1 and human NPC1 suggest a striking conservation of the secondary structure between the two proteins, in that both have 11 to 12 transmembrane domains whose spacing is conserved. Interestingly, a NPC1 homolog is present in several species of apicomplexan parasites and their predicted sequences are closely related to that of TgNCR1. Mutations in human NPC1 that cause NPC disease lay in residues conserved in TgNCR1. When expressed in mammalian cells with impaired NPC1 activity, TgNCR1 can restore the sterol and GM1 trafficking. Overall, despite the evolutionary distance of mammals and protozoa, human NPC1 and TgNCR1 are very functionally interchangeable at least for LDL-cholesterol and sphingolipid movement.
The ability of TgNCR1 to complement a mutant human NPC1 is a priori intriguing given that the core problem of the NPC disease is an accumulation of cholesterol and other lipids in certain endocytic organelles [9], [47]. T. gondii does not have a process of receptor-mediated uptake of sterols nor contains degradative lysosomes. In fact, though T. gondii is auxotrophic for cholesterol derived from plasma LDL [2], the parasite internalizes this lipid by using membrane translocators [5]. Nevertheless, the detection of cholesterol transport activity in NPC1-deficient cells expressing TgNCR1 implies that the parasite protein can bind cholesterol, perhaps by its putative SBD, and is maybe recognized by necessary accessory proteins in mammalian cells, e.g., NPC2 for cholesterol egress from endo-lysosomes. Though NPC2 proteins are highly conserved across genera that have clear NPC1 orthologs, a gene encoding a NPC2-like protein could not be identified in T. gondii. All of these intriguing observations predict that TgNCR1 would function in an unconventional way in Toxoplasma, likely distinct from a role in exogenous cholesterol transport. To explore the function of TgNCR1 in Toxoplasma, a parasite strain lacking the NCR1 gene was generated. ΔNCR1 progeny are viable, indicating that NCR1 is not required for replication. Altering the environmental cholesterol conditions by either starving Toxoplasma-infected cells for LDL or feeding infected cells with excess LDL, did not disturb the growth of knockout parasites differently than the parental population. The ΔNCR1 strain is not overloaded with free cholesterol, as are mutant mammalian NPC1 cells but instead abnormally accumulates sphingolipids and cholesteryl esters, and to a lesser extent, phosphatidylethanolamine and phosphatidylserine. Since NPC1 proteins universally function as lipid transporters, the observed accumulation of lipids in the ΔNPC1 strain is most likely the primary effect resulting from the loss of NCR1. Consequently, TgNCR1 seems to be involved in controlling the intracellular levels of specific lipids in the parasite. The yeast NPC homolog (NCR1), which is localized to the vacuole, functions in sphingolipid recycling and not in sterol transport even though yeast NCR1 complements the loss of NPC1 in mammalian cells [21]. NPC1 proteins also bear intriguing similarities to the Resistance-Nodulation Division (RND) system of bacterial permeases, which transport acriflavine and fatty acids [48]. Heterologous expression of RND from Escherichia coli in human NPC1-deficient fibroblasts results in the accumulation of acriflavine and fatty acids, but not the restoration of the NPC1 sterol accumulation phenotype. As Toxoplasma, yeast and bacteria do not internalize sterols from their external environment by endocytosis, our study here widen the concept proposed by SL Sturley [16] that in cells that do not ingest exogenous cholesterol in endocytic organelles, the NPC1 proteins do not primarily function as a cholesterol sensor/transporter, but as a regulator of the movement of other lipophilic substrates including sphingolipids. In this case, the role of NPC1 in higher eukaryotic cells as a cholesterol transporter might have arisen during evolution concomitantly to the development of receptor-mediated pathways for sterol uptake. Nevertheless, it remains possible that the accumulation of cholesterol in mammalian late endosomes may not be the primary cause of NPC disease but a secondary effect as a consequence of impaired transport of other metabolites aside from sterols.
The abnormal lipid accumulation resulting from the loss of NCR1 in T. gondii leads to secondary defects including the stimulation of daughter formation, virulence, membrane biosynthesis and lipid body biogenesis. Mutants produce more progeny parasites than controls. The accumulation of mutant parasites in S phase seems attributable to the absence of NPC1 since this phenomenon has not been observed in the parental strain or wild-type population of T. gondii. This feature may be linked to the propensity of TgNCR1-deficient parasites to form multibuds with multiple nuclear divisions between rounds of S and G2/M phase. During endopolygeny, the parasites reinitiate DNA synthesis bypassing G1 checkpoints but then re-coordinate the cell cycle as they reach the next mitosis. By analogy, mammalian tumor cells with high proliferation rate, e.g., HeLa cells are also characterized by cell cycle alterations with the observed accumulation of cells in S phase and reduction of cells in G1 phase. Such a deregulated cell cycle in cancer cells is mainly due to defective genome-integrity checkpoints, leading to abnormal DNA content. Several cell cycle checkpoints with controllers of cyclin-Cdk activity (G1, START (G1/S) checkpoints and mitotic controls) have been characterized in Toxoplasma [35]. However, the synchronization of multibud formation in the ΔNCR1 strain precludes the idea that these parasites escape from the regulatory systems of checkpoints, which complete mitosis in synchrony and control the quality of progeny.
TgNCR1 localizes to the IMC, a unique cortical system of membrane cisternae connected to the plasma membrane. As in mammalian cells, most of the parasite's cholesterol is concentrated at the plasma membrane [49]. However, it has also been documented that the IMC contains cholesterol-rich domains in which the parasite myosin motor machinery is immobilized [50]. These domains are critical for parasite gliding motility and attachment to a substrate. The location of TgNCR1 to the parasite IMC underlines the possibility that TgNCR1 may act as a catalyst to assemble cholesterol-rich. The highly dynamic properties of the IMC during parasite replication could facilitate the trafficking and mobilization of lipids necessary for daughter cell formation. Disruption of NCR1 results in endopolygeny. This suggests that the function of TgNCR1 may be relevant in some way for coordinating normal progeny number and assembly, for example by delivering appropriate amount of lipids for membrane/organelle biosynthesis. It has been reported that genetic factors or local conditions increase the tendency to form multiple daughters [42]. For example, the ISP1 protein, which is located to the apical portion of the IMC, is involved in the targeting of many proteins to the IMC and seems to play a role in synchronizing endodyogeny [51]. Interestingly, T. gondii lacking ISP1 also assemble more than two siblings per mother cell. Observations based on TgNCR1 and ISP1 mutants suggest an important contribution of the IMC to the regulation of parasite division pathways, and thus to the control of infectivity.
Deletion of TgNCR1 from parasites leads to an increase in virulence as mice infected by the mutant strain succumb slightly faster than mice infected with control parasites. The fitness of TgNCR1-deficient parasites in vivo may be due to their higher propensity to produce multiple daughters, and therefore to disseminate faster within the host. Alternatively, the high content of sphingolipids inTgNCR1-deficient parasites may lead to increased virulence for several reasons: i) these lipids may play an important role to promote the parasite's intracellular development, e.g., by the induction of long PV membranous extensions that facilitate the parasite colonization of host cells; ii) the sphingolipids may act as immunomodulators during the progression of the infection; and iii) these lipids may stimulate the expression of virulence factors. First, the excess sphingolipids may promote intracellular parasite development as in mammalian cells it is known that sphingolipids are implicated in the regulation of cell growth and differentiation via DNA stimulation [52]. These lipids act as intracellular second messengers, and when added exogenously, they elicit diverse cellular responses related to cell division. Second, in different mammalian cell types, sphingolipids and their derivatives are involved in inflammation and can initiate parts of the inflammatory process by activating pro-inflammatory transcription factors [53]. It is known that the overproduction of pro-inflammatory cytokines, particularly IL-12, INF-γ, and TNF-α by mice infected with virulent strains of T. gondii contributes to the animal death due to apoptosis in vital organs [54]. Lastly, the mutant parasites may use its sphingolipids as part of its signaling machinery to stimulate the expression of virulence factors secreted into the host cell, e.g. ROP proteins that control the host transcriptional responses by subverting STAT3 and STAT6 signaling [55].
Though there are widespread disruptions in lipid metabolism in TgNCR1-deficient parasites, the observed increase in lipid body biogenesis may represent a bypass pathway utilized by the parasite to repress the consequences of the loss of NCR1. Bypass pathways have been described in mammalian cells expressing a mutated NPC1 protein. For instance, chemicals that activate protein kinase C or elevate cytosolic calcium can revert the NPC phenotype [47]. In addition, overexpression of Rab7 (involved in early/late endosome to lysosome transport) or Rab9 (involved in late endosome to Golgi transport) results in a reduction in the amount of cholesterol stored in late endosomes. Overexpression of Rab7 or Rab9 also induces an increase in Nile Red staining, which presumably reflects an increased storage of neutral lipids, including cholesterol esters in lipid bodies. The synthesis of cholesteryl esters, which have lower biological toxicity than free sterols, provides an efficient and readily accessible sink for cholesterol, and this process can then compensate for the loss of NPC1 activity. By analogy, the accumulation of additional lipid bodies and the bulky production of cholesteryl esters in TgNCR1-deficient parasites may be viewed as a compensatory mechanism activated by the mutant parasites to circumvent the lipotoxic effects of free cholesterol accumulation. Although we do not know whether there is transient accumulation of free cholesterol in mutant parasites, the high number of cholesteryl ester-containing lipid bodies would implicate TgNCR1 in cholesterol movement e.g., delivery of cholesterol to organelles. In fact, the trafficking of cholesterol to large-size lipid bodies has been observed in human NPC fibroblasts [56]. The enlargement of lipid bodies is caused by the abnormal fusion of lipid bodies with one another, but upon correction of the NPC phenotype by ectopic expression of NPC1, cholesterol is targeted to small-sized lipid bodies. It has been proposed that the activity of NPC1 resides in the maintenance of normal sized lipid bodies, possibly by preventing their fusion. T. gondii contains two ACAT enzymes located to the ER, which are both essential for parasite development [37; Lige and Coppens, data in preparation]. Inventories of cholesteryl esters produced by Toxoplasma reveal a predominant content of cholesteryl oleate and palmitate (∼70%) as well as other esters with polyunsaturated fatty acids that are rarely found in mammalian cells. An overall increase of ∼30% in cholesteryl esters is observed in TgNCR1-deficient Toxoplasma, reflecting that the cycle of esterification/hydrolysis of cholesterol esters is severely dysregulated in mutant parasites. This phenotype may result in an impairment of cholesteryl ester hydrolysis to free cholesterol. The Toxoplasma genome contains a gene coding for a cholesteryl ester hydrolase homolog (TgME49_038200) that can be involved in this function. Alternatively, ACAT activities can be stimulated in mutant parasites in response to accumulated free cholesterol. Concomitant to the increase in cholesteryl ester content, the number and size of lipid bodies are dramatically enhanced in mutant parasites, probably to collect excess cholesteryl esters. While TgNCR1 has never been detected on parasite lipid bodies, the accumulation of lipid bodies and cholesteryl esters in the ΔNCR1 strain suggest that TgNCR1 may regulate the cycles of cholesterol esterification and lipid body content.
Lipidomic analyses reveal that Toxoplasma contains more than twenty different species of sphingolipids consisting of both saturated and unsaturated fatty acids. The origin of sphingolipids in the parasite remains unclear. Evidence for de novo sphingolipid synthesis by Toxoplasma is based on the indirect effect of drugs known to block sphingolipid biosynthesis in mammalian cells or fungi but their enzymatic targets have not been identified in the parasite [57]. Other studies support the active scavenging of host cell-derived ceramides by Toxoplasma [58; Romano and Coppens, data in preparation]. Like in NPC1-deficient mammalian cells, the homeostasis of sphingolipids is altered in TgNCR1-deficientparasites, with a selective accumulation of several species of very-long-chain fatty acids. A few species of phosphatidylethanolamine and phosphatidylserine also show increased levels in the mutant parasites. The cause of this accumulation is unknown. One possible explanation may be linked to the multiple biosynthetic pathways of serine. Besides being the precursor of ceramides, serine is involved in the formation of phosphatidylserine and phosphatidylethanolamine through ethanolamine synthesis. Increases in the levels of phosphatidylethanolamine and phosphatidylserine may be due to the enhanced synthesis of these phospholipids via the diversion of serine from sphingolipid pathways. The dysregulation of the sphingolipid levels in the ΔNCR1 strain may have an indirect effect on the metabolism of phospholipids, which use the same precursor. Interestingly, an accumulation of other phospholipids, e.g., phosphatidylcholine has been reported in NPC1-deficient hepatocytes though the mechanism has not been explored [59].
In mammalian cells, sphingolipid metabolism is closely coordinated with that of sterols as these two lipids associate physically and there is considerable cross-talk between their metabolic pathways [60]. Among the cooperative activities of cholesterol and sphingolipids is the nucleation of lipid microdomains. Imbalance in either of these lipids results in disease pathology. It is possible that perturbations in sphingolipid levels in the ΔNCR1 mutant parasites may be related to changes in cholesterol homeostasis, either as feed-forward or feed-backward sequelae of cholesterol imbalance. In addition, the accumulation of sphingolipids in the ΔNCR1 strain may be due to impaired catabolism, altered vesicular transport of these lipids and/or of cholesterol, or blockade of sphingolipid export. In eukaryotic cells, sphingolipids and their metabolites play key roles both as structural components of membranes and signaling molecules that mediate responses to physiologic cues and stresses [61]. Sphingolipids are particularly abundant on the extracellular face of the plasma membrane, where they participate in cell-cell communication and host-pathogen interactions [62], [63]. The structural features of sphingolipids can influence the order of the lipid phase, and impact membrane curvature and thickness [64], [65]. The distribution, function and regulation of sphingolipids in Toxoplasma as well as the contributions of these lipids to parasite development are unknown. Long extensions derived from the PV membrane are observed in mutant parasites. It is then tempting to propose that these extensions may result from the selective incorporation of sphingolipids, i.e. sphingomyelins in this membrane, which contributes to the stability of the extensions. It might be of interest to compare the Toxoplasma PV tubules extending into the host cytoplasm with the tubovesicular network derived from the PV of intraerythrocytic Plasmodium that also pervades the host cytosol. Interestingly, the biosynthesis of sphingolipids in the malaria parasite is an import feature of this tubovesicular network and its membranes accumulate specific sphingolipids including sphingomyelin [66], [67]. The physiological role of such extensions in Toxoplasma is still unknown. They may participate in host cell remodeling by deploying parasite proteins into the host cell and recruiting host organelles to the PV. These extensions may also serve as a route for exchange of material between two PV. The intracellular bacterium Chlamydia trachomatis forms long fibers extending from the inclusion membrane into the host cytosol [37], and it has been proposed that the fibers play a role in facilitating the generation of new infective vacuoles, and therefore promoting bacteria replication in the host cell. Some PV membranous extensions can also connect two PV located in different host cells and our preliminary studies reveal the presence of these extensions inside nanotubules connecting two mammalian cells (Romano and Coppens, data in preparation). This feature may represent an efficient means for Toxoplasma to disseminate among cells and tissues like C. trachomatis. Nonetheless, the high abundance of PV membranous extensions in virulent TgNCR1-deficient parasites leads to the assumption that they could contribute to the pathogenicity.
In conclusion, the phenotype of NPC1 deficiency in Toxoplasma mimics in part the defects that have been observed in mammalian cells and yeast lacking functional NPC1 proteins, regarding the storage of multiple sphingolipid species. The accumulation of cholesteryl esters in parasite mutants, as observed in hepatocytes but not in other mammalian cells [59], is rather intriguing but may reflect the unusual properties of cholesterol metabolism in the parasite. Our results suggest that TgNCR1 may be an important contributor to lipid homeostasis in T. gondii. How precisely this protein exerts its action, e.g. as a detector of lipid levels in membranes, lipid translocator or negative regulator of lipid production, remains to be elucidated. One challenge for the future is to clarify the source of sphingolipids for the parasite, and the regulation and coordination of sphingolipid production with regards to parasite growth and virulence. Lipid molecules play an integrated role in human disease, and when one of them is misregulated, pathology frequently ensues. The control of levels of many lipids is obviously lost in the ΔNCR1 strain, and yet these parasites can overcome these lipidic perturbations and remain infective. On the one hand, these parasites adeptly respond to NCR1 loss by producing lipid bodies for the storage of excess lipids. On the other hand, TgNCR1-deficient Toxoplasma seems proficient in using lipids to build new membranes for organelle biogenesis. The ΔNCR1 strain provides an attractive model for studying cell division in Apicomplexa. In pursuit of a deeper understanding of the coordination of this process, our mutant parasites allow observations of the dynamics of the IMC and collection of ultrastructural details about organelle partitioning between descendants.
Materials and Methods
Ethics statement
All animal procedures were approved by the Institutional Animal Care and Use Committee of the Johns Hopkins University following the National Institutes of Health guidelines for animal housing and care.
Chemicals and antibodies
All chemicals were obtained from Sigma (St Louis, MO) or Fisher (Waltham, MA) unless indicated otherwise. Solvents and standards for chromatography were of the highest analytical grade. Silica gel 60 thin-layer chromatography (TLC) plates (Merck KgAG, Darmstadt, Germany) were purchased through EM Science (Gibbstown, NJ). Radiolabeled reagent [5,6-3H] uracil (40 Ci/mmol) was purchased from Amersham Corp. (Arlington Heights, IL, USA). LysoTracker Red DND-99 was obtained from Invitrogen (Carlbad, CA). Ganglioside, sphingomyelin and ceramide standards were purchased from Avanti Polar Lipids (Alabaster, AL). Primary antibodies used in this study were: commercial mouse antibodies against HA (Roche Applied Science, Indianapolis, IN), myc (Cell Signaling Technology, Boston, MA), EEA1 (Abcam, Cambridge, MA) and LAMP1 (BD Biosciences, Palo Alto, CA), commercial rabbit antibody against myc (Santa Cruz Biotechnology, Santa Cruz, CA), rabbit polyclonal antibody against TgGRA7 [3] and mouse monoclonal antibody against TgGRA3 generously provided by J.F. Dubremetz (University of Montpellier).
Mammalian cell lines, culture conditions and parasite propagation
Mammalian cell lines used included primary human foreskin fibroblasts (HFF), Chinese hamster ovary cells (CHO cells) and the somatic 2-2 mutant CHO cells with defective NPC1 kindly provided by L. Liscum (Tufts University) [29]. All of these cells were grown as monolayers and cultivated in α-minimum essential medium (MEM) supplemented with 10% fetal bovine serum (FBS), 2 mM glutamine and penicillin/streptomycin (100 units/ml per 100 µg/ml) as described [2]. The tachyzoite RH strain of Toxoplasma gondii and modified strains were propagated in vitro by serial passage in HFF [68]. To evaluate parasite viability, the measurement of [3H]uracil incorporated into the parasites was determined as described [69].
Sequence analysis
Nucleotide and amino acid sequences were searched in the T. gondii database (www.toxodb.org) and the NCBI database using the BLAST algorithm [70]. Multiple sequence alignments were created using ClustalW, and the resulting similarities were then visualized by subjecting the alignment to BioEdit. Percent identity and similarity were calculated using standard tool for sequence analysis from NCBI (ncbi.nlm.nih.gov).
Cloning of full-length cDNA encoding TgNCR1
Based on the blast search results of the ToxoDB, the ORF of the Toxoplasma homolog for NPC1 (TgNCR1: TGME49_090870) was amplified from a T. gondii sporozoite cDNA library generously provided by M.W. White (Montana State University) by using the primers F-TgNCR1-P1 and R-TgNCR1-P2 (see Table S2 for the sequences of primers used in this study). Amplified fragments (∼3.5-kb) were subcloned into pCR2.1 using the TOPO-TA cloning protocol (Invitrogen) and the insertion was confirmed by enzymatic digestion with EcoRI and sequencing. The cDNA sequence of TgNCR1 has been deposited in GenBank under the accession number JF836804.
Plasmid constructs for expression in mammalian cells
Three plasmids were engineered for functional and localization studies of TgNCR1. The pcDNA3.1 vector allowing the expression of proteins with a myc tag at the C-terminus driven by a CMV promoter was used as a backbone. For the construction of the plasmid pTgNCR1-myc, the 5′ and 3′ ends of the TgNCR1 sequence were PCR-modified using primers TgNCR1-P1 and TgNCR1-P2. The resulting PCR fragment was digested with EcoRI and HindIII before ligation into the same restriction sites in the pcDNA3.1 vector. This construct allows the expression of TgNCR1 in mammalian cells with a C-terminal myc tag. We also expressed a hybrid construct in which the C-terminal 78 residues of TgNCR1 were replaced with amino acids 1214 to1278 of human NPC1 by fusion PCR to generate the plasmid pTgNCR1-hNPC1. After RNA extraction from HFF, the coding sequence corresponding to the amino acids 1214-1278 of human NPC1 was amplified by RT-PCR (SuperScript One-Step RT-PCR with Platinum Taq, Invitrogen) using primers F-hNPC11214-1278 and R-hNPC11214-1278. The construct TgNCR1Δ1101-1178 was created by PCR amplification using F-myc-TgNCR1 1-1101 and R-TgNCR11-1101. The two fragments were fused by PCR and amplified using the primers F-myc-TgNCR11-1101 and R-hNCR11214-1278. The fusion product was then ligated into pcDNA3.1 vector through EcoRI and HindIII to generate pTgNCR1-hNPC1. In this plasmid, a myc tag is introduced at the N-terminus of the resulting protein while the expression of the C-terminal myc tag originally present in vector pcDNA3.1. was blocked by introducing a stop codon in the primer R-hNPC1. Using the same cloning strategy, a plasmid control combining residues 938-1100 of TgNCR1 with residues 1214-1278 of human NPC1 named tr.(truncated) TgNCR1-hNPC1, was constructed with a N-terminal myc tag. Primers used for the fusion PCR were F-myc-TgNCR1938-1100, R-myc-TgNCR1938-1100, F-hNPC11214-1278 and R-hNPC11214-1278. For expression analysis in mammalian cells, cells were transfected with the different plasmids using lipofectamine method (Invitrogen).
Plasmid construction for expression in Toxoplasma
To generate a strain of T. gondii stably expressing TgNCR1 wild-type or mutants with a HA tag at the N-terminus, we used the backbone of the parasite expression vector psagCATsag_Tub2358YFP obtained from the Roos laboratory (University of Pennsylvania) to construct pHA-TgNCR1. The TgNCR1 sequence was modified by PCR using the primers F-HA-TgNCR1 that contains the coding sequence of an HA tag for expression at the N-terminus and R-YFP-TgNCR1 that includes a stop codon to block the translation of the downstream YFP. Same approach was used for the construction of four plasmids harboring punctual mutations in the TgNCR1 sequence. The mutations of the targeted residues were carried out by fusion PCR. Two flanking primers were F - HA-TgNCR1 and R-YFP-TgNCR1, and the internal primers used were: F-TgNCR1D571N and R-TgNCR1D571N to generate pHA-TgNCR1D571N; F-TgNCR1P913A and R-TgNCR1P913A for pHA-TgNCR1P913A; F-TgNCR1I957T and R-TgNCR1I957T for pHA-TgNCR1I957T; F-TgNCR1L1100V and R-TgNCR1L1100V for pHA-TgNCR1L1100V. To create a strain of T. gondii transiently expressing TgNCR1 with a HA tag at the N-terminus under the NTPase promotor, we used the backbone of the parasite expression vector pRab5-HA containing the nucleoside triphosphatase III (NTPase III) promoter obtained from the Joiner laboratory (Yale University). The coding sequence of TgNCR1 was PCR-amplified using the primers F-haNTP and R-haNTP. The resulting PCR fragment was digested with SpeI and PacI, and directly ligated into the NheI and PacI sites of the pRab5-HA vector. In this construct, TgNCR1 was cloned in frame with the upstream HA tag coding sequence in the vector. To generate a strain of T. gondii transiently expressing TgNCR1 with a HA tag at the C-terminus under the tubulin promotor, the TgNCR1 sequence was modified by PCR using the primers F-NCRHA and R-NCRHA. The resulting PCR was digested by BamHI and AvrII and ligated into the BglII and AvrII sites of the sagCATsag_Tub2358YFP vector. The reverse primer contained the coding sequence of HA tag followed by a stop codon to block the translation of the downstream YFP tag. For transient transfection in T. gondii, expression plasmids containing TgNCR1coding sequences, wild-type or mutants, were transformed in E. coli DH-5α and isolated using Qiagen Plasmid Purification Kits. Parasites were transfected by electroporation as described [6].
Genetic disruption of TgNCR1 gene
To create the ΔNCR1 strain, a fusion PCR knockout construct was generated, consisting of 3′ and 5′NCR1 genomic flanks fused on either site of the hypoxanthine-xanthine-guanine phosphoribosyltransferase (HXGPRT) selectable marker cassette (Figure S9). Genomic flanking sequences of the TgNCR1 gene were obtained from the Toxoplasma database (www.toxodb.org; version 4.3). Primers were designed to amplify ∼550-bp of each of the two flanking regions from the genomic DNA of the RH strain of Toxoplasma. Two flanks were amplified to overlap on one end with the HXGPRT selectable marker cassette, and similarly, the HXGPRT marker cassette was amplified from pminiHXGPRT plasmid to incorporate the TgNCR1 sequence on the other end. The three individual fragments corresponding to 5′ flank, 3′ flank and HXGPRT were obtained by PCR reactions using primers KOPCR-1 and KOPCR-4, KOPCR-5 and KOPCR-2, KOPCR-3 and KOPCR-6, respectively. The three PCR fragments were then used as template to generate the fusion construct using primers KOPCR-1 and KOPCR-2 as shown in Figure 5. Deletion of TgNCR1 from the parasite genome and HXGPRT arrangement were verified by PCR and Southern blot analyses. For PCR confirmation, genomic DNA was isolated from the colonies obtained through selection and used as template for PCR. Replacement of TgNCR1 by HXGPRT was monitored using a primer outside the 5′ genomic flanking region (P1) and a primer within the selectable marker HXGPRT (P2). The primer pairs P3-P4 and P5-P6 were used to assert the absence of TgNCR1/the presence of a single copy of HXGPRT in the ΔNCR1 strain and the presence of TgNCR1/the absence of HXGPRT in the ΔKu80ΔHXGPRT (ΔK80) parental strain [71], respectively. For Southern blotting, the primers F-NPC-sb-1 and R-NPC-sb-1 were used in the PCR to generate a ∼0.9-kb probe specific for the HXGPRT gene. Genomic DNA samples (80 µg) were digested with AgeI and electrophoresed through 0.7% agarose gels. The DNA was then transferred onto a Hybond N+ membrane (Biotech) and hybridized randomly with the 32P-labeled DNA probe. Blots were then washed at appropriate stringency and visualized by autoradiography.
Selection of parasite stable lines
For the generation of the ΔNCR1 strain, fusion PCR products (15–20 µg) containing 5′ UTR of TgNCR1 gene, HXGPRT cassette and 3′ UTR of TgNCR1 gene were electroporated into the parasite ΔKu80 strain [71] as described [6]. After overnight growth, transformants were placed under 25 µg/ml of mycophenolic acid and 50 µg/ml of xanthine. Transformant pools were tested by PCR to validate the deletion of NCR1 in parasites, and TgNCR1 knockout parasites were further selected by limiting dilution under drug selection. Individual knockout clones were then plated in 96-well plates and cultivate to ensure clonality.
Fusion PCR construct for complementation
To create the construct for the complementation of the ΔNCR1 strain, the same fusion PCR approach shown in Figure S9 was used. Two fragments, 5′ and 3′ flanking sequences of the NCR1 gene, were amplified using the primers KOPCR-1 and Ncomp-4, Ncomp-5 and KOPCR-2, respectively, to overlap on one end with the TgNCR1 coding sequence. The third fragment, TgNCR1 coding sequence, was amplified using primers Ncomp-3 and Ncomp-6. The three PCR fragments were then used as template to generate the fusion construct using the primers KOPCR-1 and KOPCR-2.
Negative selection of HXGPRT with 2-hydroxy-6-mercaptopurine
For the complementation of the ΔNCR1 strain, 25 µg of the fusion PCR product was electroporated into the ΔNCR1 strain. Twenty four hours post-transfection, the culture media of transformants was replaced with DMEM containing HEPES and 1% dialyzed FBS, and the tranformants were placed under the selection of 0.85 µg/ml of 2-hydroxy-6-mercaptopurine. Drug resistant transformant pools were tested by PCR to validate the replacement of HXGPRT with NCR1 in parasites, and NCR1-complemented parasites were further selected by limiting dilution in 96-well plates. Clones were then tested using drug selection to ensure that they grow in 2-hydroxy-6-mercaptopurine but no longer in mycophenolic acid and xanthine. After the selection, the 5′ integration of TgNCR1 coding sequence was confirmed by PCR using the pair of primers P1-P7, and the 3′integration was verified by PCR using the pair of primers P8-P9.
Recombinant peptide expression in E. coli and affinity purification
To generate antibodies against TgNCR1, we engineered a plasmid to produce a recombinant peptide corresponding to the sequence of TgNCR1162-501. The coding sequence corresponding to TgNCR1162-501 was PCR-amplified using the primers F - TgNCR1162-501 and R-TgNCR1162-501 and directly cloned into the BamHI and HindIII sites of the pQE-30 vector (Qiagen, Hilden, Germany) to generate N-terminal 6-His tagged fusion protein. The recombinant peptide expressed in E. coli M15 strain were purified under denatured condition on Ni2+-NTA resin according to the Qiagen protocol. After purification, the peptide was refolded by diluting the sample 10 times in the refolding buffer (50 mM Tris/HCl pH 7.5, 1 mM EDTA, 1 M L-arginine, 1 mM reduced form of glutathione, 0.8 mM of oxidized form of glutathione) at 4°C overnight before concentration and dialysis against phosphate-buffered saline (PBS). Some batches of antibodies against TgNCR1162-501 were further preadsorbed on a fibroblast lysate (overnight, 4°C) to diminish unspecific cross-reactions between the primary antibody and host cell epitopes.
Immunoblot analysis of parasites stably expressing HA-TgNCR1
For immunodetection, transgenic parasites were lysed in the M-PER mammalian protein extraction reagent (Pierce Biotechnology, Rockford, IL). The cell extracts were suspended in SDS gel-loading buffer (50 mM Tris–HCl (pH 6.8), 50 mM 2-mercaptoethanol, 2% SDS, 0.1% bromophenol blue, 10% glycerol) and lysed by boiling in a water bath. The samples were subjected to SDS-PAGE, and the proteins were then electrophoretically transferred to a membrane (Immobilon Transfer Membranes, Millipore, Bedford, MA). The membrane was immersed in blocking buffer (PBS containing 3% skim milk) for 60 min, and then incubated with anti-HA antibody (1∶5000) in the blocking buffer for 60 min. Unbound antibody was removed by washing the membrane six times with blocking buffer. Next, the membrane was incubated with horseradish peroxidase-conjugated goat anti-mouse IgG antibody (Amersham Pharmacia Biotech; dilution, 1∶1000) in blocking buffer for an additional hour, before detection by chemiluminescence.
Cell cycle analysis
Vero cells were infected with either the ΔNCR1 or parental strain at the M.O.I of 10 and washed every 2 h p.i. to synchronize the infection. Fifteen hours p.i., infected cells were exposed to the calcium ionophore A23187 at 4 µM for 10 min to induce parasites egress. Parasites in the supernatant were collected by aspiration, washed by centrifugation and fixed in ice-cold ethanol (70%), followed by incubation with propidium iodide (PI) solution (0.01 mg/ml PI; 0.01 mg/ml DNAse-free RNAse A; 0.1% Triton X-100; 1 mg/ml sodium citrate) at 4°C overnight. Uninfected VERO cells as controls were treated similarly to infected cells. Flow cytometry analysis was performed on a FACScan flow cytometer (Becton Dickinson, Mountain View, CA). Cell cycle distribution pattern was assessed using FlowJo software (Tree Star, Inc.); G1, G2 and S peaks were defined using the Dean-Jet-Fox model.
Mice and in vivo virulence assays
5 week-old female BALB/C mice were purchased from The Jackson Laboratory (Bar Harbor, ME). Groups of six mice were infected intraperitoneally with 5×104 tachyzoites of ΔNCR1 or ΔKu80ΔHXGPRT parasites. The survival of mice after infection was monitored daily. Results of three independent experiments are presented.
Measurement of lipids
Thin layer chromatography (TLC)
Total lipids from mammalian cells were extracted in chloroform/methanol (v/v, 2∶1), subsequently separated by TLC on silica plates using hexane:diethyl ether:acetic acid (v/v/v, 90∶10∶1) for free cholesterol and cholesteryl esters, or using chloroform:methanol:0.2% CaCl2 (v/v/v/, 60∶35∶8) for gangliosides, and visualized as described [31].
Mass spectrometry
Total lipids from the ΔNCR1 and parental strains were extracted using a modified Bligh and Dyer procedure as described [72]. Purified standards of lipids were added directly to homogenates. Mass spectrometry analyses of species of cholesterol, cholesteryl esters, triglycerides, sphingolipids and phospholipids were performed using a Sciex API 3000 triple stage quadruple tandem mass spectrometer (ESI/MS/MS) from Sciex Inc. (Thornhill, Ontario, Canada), using methods similar to those described in previous studies [72], [73]. Peaks were evaluated with non-parametric one-way ANOVA (Kruskal-Wallis test, with a threshold of P-value <0.05 considered as statistically significant.
Light and electron microscopy studies
Light and epifluorescence microscopy were performed on infected cells seeded on sterile coverslips in 24-well culture dishes. IFA on parasites or mammalian cells were performed as previously described [56] using primary antibodies against GRA3 (1∶100), anti-GRA7 (1∶100), myc (1∶100), HA (1∶1000), EEA1 (1∶100), LAMP1 (1∶100), and secondary antibodies (Invitrogen): anti-mouse and anti-rabbit IgG antibodies conjugated to either Alexa 488 or Alexa 594 diluted at 1∶2000. For detection of cholesterol or lipid bodies by fluorescence microscopy, intravacuolar parasites were fixed in paraformaldehyde, and treated as described [2], [56]. Slides were observed using a Nikon Eclipse E800 microscope equipped with a Spot RT CCD Camera and processed using Image-Pro-Plus software (Media Cybernetics, Silver Spring, MD) before assembly using Adobe Photoshop (Adobe Systems, Mountain View, CA). For analysis of PV membranous extensions, HFF were infected with either the ΔNCR1 or ΔHXGPRT stain for 36-h. After fixation and immunostaining for GRA7 and GRA3, parasites were imaged with a fluorescent microscope (Nikon 90i) using a Plan-Apochromat 100x/1.4 NA. Serial optical z-sections were acquired with a Hammatsu camera and Volocity software (Improvision, Waltham, MA). Using the Volocity software, images were processed by iterative restoration (deconvolution algorithm) and the length and diameter of the PV membranous extensions, as detected by GRA3 staining, were measured. To quantify the levels of filipin accumulation, images were collected using sequential scanning, processed and merged using Volocity software. For ultrastructural observation of the ΔNCR1 strain by thin-section transmission electron microscopy (EM), infected cells were fixed in 2.5% glutaraldehyde (Electron Microscopy Sciences, Hatfield, PA) and processed as described [56]. Ultrathin sections of infected cells were stained before examination with a Philips CM120 EM (Eindhoven, the Netherlands) under 80 kV. For immunoelectron microscopy, Toxoplasma-infected cells were fixed in 4% paraformaldehyde (Electron Microscopy Sciences) and processed as previously described [38]. The sections were immunolabeled with anti-HA antibodies (1∶200 in PBS/1% fish skin gelatin), then with mouse anti-mouse IgG antibodies, followed directly by 10 nm protein A-gold particles (Department of Cell Biology, Medical School, Utrecht University, the Netherlands) before microscopic examination.
Protein determination
Protein content was determined using the bicinchoninic acid assay [74] with serum bovine albumin (BSA) as a standard.
Supporting Information
Zdroje
1. RobibaroBHoppeHCYangMCoppensINgôHM 2001 Endocytosis in different lifestyles of protozoan parasitism: role in nutrient uptake with special reference to Toxoplasma gondii. Int J Parasitol 31 1343 1353
2. CoppensISinaiAPJoinerKA 2000 Toxoplasma gondii exploits host low-density lipoprotein receptor-mediated endocytosis for cholesterol acquisition. J Cell Biol 149 167 180
3. CoppensIDunnJDRomanoJDPypaertMZhangH 2006 Toxoplasma gondii sequesters lysosomes from mammalian hosts in the vacuolar space. Cell 125 261 274
4. SehgalABettiolSPypaertMWenkMRKaaschA 2005 Peculiarities of host cholesterol transport to the unique intracellular vacuole containing Toxoplasma. Traffic 6 1125 1141
5. EhrenmanKSehgalALigeBStedmanTTJoinerKA 2010 Novel roles for ATP-binding cassette G transporters in lipid redistribution in Toxoplasma. Mol Microbiol 76 1232 1249
6. LigeBJayabalasinghamBZhangHPypaertMCoppensI 2009 Role of an ancestral d-bifunctional protein containing two sterol-carrier protein-2 domains in lipid uptake and trafficking in Toxoplasma. Mol Biol Cell 20 658 672
7. VoelkerDR 2009 Genetic and biochemical analysis of non-vesicular lipid traffic. Annu Rev Biochem 78 827 856
8. ChangTYChangCCOhgamiNYamauchiY 2006 Cholesterol sensing, trafficking, and esterification. Annu Rev Cell Dev Biol 22 129 157
9. LiscumLSturleySL 2004 Intracellular trafficking of Niemann-Pick C proteins 1 and 2: obligate components of subcellular lipid transport. Biochim Biophys Acta 1685 22 27
10. RosenbaumAIMaxfieldFR 2011 Niemann-Pick type C disease: molecular mechanisms and potential therapeutic approaches. J Neurochem 116 789 795
11. ScottCIoannouYA 2004 The NPC1 protein: structure implies function. Biochim Biophys Acta 1685 8 13
12. StorchJXuZ 2009 Niemann-Pick C2 (NPC2) and intracellular cholesterol trafficking. Biochim Biophys Acta 1791 671 678
13. KwonHJAbi-MoslehLWangMLDeisenhoferJGoldsteinJL 2009 Structure of N-terminal domain of NPC1 reveals distinct subdomains for binding and transfer of cholesterol. Cell 137 1213 1224
14. WangMLMotamedMInfanteREAbi-MoslehLKwonHJ 2010 Identification of surface residues on Niemann-Pick C2 essential for hydrophobic handoff of cholesterol to NPC1 in lysosomes. Cell Metab 12 166 173
15. KuwabaraPELabouesseM 2002 The sterol-sensing domain: multiple families, a unique role? Trends Genet 18 193 201
16. HigakiKAlmanzar-ParamioDSturleySL 2004 Metazoan and microbial models of Niemann-Pick Type C disease. Biochim Biophys Acta 1685 38 47
17. WorgallTSSturleySLSeoTOsborneTFDeckelbaumRJ 1998 Polyunsaturated Fatty Acids Decrease Expression of Promoters with Sterol Regulatory Elements by Decreasing Levels of Mature Sterol Regulatory Element-binding Protein. J Biol Chem 273 25537 25540
18. WorgallTSJohnsonRASeoTGierensHDeckelbaumRJ 2002 Unsaturated fatty acid-mediated decreases in sterol regulatory element-mediated gene transcription are linked to cellular sphingolipid metabolism. J Biol Chem 277 3878 3885
19. KumagaiHChunKTSimoniRD 1995 Molecular dissection of the role of the membrane domain in the regulated degradation of 3-hydroxy-3-methylglutaryl coenzyme A reductase. J Biol Chem 270 19107 19113
20. SeegmillerACDobrosotskayaIGoldsteinJLHoYKBrownMS 2002 The SREBP Pathway in Drosophila: Regulation by Palmitate, Not Sterols. Dev Cell 2 229 238
21. MalathiKHigakiKTinkelenbergAHBalderesDAAlmanzar-ParamioD 2004 Mutagenesis of the putative sterol-sensing domain of yeast Niemann Pick C-related protein reveals a primordial role in subcellular sphingolipid distribution. J Cell Biol 164 547 556
22. WaterhouseAMProcterJBMartinDMAClampMBartonGJ 2009 Jalview Version 2 – a multiple sequence alignment editor and analysis workbench. Bioinformatics 25 1189 1191
23. IoannouYA 2000 The structure and function of the Niemann-Pick C1 protein. Mol Genet Metab 71 175 181
24. GarverWSJelinekDMeaneyFJFlynnJPettitKM 2010 The National Niemann-Pick Type C1 Disease Database: correlation of lipid profiles, mutations, and biochemical phenotypes. J Lipid Res 51 406 415
25. MillardEEGaleSEDudleyNZhangJSchafferJE 2005 The sterol-sensing domain of the Niemann-Pick C1 (NPC1) protein regulates trafficking of low density lipoprotein cholesterol. J Biol Chem 280 28581 28590
26. GreerWLDobsonMJGirouardGSByersDMRiddellDC 1999 Mutations in NPC1 highlight a conserved NPC1-specific cysteine-rich domain. Am J Hum Genet 65 1252 12560
27. ScottCHigginsMEDaviesJPIoannouYA 2004 Targeting of NPC1 to late endosomes involves multiple signals, including one residing within the putative sterol-sensing domain. J Biol Chem 279 48214 48223
28. InfanteRERadhakrishnanAAbi-MoslehLKinchLNWangML 2008 Purified NPC1 protein: II. Localization of sterol binding to a 240-amino acid soluble luminal loop. J Biol Chem 283 1064 1075
29. KwonHJAbi-MoslehLWangMLDeisenhoferJGoldsteinJL 2009 Structure of N-terminal domain of NPC1 reveals distinct subdomains for binding and transfer of cholesterol. Cell 137 1213 1224
30. DahlNKReedKLDaunaisMAFaustJRLiscumL 1992 Isolation and characterization of Chinese hamster ovary cells defective in the intracellular metabolism of LDL-derived cholesterol. J Biol Chem 267 4889 4896
31. SugimotoYNinomiyaHOhsakiYHigakiKDaviesJP 2001 Accumulation of cholera toxin and GM1 ganglioside in the early endosome of Niemann-Pick C1-deficient cells. Proc Natl Acad Sci U S A 98 12391 12396
32. D'HaeseJMehlhornHPetersW 1977 Comparative electron microscope study of pellicular structures in coccidia (Sarcocystis, Besnoitia and Eimeria). Int J Parasitol 7 505 518
33. MannTBeckersC 2001 Characterization of the subpellicular network, a filamentous membrane skeletal component in the parasite Toxoplasma gondii. Mol Biochem Parasitol 115 257 268
34. HuKJohnsonJFlorensLFraunholzMSuravajjalaS 2006 Cytoskeletal components of an invasion machine - the apical complex of Toxoplasma gondii. PLoS Pathog 2 e13
35. GubbelsMJWhiteMSzatanekT 2008 The cell cycle and Toxoplasma gondii cell division: tightly knit or loosely stitched? Int J Parasitol 38 1343 1358
36. CruzJCSugiiSYuCChangTY 2000 Role of Niemann-Pick type C1 protein in intracellular trafficking of low density lipoprotein-derived cholesterol. J Biol Chem 275 4013 4021
37. NishikawaYQuittnatFStedmanTTVoelkerDRChoiJY 2005 Host cell lipids control cholesteryl ester synthesis and storage in intracellular Toxoplasma. Cell Microbiol 7 849 867
38. QuittnatFNishikawaYStedmanTTVoelkerDRChoiJY 2004 On the biogenesis of lipid bodies in ancient eukaryotes: synthesis of triacylglycerols by a Toxoplasma DGAT1-related enzyme. Mol Biochem Parasitol 138 107 122
39. Lloyd-EvansEMorganAJHeXSmithDAElliot-SmithE 2008 Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat Med 14 1247 1255
40. WeltiRMuiESparksAWernimontSIsaacG 2007 Lipidomic analysis of Toxoplasma gondii reveals unusual polar lipids. Biochemistry 46 13882 13890
41. GuptaNZahnMMCoppensIJoinerKAVoelkerDR 2005 Selective disruption of phosphatidylcholine metabolism of the intracellular parasite Toxoplasma gondii arrests its growth. J Biol Chem 280 16345 19353
42. HuKMannTStriepenBBeckersCJRoosDS 2002 Daughter cell assembly in the protozoan parasite Toxoplasma gondii. Mol Biol Cell 13 593 606
43. HuKRoosDSAngelSOMurrayJM 2004 Variability and heritability of cell division pathways in Toxoplasma gondii. J Cell Sci 117 5697 5705
44. BrownWJSkeikyYAProbstPRockeyDD 2002 Chlamydial antigens colocalize within IncA-laden fibers extending from the inclusion membrane into the host cytosol. Infect Immun 70 5860 5864
45. RomeMEBeckJRTuretzkyJMWebsterPBradleyPJ 2008 Intervacuolar transport and unique topology of GRA14, a novel dense granule protein in Toxoplasma gondii. Infect Immun 76 4865 4875
46. MotaLJRamsdenAELiuMCastleJDHoldenDW 2009 SCAMP3 is a component of the Salmonella-induced tubular network and reveals an interaction between bacterial effectors and post-Golgi trafficking. Cell Microbiol 11 1236 1253
47. Lloyd-EvansEPlattFM 2010 Lipids on trial: the search for the offending metabolite in Niemann-Pick type C disease. Traffic 11 419 428
48. IoannouYA 2001 Multidrug permeases and subcellular cholesterol transport. Nat Rev Mol Cell Biol 2 657 668
49. CoppensIJoinerKA 2003 Host but not parasite cholesterol controls Toxoplasma cell entry by modulating organelle discharge. Mol Biol Cell 14 3804 3820
50. JohnsonTMRajfurZJacobsonKBeckersCJ 2007 Immobilization of the type XIV myosin complex in Toxoplasma gondii. Mol Biol Cell 18 3039 3046
51. BeckJRRodriguez-FernandezIACruz de LeonJHuynhMHCarruthersVB 2010 A novel family of Toxoplasma IMC proteins displays a hierarchical organization and functions in coordinating parasite division. PLoS Pathog 6 e1001094
52. SchroederJJCraneHMXiaJLiottaDCMerrillAHJr 1994 Disruption of sphingolipid metabolism and stimulation of DNA synthesis by fumonisin B1. J Biol Chem 269 3475 3481
53. PettusBJChalfantCEHannunYA 2004 Sphingolipids in inflammation: roles and implications. Curr Mol Med 4 405 418
54. MordueDGMonroyFLa ReginaMDinarelloCASibleyLD 2001 Acute toxoplasmosis leads to lethal overproduction of Th1 cytokines. J Immunol 167 4574 4584
55. ButcherBAFoxBARommereimLMKimSGMaurerKJ 2011 Toxoplasma gondii rhoptry kinase ROP16 activates STAT3 and STAT6 resulting in cytokine inhibition and arginase-1-dependent growth control. PLoS Pathog 7 e1002236
56. WiegandVChangTYStraussJF3rdFahrenholzFGimplG 2003 Transport of plasma membrane-derived cholesterol and the function of Niemann-Pick C1 Protein. FASEB J 17 782 784
57. SondaSSalaGGhidoniRHemphillAPietersJ 2005 Inhibitory effect of aureobasidin A on Toxoplasma gondii. Antimicrob Agents Chemother 49 1794 1801
58. de MeloEJde SouzaW 1996 Pathway of C6-NBD-Ceramide on the host cell infected with Toxoplasma gondii. Cell Struct Funct 21 47 52
59. KulinskiAVanceJE 2007 Lipid homeostasis and lipoprotein secretion in Niemann-Pick C1-deficient hepatocytes. J Biol Chem 282 1627 1637
60. GulatiSLiuYMunkacsiABWilcoxLSturleySL 2010 Sterols and sphingolipids: dynamic duo or partners in crime? Prog Lipid Res 49 353 365
61. BreslowDKWeissmanJS 2010 Membranes in balance: mechanisms of sphingolipid homeostasis. Mol Cell 40 267 279
62. LopezPHHSchnaarRL 2009 Gangliosides in cell recognition and membrane protein regulation. Curr Opin Struct Biol 19 549 557
63. TsaiBGilbertJMStehleTLencerWBenjaminTL 2003 Gangliosides are receptors for murine polyoma virus and SV40. EMBO J 22 4346 4355
64. LingwoodDSimonsK 2010 Lipid rafts as a membrane-organizing principle. Science 327 46 50
65. Lippincott-SchwartzJPhairRD 2010 Lipids and cholesterol as regulators of traffic in the endomembrane system. Annu Rev Biophys 39 559 578
66. ElmendorfHGHaldarK 1994 Plasmodium falciparum exports the Golgi marker sphingomyelin synthase into a tubovesicular network in the cytoplasm of mature erythrocytes. J Cell Biol 124 449 462
67. TamezPABhattacharjeeSvan OoijCHillerNLLlinásM 2008 An erythrocyte vesicle protein exported by the malaria parasite promotes tubovesicular lipid import from the host cell surface. PLoS Pathog 4 e1000118
68. RoosDSDonaldRGKMorissetteNSMoultonALC 1994 Molecular tools for genetic dissection of the protozoan parasite Toxoplasma gondii. Methods Cell Biol 45 27 63
69. NakaarVSamuelBUNgoEOJoinerKA 1999 Targeted reduction of nucleoside triphosphate hydrolase by antisense RNA inhibits Toxoplasma gondii proliferation. J Biol Chem 274 5083 5087
70. AltschulSFMaddenTLSchäfferAAZhangJZhangZ 1997 Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25 3389 3402
71. HuynhMHCarruthersVB 2009 Tagging of endogenous genes in a Toxoplasma gondii strain lacking Ku80. Eukaryot Cell 8 530 539
72. HaugheyNJCutlerRGTamaraAMcArthurJCVargasDL 2004 Perturbation of sphingolipid metabolism and ceramide production in HIV-dementia. Ann Neurol 55 257 267
73. BandaruVVMcArthurJCSacktorNCutlerRGKnappEL 2007 Associative and predictive biomarkers of dementia in HIV-1-infected patients. Neurology 68 1481 1487
74. SmithPKKrohnRIHermansonGTMalliaAKGartnerFH 1998 Measurement of protein using bicinchoninic acid. Anal Biochem 150 76 85
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2011 Číslo 12
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Controlling Viral Immuno-Inflammatory Lesions by Modulating Aryl Hydrocarbon Receptor Signaling
- Fungal Virulence and Development Is Regulated by Alternative Pre-mRNA 3′End Processing in
- Cryo Electron Tomography of Herpes Simplex Virus during Axonal Transport and Secondary Envelopment in Primary Neurons
- Epstein-Barr Virus Nuclear Antigen 3C Stabilizes Gemin3 to Block p53-mediated Apoptosis
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy