
A Major Role for the ApiAP2 Protein PfSIP2 in Chromosome End Biology
The heterochromatic environment and physical clustering of chromosome ends at the nuclear periphery provide a functional and structural framework for antigenic variation and evolution of subtelomeric virulence gene families in the malaria parasite Plasmodium falciparum. While recent studies assigned important roles for reversible histone modifications, silent information regulator 2 and heterochromatin protein 1 (PfHP1) in epigenetic control of variegated expression, factors involved in the recruitment and organization of subtelomeric heterochromatin remain unknown. Here, we describe the purification and characterization of PfSIP2, a member of the ApiAP2 family of putative transcription factors, as the unknown nuclear factor interacting specifically with cis-acting SPE2 motif arrays in subtelomeric domains. Interestingly, SPE2 is not bound by the full-length protein but rather by a 60kDa N-terminal domain, PfSIP2-N, which is released during schizogony. Our experimental re-definition of the SPE2/PfSIP2-N interaction highlights the strict requirement of both adjacent AP2 domains and a conserved bipartite SPE2 consensus motif for high-affinity binding. Genome-wide in silico mapping identified 777 putative binding sites, 94% of which cluster in heterochromatic domains upstream of subtelomeric var genes and in telomere-associated repeat elements. Immunofluorescence and chromatin immunoprecipitation (ChIP) assays revealed co-localization of PfSIP2-N with PfHP1 at chromosome ends. Genome-wide ChIP demonstrated the exclusive binding of PfSIP2-N to subtelomeric SPE2 landmarks in vivo but not to single chromosome-internal sites. Consistent with this specialized distribution pattern, PfSIP2-N over-expression has no effect on global gene transcription. Hence, contrary to the previously proposed role for this factor in gene activation, our results provide strong evidence for the first time for the involvement of an ApiAP2 factor in heterochromatin formation and genome integrity. These findings are highly relevant for our understanding of chromosome end biology and variegated expression in P. falciparum and other eukaryotes, and for the future analysis of the role of ApiAP2-DNA interactions in parasite biology.
Published in the journal:
. PLoS Pathog 6(2): e32767. doi:10.1371/journal.ppat.1000784
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1000784
Summary
The heterochromatic environment and physical clustering of chromosome ends at the nuclear periphery provide a functional and structural framework for antigenic variation and evolution of subtelomeric virulence gene families in the malaria parasite Plasmodium falciparum. While recent studies assigned important roles for reversible histone modifications, silent information regulator 2 and heterochromatin protein 1 (PfHP1) in epigenetic control of variegated expression, factors involved in the recruitment and organization of subtelomeric heterochromatin remain unknown. Here, we describe the purification and characterization of PfSIP2, a member of the ApiAP2 family of putative transcription factors, as the unknown nuclear factor interacting specifically with cis-acting SPE2 motif arrays in subtelomeric domains. Interestingly, SPE2 is not bound by the full-length protein but rather by a 60kDa N-terminal domain, PfSIP2-N, which is released during schizogony. Our experimental re-definition of the SPE2/PfSIP2-N interaction highlights the strict requirement of both adjacent AP2 domains and a conserved bipartite SPE2 consensus motif for high-affinity binding. Genome-wide in silico mapping identified 777 putative binding sites, 94% of which cluster in heterochromatic domains upstream of subtelomeric var genes and in telomere-associated repeat elements. Immunofluorescence and chromatin immunoprecipitation (ChIP) assays revealed co-localization of PfSIP2-N with PfHP1 at chromosome ends. Genome-wide ChIP demonstrated the exclusive binding of PfSIP2-N to subtelomeric SPE2 landmarks in vivo but not to single chromosome-internal sites. Consistent with this specialized distribution pattern, PfSIP2-N over-expression has no effect on global gene transcription. Hence, contrary to the previously proposed role for this factor in gene activation, our results provide strong evidence for the first time for the involvement of an ApiAP2 factor in heterochromatin formation and genome integrity. These findings are highly relevant for our understanding of chromosome end biology and variegated expression in P. falciparum and other eukaryotes, and for the future analysis of the role of ApiAP2-DNA interactions in parasite biology.
Introduction
Throughout the eukaryotic kingdom, the overall structure of chromosome ends is conserved and characterized by the telomeric tract, composed of short G-rich repeats, and an extensive subtelomeric region consisting of various types and lengths of repeats, also known as telomere-associated sequences (TAS) [1]. This conservation underscores the functional importance of these domains in genome function and maintenance. Due to the heterochromatic nature of subtelomeric regions, genes located nearby are subject to epigenetic control and variegated expression [2]–[5]. Furthermore, subtelomeric domains promote frequent recombination events driving the evolution and diversity of gene families located close to chromosome ends [1],[3]. Pathogenic microorganisms exploit this system for antigenic variation of surface antigens to evade adaptive immune responses or to respond to other changes in environmental conditions [6].
The apicomplexan parasite Plasmodium falciparum causes the most severe form of malaria in humans with up to two million deaths annually [7]. Malaria symptoms are entirely associated with the erythrocytic phase of infection where repeated rounds of intra-erythrocytic parasite multiplication take place. Sequestration of infected red blood cell aggregates in the microvasculatory system, which is mediated by the binding of P. falciparum erythrocyte membrane protein 1 (PfEMP1) to a variety of endothelial receptors [8]–[11], represents one of the main contributors to severe disease, including cerebral and placental malaria [12]–[14]. PfEMP1 is encoded by the var gene family comprising approx. 60 mostly subtelomeric members [15]–[18]. Importantly, due to mutually exclusive transcription of var genes, only one PfEMP1 variant is exposed per parasite at any time and switches in var gene expression result in antigenic variation of PfEMP1 [17],[19] facilitating immune evasion and chronic infection. Recent studies highlighted the important contribution of the specific biology and dynamics of heterochromatic chromosome ends in the regulation of var genes and additional subtelomeric gene families coding for proteins involved in host-parasite interactions [20]–[27].
P. falciparum chromosome ends consist of a stretch of telomeric GGGTT(T/C)A repeats with an average size of 1.2 kb, followed by an extensive 20 to 40 kb TAS domain [28]. This region is composed of a conserved arrangement of so-called telomere-associated repeat elements (TAREs 1 to 6), each of which consists of distinct non-coding repeat arrays of varying length and sequence [29]. On all chromosome ends, the coding part of the genome directly downstream of TARE 6 is characterized by members of multiple antigen gene families including var, rif, stevor and pfmc-2tm [18]. Similar to other unicellular eukaryotes, P. falciparum chromosome ends associate into clusters that are anchored to the nuclear periphery [30]–[32]. This structurally conserved context facilitates meiotic recombination between var genes on heterologous chromosomes [30]. Interestingly, spontaneous chromosome breakage and telomere healing events create chromosome ends lacking the entire TARE region; while such chromosomes are still tethered to the nuclear periphery, they display a reduced association with other chromosome ends implicating a role for TARE in cluster formation [33].
Expression of P. falciparum subtelomeric gene families is clonally variant and restricted to only one member (or a few) in each family [19], [34]–[36]. Transgenes inserted into TARE 6 as well as endogenous var genes are reversibly silenced in a manner reminiscent to telomere-postion effect in other eukaryotes [25]. Recent genome-wide studies highlighted the striking and exclusive association of the repressive histone 3 lysine 9 tri-methylation mark (H3K9me3) and heterochromatin protein 1 (PfHP1) throughout the TAS region and adjacent gene families on all chromosomes [23],[27],[37]. These heterochromatic marks are also important in telomere-proximal gene silencing in S. pombe and higher eukaryotes [38],[39], indicating the existence of conserved epigenetic control strategies in highly divergent eukaryotes. The epigenetic changes underlying mutually exclusive var gene transcription and switching have been studied in some detail. Active var loci are enriched in acetylated H3K9 and H3K4me2/me3 [21], and the process of activation is linked to locus repositioning into an ill-defined transcriptionally active zone at the nuclear periphery [24],[31],[40]. Silenced var loci lack these activation marks and are enriched in H3K9me3 and PfHP1 instead [21],[22],[41]. Furthermore, silencing of var and a subset of rif genes is dependent on the two P. falciparum orthologs of silent information regulator 2 (PfSIR2) [20],[25],[26].
Overall, these results show that conserved epigenetic mechanisms that are also in place in other eukaryotes dictate heterochromatic silencing in P. falciparum. However, it remains completely unknown which proteins and cis-acting sequences are involved in the recruitment and organization of P. falciparum heterochromatin, and how they contribute to the important role of chromosome end biology in this pathogen. Our understanding of sequence-specific DNA-protein interactions in P. falciparum is negligible and mostly limited to the description of upstream sequence elements and their interaction with unknown nuclear proteins, and to in silico mapping of over-represented motifs in candidate promoter sequences [42]. This lack of knowledge is related to the extreme diversity of specific DNA-binding proteins in eukaryotes and the poor representation of such factors in the apicomplexan lineage [43]–[45]. Until recently, PfMYB1 was the only sequence-specific DNA-binding protein that had been investigated to some extent in vivo [46]. New impulses were given by the discovery of the lineage-specific expansion of the ApiAP2 family of putative transcription factors in apicomplexan parasites, characterized by the presence of plant-like AP2 DNA-binding domains [47]. The binding of three parasite AP2 domains to specific cis-acting elements upstream of P. falciparum genes in vitro has recently been demonstrated [48], and another study described an essential role for PbAP2-O in stage-specific transcription in P. berghei ookinetes [49].
Here, we identified a member of the ApiAP2 family as the unknown protein binding to SPE2 arrays upstream of subtelomeric var genes at the border of the non-coding and coding parts of P. falciparum chromosomes [50]. The SPE2-interacting protein, termed PfSIP2, contains two adjacent AP2 domains and is proteolytically processed in vivo to release a 60kDa functional N-terminal domain, PfSIP2-N. We show that both AP2 domains are strictly required for binding to the bipartite SPE2 motif. In vivo, PfSIP2-N is associated with over 700 SPE2 consensus sites that cluster upstream of subtelomeric var genes and in TARE2/3, but not to single chromosome internal sites. Consistent with this striking and exclusive binding of PfSIP2-N to heterochromatic regions, we found no effect of PfSIP2-N over-expression on global gene transcription. Instead, our results imply major roles for PfSIP2-N in var gene silencing and chromosome end biology.
Results
Purification and Identification of PfSIP2, a P. falciparum ApiAP2 Protein Interacting with SPE2 Motifs Upstream of Subtelomeric var Genes
The bipartite SPE2 motif consists of two imperfect 6bp repeats separated by 4bp and its sequence-specific interaction with the unknown nuclear protein is only detectable after the onset of S-phase [50]. We used a high salt schizont stage nuclear extract, pre-cleared by incubation with single-stranded DNA, to purify the SPE2-binding activity based on its affinity to immobilized concatenated SPE2 elements. As control, mutated SPE2M motifs that are unable to interact with the protein were used [50]. EMSA monitoring showed that the SPE2-binding activity was efficiently depleted only after incubation with SPE2 but not SPE2M. Likewise, the activity was eluted only from beads carrying SPE2 but not SPE2M motifs (Figure 1A). Total protein eluted from both beads were precipitated separately, trypsinized and analyzed by LC-MS/MS. Peptide spectra were searched against a combined human and P. falciparum annotated protein database using TurboSequest software [51]. 89 and 82 P. falciparum proteins represented by two or more unique peptides were identified in the SPE2 - and the SPE2M-bound fractions, respectively (Tables S1 and S2). Interestingly, of the 18 proteins exclusively detected in the SPE2 sample (Table 1), two proteins belong to the ApiAP2 family of transcription factors carrying putative sequence-specific AP2 DNA-binding domains, including the second-ranked protein encoded by PFF0200c. Furthermore, six co-purifying proteins have predicted roles in DNA and chromatin metabolism. In contrast, the 15 proteins detected exclusively in the control sample showed no such enrichment (Table S2). Due to the high peptide coverage and a temporal expression profile matching the presence of the SPE2-binding activity in stage-specific nuclear extracts [50],[52],[53], we considered PFF0200c the most likely candidate to encode the SPE2-binding activity.


PFF0200c encodes a large 230kDa protein containing two N-terminal AP2 domains as the only annotated features (Figure 1B). However, UV-crosslinking experiments revealed a 70kDa SPE2-protein complex, which is consistent with an estimated size of 50–60kDa of the SPE2-binding protein (Figure 1C). This discrepancy, and the fact that all seven PFF0200c-derived tryptic peptides mapped to the region containing both AP2 domains (Table S1), prompted us to consider possible proteolytic processing and release of a DNA-binding N-terminal fragment. We therefore expressed epitope-tagged N-terminal fragments of PFF0200c containing both AP2 domains as recombinant proteins in both P. falciparum and E. coli (Figure 1B). Nuclear extracts from 3D7/SIP2-N-HA parasites produced a SPE2-specific complex similar to the one observed in wild-type parasites (Figure 1D), and anti-HA antibodies specifically supershifted the complex obtained with the 3D7/SIP2-N-HA-derived extract only. Similarly, E. coli lysates containing the same fragment as 6×HIS tagged version (SIP2-N-HIS_A) produced a shift of similar size and specificity that was supershifted in presence of anti-6×HIS antibodies. Consistent with the smaller size of the SIP2-N-HIS_B protein, we observed a faster migrating complex of identical specificity. For completeness, we also expressed a 150kDa protein in P. falciparum containing all three AP2 domains of the second ApiAP2 protein PF10_0075 but were unable to detect binding to SPE2 (data not shown). Together, these results unambiguously identified PFF0200c as the P. falciparum SPE2-binding protein, which we termed PfSIP2 (SPE2-interacting protein).
Proteolytic Processing of Inactive Full-Length PfSIP2 is Required to Release the SPE2-binding N-terminal Fragment PfSIP2-N
As indicated by gel shift and UV-crosslinking experiments, the sizes of the endogenous PfSIP2 activity and the N-terminal PfSIP2-N-HA protein were of similar size. To test if PfSIP2 is at all expressed as a full-length protein we generated a transgenic line expressing C-terminally tagged full-length PfSIP2 from the endogenous locus (3D7/SIP2-Ty) (Figure S1). Western analysis of nuclear extracts identified a 250kDa band, consistent with the predicted size of full-length PfSIP2-Ty, specifically in early and late schizonts (Figure 1E, top panel). An additional 150kDa C-terminal fragment (SIP2-C-Ty) appeared in late schizonts indicating that indeed a specific proteolytic event releases an N-terminal DNA-binding isoform. In line with this result, the same early and late schizont extracts produced a typical SPE2/PfSIP2-N complex in EMSA (Figure 1E, bottom panel). These results further suggested that full-length PfSIP2 is unable to interact with SPE2 in vitro. To test this, we performed SPE2 pull-down experiments and confirmed that only N-terminal PfSIP2-N-Ty bound to SPE2 beads, but not full-length PfSIP2-Ty nor the C-terminal fragment PfSIP2-C-Ty (Figure 1F). Together, these results substantiate the existence of a specific proteolytic event to activate the release of the functional DNA-binding protein PfSIP2-N during schizogony.
PfSIP2-N Co-localizes with PfHP1 to Chromosome End Clusters and Interacts with SPE2 Arrays Upstream of var Genes in vivo
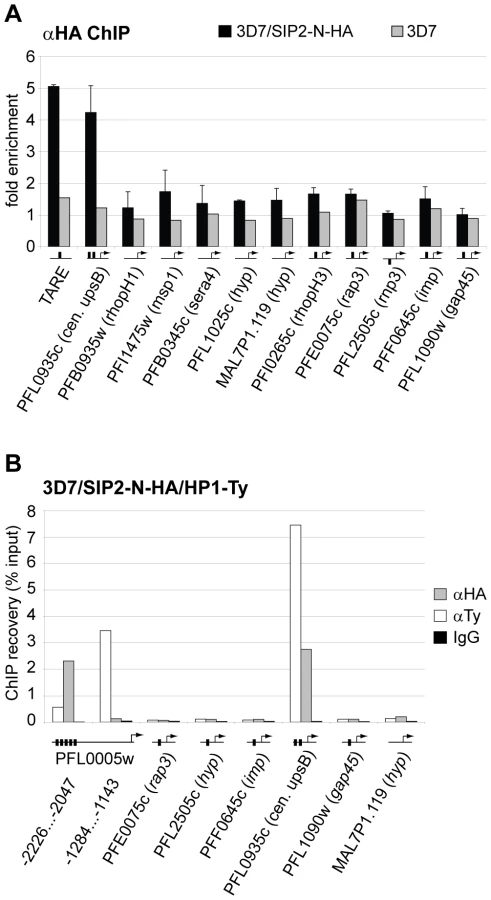
Since SPE2 arrays occur in conserved positions upstream of subtelomeric upsB var genes we expected PfSIP2-N to mark chromosome end clusters. Consistent with this assumption, indirect immunofluorescence (IFA) microscopy identified discrete PfSIP2-N-HA foci at the nuclear periphery with increasing numbers of foci in replicating stages (Figure 2A). To confirm that these signals indeed represented chromosome end clusters we compared the PfSIP2-N-HA signals with those of the heterochromatic marker PfHP1 [27] in a double transgenic line co-expressing PfSIP2-N-HA and PfHP1-Ty simultaneously. As expected, double-labeling IFAs revealed that both proteins co-localized at the nuclear periphery (Figure 2B). Next, we tested if PfSIP2-N binds to SPE2 elements in vivo by targeted chromatin immunoprecipitation (ChIP-qPCR). We observed specific enrichment of PfSIP2-N-HA at the SPE2 array upstream of the upsB var gene PFL0005w while three regions further downstream showed no association (Figure 2C). Together, these findings demonstrate that PfSIP2-N binds specifically to SPE2 arrays upstream of upsB var genes in vivo.

We recently demonstrated that var gene promoters driving expression of the drug-selectable marker hdhfr are silenced by default. Challenge with the antifolate WR99210 allowed selection for activated promoters, which displayed a ring stage-specific temporal activity profile similar to the endogenous promoters [24],[54]. To explore if PfSIP2 participates in the regulation of upsB var gene promoters, we used quantitative reverse transcriptase-PCR (qRT-PCR) to compare the activities of an episomal upsB promoter and a truncated version lacking a 500bp region containing the entire SPE2 array in transfected parasites lines 3D7/upsBR [54] and 3D7/upsBRΔSPE2, respectively, in a time course experiment (Figure S2). As expected, the default state of the wild-type upsB promoter in 3D7/upsBR was silenced. Interestingly, deletion of the region including the SPE2 array resulted in a ten-fold increase in default activity in all three ring stage samples, which is in line with previous results obtained by transient transfection [50]. In their activated states, however, both promoters displayed strong and almost identical activities and temporal profiles (Figure S2). These findings indicate that PfSIP2 may contribute to, but is not the only determinant of, upsB promoter-mediated silencing, and has no role in stage-specific var promoter activity.
The Interaction of PfSIP2-N with SPE2 is Highly Sequence-Specific and Dependent on the Bipartite Nature of Both Interacting Partners
As an important step towards understanding the role of PfSIP2-N in parasite biology, we were interested in scrutinizing the specificity of the PfSIP2-N/SPE2 interaction. Our earlier work demonstrated that two point mutations in either half of the bipartite SPE2 sequence abrogated binding [50]. Intriguingly, a recent study identified GTGCA (which is identical to the first half of the bipartite SPE2 sequence) as consensus motif for PfSIP2-N [48]. These authors also argued that the first AP2 domain alone was sufficient for binding. To clarify these conflicting results, we compared the ability of PfSIP2-N to bind to a bona fide SPE2 motif and to a DNA sequence of identical length carrying the GTGCA motif only. Figure 3A shows that under identical conditions PfSIP2-N binds only to SPE2 but not GTGCA. We next speculated that the bipartite nature of both the SPE2 element and PfSIP2-N with two adjacent AP2 domains reflects the strict requirement of both intact modules for successful interaction. We expressed both AP2 domains separately or in combination as GST-fusions in E. coli and used gel shift assays to confirm that indeed both adjacent AP2 domains are required for binding (Figure 3B, right panel).

Next, we used gel shift competition assays to pinpoint as accurately as possible the minimal sequence requirements for a functional SPE2 consensus motif (Figure 3C and S3). A first set of 16 competitors incorporated single or multiple base deviations within the first and/or second half, and tested the importance of spacing between the half sites. The only changes tolerated were G to C at position one or two in the first or second repeat, respectively; all other changes completely averted binding. The spacing between the half sites was also critical with only four base pairs tolerated. A second set of competitors included eleven SPE2-like motifs naturally occurring upstream of genes coding for invasion-related proteins [55], and two untested SPE2 versions naturally present in SPE2 arrays upstream of var genes (M1.C1, M1.A1C2). Only three motifs competed efficiently (rap3, rhoph3, M1.A1C2) and two competed moderately (MAL6P1.292, M1.C1). These motifs are most closely related to the original SPE2 sequence. Furthermore, in two instances a 5bp spacer was tolerated. Interestingly, the rap2 (non-competing) and rap3 (competing) sequences are identical except for the fifth base in the spacer, indicating that these positions can also contribute to specificity. The competition EMSAs were repeated several times with independent batches of competitors and input protein (both nuclear extracts and E. coli lysates) and yielded identical results (Figure S3 and data not shown). This high degree of sequence-specificity of the PfSIP2-N/SPE2 interaction allowed us to deduce a functional SPE2 consensus motif (Figure 3C). Genome-wide in silico prediction using the consensus motif as query revealed a striking distribution of 777 putative PfSIP2-N binding sites throughout the genome (Tables S3 and S4). 330 sites (42.5%) are associated with the full set of 24 upsB var genes encoded in the genome. The majority of these (262) occur in sense-oriented tandem arrays approx. 2.2kb upstream of 23 subtelomeric upsB loci with an average of eleven motifs spaced by 12bp per locus, and the only chromosome-internal upsB var gene contains two upstream SPE2 sites. 66 sites define a second highly conserved cluster of upsB-associated SPE2 sites approx. 2.7 kb upstream of every subtelomeric locus. Interestingly, 393 sites (51.7%) are located in TAS concentrated in the TARE2/3 region on every chromosome end, with conserved position and orientation and an average of 18 motifs per chromosome end, most of which are spaced by 120bp. Figure 3D shows the right end of chromosome three as a representative example for the conserved arrangement of subtelomeric SPE2 sites on all chromosome ends. Of the remaining 45 sites (5.8%), 30 are located as single motifs in sense orientation upstream of mainly centrally located single copy genes coding for hypothetical proteins, and 15 sites map to coding regions or introns. In summary, this analysis identified a surprising pattern of putative PfSIP2-N-binding sites throughout the genome, with 94% of all predicted motifs confined to two major landmark regions in the subtelomeric domains of P. falciparum chromosomes.
In vivo Binding of PfSIP2-N is Restricted to Subtelomeric SPE2 Landmarks in TAS and Upstream of UpsB-type var Genes
To test our in silico prediction and to identify potential additional PfSIP2-N target sites we performed genome-wide ChIP (ChIP-on-chip) on a high-density whole genome tiling array (NimbleGen Systems Inc.) [27],[37]. Comparison of the genome-wide PfSIP2-N-HA occupancy pattern with the in silico prediction revealed a high degree of overlap (Figure 4 and Table S3). Strikingly, the in vivo association of PfSIP2-N-HA was restricted to SPE2 sites in the predicted landmarks in TARE2/3 and upstream of subtelomeric var genes, both of which are located within H3K9me3/PfHP1-enriched heterochromatin [23],[27],[37]. In contrast, none of the chromosome-internal sites was bound by PfSIP2-N-HA and we observed no enrichment of PfSIP2-N-HA at non-SPE2 loci.

To validate the ChIP-on-chip data we performed ChIP-qPCR on selected loci in 3D7/SIP2-N-HA schizont stage parasites. Figure 5A shows that in addition to SPE2 upstream of upsB var genes (see Figure 2C), PfSIP2-N-HA was also bound to TARE2/3, and interestingly also to the only internal upsB var locus PFL0935c containing two juxtaposed SPE2 motifs. However, we found no specific enrichment at five promoters of internal genes carrying a single SPE2 site. As negative control, we tested the promoters of five genes that are not associated with SPE2. To confirm these results, and to test the anticipated co-occupancy of PfSIP2-N with PfHP1, which was previously shown to be enriched at upsB loci [27], we performed parallel ChIP using chromatin isolated from 3D7/SIP2-N-HA/HP1-Ty schizonts. Indeed, PfHP1 and PfSIP2-N were both enriched at a subtelomeric and the internal upsB locus (Figure 5B), corroborating the findings presented in Figures 2C and 5A. In independent experiments, we used ChIP-re-ChIP to directly confirm the co-occupancy of PfSIP2-N and PfHP1 on the same chromatin fragments in 3D7/SIP2-N-HA/HP1-Ty parasites (Figure S4). Interestingly, in both instances PfHP1 showed a marked reduction directly over the SPE2 array at the PFL0005w locus indicating that in vivo occupancy by PfSIP2-N might be incompatible with local nucleosome formation.
In summary, our combination of in vitro, in silico and in vivo characterisation uncovers a highly specific association of PfSIP2-N with SPE2 consensus motifs on a genome-wide level. The exclusive local restriction of this interaction to telomere-proximal non-coding regions implies important structural and functional roles for PfSIP2-N in subtelomeric heterochromatin formation and chromosome end biology.
Over-expression of PfSIP2-N has no Effect on Transcriptional Regulation of Putative Target Genes
To test a possible role for PfSIP2-N in transcriptional regulation by alternative means we compared the global transcript levels in 3D7/SIP2-N-HA parasites to a mock-transfected line at four stages during the intra-erythrocytic developmental cycle (IDC). Over-expression of PfSIP2-N-HA was evident by up to eight-fold higher levels of pfsip2 transcripts in 3D7/SIP2-N-HA compared to the control (Figure 6). The overall effect of PfSIP2-N-HA over-expression was surprisingly minor with only 21 genes up - or down-regulated by more than three-fold in at least one time point. None of the de-regulated genes is associated with an upstream SPE2 motif and none of the putative PfSIP2-N target genes identified here or by others [48],[55] was affected, arguing strongly against a role of PfSIP2-N in transcriptional activation. Most of the affected genes, including seven non-upsB var genes, are located in heterochromatic regions, which can be explained by stochastic variation in the expression of heterochromatic genes between different isogenic cell lines.

Discussion
Sequence-specific DNA-protein interactions serve to target specific activities to specific sites in the genome and are instrumental in genome organization and gene regulation. Here, we identified PfSIP2, a member of the ApiAP2 family of putative transcription factors, as the unknown nuclear protein binding to SPE2 tandem arrays upstream of subtelomeric var genes. Our comprehensive analysis reveals important novel insights into the nature and specificity of the PfSIP2/SPE2 interaction and provides compelling evidence for a major role of PfSIP2 in chromosome end biology.
Surprisingly, we found that the SPE2-binding activity is not exerted by the full-length protein but rather by a processed N-terminal fragment, PfSIP2-N. The inability of full-length PfSIP2 to interact with SPE2 in vitro indicates that the release of PfSIP2-N does not occur accidentally during extraction but reflects a true proteolytic cleavage event required to activate the DNA-binding activity. Our thorough re-definition of the specificity of the PfSIP2-N/SPE2 interaction in gel shift competition assays allowed us to determine a highly specific SPE2 consensus motif. Both 6bp half sites of the bipartite SPE2 element are strictly required for binding, which is in agreement with our earlier findings [50]. We also show that both adjacent AP2 domains are necessary for binding, probably through reinforcing interactions of each domain with each half of the bipartite motif. Such a scenario is consistent with the binding of single AP2 domains to single 5–6bp motifs in Plasmodium [48],[49] and plants [56],[57], and the specific interaction of the Arabidopsis tandem AP2-domain protein AINTEGUMENTA to a 16bp sequence, which also requires both AP2 domains for binding [58]. These findings challenge the previously proposed role of PfSIP2 in transcriptional activation of genes carrying an upstream GTGCA motif [48]. We clearly show that PfSIP2-N does not bind to GTGCA neither in vitro nor in vivo and, for that matter, consider PfSIP2-N unlikely to act as transcriptional activator of GTGCA-associated genes. We explain these conflicting results by fact that the protein binding microarray technology used in the former study was limited to the screening of random 10mers only, which precluded identification of the 16bp SPE2 element as high affinity binding motif. Our results are highly relevant for our understanding of the role of PfSIP2 in parasite biology, and provide important information for the future investigation of specific ApiAP2-DNA interactions.
Our genome-wide in silico prediction identified 777 PfSIP2-N target sites, 94% of which are located in two distinct landmark regions within subtelomeric heterochromatin on all chromosomes. One cluster corresponds to the previously described tandem arrays upstream of subtelomeric var genes [50], and a second newly identified cluster lies within TARE2/3. In addition, we detected 30 internal SPE2 sites located upstream of single copy genes. Importantly, genome-wide and targeted ChIP analysis of PfSIP2-N occupancy correlated strongly with the in silico prediction of subtelomeric target sites, showing that both subtelomeric landmark regions were bound by PfSIP2-N in vivo. In contrast, PfSIP2-N was absent at internal sites, except for the only internal upsB locus with two upstream SPE2 motifs. Therefore, binding of PfSIP2-N is restricted to heterochromatic regions including the entire subset of upsB var genes. The co-localization of PfSIP2-N with PfHP1 at perinuclear chromosome end clusters and upstream of upsB var genes corroborates this exclusive association. The observed increase in default upsB promoter activity upon deletion of the SPE2 array suggests a role for PfSIP2-N in var gene silencing, possibly through direct or indirect recruitment of effector proteins. However, the overall distribution pattern of PfSIP2-N implies important roles in structural and functional organization and maintenance of P. falciparum chromosome ends that go beyond regulating var gene expression.
In contrast to the well-established and conserved roles of telomere repeat-binding proteins ScRAP1, SpTAZ1, HsTRF1/2 in telomere position effect, telomere length regulation and heterochromatin formation in yeasts and humans, respectively [2], [59]–[64], specific DNA-protein interactions in TAS are hardly known and have only been investigated in detail in S. cerevisiae. One such factor is ABF1, which is involved in silencing, initiation of DNA replication, alteration of chromatin structure and nucleotide excision repair [65]–[70]. Our results are in agreement with similar roles of PfSIP2-N in P. falciparum. First, multiple attempts to generate a PfSIP2 knockout line failed due to refractoriness of the pfsip2 locus to disruption (data not shown). Since pfsip2 was readily accessible for 3′ replacement, we believe PfSIP2 is essential for parasite survival. Second, given the close connection between DNA replication and heterochromatin formation, the specific co-purification of several DNA replication/repair and chromatin remodeling factors (RFC, DNA pol ε, SNF2L, PRS) with PfSIP2-N (Table 1) supports a role in chromosome end maintenance. DNA pol ε and RFC, as well as members of the ATP-dependent chromatin remodeling complexes SWI/SNF, are central players in chromosome end replication and repair [71]–[75] and have important roles in silencing [76],[77]. Interestingly, these factors are also associated with DNA repair at stalled replication forks [78],[79], which can be induced by tight DNA-protein interactions and are crucially involved in recombination at rDNA repeats and mating type switching in S. cerevisiae and S. pombe, respectively [80]. Third, PfSIP2 expression and subsequent release of PfSIP2-N correlate with the DNA replication and nuclear division cycles during schizogony, and it is tempting to speculate that proteolytic activation of PfSIP2-N may occur in a cell cycle-dependent manner. A similar process has been described in activation of the CDP/Cut transcription factor by S phase-specific cleavage [81], and proteolytic processing of various targets, including cyclins, DNA replication factors and cohesin is an important regulatory strategy in cell cycle progression [82]. In summary, these results and observations are consistent with a potential multifunctional role of PfSIP2-N in chromosomal replication and/or segregation and in the nucleation of subtelomeric heterochromatin on newly replicated chromosomes.
ApiAP2 factors have been proposed to act as regulators of stage-specific expression [47],[48], and this was experimentally demonstrated for AP2-O in P. berghei ookinetes [49]. We mapped 45 SPE2 consensus sites in internal regions, mostly located upstream of single copy genes that are transcribed late during the IDC (Table S5). Another study identified SPE2-like motifs upstream of eleven genes coding for invasion-related proteins with similar expression profiles [55]. Interestingly, our ChIP experiments failed to reveal binding of PfSIP2-N to any of these sites. Although we cannot exclude that this lack of association is due to insufficient ChIP sensitivity or physical masking of the HA epitope at internal loci, we believe this finding reflects the true absence of this protein since PfSIP2-N was undoubtedly bound to the only chromosome-central upsB var locus carrying two SPE2 motifs. In addition, over-expression of PfSIP2-N had no effect on transcription of any of these genes. These observations are clearly inconsistent with a role for PfSIP2-N in transcriptional activation. However, we do not rule out a possible function of full-length PfSIP2 in regulation of target loci. First, given the overlap in expression of PfSIP2 with that of SPE2-asscociated genes, a role for PfSIP2 in their activation is conceivable. Although full-length PfSIP2 did not bind to SPE2 in vitro, it is still possible that PfSIP2 binds to and regulates target sites in vivo, possibly in association with other factors. Second, deletion of the SPE2 motif from the rap3 promoter resulted in reduced activity [55] and, conversely, introduction of SPE2 into a heterologous promoter activated transcription in late-stage parasites [54]. Third, PfSIP2 orthologs exist in a subset of other apicomplexan parasites including all sequenced Plasmodium species, yet subtelomeric SPE2 arrays are unique to P. falciparum. In the P. vivax and P. knowlesi genomes, for instance, we predicted 120 and 80 SPE2 consensus sites, respectively, all of which occur as single sites mostly in chromosome-internal regions (data not shown). Hence, if SIP2 has the same binding specificity in other species (which is likely due to the remarkable sequence identity in their DNA-binding domains) then SIP2 must have a function other than being involved in subtelomere biology. It will be interesting to test if processing of PfSIP2 reflects a specific gain-of-function process during evolution of the P. falciparum lineage in order to cope with the massive expansion and control of subtelomeric virulence gene families.
In conclusion, we have identified the first sequence-specific component of subtelomeric regions in P. falciparum. To the best of our knowledge, PfSIP2-N/SPE2 represents a novel type of sequence-specific interaction at chromosome ends that has not been reported in any other eukaryote. Our results are highly relevant in the dissection of the specific biology of P. falciparum chromosome ends, which is key to the evolution and variable expression of subtelomeric virulence gene families, and will help to understand similar processes in other systems. Efforts to analyze a loss-of-function phenotype and to identify PfSIP2 interaction partners will be important future steps into this direction.
Materials and Methods
Parasite culture and transfection
P. falciparum 3D7 parasites were cultured as described previously [83]. Growth synchronisation was achieved by repeated sorbitol lysis [84]. Transfection constructs are described in Protocol S2. Transfections were performed as described [24] and selected on either 5µg/ml blasticidin-S-HCl or 4nM WR99210, or both. To obtain C-terminally tagged endogenous PfSIP2 parasites transfected with pSIP2-2×Ty_3′RP were subject to 3 cycles of growth in presence and absence of WR99210. Plasmid integration was verified by Southern analysis.
Nuclear extracts and EMSA
High salt nuclear extracts and EMSAs were prepared and carried out as described [50] (see also Protocol S1). E. coli lysates were diluted to avoid excess input of recombinant protein. Competition EMSAs were performed in presence of non-specific competitor DNA (1µg sheared salmon sperm DNA and 200fmol random 30 base ss oligonucleotide per reaction) and a 50 - to 100-fold molar excess of specific ds competitors.
UV-crosslinking
Protein samples were incubated for 20min in EMSA buffer with 20fmol 32P-labeled SPE2 probe in presence or absence of a 25-fold molar excess of SPE2 or SPE2M competitors. DNA-protein interactions were UV-crosslinked for 60min (107 Joule) in a Stratalinker 1800 (Stratagene) and separated by SDS-PAGE. Gels were directly exposed to X-ray film.
Affinity purification and identification of the SPE2-binding protein
The complete protocol is explained in detail in Protocol S1. Briefly, the SPE2-binding activity was purified by incubation of schizont stage nuclear extracts with streptavidin magnetic beads carrying immobilized biotinylated SPE2 or SPE2M elements (see Protocol S4 for oligonucleotide sequences). Bound proteins were eluted with 2M KCl, precipitated with 10% TCA and dissolved in 50µl 100 mM Tris-HCl, pH 8.0. Proteins were digested with trypsin and analysed by capillary liquid chromatography tandem mass spectrometry (LC-MS/MS) using an Orbitrap FT hybrid instrument (Thermo Finnigan, San Jose, CA, USA). MS/MS spectra were searched against a combined P. falciparum/human annotated protein database.
Southern blot analysis
To verify successful 3′ replacement at the PFF0200c locus, gDNA from 3D7 wild-type parasites and drug-cycled 3D7/SIP2-Ty parasites was digested with BamHI and HindIII and analysed by Southern blot. The blot was probed with a 32P-dATP-labeled 702bp fragment derived from the 3′ end of PFF0200c.
Recombinant protein expression
Plasmids are described in Protocol S2. Recombinant proteins were expressed in E. coli Tuner (DE3) cells (Novagen) replicating pMICO [85]. After 4h induction using 1mM IPTG at 30°C, bacteria were pelleted and resuspended in 50mM Tris-HCl (pH7.5) containing protease inhibitors (Roche Diagnostics). The suspension was frozen, thawed and sonicated. NaCl, Triton X-100, β-ME and glycerol were added to final concentrations of 0.3M, 0.5%, 10mM and 5%, respectively, followed by centrifugation for 15min at 15,000g and 4°C. Supernatants were used in gel shift assays without further purification.
Western blot analysis and protein pulldown
Primary antibody dilutions were: anti-HA 3F10 (Roche Diagnostics) 1∶2,000; anti-Ty BB2 (kind gift of K. Gull) 1∶10,000; anti-6×HIS (R&D Systems) 1∶5,000. High salt nuclear extracts from 3D7/SIP2-Ty and 3D7/SIP2-N-Ty schizonts were incubated for 1hr with streptavidin agarose beads carrying immobilized SPE2 or mutated SPE2M motifs in EMSA buffer supplemented with non-specific competitor DNA. After centrifugation at 2000rpm the supernatant was saved and beads washed three times in binding buffer. Bound proteins were eluted with 2M KCl.
Immunofluorescence assays
Methanol-fixed cells were analysed using rat anti-HA 3F10 (1∶100) or mouse anti-Ty BB2 (1∶1,000) antibodies. Alexa-Fluor® 568-conjugated anti-rat IgG (Molecular Probes) 1∶500; FITC-conjugated anti-mouse IgG (Kirkegaard Perry Laboratories) 1∶300; TexasRed-conjugated anti-mouse IgG (Molecular Probes) 1∶500. Images were taken on a Leica DM 5000B microscope with a Leica DFC 300 FX camera and acquired via the Leica IM 1000 software and processed and overlayed using Adobe Photoshop CS2.
Genome-wide SPE2 prediction
To identify the full complement of SPE2 consensus elements in P. falciparum, P. vivax and P. knowlesi, genome sequences available at PlasmoDB (version5.5) were searched using the PfSIP2-N-binding consensus sequence determined in competition gel shift assays (Figure 3C and Figure S3). A regular expression search engine (DREG) from the EMBOSS software package was used.
ChIP and ChIP-on-CHIP
ChIP and ChIP-on-chip using formaldehyde-crosslinked chromatin was performed as described in detail elsewhere [27]. PfSIP2-N-HA enrichment at selected loci was tested by ChIP-qPCR using three independent chromatin preparations isolated from 3D7/SIP2-N-HA parasites, and by ChIP - and ChIP-re-ChIP-qPCR using two independent chromatin preparations isolated from the double-transfectant 3D7/SIP2-N-HA/HP1-Ty (qPCR primers are listed in Protocol S4). Enrichment values were calculated by dividing the recovery values obtained by ChIP with anti-HA 3F10 (Roche Diagnostics)/anti-Ty BB2 antibodies with that of the non-specific control IgG antibody. For ChIP-re-ChIP, chromatin fragments immuno-precipitated with anti-Ty BB antibodies were eluted with 50µl elution buffer (1% SDS and 0.1M NaHCO3). After incubation at 65°C for 10min to inactivate the BB2 antibody, the eluate was diluted 6× in incubation buffer lacking SDS (resulting in SDS concentration comparable to that of the first ChIP). Re-ChIP reactions were carried out in the presence of 0.2mg/ml Ty peptide to avoid immuno-precipitation by the BB2 antibody carried over from the first ChIP. Recovery values were defined in the percentage of the input material of the first ChIP. For genome-wide analysis, immunoprecipitated DNA was amplified by a modified T7 linear amplification method and analyzed on a tiling array (based on the May 2005 NCBI sequence of the P. falciparum genome; 385,000 probes with a median spacing of 48bp, Roche NimbleGen) [37].
Quantitative reverse transcriptase PCR
qPCR was performed on reverse transcribed total RNA and gDNA isolated from synchronous parasite cultures at six timepoints across the IDC. A detailed protocol, relative transcript calculation and primer sequences are provided in Protocols S3 and S4.
Transcriptional profiling and data analysis
Growth of 3D7/SIP2-N-HA parasites was tightly synchronized in parallel three times by sorbitol treatment to achieve a ten-hour growth window. Total RNA was isolated at four timepoints across the IDC at early ring stages (4–14 hours post-invasion (hpi)), late ring stages (14–24 hpi), trophozoites (24–34 hpi) and schizonts (32–42 hpi) by lysis of pelleted RBCs in TriReagent (Sigma). Transcript levels in 3D7/SIP2-N-HA were compared to those of the control line 3D7/camHG [27]. RNA samples were analyzed using a P. falciparum microarray as previously described [86]. RNA from each time point was hybridized against a RNA pool assembled from equal amounts of total RNA collected from the 3D7 strain at every eight hours across the IDC.
Accession numbers
PlasmoDB (www.plasmoDB.org) accession numbers for genes and proteins discussed in this publication are: PfSIP2 (PFF0200c); PfHP1 (PFL1005c); upsB var genes (PFL0005w, PFL0935c); rhoph1/clag2 (PFB0935w); rhoph1/clag3.1 (PFC0120w); rhoph1/clag9 (PFI1730w); rhoph2 (PFI1445w); rhoph3 (PFI0265c); rap1 (PF14_0102); rap2 (PFE0080c); rap3 (PFE0075c); rama (MAL7P1.208); rnp3 (PFL2505c); imp, putative (PFF0645c/MAL6P1.292); gap45 (PFL1090w); asp (PFD0295c); msp1 (PFI1475w); sera4 (PFB0345c); conserved Plasmodium proteins (PFL1025c, MAL7P1.119); RFC, subunit 2 (PFB0840w); PfSNF2L (PF11_0053); DNA pol ε, subunit A (PFF1470c); transcription factor with AP2 domains (PF10_0075).
Supporting Information
Zdroje
1. PrydeFE
GorhamHC
LouisEJ
1997 Chromosome ends: all the same under their caps. Curr Opin Genet Dev 7 822 828
2. TaddeiA
HedigerF
NeumannFR
GasserSM
2004 The function of nuclear architecture: a genetic approach. Annu Rev Genet 38 305 345
3. LouisEJ
VershininAV
2005 Chromosome ends: different sequences may provide conserved functions. Bioessays 27 685 697
4. ThamWH
ZakianVA
2002 Transcriptional silencing at Saccharomyces telomeres: implications for other organisms. Oncogene 21 512 521
5. MoazedD
2001 Common themes in mechanisms of gene silencing. Mol Cell 8 489 498
6. BarryJD
GingerML
BurtonP
McCullochR
2003 Why are parasite contingency genes often associated with telomeres? Int J Parasitol 33 29 45
7. SnowRW
GuerraCA
NoorAM
MyintHY
HaySI
2005 The global distribution of clinical episodes of Plasmodium falciparum malaria. Nature 434 214 217
8. BaruchDI
GormelyJA
MaC
HowardRJ
PasloskeBL
1996 Plasmodium falciparum erythrocyte membrane protein 1 is a parasitized erythrocyte receptor for adherence to CD36, thrombospondin, and intercellular adhesion molecule 1. Proc Natl Acad Sci U S A 93 3497 3502
9. GardnerJP
PinchesRA
RobertsDJ
NewboldCI
1996 Variant antigens and endothelial receptor adhesion in Plasmodium falciparum. Proc Natl Acad Sci U S A 93 3503 3508
10. RoweJA
MouldsJM
NewboldCI
MillerLH
1997 P. falciparum rosetting mediated by a parasite-variant erythrocyte membrane protein and complement-receptor 1. Nature 388 292 295
11. ReederJC
CowmanAF
DavernKM
BeesonJG
ThompsonJK
1999 The adhesion of Plasmodium falciparum-infected erythrocytes to chondroitin sulfate A is mediated by P. falciparum erythrocyte membrane protein 1. Proc Natl Acad Sci U S A 96 5198 5202
12. PongponratnE
RigantiM
PunpoowongB
AikawaM
1991 Microvascular sequestration of parasitized erythrocytes in human falciparum malaria: a pathological study. Am J Trop Med Hyg 168 175
13. MacPhersonGG
WarrellMJ
WhiteNJ
LooareesuwanS
WarrellDA
1985 Human cerebral malaria. A quantitative ultrastructural analysis of parasitized erythrocyte sequestration. Am J Pathol 119 385 401
14. BeesonJG
DuffyPE
2005 The immunology and pathogenesis of malaria during pregnancy. Curr Top Microbiol Immunol 297 187 227
15. BaruchDI
PasloskeBL
SinghHB
BiX
MaXC
1995 Cloning the P. falciparum gene encoding PfEMP1, a malarial variant antigen and adherence receptor on the surface of parasitized human erythrocytes. Cell 82 77 87
16. SuXZ
HeatwoleVM
WertheimerSP
GuinetF
HerrfeldtJA
1995 The large diverse gene family var encodes proteins involved in cytoadherence and antigenic variation of Plasmodium falciparum-infected erythrocytes. Cell 82 89 100
17. SmithJD
ChitnisCE
CraigAG
RobertsDJ
Hudson-TaylorDE
1995 Switches in expression of Plasmodium falciparum var genes correlate with changes in antigenic and cytoadherent phenotypes of infected erythrocytes. Cell 82 101 110
18. GardnerMJ
HallN
FungE
WhiteO
BerrimanM
2002 Genome sequence of the human malaria parasite Plasmodium falciparum. Nature 419 498 511
19. ScherfA
Hernandez-RivasR
BuffetP
BottiusE
BenatarC
1998 Antigenic variation in malaria: in situ switching, relaxed and mutually exclusive transcription of var genes during intra-erythrocytic development in Plasmodium falciparum. EMBO J 17 5418 5426
20. Freitas-JuniorLH
Hernandez-RivasR
RalphSA
Montiel-CondadoD
Ruvalcaba-SalazarOK
2005 Telomeric heterochromatin propagation and histone acetylation control mutually exclusive expression of antigenic variation genes in malaria parasites. Cell 121 25 36
21. Lopez-RubioJJ
GontijoAM
NunesMC
IssarN
HernandezRR
2007 5′ flanking region of var genes nucleate histone modification patterns linked to phenotypic inheritance of virulence traits in malaria parasites. Mol Microbiol 66 1296 1305
22. Perez-ToledoK
Rojas-MezaAP
Mancio-SilvaL
Hernandez-CuevasNA
DelgadilloDM
2009 Plasmodium falciparum heterochromatin protein 1 binds to tri-methylated histone 3 lysine 9 and is linked to mutually exclusive expression of var genes. Nucleic Acids Res 37 2596 2606
23. Lopez-RubioJJ
Mancio-SilvaL
ScherfA
2009 Genome-wide analysis of heterochromatin associates clonally variant gene regulation with perinuclear repressive centers in malaria parasites. Cell Host Microbe 5 179 190
24. VossTS
HealerJ
MartyAJ
DuffyMF
ThompsonJK
2006 A var gene promoter controls allelic exclusion of virulence genes in Plasmodium falciparum malaria. Nature 439 1004 1008
25. DuraisinghMT
VossTS
MartyAJ
DuffyMF
GoodRT
2005 Heterochromatin silencing and locus repositioning linked to regulation of virulence genes in Plasmodium falciparum. Cell 121 13 24
26. TonkinCJ
CarretCK
DuraisinghMT
VossTS
RalphSA
2009 Sir2 paralogues cooperate to regulate virulence genes and antigenic variation in Plasmodium falciparum. PLoS Biol 7 e84 doi:10.1371/journal.pbio.1000084
27. FlueckC
BartfaiR
VolzJ
NiederwieserI
Salcedo-AmayaAM
2009 Plasmodium falciparum heterochromatin protein 1 marks genomic loci linked to phenotypic variation of exported virulence factors. PLoS Pathog 5 e1000569 doi:10.1371/journal.ppat.1000569
28. ScherfA
FigueiredoLM
Freitas-JuniorLH
2001 Plasmodium telomeres: a pathogen's perspective. Curr Opin Microbiol 4 409 414
29. FigueiredoLM
PirritLA
ScherfA
2000 Genomic organisation and chromatin structure of Plasmodium falciparum chromosome ends. Mol Biochem Parasitol 106 169 174
30. Freitas-JuniorLH
BottiusE
PirritLA
DeitschKW
ScheidigC
2000 Frequent ectopic recombination of virulence factor genes in telomeric chromosome clusters of P. falciparum. Nature 407 1018 1022
31. MartyAJ
ThompsonJK
DuffyMF
VossTS
CowmanAF
2006 Evidence that Plasmodium falciparum chromosome end clusters are cross-linked by protein and are the sites of both virulence gene silencing and activation. Mol Microbiol 62 72 83
32. GasserSM
HedigerF
TaddeiA
NeumannFR
GartenbergMR
2004 The function of telomere clustering in yeast: the circe effect. Cold Spring Harb Symp Quant Biol 69 327 337
33. FigueiredoLM
Freitas-JuniorLH
BottiusE
Olivo-MarinJC
ScherfA
2002 A central role for Plasmodium falciparum subtelomeric regions in spatial positioning and telomere length regulation. EMBO J 21 815 824
34. NiangM
YanY, X
PreiserPR
2009 The Plasmodium falciparum STEVOR multigene family mediates antigenic variation of the infected erythrocyte. PLoS Pathog 5 e1000307 doi:10.1371/journal.ppat.1000307
35. LavazecC
SanyalS
TempletonTJ
2007 Expression switching in the stevor and Pfmc-2TM superfamilies in Plasmodium falciparum. Mol Microbiol 64 1621 1634
36. MokBW
RibackeU
WinterG
YipBH
TanCS
2007 Comparative transcriptomal analysis of isogenic Plasmodium falciparum clones of distinct antigenic and adhesive phenotypes. Mol Biochem Parasitol 151 184 192
37. Salcedo-AmayaAM
van DrielMA
AlakoBT
TrelleMB
van den ElzenAM
2009 Dynamic histone H3 epigenome marking during the intraerythrocytic cycle of Plasmodium falciparum. Proc Natl Acad Sci U S A 106 9655 9660
38. GrewalSI
JiaS
2007 Heterochromatin revisited. Nat Rev Genet 8 35 46
39. SinghPB
GeorgatosSD
2002 HP1: facts, open questions, and speculation. J Struct Biol 140 10 16
40. RalphSA
Scheidig-BenatarC
ScherfA
2005 Antigenic variation in Plasmodium falciparum is associated with movement of var loci between subnuclear locations. Proc Natl Acad Sci U S A 102 5414 5419
41. ChookajornT
DzikowskiR
FrankM
LiF
JiwaniAZ
2007 Epigenetic memory at malaria virulence genes. Proc Natl Acad Sci U S A 104 899 902
42. HorrocksP
WongE
RussellK
EmesRD
2009 Control of gene expression in Plasmodium falciparum - Ten years on. Mol Biochem Parasitol 164 9 25
43. CoulsonRM
HallN
OuzounisCA
2004 Comparative genomics of transcriptional control in the human malaria parasite Plasmodium falciparum. Genome Res 14 1548 1554
44. AravindL
IyerLM
WellemsTE
MillerLH
2003 Plasmodium biology: genomic gleanings. Cell 115 771 785
45. IyerLM
AnantharamanV
WolfMY
AravindL
2008 Comparative genomics of transcription factors and chromatin proteins in parasitic protists and other eukaryotes. Int J Parasitol 38 1 31
46. GissotM
BriquetS
RefourP
BoschetC
VaqueroC
2005 PfMyb1, a Plasmodium falciparum transcription factor, is required for intra-erythrocytic growth and controls key genes for cell cycle regulation. J Mol Biol 346 29 42
47. BalajiS
BabuMM
IyerLM
AravindL
2005 Discovery of the principal specific transcription factors of Apicomplexa and their implication for the evolution of the AP2-integrase DNA binding domains. Nucleic Acids Res 33 3994 4006
48. De SilvaEK
GehrkeAR
OlszewskiK
LeonI
ChahalJS
2008 Specific DNA-binding by apicomplexan AP2 transcription factors. Proc Natl Acad Sci U S A 105 8393 8398
49. YudaM
IwanagaS
ShigenobuS
MairGR
JanseCJ
2009 Identification of a transcription factor in the mosquito-invasive stage of malaria parasites. Mol Microbiol 71 1402 1414
50. VossTS
KaestliM
VogelD
BoppS
BeckHP
2003 Identification of nuclear proteins that interact differentially with Plasmodium falciparum var gene promoters. Mol Microbiol 48 1593 1607
51. GatlinCL
EngJK
CrossST
DetterJC
YatesJRIII
2000 Automated identification of amino acid sequence variations in proteins by HPLC/microspray tandem mass spectrometry. Anal Chem 72 757 763
52. BozdechZ
LlinasM
PulliamBL
WongED
ZhuJ
2003 The transcriptome of the intraerythrocytic developmental cycle of Plasmodium falciparum. PLoS Biol 1 e5 doi:10.1371/journal.pbio.0000005
53. LlinasM
BozdechZ
WongED
AdaiAT
DeRisiJL
2006 Comparative whole genome transcriptome analysis of three Plasmodium falciparum strains. Nucleic Acids Res 34 1166 1173
54. VossTS
TonkinCJ
MartyAJ
ThompsonJK
HealerJ
2007 Alterations in local chromatin environment are involved in silencing and activation of subtelomeric var genes in Plasmodium falciparum. Mol Microbiol 66 139 150
55. YoungJA
JohnsonJR
BennerC
YanSF
ChenK
2008 In silico discovery of transcription regulatory elements in Plasmodium falciparum. BMC Genomics 9 70
56. Ohme-TakagiM
ShinshiH
1995 Ethylene-inducible DNA binding proteins that interact with an ethylene-responsive element. Plant Cell 7 173 182
57. BakerSS
WilhelmKS
ThomashowMF
1994 The 5′-region of Arabidopsis thaliana cor15a has cis-acting elements that confer cold-, drought - and ABA-regulated gene expression. Plant Mol Biol 24 701 713
58. Nole-WilsonS
KrizekBA
2000 DNA binding properties of the Arabidopsis floral development protein AINTEGUMENTA. Nucleic Acids Res 28 4076 4082
59. ConradMN
WrightJH
WolfAJ
ZakianVA
1990 RAP1 protein interacts with yeast telomeres in vivo: overproduction alters telomere structure and decreases chromosome stability. Cell 63 739 750
60. ZakianVA
1996 Structure, function, and replication of Saccharomyces cerevisiae telomeres. Annu Rev Genet 30 141 172
61. SadaieM
NaitoT
IshikawaF
2003 Stable inheritance of telomere chromatin structure and function in the absence of telomeric repeats. Genes Dev 17 2271 2282
62. KanohJ
SadaieM
UranoT
IshikawaF
2005 Telomere binding protein Taz1 establishes Swi6 heterochromatin independently of RNAi at telomeres. Curr Biol 15 1808 1819
63. CooperJP
NimmoER
AllshireRC
CechTR
1997 Regulation of telomere length and function by a Myb-domain protein in fission yeast. Nature 385 744 747
64. van SteenselB
de LangeT
1997 Control of telomere length by the human telomeric protein TRF1. Nature 385 740 743
65. DiffleyJF
StillmanB
1989 Similarity between the transcriptional silencer binding proteins ABF1 and RAP1. Science 246 1034 1038
66. YuS
SmirnovaJB
FriedbergEC
StillmanB
AkiyamaM
2009 ABF1-binding sites promote efficient global genome nucleotide excision repair. J Biol Chem 284 966 973
67. ReedSH
AkiyamaM
StillmanB
FriedbergEC
1999 Yeast autonomously replicating sequence binding factor is involved in nucleotide excision repair. Genes Dev 13 3052 3058
68. DiffleyJF
StillmanB
1988 Purification of a yeast protein that binds to origins of DNA replication and a transcriptional silencer. Proc Natl Acad Sci U S A 85 2120 2124
69. VendittiP
CostanzoG
NegriR
CamilloniG
1994 ABFI contributes to the chromatin organization of Saccharomyces cerevisiae ARS1 B-domain. Biochim Biophys Acta 1219 677 689
70. PrydeFE
LouisEJ
1999 Limitations of silencing at native yeast telomeres. EMBO J 18 2538 2550
71. BurgersPM
1998 Eukaryotic DNA polymerases in DNA replication and DNA repair. Chromosoma 107 218 227
72. MoserBA
SubramanianL
ChangYT
NoguchiC
NoguchiE
2009 Differential arrival of leading and lagging strand DNA polymerases at fission yeast telomeres. EMBO J 28 810 820
73. MossiR
HubscherU
1998 Clamping down on clamps and clamp loaders–the eukaryotic replication factor C. Eur J Biochem 254 209 216
74. ConawayRC
ConawayJW
2009 The INO80 chromatin remodeling complex in transcription, replication and repair. Trends Biochem Sci 34 71 77
75. CollinsN
PootRA
KukimotoI
Garcia-JimenezC
DellaireG
2002 An ACF1-ISWI chromatin-remodeling complex is required for DNA replication through heterochromatin. Nat Genet 32 627 632
76. Ehrenhofer-MurrayAE
KamakakaRT
RineJ
1999 A role for the replication proteins PCNA, RF-C, polymerase epsilon and Cdc45 in transcriptional silencing in Saccharomyces cerevisiae. Genetics 153 1171 1182
77. DrorV
WinstonF
2004 The Swi/Snf chromatin remodeling complex is required for ribosomal DNA and telomeric silencing in Saccharomyces cerevisiae. Mol Cell Biol 24 8227 8235
78. FrancoAA
LamWM
BurgersPM
KaufmanPD
2005 Histone deposition protein Asf1 maintains DNA replisome integrity and interacts with replication factor 1. Genes Dev 19 1365 1375
79. Van AttikumH
GasserSM
2005 ATP-dependent chromatin remodeling and DNA double-strand break repair. Cell Cycle 4 1011 1014
80. LabibK
HodgsonB
2007 Replication fork barriers: pausing for a break or stalling for time? EMBO Rep 8 346 353
81. MoonNS
PremdasP
TruscottM
LeduyL
BerubeG
2001 S phase-specific proteolytic cleavage is required to activate stable DNA binding by the CDP/Cut homeodomain protein. Mol Cell Biol 21 6332 6345
82. ClarkeDJ
2002 Proteolysis and the cell cycle. Cell Cycle 1 233 234
83. TragerW
JensonJB
1978 Cultivation of malarial parasites. Nature 273 621 622
84. LambrosC
VanderbergJP
1979 Synchronization of Plasmodium falciparum erythrocytic stages in culture. J Parasitol 65 418 420
85. CinquinO
ChristophersonRI
MenzRI
2001 A hybrid plasmid for expression of toxic malarial proteins in Escherichia coli. Mol Biochem Parasitol 117 245 247
86. HuG
LlinasM
LiJ
PreiserPR
BozdechZ
2007 Selection of long oligonucleotides for gene expression microarrays using weighted rank-sum strategy. BMC Bioinformatics 8 350
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2010 Číslo 2
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Caspase-1 Activation via Rho GTPases: A Common Theme in Mucosal Infections?
- Kaposi's Sarcoma Associated Herpes Virus (KSHV) Induced COX-2: A Key Factor in Latency, Inflammation, Angiogenesis, Cell Survival and Invasion
- IL-1β Processing in Host Defense: Beyond the Inflammasomes
- Reverse Genetics in Predicts ARF Cycling Is Essential for Drug Resistance and Virulence
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy