
Mutational Profile of Metastatic Breast Cancers: A Retrospective Analysis
Fabrice Andre and colleagues describe the mutations occurring in a large group of patients with metastatic breast cancer.
Published in the journal:
. PLoS Med 13(12): e32767. doi:10.1371/journal.pmed.1002201
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pmed.1002201
Summary
Fabrice Andre and colleagues describe the mutations occurring in a large group of patients with metastatic breast cancer.
Introduction
Major efforts have been made to characterize early breast cancer at the genomic level [1,2]. These efforts have led to extensive description of genomic alterations involved in tumorigenesis or tumor progression of early breast cancer. These studies report that early breast cancer includes a large number of rare segments characterized by actionable genomic alterations such as PIK3CA mutations, ERBB2 amplification, FGFR1 amplification, CCND1 amplification, AKT1 mutations, and GATA3 mutations [1,2]. Follow-up studies report that C>T mutations at CpG sites are the major mutational pattern in early breast cancer [3]. Although sequencing of primary breast cancer has provided insight into the biology of early malignancy, around 80% of the patients presenting with such a disease will never relapse after conventional therapy. Therefore, understanding the biology of early breast cancer will not help in deciphering the specificities of the lethal disease or translate into treatment advances. Recent data from different types of cancer have suggested that there is a strong heterogeneity between primary tumors and metastases and that genomic profiles of metastases could dramatically differ from primary tumors. Gerlinger and colleagues have shown that only 30% of the mutations are shared between different tumor sites of kidney cancers [4]. Also, Haffner and colleagues have shown that lethal prostate cancer can derive from a minority subclone of the primary tumor [5]. There is therefore a need to extensively describe the genomic alterations observed in metastatic breast cancers in order to identify pathways involved in drug resistance and metastatic processes and to generate new strategies to treat these patients. To this end, we have performed whole-exome sequencing of 216 pairs of metastatic breast cancers and blood and report on the mutational landscape associated with lethal malignancy.
Materials and Methods
The following methodology was specifically developed for this analysis and did not follow an established protocol or analysis plan.
Patients
Metastatic breast cancer patients who underwent a biopsy in the context of the SAFIR01 [6] (NCT01414933), SAFIR02 (NCT02299999), SHIVA [7] (NCT01771458), and MOSCATO (NCT01566019) prospective trials were potentially eligible for this study. These French multicenter trials used high-throughput genome analysis on fresh frozen tumor biopsies as a therapeutic decision tool for metastatic cancer patients, with solid cancers (SHIVA and MOSCATO) or specifically with breast cancer (SAFIR01 and SAFIR02). SAFIR01 included patients with metastatic breast cancers resistant to therapy, and SHIVA and MOSCATO included patients with metastatic cancers eligible for phase I trial, while SAFIR02 included patients with metastatic breast cancers who were starting first - or second-line chemotherapy. Details of each trial are given in S1 Text. Exclusion criteria for the whole-exome sequencing analysis were defined as follows: small or no quantity of tumoral DNA, <30% cancer cells on the biopsy sample (from frozen tissue), and no blood sample available. With these criteria, we identified 86 tumor-normal pairs from patients included in the SAFIR01 trial, 80 pairs in the SAFIR02 trial, 35 pairs in the SHIVA trial, and 15 pairs in the MOSCATO trial (S1 Table). All patients gave their informed consent for translational research and genetic analyses of their somatic DNA. All the studies were approved by the relevant IRBs. Overall, whole-exome sequencing for a total of 216 pairs of metastatic tumor and unmutated DNA derived from corresponding blood samples was performed using Illumina technology. Estrogen (ER) and progesterone (PR) receptors were considered positive if >1% of the cancer cells were stained or when the case was reported positive in the case report form of the trial. HER2 status was determined locally.
Statistical Consideration
Data were summarized by frequency and percentage for categorical variables and by median and range for continuous variables. Comparisons between groups were performed using the Mann-Whitney rank sum test for continuous variables and Chi square or Fisher's exact test for categorical variables. Overall survival (OS) was estimated by using the Kaplan-Meier method, and univariate analyses were performed using the log-rank test. OS was defined as the delay between the inclusion in the trial and death. Patients who were alive were censored at last follow-up news. The Cox proportional hazard regression model was used for multivariate analysis. All variables associated with p < 0.05 on univariate analysis were included in the model. All statistical tests were two sided, and differences were considered statistically significant when p < 0.05. Stata 13.0 software (StatCorp, College Station, Texas) or R version 3.2.2 were used for the statistical analyses. False discovery rate (FDR), used for correcting p-values for multiple hypothesis testing, was computed using the Benjamini-Hochberg procedure.
Whole-Exome Sequencing
Genomic DNA was captured using Agilent in-solution enrichment methodology with their biotinylated oligonucleotides probes library (SureSelect All Exon V5, Agilent, or SureSelect Clinical Research Exome, Agilent), followed by 75-base paired-end massively parallel sequencing on Illumina HiSeq2500, HiSeq4000, or NextSeq500 (S2 Table). For detailed explanations of the process, we refer the reader to the publication by A. Gnirke and colleagues [8]. Sequence capture, enrichment, and elution were performed according to the manufacturer’s instruction and protocols (SureSelect, Agilent) without modification. Briefly, 600 ng of each genomic DNA was fragmented by sonication and purified to yield fragments of 150–200 bp. Paired-end adaptor oligonucleotides from Illumina were ligated on repaired, A-tailed fragments and then purified and enriched by 4–6 PCR cycles. Five hundred ng of these purified libraries was then hybridized to the SureSelect oligo probe capture library for 24 h. After hybridization, washing, and elution, the eluted fraction was PCR amplified for 10–12 cycles, purified, and quantified by qPCR to obtain sufficient DNA template for downstream applications. Each eluted-enriched DNA sample was then sequenced on an Illumina HiSeq2500/4000 or NextSeq500 as paired-end 75 b reads. Image analysis and base calling were performed using Illumina Real Time Analysis Pipeline version 1.12.4.2 with default parameters. Mean coverage was 83 +/ − 18X for normal blood samples and 122 +/ − 15X for tumor samples, with respectively 87% (77%–93%) and 90% (85%–95%) of the targeted regions covered at 20X or more (S2 Table).
Somatic Mutation Calling
Fastq files were aligned to the reference genome hg19 with the BWA mem algorithm [9]. After alignment, the BAM files were treated for PCR duplicate removal and then sorted and indexed with Picard for further analyses. Base recalibration and local realignment around indels were done with GATK [10]. For defining somatic mutations, we used the Mutect [11] (version 1.1.7) algorithm for identifying substitutions and the Scalpel [12] algorithm (version 0.5.2) for identifying small insertions and deletions (indels). Indels occurring in regions with a high number of point mutations detected by Scalpel were filtered out using the GATK VariantFiltration tool with parameters set to 3 mutations in a window of 35 bp. We kept indels of a size lower than 35 bp. We then merged the output of Mutect and Scalpel and further filtered for mutations organized in a cluster of 3 mutations or more in a window of 35 bp using the GATK VariantFiltration tool. We defined the final list of somatic mutations with the following filters: frequency of the reads with the altered base in the tumor > 10%; number of reads with the altered base in the tumor sample ≥ 5; frequency of the reads with the altered base in the normal sample < 2%; number of reads with the altered base in the normal sample ≤ 3; and total coverage in normal and tumor samples ≥ 10. The resulting somatic mutations were annotated with the snpEff and snpSift algorithms [13], and we selected somatic mutations occurring in coding regions only. We removed variants that were also detected in at least one normal sample in our cohort or annotated as known polymorphisms (reported by 1000 Genomes or the ESP databases) unless the variant was also reported in Catalogue of Somatic Mutations in Cancer (COSMIC) [14] or ClinVar (http://www.ncbi.nlm.nih.gov/clinvar/). In order to control for possible biases due to the difference in bait territories from the two capture kits, we verified the mutations that were unique to one bait territory and found that 50 mutations involving 39 genes were unique to one of the bait but none of these mutations affected significantly mutated genes. We filtered six indels after manual inspection. We manually added 2 hotspot mutations (1 His1047Arg PIK3CA [COSM94986] and 1 Glu349* TP53 [COSM140784]) that were originally identified in the tumor in the clinical trial screenings and that were filtered by the somatic mutation filters because of the high frequency of the altered allele in the blood samples (respectively four supporting reads for an allele frequency of 0.022 and seven supporting reads for an allele frequency of 0.11), probably due to circulating tumor DNA. The list of mutations is reported in S3 Table. We computed the cancer cell fraction (CCF) of each mutation using the following steps: we first estimated the tumor purity with Sequenza [15] as well as the copy number at the mutated locus and the number of mutated alleles, as estimated by the altered reads allelic fraction [15]. We then computed the CCF of each mutation using the predicted tumor cellularity by Sequenza, the reference and variant allele read counts at the corresponding chromosomal position, and the estimated copy number at the locus following the framework previously proposed by Carter and colleagues [16]. Mutations were classified as clonal if the 95% confidence interval of the CCF overlapped 1 and as subclonal otherwise. To identify significantly mutated genes, we used the Mutation Significance (MutSig) [17], Mutational Significance in Cancer (MuSiC) [18], and Driver Genes and Pathways (drGAP) [19] algorithms. We defined significantly mutated genes as those with an FDR < 0.1 according to the MutSig algorithm that takes into account more parameters for identifying drivers than the other two algorithms, including gene size, background mutation rate, and replication timing.
Copy Number Analysis
For deriving somatic copy number variations from whole-exome sequence data, we used the following strategy: we first computed the normalized ratio of reads between each tumor and corresponding normal sample using the package ExomeCNV in R and created the segmented profiles with the DNAcopy package. For defining amplifications and deletions, we used the Gistic2 algorithm [20] with the following thresholds for the log2 ratios: amp > 0.3 and del < −0.3. Gistic2 was run including all the samples and specifically for the HR+/HER2 − samples and for the HR−/HER2 − samples in order to control for disease subtypes. Focal peaks are listed in S4 Table.
Mutational Processes
De novo mutational signature analysis was done using the Matlab Welcome Trust Sanger Institute’s signature framework. We used the deconstructSigs R package [21] to determine the contribution of the known signatures that explain each sample mutational profile with more than 50 somatic mutations. We considered the 13 signatures (Signatures 1, 2, 3, 5, 6, 8, 10, 13, 17, 18, 20, 26, and 30) operative in breast cancer as defined in COSMIC (http://cancer.sanger.ac.uk/signatures/matrix.png). A signature was defined as operative or predominant if its contribution to the mutational pattern was respectively >25% (or >100 mutations) or >50%.
{kind=link}
The Cancer Genome Atlas (TCGA) Data
Somatic mutations for breast cancer TCGA cohort were extracted from the genome.wustl.edu_BRCA.IlluminaGA_DNASeq.Level_2.5.3.0.somatic.maf file available for download on the TCGA data matrix website, with somatic mutations available for primary tumors of 772 patients. We extracted ER, PR, and HER2 status from the clinical file downloaded from the TCGA data matrix, retrieving 419 HR+/HER2−, 100 HR−/HER2−, and 145 HER2+. In order to fairly compare mutational loads between TCGA and the metastatic cohort, we downloaded raw data for 33 randomly selected TCGA patients and processed the BAM files with the same pipeline described in this manuscript. We found that the number of mutations identified by our pipeline and by the TCGA pipeline was very similar (S1 Fig, linear regression R2 = 0.98, p < 2e-16). We also verified the identity of mutations called by the two pipelines and found that 80% of the mutations were common to the two pipelines (S2 Fig). Therefore, we used the somatic mutations as defined in the TCGA maf file for comparing the mutation frequencies of the genes.
Results
Patient Characteristics
The population analyzed in the current study included 216 pairs of tumor and normal blood DNA from patients with metastatic breast cancer. Patients were classified in three subgroups according to hormone receptors (HRs; estrogen and progesterone receptors) and HER2 status (Table 1). One hundred and forty-three patients (66%) presented with HR+/HER2 − breast cancer, 51 (24%) with triple-negative breast cancer, and 14 (6%) with HER2-overexpressing breast cancer. Ninety-four percent of the patients had received prior chemotherapy, and 120 (84%) of the patients with HR+/HER2 − disease had received prior endocrine therapy.

Genes Mutated in Metastatic Breast Cancers
We identified 12 driver genes using the MutSig algorithm (FDR < 0.1) (Fig 1, S5 Table). Ten of these genes (TP53, PIK3CA, GATA3, MAP3K1, CDH1, AKT1, MAP2K4, PTEN, CBFB, and CDKN2A) have been previously shown to be frequently mutated in primary breast cancers (>2%, TCGA). In particular, TP53 was mutated in 27% of HR+/HER2 − metastatic breast cancer (mBC) as compared to 20% in HR+/HER2 − early breast cancer (eBC) (Fisher Exact Test p = 0.13) while PIK3CA was mutated in 37% of the HR+/HER2 − mBC and in 40% in eBC.

Two of the driver genes observed in mBC (ESR1 and RB1) were infrequently mutated in primary tumors (<1% of HR+/HER2 − eBC [TCGA]). Twenty-four mutations of ESR1 were identified (1 synonymous, 2 indels, and 21 missense mutations) for a total of 22 mBCs, and these included 22 mutations in 20 out of 143 HR+/HER2 - mBCs (14%). All ESR1 mutations occurred in the hormone receptor domain (S3 Fig) and included mutations in previously reported hotspots [22–24], as well as 2 new mutations (S3 Table). All of these 22 patients had received prior endocrine therapy. RB1 was mutated in 7 out of 143 HR+/HER2 − mBCs (5%) and 3 out of 51 HR−/HER2 − mBCs (6%). Most of the mutations were disruptive, leading to truncated proteins (5 nonsense mutations, 3 splice sites, 1 indel, and 2 missense mutations, S4 Fig). When considering the estimation of the percentage of tumor cells harboring the mutation, i.e., CCF, we found that ESR1 and RB1 mutations were mostly identified as subclonal (ESR1 : 14/21 mutations [67%]; RB1 : 5/10 mutations [50%]). In comparison, PIK3CA and TP53 mutations were identified as subclonal for respectively 32% and 37% of their nonsynonymous mutations.
Mutations Enriched in mBCs
Using a FDR < 0.1, 199 genes out of 1,569 genes tested were more frequently mutated in mBC (n = 216) as compared to eBC (TCGA) (S6 Table). When a FDR < 0.01 was applied, 8 genes (ESR1, FSIP2, AGRN, FRAS1, IGFN1, EDC4, OSBPL3, and PALB2) were found to be more frequently mutated in mBC as compared to eBC (Fig 2). None of these genes, except ESR1, were identified as a driver using MutSig. However, OSBPL3 and PALB2 were both identified as drivers by MuSiC and drGAP at an FDR < 0.1 (S5 Table). PALB2 was mutated in eight (4%) samples, while only one (0.1%) eBC was mutated in TCGA (FDR for enrichment in mBC = 0.006). Out of the 8 PALB2 mutations, 5 were found in HR+/HER2 − mBC. None of the cases with PALB2 somatic mutations presented with a PALB2 deleterious germline polymorphism in the other allele. We analyzed outcome data for comparing the OS of patients with metastatic tumors carrying at least one of the mutations enriched in the metastatic setting (n = 76) to the rest of the population (n = 140). Results of the univariate and multivariate analyses are reported in S7 and S8 Tables. In a multivariate analysis, mBC with at least one mutation in the 8 genes enriched in the metastatic setting presented a 2-fold increase in the hazard of death (hazard ratio = 1.97, 95% CI: 1.34–2.89, p = 0.001). Survival curves are reported in Fig 3.


As this analysis might be biased by the difference in distribution of HR and HER2 subtypes between eBC and mBC, we also focused the analysis on the HR+/HER2 − subtype (n = 143), in which 278 genes were more frequently mutated in mBC as compared to eBC (FDR < 0.1, S6 Table). Several of these genes were considered actionable. TSC1 and TSC2 were mutated in five (3.5%) and four (2.8%) samples, respectively (Fig 4). Overall, 6.3% of HR+/HER2 − mBC presented an alteration in TSC1/2 as opposed to 0.7% of HR+/HER2 − eBC (TCGA, p = 0.0004). Other actionable genes were more frequently mutated in HR+ mBC with an FDR < 0.1. These include ERBB4 (nine missense mutations, including the two mutations COSM4764538 and COSM1015992, involving eight mBCs [five HR+/HER2−]), NOTCH3 (eight missense and one splice site mutation(s) involving seven mBCs [four HR+/HER2−]), ALK (five missense and two splice site mutations in seven mBCs [six HR+/HER2−]), EZH2 (two missense and one splice site mutation(s), including COSM220530, involving three HR+/HER2 − mBCs) and BRAF (four missense mutations, including one COSM476, involving four mBCs [three HR+/HER2−]). The consequence of these mutations on the activity of the encoded proteins was difficult to assess as, even though ERBB4 and NOTCH3 mutations were all missense mutations (except for one splice site mutation in NOTCH3), they were located in different protein domains with no apparent hotspot (Fig 4).

Mutational Signatures in mBCs
First, in order to identify a potential metastatic-specific mutational signature, we performed de novo mutational signature analysis that revealed five signatures operative in metastatic and primary breast cancer [25], but none of these signatures were specific to the metastatic setting (S1 Text, S9 Table and S5 and S6 Figs). We then assessed the contribution of 13 mutational signatures [25] in 118 metastatic samples and 278 primary tumors from TCGA presenting >50 mutations (S10 Table). Among the 13 signatures previously identified as operative in primary breast cancer, the most represented signatures in the metastatic samples were signature 1, related to aging; signatures 2 and 13, related to APOBEC3B activity; signature 3, associated with failure of DNA double-strand break-repair by homologous recombination; and signature 6, associated with defective DNA mismatch repair (Fig 5). While the identity of the signatures remained the same between primary and metastatic samples, their contribution dramatically changed, especially in the HR+/HER2 − subtype (Fig 6 and S7 and S8 Figs). Of note, the signatures related to the APOBEC3B enzyme (signatures 2 and 13) contributed to 58.8% of the mutations of the HR+/HER2 − metastatic tumors as compared to only 31.9% in the primary TCGA samples (p < 2e-16), confirming previous work demonstrating a link between APOBEC-mediated mutagenesis and the acquisition of subclonal mutations [26].
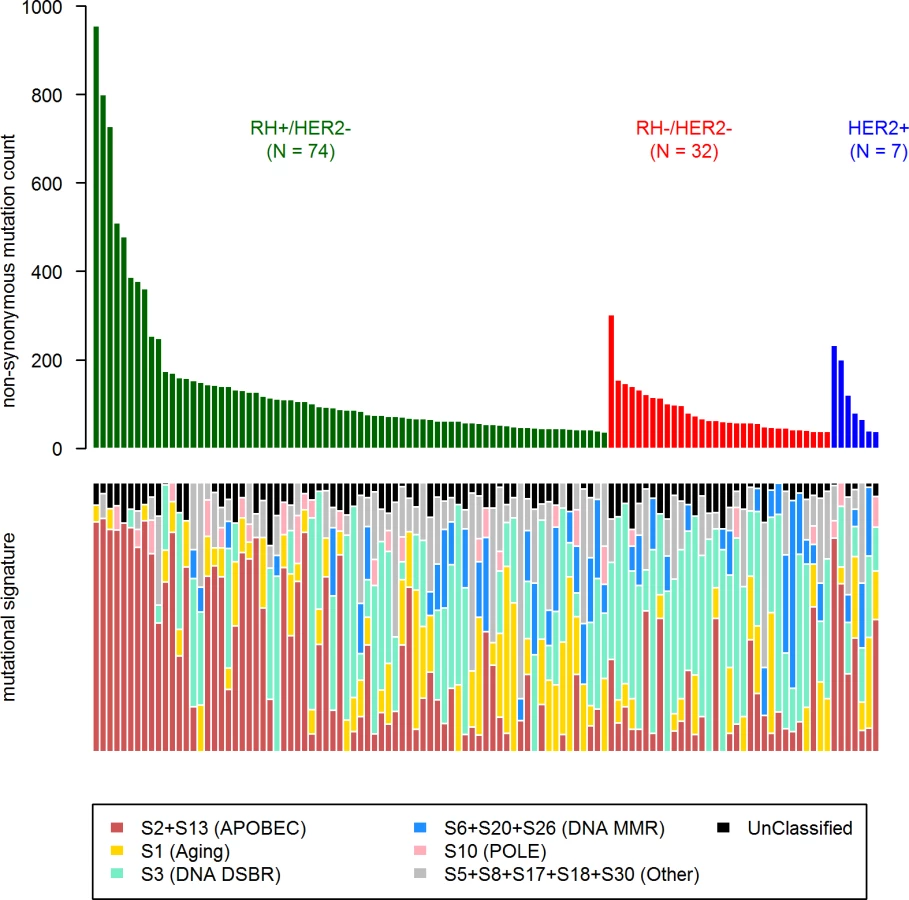

Copy Number Alterations in mBCs
Gistic2 analysis using sequence-based levels reported regions previously described to drive oncogenesis of primary tumors including amplified genes CCND1, ERBB2, and MYC and lost gene PTEN (S4 Table). In addition to these previously reported gene amplifications and deletions, the current study identifies a focal amplification of the ESR1 locus confirming the mutational pattern of the gene. ESR1 was amplified in 7 HR+/HER2 − mBCs for a total of 27 HR+/HER2 − mBCs (19%) with either ESR1 mutation or amplification (Fig 1). Additionally, RB1 was lost in 2 HR+/HER2 − mBCs for a total of 9 samples (6%) with either RB1 mutation or homozygous deletion.
Finally, we computed two indices to describe the chromosomal instability of the metastatic samples based on the copy number analysis as previously described [27]: a global genomics index (GGI) and the number of breakpoints per sample (S1 Table). We found that the number of mutations per tumor did not correlate with either the GGI or the number of break points (S9 Fig). We also verified that the mutational load and the chromosomal instability were not affected by the tumor cell content of the samples. While we found that there was no correlation between cellularity and estimated chromosomal instability, we found a positive correlation between the percentage of tumor cells and the number of mutations (Pearson’s cor = 0.16, p = 0.02). However, among the five samples with no nonsynonymous mutations, only two samples had <50% tumor cells, while the other three had >70% tumor cells.
Discussion
In the present manuscript, we have described the mutational landscape of 216 mBCs. This study reported genes significantly mutated in mBC and genes significantly more mutated in mBC as compared to eBC. HR+/HER2 − mBC presented the most differences with HR+/HER2 − eBC, including an increased mutational signature linked to APOBEC3B activity and a higher prevalence of actionable genes that may represent new strategies for mBC treatment.
Using a stringent definition (MutSig, FDR < 0.1), the current study identified ESR1 and RB1 as driver genes that are specific to mBCs. Previous studies [23,24,28] have already reported that ESR1 mutations could be acquired during the disease evolution and could mediate resistance to endocrine therapy. In the present study, we confirm that mutation of ESR1 is the most frequent “metastasis-specific” mutation observed in mBC. As expected, all the 22 patients who presented ESR1 mutations were ER+ and resistant to endocrine therapy. ESR1-mutated mBC could be a genomic segment defining an unmet medical need, for which fast-track approval of new agents is required. Rb1 is a tumor suppressor protein involved in cell cycle and phosphorylated by CDK4. The protein is required for the bioactivity of palbociclib (CDK4 inhibitor), a drug recently approved to treat HR+/HER2 − mBC [29]. The present study identifies RB1 mutations, most of them pointing to a loss of function of the protein, as driver alterations in mBCs; while this gene is almost never mutated in HR+/HER2 − eBC (<1%), it was found mutated in 5% of the HR+/HER2 − mBC (p = 0.008, FDR = 0.09). This finding suggests that a subset of HR+/HER2 − mBC is deficient for RB1 and could present a primary resistance to CDK4 inhibitors. If validated, this finding suggests that RB1 mutations should be assessed on metastatic samples before starting CDK4 inhibitors.
Several genes were more frequently mutated in mBC as opposed to eBC but did not meet the criteria for drivers using the MutSig algorithm. PALB2 is a partner of BRCA1/2 and is involved in Fanconi anemia. Heterozygous loss-of-function mutations in PALB2 have been shown to be a risk factor for breast cancer, while PALB2 germline mutations have recently been associated with a poor outcome [30]. Several studies have suggested that PALB2-deficient cancers could be sensitive to PARP inhibitors. In the present study, PALB2 somatic mutations were found in 4% of metastatic samples (n = 8), while the gene is mutated in only 0.1% of eBC (FDR = 0.006). The present results suggest that there is a population of PALB2-deficient mBC in which PARP inhibitors could be evaluated. Genes located on the mTOR pathway (TSC1 and TSC2) were more frequently mutated in HR+/HER2 − mBC (6%) as opposed to HR+/HER2 − eBC (0.7%). All these mutations were observed in patients previously treated with endocrine therapy, suggesting that it could be a mechanism of resistance. mTOR inhibitors (everolimus) have been approved in HR+/HER2 − mBC [31]. While this drug prolongs progression-free survival (PFS) for a majority of patients, only a few percent of them are outlier responders to this drug, and there is currently no molecular alteration that explains such cases. Further studies should evaluate whether the subset of patients with genomic alterations on mTOR pathways (TSC1 and TSC2) could be outlier responders to everolimus. Other actionable genes were more frequently mutated in HR+ mBC, including ERBB4, NOTCH3, and ALK.
Analysis of mutational processes did not identify any signature specific to the metastatic setting but revealed a high increase in APOBEC-mediated mutations in HR+/HER2 − mBC as compared to eBC. As for metastatic-specific mutations identified, this might also present a mechanism of resistance to therapy that needs further careful investigation.
The study included 216 sample pairs classified in three different classes based on hormonal receptor expression in the tumors, the largest group being HR+/HER2 − (n = 143). Although this is the largest effort for profiling the mutational landscape of mBC to this day, the size of the cohort presents a limitation to the identification of rare events, especially in the triple-negative and HER2+ groups. Our ability to provide an exhaustive picture of mutational profiles of mBC may be limited by two main biases. The first bias comes from the absence of bone metastases in the study, due to the difficulty of extracting DNA from these lesions. A second potential bias may come from our inability to identify those mutations leading to a disease so aggressive that the patients will not be eligible for trial recruitment, preventing an exhaustive picture of the mutational profiles of mBC. However, recent studies on first-line therapies for advanced breast cancer have shown that early death is limited [32,33], and it is therefore unlikely that it has dramatically impacted the analysis. Additionally, there is a chance that mutations enriched in metastasis might be subclonal driver events and therefore might not be such good drug targets. The comparison of gene mutational prevalence between mBC and eBC suffers from two limitations. First, it would have been ideal to directly compare the mBCs with their corresponding primary tumor profiles, but this was not possible because of the obvious reason of sample availability. Second, we used mutational profiles of TCGA tumors that were identified by the TCGA team, whereas it would have been ideal to run the pipeline used for mBC on the TCGA data. Although we controlled for major biases, the use of different bioinformatics pipelines may have some unexpected consequences. It should also be noted that the copy number analysis is limited by the nature of the sequencing data, which does not allow for a uniform coverage of the genome. Finally, the survival analysis based on the mutational status of the eight metastasis-specific genes was independent from any other parameters such as mutational load or mutational signature contribution, making it difficult to establish a causal link between mutated genes and prognosis.
The dataset and the accompanying analysis described in this study provide a better understanding of the genetic basis of mBC and how much it differs from that of primary breast tumors. This study demonstrated that profiling metastatic cancer can be a major step in defining optimal treatments for patients, as new mutation events and processes may arise during cancer treatment. Follow-up studies will be essential for validating resistance mechanisms identified in this study.
Supporting Information
Zdroje
1. Ellis MJ, Ding L, Shen D, Luo J, Suman VJ, Wallis JW, et al. Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature. 2012;486(7403):353–60. PubMed Central PMCID: PMCPMC3383766. doi: 10.1038/nature11143 22722193
2. The Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. doi: 10.1038/nature11412 23000897
3. Alexandrov LB, Nik-Zainal S, Wedge DC, Campbell PJ, Stratton MR. Deciphering signatures of mutational processes operative in human cancer. Cell Rep. 2013;3(1):246–59. PubMed Central PMCID: PMCPMC3588146. doi: 10.1016/j.celrep.2012.12.008 23318258
4. Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366(10):883–92. doi: 10.1056/NEJMoa1113205 22397650
5. Haffner MC, Mosbruger T, Esopi DM, Fedor H, Heaphy CM, Walker DA, et al. Tracking the clonal origin of lethal prostate cancer. J Clin Invest. 2013;123(11):4918–22. PubMed Central PMCID: PMC3809798. doi: 10.1172/JCI70354 24135135
6. Andre F, Bachelot T, Commo F, Campone M, Arnedos M, Dieras V, et al. Comparative genomic hybridisation array and DNA sequencing to direct treatment of metastatic breast cancer: a multicentre, prospective trial (SAFIR01/UNICANCER). Lancet Oncol. 2014;15(3):267–74. doi: 10.1016/S1470-2045(13)70611-9 24508104
7. Le Tourneau C, Delord JP, Gonçalves A, Gavoille C, Dubot C, Isambert N, et al. Molecularly targeted therapy based on tumour molecular profiling versus conventional therapy for advanced cancer (SHIVA): a multicentre, open-label, proof-of-concept, randomised, controlled phase 2 trial. Lancet Oncol. 2015;16(13):1324–34. doi: 10.1016/S1470-2045(15)00188-6 26342236
8. Gnirke A, Melnikov A, Maguire J, Rogov P, LeProust EM, Brockman W, et al. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nat Biotechnol. 2009;27(2):182–9. PubMed Central PMCID: PMCPMC2663421. doi: 10.1038/nbt.1523 19182786
9. Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26(5):589–95. PubMed Central PMCID: PMC2828108. doi: 10.1093/bioinformatics/btp698 20080505
10. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–303. PubMed Central PMCID: PMC2928508. doi: 10.1101/gr.107524.110 20644199
11. Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31(3):213–9. PubMed Central PMCID: PMC3833702. doi: 10.1038/nbt.2514 23396013
12. Narzisi G, O'Rawe JA, Iossifov I, Fang H, Lee YH, Wang Z, et al. Accurate de novo and transmitted indel detection in exome-capture data using microassembly. Nat Methods. 2014;11(10):1033–6. PubMed Central PMCID: PMCPMC4180789. doi: 10.1038/nmeth.3069 25128977
13. Cingolani P, Platts A, Wang le L, Coon M, Nguyen T, Wang L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly. 2012;6(2):80–92. PubMed Central PMCID: PMC3679285. doi: 10.4161/fly.19695 22728672
14. Forbes SA, Beare D, Gunasekaran P, Leung K, Bindal N, Boutselakis H, et al. COSMIC: exploring the world's knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015;43(Database issue):D805–11. doi: 10.1093/nar/gku1075 25355519
15. Favero F, Joshi T, Marquard AM, Birkbak NJ, Krzystanek M, Li Q, et al. Sequenza: allele-specific copy number and mutation profiles from tumor sequencing data. Ann Oncol. 2015;26(1):64–70. PubMed Central PMCID: PMCPMC4269342. doi: 10.1093/annonc/mdu479 25319062
16. Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, et al. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol. 2012;30(5):413–21. PubMed Central PMCID: PMCPMC4383288. doi: 10.1038/nbt.2203 22544022
17. Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499(7457):214–8. PubMed Central PMCID: PMCPMC3919509. doi: 10.1038/nature12213 23770567
18. Dees ND, Zhang Q, Kandoth C, Wendl MC, Schierding W, Koboldt DC, et al. MuSiC: identifying mutational significance in cancer genomes. Genome Res. 2012;22(8):1589–98. PubMed Central PMCID: PMCPMC3409272. doi: 10.1101/gr.134635.111 22759861
19. Hua X, Xu H, Yang Y, Zhu J, Liu P, Lu Y. DrGaP: a powerful tool for identifying driver genes and pathways in cancer sequencing studies. Am J Hum Genet. 2013;93(3):439–51. PubMed Central PMCID: PMCPMC3769934. doi: 10.1016/j.ajhg.2013.07.003 23954162
20. Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, Getz G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011;12(4):R41. PubMed Central PMCID: PMCPMC3218867. doi: 10.1186/gb-2011-12-4-r41 21527027
21. Rosenthal R, McGranahan N, Herrero J, Taylor BS, Swanton C. deconstructSigs: delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol. 2016;17(1):31. PubMed Central PMCID: PMCPMC4762164.
22. Li S, Shen D, Shao J, Crowder R, Liu W, Prat A, et al. Endocrine-therapy-resistant ESR1 variants revealed by genomic characterization of breast-cancer-derived xenografts. Cell Rep. 2013;4(6):1116–30. PubMed Central PMCID: PMCPMC3881975. doi: 10.1016/j.celrep.2013.08.022 24055055
23. Toy W, Shen Y, Won H, Green B, Sakr RA, Will M, et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat Genet. 2013;45(12):1439–45. PubMed Central PMCID: PMCPMC3903423. doi: 10.1038/ng.2822 24185512
24. Robinson DR, Wu YM, Vats P, Su F, Lonigro RJ, Cao X, et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat Genet. 2013;45(12):1446–51. PubMed Central PMCID: PMCPMC4009946. doi: 10.1038/ng.2823 24185510
25. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature. 2013;500(7463):415–21. Epub 2013/08/16. PubMed Central PMCID: PMCPmc3776390. doi: 10.1038/nature12477 23945592
26. McGranahan N, Favero F, de Bruin EC, Birkbak NJ, Szallasi Z, Swanton C. Clonal status of actionable driver events and the timing of mutational processes in cancer evolution. Sci Transl Med. 2015;7(283):283ra54. PubMed Central PMCID: PMCPMC4636056. doi: 10.1126/scitranslmed.aaa1408 25877892
27. Orsetti B, Selves J, Bascoul-Mollevi C, Lasorsa L, Gordien K, Bibeau F, et al. Impact of chromosomal instability on colorectal cancer progression and outcome. BMC Cancer. 2014;14 : 121. PubMed Central PMCID: PMCPMC4233623. doi: 10.1186/1471-2407-14-121 24559140
28. Jeselsohn R, Buchwalter G, De Angelis C, Brown M, Schiff R. ESR1 mutations—a mechanism for acquired endocrine resistance in breast cancer. Nat Rev Clin Oncol. 2015;12(10):573–83. doi: 10.1038/nrclinonc.2015.117 26122181
29. Turner NC, Huang Bartlett C, Cristofanilli M. Palbociclib in Hormone-Receptor-Positive Advanced Breast Cancer. N Engl J Med. 2015;373(17):1672–3.
30. Cybulski C, Kluzniak W, Huzarski T, Wokolorczyk D, Kashyap A, Jakubowska A, et al. Clinical outcomes in women with breast cancer and a PALB2 mutation: a prospective cohort analysis. Lancet Oncol. 2015;16(6):638–44. doi: 10.1016/S1470-2045(15)70142-7 25959805
31. Baselga J, Campone M, Piccart M, Burris HA, Rugo HS, Sahmoud T, et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med. 2012;366(6):520–9. doi: 10.1056/NEJMoa1109653 22149876
32. Hortobagyi GN, Stemmer SM, Burris HA, Yap YS, Sonke GS, Paluch-Shimon S, et al. Ribociclib as First-Line Therapy for HR-Positive, Advanced Breast Cancer. N Engl J Med. 2016;375(18):1738–48. doi: 10.1056/NEJMoa1609709 27717303
33. Lobbezoo DJ, van Kampen RJ, Voogd AC, Dercksen MW, van den Berkmortel F, Smilde TJ, et al. In real life, one-quarter of patients with hormone receptor-positive metastatic breast cancer receive chemotherapy as initial palliative therapy: a study of the Southeast Netherlands Breast Cancer Consortium. Ann Oncol. 2016;27(2):256–62. doi: 10.1093/annonc/mdv544 26578730
Štítky
Interní lékařstvíČlánek vyšel v časopise
PLOS Medicine
2016 Číslo 12
- Ivabradin zlepšuje kvalitu života starších pacientů se srdečním selháním
- Fixní kombinace kandesartan/amlodipin v terapii arteriální hypertenze
- Inovace v hojení ran: krytí Zetuvit Plus Silicone Border pro optimální management exsudátu z ran
- Rána vizitkou (nejen) chirurga
- Patogeneze vzniku keloidní jizvy
Nejčtenější v tomto čísle
- Genomic Analysis of Uterine Lavage Fluid Detects Early Endometrial Cancers and Reveals a Prevalent Landscape of Driver Mutations in Women without Histopathologic Evidence of Cancer: A Prospective Cross-Sectional Study
- Overcoming Steroid Resistance in T Cell Acute Lymphoblastic Leukemia
- Clonal Evolutionary Analysis during HER2 Blockade in HER2-Positive Inflammatory Breast Cancer: A Phase II Open-Label Clinical Trial of Afatinib +/- Vinorelbine
- Predictors of Chemosensitivity in Triple Negative Breast Cancer: An Integrated Genomic Analysis
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy