
JAK/STAT and Hox Dynamic Interactions in an Organogenetic Gene Cascade
Organogenesis is controlled by gene networks activated by upstream selector genes. To address how the network organization changes during development and how the target genes integrate the genetic information it provides, we analyze in Drosophila the induction of posterior spiracle organogenesis by the Hox gene Abdominal-B (Abd-B). Initially, Abd-B activates in the spiracle primordium a cascade of transcription factors and signalling molecules including the JAK/STAT pathway. We find that at later stages STAT activity feeds back into Abd-B, initiating the transformation of the Hox cascade into a gene-network. Focusing on a spiracle downstream target gene of Abd-B, we analyze how its cis regulatory elements integrate the dynamic network information set by Abd-B and the JAK/STAT signalling pathway during development. Our results also show that the well known transcription factor STAT can control gene expression as a “counter-repressor”, uncovering an alternative novel mode for STAT directed transcriptional regulation.
Published in the journal:
. PLoS Genet 11(7): e32767. doi:10.1371/journal.pgen.1005412
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1005412
Summary
Organogenesis is controlled by gene networks activated by upstream selector genes. To address how the network organization changes during development and how the target genes integrate the genetic information it provides, we analyze in Drosophila the induction of posterior spiracle organogenesis by the Hox gene Abdominal-B (Abd-B). Initially, Abd-B activates in the spiracle primordium a cascade of transcription factors and signalling molecules including the JAK/STAT pathway. We find that at later stages STAT activity feeds back into Abd-B, initiating the transformation of the Hox cascade into a gene-network. Focusing on a spiracle downstream target gene of Abd-B, we analyze how its cis regulatory elements integrate the dynamic network information set by Abd-B and the JAK/STAT signalling pathway during development. Our results also show that the well known transcription factor STAT can control gene expression as a “counter-repressor”, uncovering an alternative novel mode for STAT directed transcriptional regulation.
Introduction
Organogenesis is controlled by the activation of complex gene regulatory networks in precise positions of the organism [1]. Selector genes, or master regulator genes, encode transcription factors required for the expression of entire organogenetic gene regulatory networks and, when expressed ectopically, can induce the formation of additional organs at new locations [2,3]. Prominent examples are Eyeless (Ey) capable of inducing ectopic eyes; or the Hox proteins capable of inducing segment specific organs [1,4]. However, these genes by themselves are unable to provide all the information required to specify an organ. For example, the organ specified by a particular Hox protein varies at different positions of the segment depending on its interaction with tissue-specific transcription factors and signalling pathway effectors active in each region. Similarly, ectopic Ey expression can only induce the formation of additional eyes at certain locations of the imaginal discs showing that the functional outcome of these proteins is locally modulated. Once a regulatory gene network is selected, each network gene has to be precisely activated in time and space and this is usually controlled through the gene’s non-coding cis-regulatory modules (CRMs). Thus, CRMs play a critical role since they act as integrators of specific combinations of transregulatory transcription factors and signalling pathway effectors, resulting in localized transcriptional activity.
Examples of Hox-induced organogenetic networks include the formation of the corpora allata or the maxillary cirri by Deformed (Dfd); the development of the prothoracic or the salivary glands by Sex combs reduced (Scr); or the formation of the posterior spiracles by Abdominal-B (Abd-B) [5–8]. In all these cases the Hox protein is the most upstream activator of an organogenetic gene-cascade. This simple and linear view of the role of Hox genes in the establishment of gene networks, contrasts with what is known for other selector genes. Eye development shows a more complex situation where Ey is not simply upstream of a linear cascade but forms part of the gene network itself, collaborating with other transcription factors as Eyes absent, Sine oculis or Dachshund that are equally important to induce eye organogenesis [9].
The formation of the posterior spiracles of Drosophila is an excellent model to study how a Hox protein controls organogenesis [8,10]. The posterior spiracles constitute the only external opening of the trachea when the larva hatches so their development has to be completed during embryogenesis. Posterior spiracle organogenesis is induced in the eighth abdominal (A8) segment when the Abdominal-B (Abd-B) Hox selector protein activates a series of primary targets that include the transcription factor genes cut (ct), empty spiracles (ems) and spalt (sal) as well as the unpaired (upd) and upd2 genes encoding ligands for the JAK/STAT pathway receptor. Downstream of these primary targets, a number of secondary target genes are activated which require the activity of the primary targets to be expressed. Secondary targets include other transcription factors, but also genes encoding realizators of organogenesis, like non-classic cadherins involved in cell adhesion, RhoGAP and RhoGEF GTPase regulatory proteins influencing cytoskeletal organization, and cell polarity determinants [11]. Central among the cell polarity genes is crumbs (crb), one of the major determinants of apico-basal polarity [12,13]. Crb is a transmembrane protein that localizes to the subapical region of all ectodermal cells where it is required to maintain the epithelial structure by interaction with cortical proteins also involved in apico-basal polarity regulation. Although Crb is ubiquitous in all ectodermal cells, its expression is increased in the cells that invaginate to form the posterior spiracles (S1A and S1B Fig and [11]). The enhancement of crb transcription in the spiracle primordium is controlled by a posterior spiracle specific CRM regulated by JAK/STAT signalling (S1D and S1E Fig). In turn, JAK/STAT signalling in the posterior spiracles depends on upd transcription that is controlled by Abd-B (S1C and S1F Fig and [11]). Although this simple linear model, where the crb-spiracle enhancer expression is solely dependent on JAK/STAT activation, can explain why it is expressed in the spiracle primordia of A8 (S1C and S1D Fig), it does not explain why this enhancer is not also activated in the tracheal pits, where upd is transcribed and the JAK/STAT signalling cascade is also active (S1C and S1G–S1G” Fig). Thus, the regulation of the crb posterior spiracle CRM must be more complex to attain organ specificity.
To understand how organ specific gene expression is regulated in a Hox-induced morphogenetic cascade during organogenesis, we have studied the transcriptional activation of crb in the posterior spiracles. Our results show that spiracle specific expression of this downstream secondary target requires the cooperation of the primary targets and the Abd-B Hox selector protein. Moreover, we show that the simple linear cascade initiated by Abd-B early in embryogenesis, soon becomes a complex network due to feedback loops set up by the primary targets. Furthermore, we find evidence that STAT can influence gene expression in a novel way, not just as a positive transcription factor, but also as a counter-repressor. The crb posterior spiracle expression is controlled by an unanticipated number of interactions between modular elements of the CRM that influence the final transcriptional outcome.
Results
A crb518 minimal enhancer directs expression to the posterior spiracle in a JAK/STAT dependent manner
The reporter construct crb43.2-lacZ contains a 2kb crb intronic region driving posterior spiracle expression. It was shown that the activity of this reporter depends on activation of JAK/STAT signalling and that mutation of the STAT binding sites present in this element downregulate its activity [[11,14] and S1 Fig, compare E and F]. These initial studies suggested that the crb43.2 enhancer is a direct target of the JAK/STAT pathway. To confirm this observation, we expressed activated STAT-GFP in S2 cells and performed Chromatin Immunoprecipitation (ChIP) using an anti-GFP antibody (S2A Fig). The enrichment of input recovery shows that STAT is able to bind directly to the crb enhancer, reinforcing the view that crb is a direct target of the JAK/STAT pathway.
To identify the minimal sequence requirements for crb posterior spiracle specific expression we compared crb43.2 sequence among several Drosophila species (S2B Fig). As the initial characterization of crb43.2 indicated the requirement of two low affinity STAT binding sites [14], we tested a highly conserved 518 bp genomic fragment centred around these sites (S2B Fig). The 518 bp element was cloned into a lacZ reporter transformation plasmid containing a minimal promoter and lacZ expression was analyzed in transgenic flies. Like the original crb43.2 enhancer, crb518 directs expression of the transgene specifically in the posterior spiracles (S2C Fig). Moreover, the activity of crb518 is also regulated by JAK/STAT signalling as both inactivation of the pathway or mutation of the STAT binding sites result in the downregulation of the enhancer’s activity (S2E and S2F Fig).
These results show that the crb518 enhancer constitutes a minimal element able to direct specific expression of crumbs to the posterior spiracle in a JAK/STAT dependent manner.
A posterior spiracle specificity element can be separated from the STAT binding sites
To understand the molecular mechanisms by which STAT activates crumbs in the posterior spiracles, we further dissected the crb518 minimal enhancer. Based on the position of the STAT binding sites, the crb518 enhancer can be subdivided into three regions (Fig 1A): a region encompassing the first 204 nucleotides (1–204, yellow), a 101 bp region containing the STAT binding sites (205–305 bp, red) and a region including the last 213 nucleotides (306–518 bp, green). Transgenes were designed with different combinations of these regions and their expression patterns characterized. When the 1–204 bp region is deleted (crb313), spiracle expression is lost (Fig 1C). This result suggests that the STAT binding sites although required, are not sufficient to direct expression to the posterior spiracles and that additional factors binding to the 1–204 fragment (yellow) are crucial. This conclusion is further supported by the inability of the crb101 construct, which contains the 205–305 fragment (red) including both STAT sites, to direct expression in the spiracles (Fig 1D).

These observations are in accordance with our finding that the crb305 enhancer, where the 306–518 fragment (green) is deleted, is able to direct expression to the posterior spiracle (Fig 1E). Moreover, the activity of crb305 is still dependent on the JAK/STAT pathway as its expression is downregulated in the absence of all three unpaired ligands (Fig 1F). Therefore the crb305 enhancer seemed to contain all the cis-regulatory elements required to direct expression to the posterior spiracle in a JAK/STAT dependent manner.
Finally, we tested the expression of the crb204 construct, which contains the 1–204 fragment (yellow) but lacks the STAT binding sites. Surprisingly, the crb204 is able to direct expression to the posterior spiracles (Fig 1G). Moreover, this expression is dependent on JAK/STAT pathway activity as in embryos mutant for the unpaired ligands the enhancer is downregulated (Fig 1H).
Taken together, these results show that crb204 contains the crb posterior spiracle specificity element. Moreover, the expression of this construct suggests that although the activity of the JAK/STAT pathway is required to activate the crb spiracle expression, the STAT binding sites are dispensable. This seems to contradict our previous observations where mutation of the STAT sites resulted in the downregulation of the larger crb43.2 and crb518 enhancers.
The spiracle specific crb204 element does not contain cryptic STAT binding sites
The spiracle expression of the crb204 enhancer was unexpected not only because it did not contain the STAT binding sites known to be required for the larger enhancers’ activity but also because despite the absence of STAT sites the enhancer is still regulated by the JAK/STAT pathway. These results prompted us to consider the possibility of cryptic STAT binding sites in crb204 that could account for these observations. Therefore, we searched for putative additional STAT binding sites that differed in one or two nucleotides from the consensus TTCNNN(N)GAA sequence [14,15]. Using this relaxed criterion, we identified fifteen possible cryptic binding sites spread along crb305 (S3A Fig, asterisks), twelve of them in crb204. To determine if STAT is able to bind in vitro to any of these putative sites, EMSAs were performed with activated STAT92E protein incubated with oligonucleotides containing the different cryptic sites (S3B Fig). As controls we used oligonucleotides containing the STAT consensus binding sites as well as mutated versions of these sites (Materials and Methods). In these conditions, STAT92E is only able to bind the previously identified consensus sites, while it fails to bind the mutated STAT sites or any of the putative cryptic sites (S3B Fig). Therefore, these experiments exclude the possibility of cryptic binding sites being responsible for the activity of crb204.
Taken together, these results suggest that the JAK/STAT pathway is required to specify expression in the posterior spiracles most likely by indirectly regulating factors binding to the 1–204 specificity element (yellow). This detailed analysis reveals that STAT does not function as a crb spiracle enhancer coactivator, but has a different function in the context of the crb enhancer.
The binding of STAT92E to crb518 is required to counteract a repressor element
Mutation of the STAT sites in both the original crb43.2 enhancer [11,14] and in the crb518 enhancer indicated that the STAT sites are required for full activation (S1E and S1F and S2C–S2F Figs). However, the spiracle activity of crb204 excluded the possibility that the direct binding of STAT is necessary to direct expression to the posterior spiracles (Fig 1G). These contradictory results can be accommodated in a model where the specificity element is silenced by a nearby repressor element; the function of STAT being to prevent the repression, thereby, allowing spiracle specific activation (Fig 2A and 2A’). In this hypothesis the JAK/STAT pathway would play a dual role: first, it would be required indirectly to activate the specificity element by regulating activator factors; and second, it would be required directly to overcome the repression of the enhancer by counteracting a repressor element present in the CRM. Mutation of the STAT sites would prevent the binding of STAT to the CRM disabling this second counter-repression role resulting in the downregulation of expression controlled by the specificity element.

As our model predicts the existence of a repressor element, we set to identify its location by deletion of DNA in a crb518 enhancer with mutated STAT sites (Fig 2B and 2C). The logic behind this experiment was that the deletion of the repressor element should allow the activity of the specificity element in the absence of STAT binding sites. We found that removal of the 306–518 fragment (crb305-STAT-mt) results in the expression of the enhancer in the posterior spiracles (Fig 2B’ and 2C’) confirming the existence of a repressor element in the 306–518 distal region (green region).
To further support our findings we generated an additional construct fusing the putative repressor module to the crb spiracle specificity module (Fig 2D). As expected from the presence of a repressor element, the expression of this transgene in the posterior spiracles is downregulated (Fig 2D’).
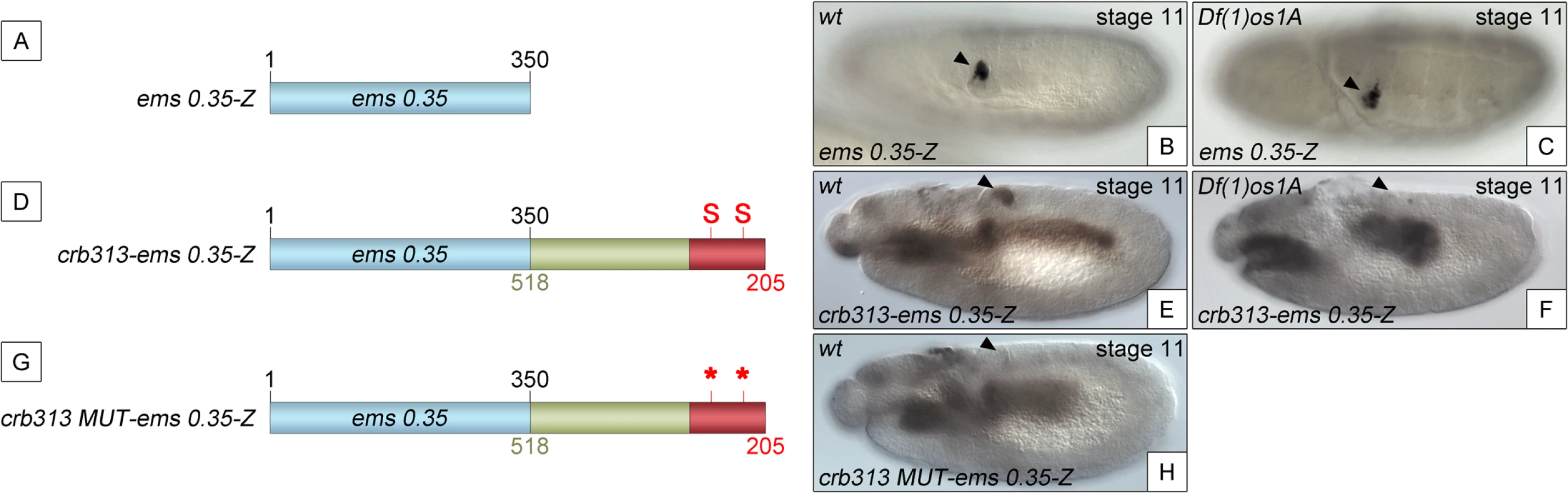
To determine whether the STAT binding sites and the repressor element present in the 205–518 fragment are able to function as regulatory modules independent of the specific context of the crb spiracle specificity module, we fused the crb313 fragment (red and green in Fig 1A), which does not drive expression in the posterior spiracles (Fig 1C), to ems0.35 (Fig 3A and 3B), an unrelated posterior spiracle enhancer of the empty spiracles gene [16]. Although the crb313-ems0.35 fusion construct generates a novel gut pattern of expression not present in any of the original constructs, ems0.35 spiracle activity is still present when fused to the crb313 element (Fig 3A and 3B and 3D and 3E). In contrast, fusion of the crb313 element with the STAT sites mutated results in the loss of the posterior spiracle expression driven by ems0.35 (Fig 3G and 3H). These results further support the presence of a repressor element in the 306–518 fragment that would be counteracted by the STAT sites, and reveal the modular nature of the crb518 CRM. Moreover, while the ems0.35 enhancer by itself is expressed independently of STAT function (Fig 3B and 3C), it becomes STAT dependent when fused to the crb313 element (Fig 3E and 3F). Taken together, these data show that crb518 is structured into three different regions, each with a specific function: an activator element driving spiracle specificity (1–204, yellow), a STAT-binding element that contains the conserved STAT binding sites (205–305, red) and a repressor element (306–518, green).
Analysis of the 306–518 DNA sequence did not provide any strong indication regarding the nature of the repressor protein. To fine map the repressor region, we constructed transgenes where we added increasing size fragments distal to crb305-STAT-mt (S4B Fig). In these constructs the position of the repressor element should be identifiable by its ability to inhibit the specificity spiracle element, which cannot be counteracted by STAT binding. Based on the sequence conservation between D. melanogaster and D. virilis (S4A Fig) the repressor containing region was divided into four elements: two highly conserved elements (CE), CE1 (345–385 bp) and CE2 (492–518 bp), intercalated by two non-conserved (NC) elements, NC1 (305–344 bp) and NC2 (386–491 bp). Addition of NC1 and CE1 has no effect on the activity of the enhancer (S4B–S4D Fig). However, when the NC2 element is included, the spiracle expression is downregulated (S4E Fig).
Taken together, these results map the position of the repressor element around the NC2 region distal to the STAT binding sites.
The repressor element regulates the temporal activation of the crumbs spiracle enhancer
The identification of a repressor element in the crb518 enhancer prompted us to determine its role in the regulation of crb in the posterior spiracle. The repressor domain does not seem to be involved in tissue-specific regulation as the specificity element (crb204, Fig 1G) is sufficient to direct spiracle activation. In fact, we were unable to observe any ectopic activation in constructs where the repressor element was deleted. Therefore, it seemed plausible that the repressor element could be required for temporal regulation of crumbs expression in the posterior spiracles. To test this possibility, we analyzed the onset of expression directed by the different crb CRM variants (Fig 4). While crb43.2 and crb518, induce reporter transcription from stage 11 (Fig 4A'–4A”, 4B’–4B”), we observe that crb305, which lacks the repressor element, induces earlier transcription starting at stage 10 (Fig 4C–4C”).

These results demonstrate that the repressor element does not have a major function on tissue-specificity as expression is still detected in the posterior spiracles, but it does affect the temporal regulation of the enhancer with the onset of transcription occurring at earlier stages of development.
Identification of trans-activating factors controlling crb spiracle specific activation
Our finding that STAT is not involved in the direct activation of the crb spiracle enhancer, made us look into the posterior spiracle gene cascade for alternative activators. To this end, using the 69B-Gal4 line, we tested whether any of the Abd-B primary targets expressed throughout the epidermis could activate ectopic crb305 expression. While ectopic expression of spalt (sal), empty spiracles (ems) or cut (ct) have no effect, ectopic expression of unpaired resulted in ectopic activation of the spiracle enhancer (Figs 5E and S5), consistent with the requirement of JAK/STAT for crb expression in the posterior spiracles. Interestingly, despite 69B-Gal4 expressing unpaired throughout the embryonic epidermis, ectopic activation of the enhancer is mostly confined to the posterior segments where Abd-B is also expressed. This result raised the possibility of Abd-B having a direct input on the activation of the crb specificity element. To test this possibility, we determined the ability of Abd-B to bind to the crb CRM in S2 cells using ChIP analysis. We obtained a 9-fold increase in input recovery when performing ChIP with Abd-B confirming that Abd-B is able to bind directly to the crb enhancer (Fig 6A).


Analysis of the crb305 sequence shows the existence of eight putative Abd-B binding sites (Fig 6B, asterisks). To determine if Abd-B could bind them directly, EMSAs were performed with oligonucleotides covering regions containing the predicted sites (Fig 6B). As a control, we designed oligonucleotides containing point mutations abolishing Abd-B binding. Abd-B was expressed in S2 cells and cell extracts prepared and used in these assays. As seen in EMSA, all eight sites are bound by Abd-B with different affinities (Fig 6C). Moreover, binding to oligonucleotides containing more than one Abd-B site, is only abolished when both sites are mutant. A similar additive binding has been described for other direct Abd-B targets [16–18].
JAK/STAT and Abd-B collaborate for crb spiracle enhancer activation
Our results suggest that Abd-B may be required both directly and indirectly for the activation of crb in the posterior spiracles. Directly, through Abd-B’s binding to the crb specificity element and indirectly through its activation of upd transcription and thus JAK/STAT signalling.
Similarly, STAT is also required directly and indirectly for crb’s spiracle expression: directly, to block the repressor’s element activity; and indirectly, because JAK/STAT signalling is also required for the activation of the crb specificity element (Fig 1H).
To study if Abd-B can induce the activation of the specificity domain in the absence of JAK/STAT signalling and vice versa, we analyzed the effect of ectopically expressing one in embryos mutant for the other. We performed these experiments using the crb305 enhancer that lacks the repressor domain, avoiding the interference of the repressor element. The ectopic crb305 expression in segments A8-A9 caused by ectopic upd is not present in Abd-B mutants (compare Fig 5E and 5F) showing that Abd-B provides an additional input in the activation of the crb305 enhancer. Similarly, ectopic Abd-B expression in Df(1)os1A embryos lacking all three upd ligands is not capable of activating the crb305 enhancer (compare Fig 5C and 5D). Therefore, these results indicate that both STAT and Abd-B inputs are necessary in parallel for spiracle specificity rather than all the Abd-B activity being mediated by its activation of upd and JAK/STAT signalling.
JAK/STAT activates expression of Abd-B via a feedback loop
The above data show that both STAT and Abd-B are required for crb specificity element expression. As the function of STAT on the specificity element is indirect, it is likely to be mediated through the regulation of another transcription factor expressed in the posterior spiracles. This means that a still unidentified STAT target mediates STAT’s indirect function on the crb spiracle enhancer. Alternatively, Abd-B itself could be regulated by STAT via a feedback mechanism.
If a feedback loop mechanism existed in the posterior spiracles, overexpressing Abd-B should result in the activation of endogenous Abd-B. To determine the existence of such feedback loop, the 69B-Gal4 line was used to ectopically activate UAS-Abd-Bm in the ectoderm of embryos containing the BAC-Abd-B-GFP (Fig 7A). In this BAC element GFP-tagged Abd-B is regulated by the endogenous upstream Abd-B cis-regulatory sequences that are sufficient to drive normal expression in the A8 and A9 segments (Figs 7B and 7C and S6A–S6A”). This set-up allows discriminating between the ectopically expressed Abd-Bm protein and any feedback induced Abd-B-GFP protein from the BAC element. Ectopic UAS-Abd-Bm induction results in ectopic expression of Abd-B-GFP, indicating that a feedback loop mechanism maintaining Abd-B expression does exist (Fig 7D). We next tested whether this feedback loop could be mediated by the activation of the JAK/STAT pathway. To determine this, we repeated the experiment in a Df(1)os1A mutant background and found that most feedback-induced ectopic expression disappeared (Fig 7E) while feedback-independent expression in A8-A9 is maintained. We also ectopically expressed upd throughout the ectoderm using the 69B-Gal4 driver line and analyzed Abd-B-GFP expression. As with ectopic expression of Abd-Bm, Upd also results in the ectopic activation of Abd-B-GFP expression (Fig 7F). As these results suggest the existence in Abd-B of an enhancer regulated by JAK/STAT signalling, we analyzed the Stark Lab genome collection of enhancers made from the Abd-B locus [19]. We found that a 2.2 kb fragment (VT42855) drives expression in the posterior spiracles and contains a high density of STAT sites (Fig 7A and 7G and 7I–7K). Chromatin immunoprecipitation with STAT-GFP shows an enrichment of input recovery of this fragment suggesting that STAT is able to bind directly to the Abd-B cis regulatory region (Fig 7L). Moreover, VT42855 expression in the posterior spiracles is abolished in Df(1)os1A mutants (Fig 7G and 7H) confirming the existence of a JAK/STAT regulated enhancer in the Abd-B locus. Taken together, these results indicate the existence of a feedback loop mechanism used to maintain spiracle expression of Abd-B via activation of the JAK/STAT pathway.

Discussion
Using the posterior spiracles as a model, we have analysed how an organogenetic gene network develops and how a particular gene of this network integrates the changing genetic information.
Dynamics of the global genetic regulatory interactions during organogenesis: transformation of a genetic cascade into a gene network
Two main regulatory structures have been described during organogenesis: genetic cascades and genetic networks. Genetic cascades are characterized by the hierarchical interactions between its components, with one gene setting the stage and the others responding to it. In this system, the type of organ to develop is chosen by the upstream selector gene, which controls the different cell behaviours via intermediate transcription factors [8,20–22].
Gene networks differ by the absence of a single master gene. In a gene network, like that controlling eye development, not only several genes can trigger organogenesis but also all of these genes are necessary for organogenesis. In the absence of anyone of them, organogenesis cannot proceed, even if forced expression of the other master regulators is induced. [23–25]. This behaviour suggests that none of the regulators is exclusively upstream of the others and that activation of any of the master genes results in the activation of the other master genes through cross-regulatory loops [9]. Cross-regulatory feedback loops confer robustness to a regulatory system and their absence in early Hox induced organogenesis could be due to the fast development of the Drosophila embryo that precludes Hox genes to become integrated by feedback loops into the cascades they activate.
The formation of the posterior spiracle is a typical example of organogenesis induced by a Hox gene cascade [11]. Here, Abd-B activates the primary targets that, in turn, activate secondary targets like Cad96C, Cad88C, Gef64C and Cad74A. The hierarchical organization of these targets was demonstrated by the observation that in Abd-B mutants the simultaneous activation of the four Abd-B primary targets can activate the expression of Cad96C, Cad88C, Gef64C and Cad74A [11]. Despite of this, here we have found evidence that during embryonic development, the Abd-B spiracle cascade is already transforming into a gene network. We have observed that Abd-B activates the JAK/STAT signalling pathway in the posterior spiracles through upd and that the JAK/STAT pathway feedbacks to enhance Abd-B expression. Two observations show that this is important to maintain robust crb expression: first, JAK/STAT pathway signalling only weakly activates the expression of the crb posterior spiracle enhancer in segments that do not express Abd-B; and second, JAK/STAT pathway activation is insufficient to activate the crb enhancer in the A8 segment of Abd-B mutant embryos (Fig 5). An indirect feedback loop affecting the Ubx Hox gene has been reported [26]. In the visceral mesoderm Ubx activates in PS7 the signalling molecule dpp. Dpp signals to the neighbouring PS8 cells to activate with Abd-A wingless expression. In turn, Wg signals from PS8 back to PS7 where it is required for the maintenance of Ubx expression. This case and the JAK/STAT—Abd-B interaction we describe here may represent the earliest examples of indirect autoregulatory loops transforming a Hox-cascade into a Hox-network. It would be interesting to analyze other organogenetic cascades to find out if the establishment of feedback loops is a common theme during development and at what stage they become active.
Integrating the global genetic regulatory information into single target genes: the posterior spiracle regulation of crb
During organogenesis the complex patterning information set by the transcription factors and signalling molecules upstream in the cascade has to be integrated to activate the realizator molecules modulating the organogenetic cell behaviours [27]. This is mediated through the cis regulatory modules of each realizator gene. In this study, we have focused on how the posterior spiracle gene cascade activates the crb polarity gene through a CRM that responds to JAK/STAT signalling. The observation that the spiracle enhancer requires the activity of upd, an Abd-B primary target, suggested that Abd-B activates crb indirectly, by setting STAT activity [11]. Our dissection of the crb-spiracle CRM has revealed that a spiracle specificity element can be separated from the STAT binding sites and that the “minimal” CRM is modular in nature. The crb-spiracle CRM studied here is composed by, at least, three independently acting elements: an activator element where the spiracle specificity resides; a STAT binding element that is only required to counteract a neighbouring repressor element; and a repressor element that interferes with the activity of the specificity module.
We have found that the spiracle specificity element is bound by Abd-B suggesting that crb is a direct target of Abd-B. However, crb is different from other Abd-B primary targets, since the crb specificity element also requires for its expression STAT activation, itself a target of Abd-B. Because of its mixed character as a direct and an indirect target, we term crb a delayed-primary target: primary to reflect its direct regulation by Abd-B, and delayed because it can only respond to the presence of Abd-B after other Abd-B primary targets are active. The requirement of both Abd-B and JAK/STAT signalling for crb activation explains why the crb-spiracle enhancer is only active in A8 and not in more anterior segments where JAK/STAT signalling is also active, as well as why crb-spiracle enhancer expression in A8 is restricted dorsally despite Abd-B being expressed throughout this segment.
The repressor element we have identified is “transplantable” and can function autonomously on an independent posterior spiracle enhancer. In fact, we have shown that the ems spiracle enhancer, that is STAT independent (Fig 3C), can be engineered to become STAT dependent by the inclusion of the STAT binding and repressor modules found in crb (Fig 3E and 3F). The observation that the specificity element in crb is sufficient to drive the activity of the enhancer, begs the question of why is there any need for the presence of further modules in the enhancer. As the specificity element cannot function until the repressor activity is counteracted by Abd-B induction of JAK/STAT signalling in the spiracle, the repressor module provides the CRM with a subtle system to delay the crb spiracle expression during development.
The integration of the results in this work uncovers the existence of complex Abd-B and JAK/STAT dynamic interactions that are described by the model shown in Fig 7M. Early Abd-B binding to the crb specificity module cannot activate transcription due to the presence of the neighbouring repressor module. This repression is relieved when Abd-B activation of upd in the posterior spiracles activates STAT. Direct binding of STAT to the crb CRM relieves the repression. We hypothesize that STAT induces the function of another activator protein (A) that also binds to the specificity module (Fig 7M). The existence of such activator driven downstream of STAT is necessary to explain why ubiquitous ectodermal Abd-B expression with the 69B-Gal4 line is unable to activate the crb305 enhancer in upd mutant embryos despite the absence of the repressor module (Fig 5D). Besides these functions, our data show that STAT reinforces Abd-B expression in the spiracles through a feed back loop. The model in Fig 7M can explain why the inactivation of JAK/STAT signalling in Df(1)os1A embryos has a stronger effect on crb518 enhancer expression than the deletion of the STAT binding sites (compare S2E and S2F Fig and similar observations using crb43.2 in Lovegrove et al. [11]). Mutation of the two STAT binding sites decreases crb expression because the repressor module is free to interfere with the specificity module. Mutation of Df(1)os1A has a larger influence on expression because it affects crb regulation at three levels: First, because, as in the previous case, the repressor module is free to interfere with the specificity module in the absence of active STAT; second, because the proposed Activator downstream of JAK/STAT will not be activated; and third, because the STAT feedback loop over Abd-B will not be activated. Although we still do not know all the players involved in the regulation of the crb posterior spiracle enhancer, this case provides an example of how gene networks and CRMs dynamically interact during development.
The complex CRM controlling crb spatial and temporal regulation in the posterior spiracles provides a plastic regulatory platform that could be subtly modulated during evolution. Earlier crb activation could be obtained by loss of the repressor module and lower or delayed expression by removal of Abd-B or STAT binding sites. Such quantitative and temporal regulation changes in either direction could be rapidly established by selection of minor mutations in these elements.
Counter-repression: an alternative way for STAT to control cell specific transcriptional regulation
STAT proteins in vertebrates and invertebrates are considered to be a family of latent transcription factors activated by phosphorylation. After their activation, the cytoplasmic proteins dimerize and accumulate in the nucleus where they bind DNA through TTC(3-4N)GAA sites to activate transcription [28,29]. STAT specific transcriptional activation has been suggested to require either cooperative interaction with other enhancer binding proteins, or interaction with co-activator proteins that stabilize or recruit RNA polymerase II complexes or destabilize chromatin [30,31]. In this view, transcription of specific downstream targets is due to the direct interaction of STAT with other activators expressed in the same cells. Initially, we expected the STAT dependent up-regulation of crb in the spiracles to be controlled using such a mechanism, however, we have found this not to be the case. Our experiments show that direct STAT binding to the posterior spiracle’s specificity element is not required for transcription. This result suggests that STAT does not function as a classical transcription factor stabilizing RNA polymerase II in the posterior spiracles, but rather acts as a counter-repressor protein. STAT function as a counter-repressor is to prevent that proteins binding to the repressor element interfere with the binding or function of transcriptional activators in the specificity module. Thus, we propose that besides its function as a transcriptional activator, STAT can control gene specific expression through a novel function: counter-repression. Although future work should identify the STAT92E domain controlling counter-repression, we can predict that if the counter-repression domain were different from the transactivation domain, deletion of the transactivation element would still result in a STAT92E protein capable of controlling transcription through counter-repression. The existence of an independent domain with such new function could explain published results showing that STAT proteins with a deleted transactivation domain are still active proteins and do not act as dominant negative transcription factors [32]. Counter-repression is not related to the non-canonical JAK/STAT control of heterochromatin stability described by Li and collaborators as this does not require direct STAT binding to its consensus DNA sites [33–35], while we show that STAT counter-repression requires the presence of its binding sites.
In summary, we have shown that the Hox protein Abd-B initiates a simple embryonic posterior spiracle cascade that soon evolves into a gene network. In this network, Abd-B and the JAK/STAT signalling pathway (which is activated downstream of Abd-B in the spiracles) interact to precisely control the temporal activation of the crb realizator gene. Importantly, our study uncovered a novel way for STAT proteins to control cell specific gene expression: permissive counter-repression.
Materials and Methods
Fly strains
The following Gal4 driver and UAS lines were used: 69B-Gal4 to drive expression in the whole ectoderm, UAS-Abd-Bm, UAS-upd, UAS-ct, UAS-sal, UAS-ems, UAS-grn. We used the Abd-BM1 null allele, the Df(1)os1A [deleting all three Upd ligands, [36]]. As reporter spiracle lines we used the crb43.2.1-lacZ [11] and ems0.35-lacZ [16], while the 10xSTAT-dGFP reporter was used to detect global JAK/STAT activation [37]. The VT42855-Gal4 line was obtained from the Stark lab fly transcriptional enhancer collection http://enhancers.starklab.org/. The [CH321-91P18] BAC-Abd-B-GFP stock is a gift from Rebecca Spokony, modENCODE consortium. To test if the BAC can rescue Abd-B function, we combined BAC-Abd-B-GFP with a UbxMX12, abd-AM1, Abd-BM8 triple mutant [38] (S6 Fig).
Immunohistochemistry and RNA in situ hybridization
The embryos were grown at 25°C except the 69B-Gal4 UAS overexpression experiments that were grown at 29°C to increase Gal4 activity. The following primary antibodies were used: mouse anti-AbdB 1A2E, anti-Crb and anti-Ct 2B10 (Developmental Studies Hybridoma Bank); rabbit anti-GFP (Invitrogen) and mouse anti-βGal (Promega). Secondary antibodies were conjugated to Alexa Fluor 488, 555 (Invitrogen). Confocal images were obtained with a Leica SP2-AOBS microscope and processed using ImageJ and Adobe Photoshop CS5. We used antisense RNA probes for upd, Gal4 and lacZ in situ.
Constructs
The design of the constructs was based on the sequence conservation analysis of crb43.2 by comparison of the twelve Drosophila species and A. gambiae, A. mellifera and T. casteneum genomes using the UCSC Genome Bioinformatics (http://genome.ucsc.edu/). All crb fragments were subcloned into the pCasper-hs43-lacZ transformation plasmid. To this end, pCasper-hs43-lacZ was digested first with EcoRI, the 5’overhang filled with Klenow and finally digested with EclXI. Unless stated otherwise, all PCR reactions were performed using as DNA template the 43.2-pBluescript-SK plasmid containing the crb43.2 genomic region [11]. The primers used to generate the different constructs are listed in S1 Table.
To generate the crb518-Z construct, the 43.2-pBluescript-SK plasmid was digested with DraII, the resulting 5’overhangs filled with Klenow and subsequently digested with EclXI. A 518 bp fragment was isolated, purified and subcloned into pCasper-hs43-lacZ.
To construct crb518-STAT-mt-Z, mutation of the STAT binding sites was carried out in a two-step process. The first STAT site was mutated by site-directed mutagenesis as described in [39] using the stat4n1-mt primer. The resulting plasmid with the mutated first STAT binding site was then used as a template to perform the mutation of the second STAT site by PCR mutagenesis. A first PCR reaction was performed using the primer pair crb-stat2-mt and rev-crb. The product was purified and used as a reverse primer in a second PCR reaction using fw-crb as forward primer. The resulting PCR product containing both mutated STAT binding sites was digested with EclXI and subcloned into pCasper-hs43-lacZ.
The crb305-Z, crb313-Z, crb101-Z, crb204-Z, crb305-STAT-mt-Z, crbΔ25-Z, crbΔ133-Z and crbΔ174-Z constructs were generated as follows: the region of interest was amplified by PCR using either 43.2-pBluescript-SK (for crb305-Z, crb313-Z, crb101-Z and crb204-Z) or crb518-STAT-mt (for crb305-STAT-mt-Z, crbΔ25-Z, crbΔ133-Z and crbΔ174-Z) as DNA template; the PCR products were digested wit EclXI and subcloned into pCasper-hs43-lacZ. The primer pairs used are as follow: fw-crb and rev-stat (crb305-Z and crb305-STAT-mt), fw-stat and rev-crb (crb313-Z), fw-stat and rev-stat (crb101-Z), fw-crb and rev-crb204 (crb204-Z), fw-crb and crbΔ25-rev (crbΔ25-Z), fw-crb and crbΔ133-rev (crbΔ133-Z) and fw-crb and crbΔ174-rev (crbΔ174-Z).
To generate crb518Δ101-Z, a first PCR was performed using the primer pair fw-Δ101 and rev-crb. The resulting product was purified and used as a primer in a second PCR with the fw-crb primer. The resulting PCR product was digested with EclXI and subcloned into pCasper-hs43-lacZ.
To generate the crb313-ems0.35-Z constructs PCR reactions were performed using the reverse primer rev-crb with fw-crb205-BHI. The PCR products were purified and digested with EclXI and cloned into pCasper-ems 0.35 [16] digested with EclXI and BamHI.
All constructs were randomly inserted using P-element transformation. We analysed between three and ten independent lines to discard insertion-site position-effects. Injection was performed by the Drosophila Consolider-Ingenio (CBM-SO, Madrid) platform or by BestGene.
Chromatin Immunoprecipitation (ChIP) assays
This protocol, based on [40], was done as previously described [14,16]. ChIP was performed using transiently transfected S2 cells. 1 x 107 cells were seeded in 10 cm cell culture dishes one day before transfection. For Abd-B ChIP, cells were transfected with either (1) 5 μg pUAST-Abd-Bm-HA and 5 μg pAC-GAL4 or (2) 5 μg of empty pUAST and 5 μg pAC-GAL4 plasmids. For STAT92E-GFP ChIP, cells were transfected with either (1) 3.5 μg pUAST-STAT92E-GFP, 3.5 μg pAC-HopTum-l and 3.5 μg pAC-GAL4 plasmids or (2) 3.5 μg empty pUAST, 3.5 μg pAC-HopTum-l and 3.5 μg pAC-GAL4 plasmids. 1/10 of cells were collected to monitor the protein expression by Western blot. Remaining cells were cross-linked, lysed and sheared to 350–1000 bp. Immunoprecipitations were performed using 6 μl of anti-HA antibody (Abcam) or 50 μl of anti-GFP conjugated magnetic beads (MBL) per 100 μg sheared chromatin. qRT-PCR was performed with primers crbQPCR1for (5’-TTCATTCATTTCCATGAACACA-3’) and crbQPCR1rev (5’-ATTCGTCGGTTTTCCTTGTC-3’) amplifying inside the crb43.2 enhancer sequence; AbdB Bac1for (5’-TTGGACAAATTCACATGCAA-3’) and AbdB Bac1rev (5’-GGCCAATGAACTTCCCTCTA-3’) amplifying inside the VT42855 enhancer sequence; and, as a control, with AbdB BacC-Afor (5’-TGAACTTAAATGCCGAATCAA-3’) and AbdB BacC-Arev (5’-CACAAGAAGTGCGTGACTGA-3’) amplifying in a sequence lacking STAT binding sites. The data are represented as recovered percentage from the input in HA-Abd-B-transfected cells or GFP-transfected cells against pUASt-empty-transfected cells.
Electrophoretic mobility shift assays (EMSA)
The complementary oligonucleotides (Sigma Aldrich) used to generate the radiolabelled probes in EMSAs to determine STAT92E and Abd-B binding are listed in S2 and S3 Tables respectively. Radioactively labelled probes were generated by annealing and subsequent end filling with [α-32P]dCTP. The conditions used were similar to those described previously [16]. Briefly, double-stranded, end-labelled DNA (50,000 cpm/binding reaction; 10 nM) was incubated for 30 min at 4°C with 2 μl of cell extract lysate expressing each tested protein or 2 μl of the cell extract control and 50 mM NaCl, 5 mM EDTA, 0,5 mM DTT, 10 mM Tris-HCl (pH 7.8), 4% glycerol, 1 mM MgCl2, and 1 mg of poly dI-dC as nonspecific competitors, in a final reaction volume of 20 μl. The reactions were run on a 5% polyacrylamide gel, in 0.5x Tris-borate-EDTA buffer to visualize complex formation by retardation of the 32P-labeled target DNA. In some experiments monoclonal anti-Abd-B or anti-GFP were incubated with aliquots of the reaction mixture for an additional 30 min. For each gel shift reaction, a control with cell extract of non-trans00FEcted cells was used to detect possible DNA binding by endogenous lysate factors. Gels were dried at 80°C in vacuum, exposed to a phosphorimager screen and detected by a typhoon scanner.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Hombria JC, Lovegrove B (2003) Beyond homeosis—HOX function in morphogenesis and organogenesis. Differentiation 71 : 461–476. 14641327
2. Hueber SD, Lohmann I (2008) Shaping segments: Hox gene function in the genomic age. Bioessays 30 : 965–979. doi: 10.1002/bies.20823 18798525
3. Pearson JC, Lemons D, McGinnis W (2005) Modulating Hox gene functions during animal body patterning. Nat Rev Genet 6 : 893–904. 16341070
4. Gehring WJ, Ikeo K (1999) Pax 6: mastering eye morphogenesis and eye evolution. Trends Genet 15 : 371–377. 10461206
5. O'Hara E, Cohen B, Cohen SM, McGinnis W (1993) Distal-les is a downstream gene of Deformed required for ventral maxillary identity. Development 117 : 847–856. 8100764
6. Sanchez-Higueras C, Sotillos S, Castelli-Gair Hombria J (2014) Common origin of insect trachea and endocrine organs from a segmentally repeated precursor. Curr Biol 24 : 76–81. doi: 10.1016/j.cub.2013.11.010 24332544
7. Abrams EW, Vining MS, Andrew DJ (2003) Constructing an organ: the Drosophila salivary gland as a model for tube formation. Trends Cell Biol 13 : 247–254. 12742168
8. Castelli Gair Hombria J, Rivas ML, Sotillos S (2009) Genetic control of morphogenesis—Hox induced organogenesis of the posterior spiracles. Int J Dev Biol 53 : 1349–1358. doi: 10.1387/ijdb.072421jc 19247941
9. Desplan C (1997) Eye development: governed by a dictator or a junta? Cell 91 : 861–864. 9428507
10. Sotillos S, Aguilar M, Hombria JC (2013) Forces shaping a Hox morphogenetic gene network. Proc Natl Acad Sci U S A 110 : 4303–4308. doi: 10.1073/pnas.1212970110 23440219
11. Lovegrove B, Simoes S, Rivas ML, Sotillos S, Johnson K, et al. (2006) Coordinated control of cell adhesion, polarity, and cytoskeleton underlies Hox-induced organogenesis in Drosophila. Curr Biol 16 : 2206–2216. 17113384
12. Tepass U (2012) The apical polarity protein network in Drosophila epithelial cells: regulation of polarity, junctions, morphogenesis, cell growth, and survival. Annu Rev Cell Dev Biol 28 : 655–685. doi: 10.1146/annurev-cellbio-092910-154033 22881460
13. Tepass U, Theres C, Knust E (1990) crumbs encodes an EGF-like protein expressed on apical membranes of Drosophila epithelial cells and required for organization of epithelia. Cell 61 : 787–799. 2344615
14. Rivas ML, Cobreros L, Zeidler MP, Hombria JC (2008) Plasticity of Drosophila Stat DNA binding shows an evolutionary basis for Stat transcription factor preferences. EMBO Rep 9 : 1114–1120. doi: 10.1038/embor.2008.170 18802449
15. Yan R, Small S, Desplan C, Dearolf CR, Darnell JE Jr. (1996) Identification of a Stat gene that functions in Drosophila development. Cell 84 : 421–430. 8608596
16. Rivas ML, Espinosa-Vazquez JM, Sambrani N, Greig S, Merabet S, et al. (2013) Antagonism versus cooperativity with TALE cofactors at the base of the functional diversification of Hox protein function. PLoS Genet 9: e1003252. doi: 10.1371/journal.pgen.1003252 23408901
17. Jeong S, Rokas A, Carroll SB (2006) Regulation of body pigmentation by the Abdominal-B Hox protein and its gain and loss in Drosophila evolution. Cell 125 : 1387–1399. 16814723
18. Williams TM, Selegue JE, Werner T, Gompel N, Kopp A, et al. (2008) The regulation and evolution of a genetic switch controlling sexually dimorphic traits in Drosophila. Cell 134 : 610–623. doi: 10.1016/j.cell.2008.06.052 18724934
19. Kvon EZ, Kazmar T, Stampfel G, Yanez-Cuna JO, Pagani M, et al. (2014) Genome-scale functional characterization of Drosophila developmental enhancers in vivo. Nature 512 : 91–95. doi: 10.1038/nature13395 24896182
20. Bradley PL, Haberman AS, Andrew DJ (2001) Organ formation in Drosophila: specification and morphogenesis of the salivary gland. Bioessays 23 : 901–911. 11598957
21. Chung S, Chavez C, Andrew DJ (2011) Trachealess (Trh) regulates all tracheal genes during Drosophila embryogenesis. Dev Biol 360 : 160–172. doi: 10.1016/j.ydbio.2011.09.014 21963537
22. Kerman BE, Cheshire AM, Andrew DJ (2006) From fate to function: the Drosophila trachea and salivary gland as models for tubulogenesis. Differentiation 74 : 326–348. 16916373
23. Bonini NM, Bui QT, Gray-Board GL, Warrick JM (1997) The Drosophila eyes absent gene directs ectopic eye formation in a pathway conserved between flies and vertebrates. Development 124 : 4819–4826. 9428418
24. Chen R, Amoui M, Zhang Z, Mardon G (1997) Dachshund and eyes absent proteins form a complex and function synergistically to induce ectopic eye development in Drosophila. Cell 91 : 893–903. 9428513
25. Pignoni F, Hu B, Zavitz KH, Xiao J, Garrity PA, et al. (1997) The eye-specification proteins So and Eya form a complex and regulate multiple steps in Drosophila eye development. Cell 91 : 881–891. 9428512
26. Thuringer F, Bienz M (1993) Indirect autoregulation of a homeotic Drosophila gene mediated by extracellular signaling. Proc Natl Acad Sci U S A 90 : 3899–3903. 8097881
27. García-Bellido A (1975) Genetic control of wing disc development in Drosophila. Amsterdam: Elsevier. 161–182 p.
28. Levy DE, Darnell JE Jr. (2002) Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol 3 : 651–662. 12209125
29. Hombria JC, Sotillos S (2013) JAK-STAT pathway in Drosophila morphogenesis: From organ selector to cell behavior regulator. JAKSTAT 2: e26089. doi: 10.4161/jkst.26089 24069568
30. Goenka S, Kaplan MH (2011) Transcriptional regulation by STAT6. Immunol Res 50 : 87–96. doi: 10.1007/s12026-011-8205-2 21442426
31. Horvath CM (2000) STAT proteins and transcriptional responses to extracellular signals. Trends Biochem Sci 25 : 496–502. 11050435
32. Ekas LA, Cardozo TJ, Flaherty MS, McMillan EA, Gonsalves FC, et al. (2010) Characterization of a dominant-active STAT that promotes tumorigenesis in Drosophila. Dev Biol 344 : 621–636. doi: 10.1016/j.ydbio.2010.05.497 20501334
33. Shi S, Larson K, Guo D, Lim SJ, Dutta P, et al. (2008) Drosophila STAT is required for directly maintaining HP1 localization and heterochromatin stability. Nat Cell Biol 10 : 489–496. doi: 10.1038/ncb1713 18344984
34. Li WX (2008) Canonical and non-canonical JAK-STAT signaling. Trends Cell Biol 18 : 545–551. doi: 10.1016/j.tcb.2008.08.008 18848449
35. Shi S, Calhoun HC, Xia F, Li J, Le L, et al. (2006) JAK signaling globally counteracts heterochromatic gene silencing. Nat Genet 38 : 1071–1076. 16892059
36. Hombria JC, Brown S, Hader S, Zeidler MP (2005) Characterisation of Upd2, a Drosophila JAK/STAT pathway ligand. Dev Biol 288 : 420–433. 16277982
37. Bach EA, Ekas LA, Ayala-Camargo A, Flaherty MS, Lee H, et al. (2007) GFP reporters detect the activation of the Drosophila JAK/STAT pathway in vivo. Gene Expr Patterns 7 : 323–331. 17008134
38. Casanova J, Sanchez-Herrero E, Busturia A, Morata G (1987) Double and triple mutant combinations of bithorax complex of Drosophila. EMBO J 6 : 3103–3109. 14650432
39. Makarova O, Kamberov E, Margolis B (2000) Generation of deletion and point mutations with one primer in a single cloning step. Biotechniques 29 : 970–972. 11084856
40. Zhai Z, Yang X, Lohmann I (2011) Functional dissection of the Hox protein Abdominal-B in Drosophila cell culture. Biochem Biophys Res Commun 414 : 761–766. doi: 10.1016/j.bbrc.2011.09.154 22005458
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2015 Číslo 7
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Functional Constraint Profiling of a Viral Protein Reveals Discordance of Evolutionary Conservation and Functionality
- Reversible Oxidation of a Conserved Methionine in the Nuclear Export Sequence Determines Subcellular Distribution and Activity of the Fungal Nitrate Regulator NirA
- Modeling Implicates in Nephropathy: Evidence for Dominant Negative Effects and Epistasis under Anemic Stress
- Nutritional Control of DNA Replication Initiation through the Proteolysis and Regulated Translation of DnaA
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy