
Emergence, Retention and Selection: A Trilogy of Origination for Functional Proteins from Ancestral LncRNAs in Primates
Although gene duplication has been believed as a predominant mechanism for creating new genes, recent reports suggested that new proteins could evolve “de novo” from non-coding DNA regions. These de novo genes are also named as “motherless” genes due to their lack of ancestral proteins as precursors, while recently we and others found that lncRNAs may represent an intermediate stage of their origination. To further elucidate this lncRNA-protein transition process, here we identified 64 hominoid-specific de novo genes and report a new mechanism for the origination of functional de novo proteins from ancestral non-coding transcripts: These non-coding “precursors” are generally not more selectively constrained than other lncRNA loci; and the existence of these de novo proteins is not beyond anticipation under neutral expectation; however, population genetics study in 67 human individuals and 82 macaque animals revealed signatures of purifying selection on these genes specifically in human population, indicating a proportion of these newly-originated proteins are already functional in human. We thus propose a mechanism for creation of functional de novo proteins from ancestral lncRNAs during the primate evolution.
Published in the journal:
. PLoS Genet 11(7): e32767. doi:10.1371/journal.pgen.1005391
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1005391
Summary
Although gene duplication has been believed as a predominant mechanism for creating new genes, recent reports suggested that new proteins could evolve “de novo” from non-coding DNA regions. These de novo genes are also named as “motherless” genes due to their lack of ancestral proteins as precursors, while recently we and others found that lncRNAs may represent an intermediate stage of their origination. To further elucidate this lncRNA-protein transition process, here we identified 64 hominoid-specific de novo genes and report a new mechanism for the origination of functional de novo proteins from ancestral non-coding transcripts: These non-coding “precursors” are generally not more selectively constrained than other lncRNA loci; and the existence of these de novo proteins is not beyond anticipation under neutral expectation; however, population genetics study in 67 human individuals and 82 macaque animals revealed signatures of purifying selection on these genes specifically in human population, indicating a proportion of these newly-originated proteins are already functional in human. We thus propose a mechanism for creation of functional de novo proteins from ancestral lncRNAs during the primate evolution.
Introduction
Although it is a generally accepted notion that gene duplication is the major way to create new genes [1–3], numerous cases have been reported in recent years demonstrating in multiple different species the creation of new proteins out of ancestral non-coding DNAs [4–18]. Recent studies further suggest that this de novo mechanism for gene origination may account for a significant proportion of new genes [2,14] and contribute to lineage-specific genetic novelties [18–20].
Currently, several comparative transcriptome studies have proposed that a proportion of de novo genes may originate from ancestral long non-coding RNAs (lncRNAs) [7,16,21], while the evolutionary mechanism underlying this lncRNA-protein transition remains elusive. First, it is unknown whether the differences in functional significance of lncRNAs, or some other sequence features, could explain the biased origination process of de novo genes from a specific subset of lncRNAs. Specifically, given that ancestral lncRNAs have precise splicing structures and tissue expression profiles similar to those of de novo proteins in human [16], it is unclear whether they have already obtained certain biological functions on the RNA level: one reason for us to hypothesize that functional non-coding genes may be favorable precursors is because they might survive longer during evolution, providing a wider time window for the emergence and stabilization of ORF, assuming that the emergence of the protein coding part does not interfere with the original function.
Second, it is unclear whether the human de novo genes have gained functional significance. Although it has been an established notion that de novo protein-coding genes could have important functions in Drosophila [17,18,22], the functional significance of de novo genes in hominoid lineage is still controversial–given the smaller effective population size in hominoids [23], the detection of these genes may be largely due to the weaker selection for removing the translational noises. Actually, for the dozens of human-specific de novo genes identified, only a few genes were linked to human diseases and regulations by circumstantial evidence [11,24–26]. While functional studies with transgenic monkeys could potentially characterize the functions of these hominoid-specific proteins, it is still technically challenging and could not provide a global view of the extent to which these genes are functional. Alternatively, a comparative population genetics approach, i.e. characterizing polymorphisms in the gene locus and comparing the pattern to that of the orthologous region in a closely related species, could provide evolutionary clues to the functional significance of the de novo genes.
We thus performed a population genetics study in human and rhesus macaque to interrogate the origin and functional significance of these newly-originated de novo protein-coding genes. We noted that these proteins in human seem to have originated from ancestral GC-rich lncRNAs. Although these lncRNAs generally are not more selectively constrained than other lncRNA loci, and the existence of these newly-originated proteins is not beyond anticipation under neutral expectation, our results showed that at least a proportion of these de novo proteins should have acquired protein-level functions, based on the signatures of purifying selection detected specifically in human populations. We thus propose a mechanism for creation of functional de novo proteins from ancestral lncRNAs during the primate evolution.
Results
Identification of 64 hominoid-specific de novo proteins originated from lncRNAs
To interrogate the genesis and functional implications of de novo proteins in primates, we firstly performed a comprehensive survey for newly-originated de novo protein-coding genes in the hominoid lineage. We devised a genome-wide pipeline integrating ab initio identifications [16] and meta-analysis of public datasets [9–11,13,16] (Fig 1A; Materials and Methods). Briefly, we first inferred the locus ages on the basis of the syntenic genomic alignment generated by UCSC, and only retained human genes with high-quality alignments in the out-group species (Discussion). With this approach, the potential bias in de novo gene identification introduced by blast-like alignments is well controlled [27]. Then, for each locus, the existence of the ORF in multiple out-group species was inferred separately (Materials and Methods). Candidate de novo genes were then identified based on age assignments of ORFs, by summing up the information on the presence and absence of orthologous ORFs in vertebrate phylogeny with the principle of parsimony [2,16]. We further performed sequence similarity study to analyze these candidates against all annotated human proteins, further verifying that they originated through de novo evolution, rather than other mechanisms such as gene duplication (Materials and Methods).
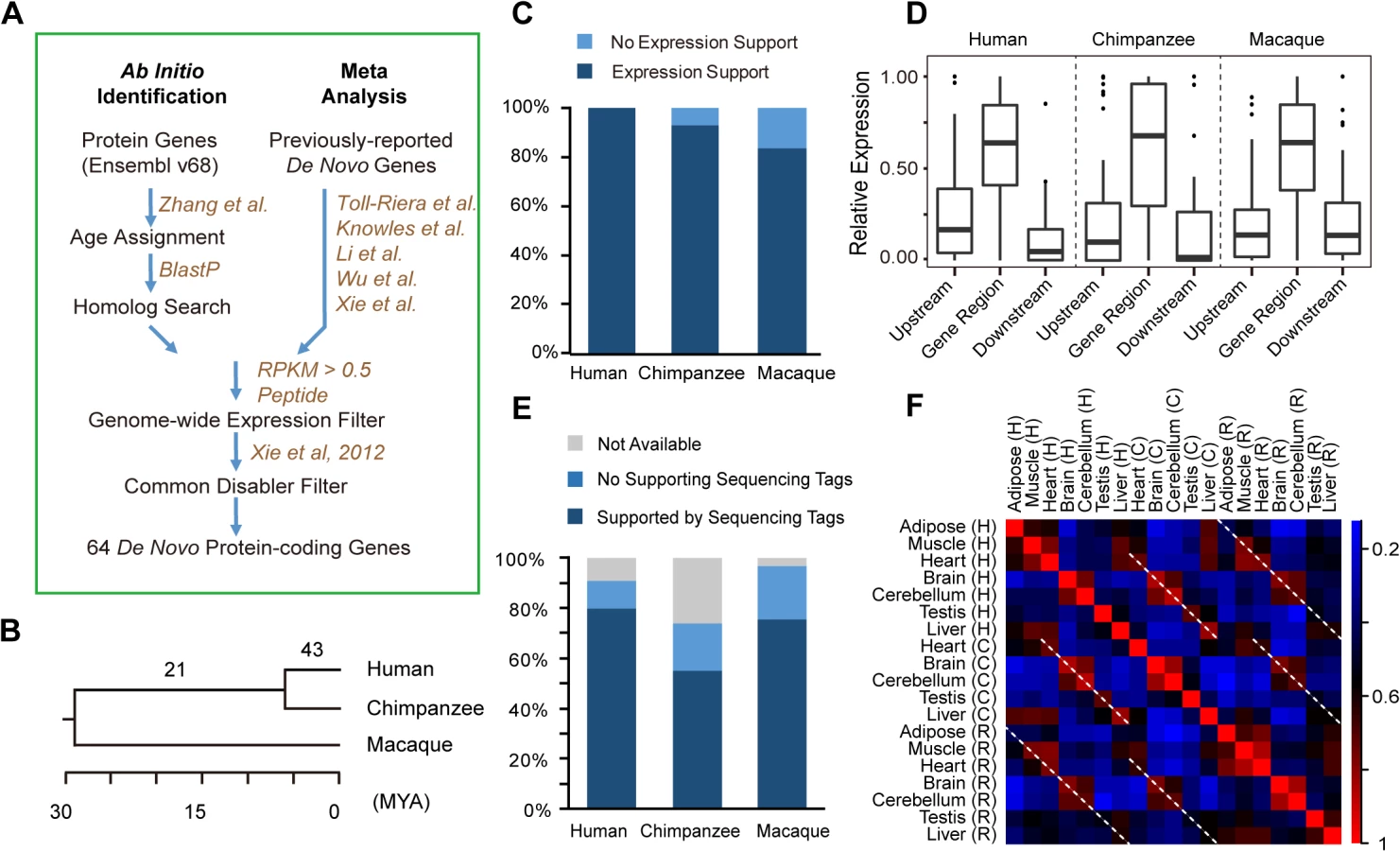
The resulting 56 candidates, together with 99 literature-documented primate-specific de novo genes [9–11,13,16], were then subjected to additional inclusion criteria (Materials and Methods). Consequently, only genes with 1) reliable evidence for transcriptional and translational activities in human (S1 Fig; Materials and Methods), and 2) detectable common ancestral “disablers”, disrupting the ORFs in all out-group species at the same sequence position [9,11], as indication for newly-created but not old dying genes, were included (Fig 1A; Materials and Methods).
In total, 64 protein-coding genes were identified with recent origination in the hominoid lineage through de novo evolution (Fig 1B; Tables 1 and S1), with 43 encoding human-specific proteins (Class I, the younger proteins), and another 21 encoding similar proteins in human and chimpanzee but not in rhesus macaque (Class II, the older proteins).

The transcript structure and expression of these genes at the transcriptional or translational levels in human are strongly supported by public genomics data. The transcriptional structure for all of these genes were supported by full-length mRNA or spliced EST evidence (S2 Table), with 88% of the splicing junctions also supported by short RNA-Seq reads (Fig 1E; Materials and Methods); the full-length transcript structure for 17 of these genes were also verified by the Iso-Seq data, generated recently through the PacBio transcriptome sequencing (S2 Table; Materials and Methods). In addition, the protein expressions for all of these genes were supported by large-scale mass spectrometry studies in human (Tables 1 and S2; Materials and Methods).
To infer the transcriptional capacity of the 64 de novo genes in the common ancestor of human and closely related species, we performed cross-species transcriptome analysis in human, chimpanzee and rhesus macaque. First, we found that 83.9% of the 64 genes, and 92.9% of the 43 human-specific genes transcribed in at least one tissue in rhesus macaque or chimpanzee as lncRNAs (Fig 1C), with the expression levels significantly higher than the background expression levels (S1 Fig; Materials and Methods). Second, the expression levels of the genic regions relative to upstream and downstream regions were comparable among the three species (Fig 1D), and the majority of human splicing junctions were also detectable in chimpanzee and rhesus macaque orthologous regions (Fig 1E). Third, the non-coding orthologs of human de novo genes in rhesus macaque and chimpanzee also show tissue expression profiles similar to human (Fig 1F). The inter-species similarity of tissue expression profiles was further supported by clustering analysis, with the same tissue types from different species clustered together (S2 Fig and S3 Table).
By the parsimony principle, we conclude that the transcription structure and expression profile of these de novo genes had been shaped in the common ancestor prior to the acquisition of coding potential in the human lineage. It is thus interesting to investigate whether these lncRNA precursors with precise splicing structures and tissue expression profiles have already obtained certain biological functions on the RNA level, and may thus represent favorable precursors for new de novo proteins.
lncRNA precursors are not more selectively constrained than other lncRNA loci
Because lncRNAs could have a variety of functions, not all of which can be easily assayed, as an alternative, we sought an evolutionary approach by quantifying the level of selective constraints in the orthologous lncRNA loci of these de novo genes in rhesus macaque as a proxy for determining the functional status in the ancestor. This assumes that the selective constraints on these loci have remained unchanged in the macaque lineage since it had a common ancestor with human.
We first compiled the whole lncRNAome in rhesus macaque using a similar strategy as described previously [31], on the basis of strand-specific RNA-Seq in ten tissues from the same macaque animal [16,32] (Fig 2A; Materials and Methods). A total of 5,641 lncRNA transcripts were assembled, verifying known features of transcripts such as the tight association with epigenetic markers and CpG islands [33,34] (Figs 2B and 2C and S3). Moreover, as positive control, we compiled a list of 89 non-coding genes in rhesus macaque. These non-coding genes are reportedly functional in human, as supported by experimental evidence [35]. Given the existence of the similar lncRNA transcripts in rhesus macaque, we assumed that these macaque lncRNA transcripts may also have functions and are under similar selective constraints as annotated non-coding genes in human (Materials and Methods).

To quantify the level of selective constraints, we then performed whole genome sequencing of 24 independent macaque animals and generated 23.7 billion paired-end reads with high quality, yielding high sequencing coverage of the macaque genome (ranging from 26 - to 70-fold). On the basis of these sequencing data, as well as seven public datasets for macaque genomes [36–38], we profiled 54,079,575 single nucleotide polymorphic sites across the macaque genome, yielding a considerable number of polymorphisms located on the lncRNA loci (S4 Table; Materials and Methods).
On the basis of the polymorphism data in the population of 31 unrelated macaque animals, we measured the level of polymorphisms in the subset of macaque lncRNA loci, whose orthologous regions in human were de novo genes, and compared that to the same measures in all macaque lncRNAs, as well as the list of 89 established non-coding genes as a reference [35] (Materials and Methods).
As expected, we found that the list of 89 established non-coding genes are selectively constrained in rhesus macaque on the basis of the significantly decreased nucleotide diversity (π), compared with the synonymous sites of known macaque coding genes as a neutral control (Wilcoxon one tail test, p-value = 0.008, Fig 2D). The selection on these non-coding genes seems to be moderate, as compared with that of the non-synonymous sites of known macaque coding genes as a benchmark (Fig 2D). In contrast to this small repertoire of non-coding genes with functions, it seems that lncRNA transcripts are in general not selectively constrained in rhesus macaque, with the nucleotide diversity comparable with that of the synonymous sites across the macaque genome as a neutral control (Wilcoxon test, p-value = 0.917, Fig 2D). In addition, the orthologous loci for the lncRNA precursors of human de novo genes are not subjected to strong selective constraints as those non-coding genes, with the population genetics feature indistinguishable from that of the synonymous sites (Wilcoxon test, p-value = 0.570; Fig 2D), as well as that of the whole lncRNA pool (Wilcoxon test, p-value = 0.449; Fig 2D; Materials and Methods). In conclusion, we didn't find evidence for higher selective constraints for the orthologous lncRNA loci for human de novo gene precursors. Hence, it seems likely that the ancestor of de novo genes may not be particularly distinct in terms of functional importance before the proteins arise.
The existence of de novo proteins is not beyond anticipation in terms of their theoretical lifespan
Given that the lncRNA precursors for human de novo genes did not display particularly distinct functional status, it is interesting to investigate whether other features, such as sequence features, may explain why de novo genes originate from some lncRNAs but not the others. In addition, given the smaller effective population size in hominoids, the detection of de novo proteins might arise from translational noise that is not acted upon or not yet removed by purifying selection, rather than being positively selected for due to their newly-acquired protein-level functions. We thus performed comprehensive sequence analysis of these de novo genes to investigate whether any sequence features could underlie the biased origination process of de novo genes from a subset of lncRNAs, and whether the existence of these de novo proteins is beyond anticipation in terms of their theoretical lifespan.
Interestingly, when analyzing the sequence features of these orthologous lncRNA precursors, we found that they have significantly higher GC contents in comparison with other lncRNAs and non-genic regions (Fig 3A and 3B; Wilcoxon one-tailed tests, p-value<1.0e-7 for both comparisons). The ORFs of de novo genes derived from GC-rich lncRNAs were also observed to have significantly higher GC content when compared with functional proteins in RefSeq (Fig 3B and 3C; Median of GC content = 0.57 vs. 0.53; Wilcoxon one-tailed test, p-value = 6.6e-6).

Further investigation revealed that such a GC-rich property endows these newly-originated ORFs with longer theoretical lifespan, even longer than their current age. As the stop codons are AT-rich, the higher GC content usually supports relatively stable ORF compared with GC-poor sequences [39,40] (S4 Fig). As expected, compared with RefSeq proteins, these newly originated ORFs have less content of fragile codons–codons convertible to stop codon by a single point-mutation, and are thus less susceptible to non-sense mutations (Fig 3D; Wilcoxon one-tailed test, p-value = 0.002). Accordingly, we found that these ORFs have long half-life time under neutrality (Materials and Methods), even significantly longer than other functional proteins in RefSeq (Fig 3E; Wilcoxon one-tailed test, p-value<2.2e-16). Especially, compared with the younger de novo genes, the older de novo genes have higher GC content (Fig 3C), less content of fragile codons (Fig 3D) and longer half-life time (Fig 3E). Overall, the theoretical lifespan of these newly-originated proteins is generally longer than their current age (Figs 1B and 3E), thus indicating that the existence of these de novo proteins is not beyond anticipation even under neutral expectation (Fig 3F).
Overall, the de novo gene repertoire we identified in the hominoid lineage actually represents a snapshot for the steady-state representation of a dynamic turnover process of ORFs. The detection of these GC-rich de novo proteins with stable ORFs, together with the previous reports that many de novo genes have stable expression profiles possibly by sharing the transcriptional context with nearby protein-coding genes through cis-natural antisense or bi-directional promoters [16,41], seems to favor the notion that a significant portion of the turnover is probably driven by genetic drift and those GC-rich "survivors" with long ORF lifespan and stable expression profile were retained and detected during a birth-and-death process.
The new de novo proteins are under selective constraints in human population
What we found above suggest that the emergence and retention of de novo genes are likely under neutral forces. However, considering these GC-rich “survivors” have been exposed to natural selection for relatively long time due to their theoretically longer lifespan, it is interesting to investigate whether some of these newly originated ORFs have been maintained by selective constraint in the current population, due to their newly-acquired protein-level functions. We thus performed population genetics study in human and rhesus macaque populations to assess whether selective constraints are applied to these ORF regions in human populations but not their non-coding counterparts in rhesus macaque.
We first profiled a set of polymorphism sites in human populations, by re-analyzing whole genome sequencing data in 67 individuals from different sub-populations (Fig 4A and S5 Table; Materials and Methods). We expect that if the de novo genes encode functional proteins and are maintained by purifying selection, the polymorphism level for exonic regions of these genes will be lower than intronic regions. The polymorphism level for non-synonymous sites should also be significantly lower than that of synonymous sites, as the former will be under much stronger selection. Moreover, we expect to find a difference in the frequency spectra at nonsynonymous vs. synonymous sites, resulting in a skew towards low frequency variants compared to the latter. These are indeed what we found: 1) The θw and π measures were significantly lower in the exonic region of the de novo genes compared to the intronic region of the same locus (Monte Carlo p-values<1e-4; Figs 5A and S5; Materials and Methods). In addition, the UTR regions of these de novo genes showed θw and π measures that are lower than the intronic regions, while slightly higher than CDS regions (Figs 5A and S5). 2) Compared with synonymous sites, the nucleotide diversity for non-synonymous sites was significantly lower (Wilcoxon one-tail test, p-value = 0.019; S6 Fig). Accordingly, the ratio of the nucleotide diversity for non-synonymous sites to synonymous sites was generally smaller than 1 (Fig 5B). 3) The frequency spectrum of the derived alleles had an excess of low-frequency variants at the non-synonymous sites in the de novo genes compared to that at the synonymous sites (Fig 5C), which is similar to known protein-coding genes (Fig 5E). As a control, we classified mutations in human lncRNAs into synonymous or non-synonymous sites within the longest pseudo-ORFs, and didn’t observe any difference in their respective frequency spectrum (Fig 5F; Materials and Methods).


Accordingly, as a negative control, we performed population genetics study on macaque orthologous regions (without coding potential) of human de novo genes, in a population of 82 unrelated rhesus macaque animals (Materials and Methods). Custom library with >135,000 120-bp DNA oligos were designed to capture the macaque orthologous regions. Ultra-deep sequencing was then performed (Materials and Methods) and 222 million 150-bp paired-end reads were generated and uniquely located on the macaque genome (NCBI SRA accession number: SRP052932; S6 Table). The average effective coverage of the targeting regions reached to 94% in each sample (Figs 4B and 4C and S7), and a total of 10,162 single nucleotide polymorphisms were identified across the target genomic regions with high sensitivity and specificity, as verified by a follow-up whole genome sequencing with 30× coverage in one of these macaque animals (Fig 4D and 4E; Materials and Methods).
On the basis of the polymorphism data of the macaque orthologous regions, we found that both the θw and π measures were uniform across the length of these regions, in contrast to the clear differences observed in human (Figs 5A and S5). We further classified the polymorphism sites on macaque lncRNAs into synonymous or non-synonymous sites, according to codon-level alignments between human de novo proteins and their orthologous lncRNAs in rhesus macaque. No significant difference was detected for the nucleotide diversity of pseudo-non-synonymous and pseudo-synonymous sites in rhesus macaque (Wilcoxon one-tail test, p-value = 0.607; S6 Fig), with the ratio of the nucleotide diversity between these two groups comparable to 1 (Fig 5B). In addition, the resulting frequency spectrum of derived alleles at the pseudo-non-synonymous sites is indistinguishable from that at the pseudo-synonymous sites (Fig 5D). The population genetics analyses thus suggest that these newly-originated de novo genes have gained new functions specifically in human.
Taken together, although the de novo proteins seem to emerge from lncRNA precursors with no bias towards those functionally-constrained lncRNAs, and their existence is not beyond the anticipation under neutral expectation, at least a proportion of these proteins should have acquired protein-level functions specifically in human, as revealed by the species-specific signatures of purifying selection on these newly-originated de novo genes. We thus depicted a new mechanism for the origination of functional proteins from ancestral non-coding transcripts with precise splice structures and specific tissue expression profiles during the primate evolution.
Discussion
De novo origination of protein-coding genes could be more pervasive in primates
Although our current study identified a list of 64 hominoid-specific de novo genes, more proteins are expected to originate through this de novo mechanism [42]. Several types of de novo genes might be underrepresented due to the computational pipelines currently used to identify these genes: 1) Genes with shorter ORF. Automatic gene annotation pipeline typically neglect genes with short ORFs by using arbitrary criteria to define the minimal ORF length. Consequently, considering de novo proteins generally have short ORFs [43–45], a large proportion of de novo proteins with short ORFs were removed. 2) Genes without orthologous regions in out-group species were not included, due to the requirements of the high-quality alignments for accurate age assignments of ORFs in vertebrate phylogeny, as well as of the detection of common ancestral “disablers” as indication for newly-created rather than old dying genes. Although such a design effectively lowered the false-positives and potential bias introduced by blast-like alignments in de novo gene identification [27], some false-negatives could still result from our stringent criteria. 3) Genes with non-stable accession numbers. Considering the difficulties in defining the coding potential, the Ensembl accession numbers assigned to de novo genes are typically not stable. Although our study has combined multiple versions of Ensembl databases to identify de novo genes, some genes may still be overlooked. 4) Genes with low expression. De novo genes typically express in low abundance [13,16,18]. As reliable evidence for transcriptional and translational expression is needed to define a de novo gene, especially considering the relatively low sensitivity of mass spectrometry technology to reliably detect peptides of low abundance, these genes may be missed. 5) Additionally, the current identification pipeline by comparative genomics approaches typically focuses on in-group ORFs that are missed in other out-group species. In such an occasion, a considerable proportion of de novo genes originated through lineage-specific expression of pre-existing ORFs might be neglected [17,18].
Overall, although these 64 genes may not fully recapitulate the true repertoire of de novo genes in the hominoid lineage, they should constitute a representative group for further analysis and elucidation of the evolving process of de novo genes from precursor lncRNAs.
Origination of functional proteins from ancestral lncRNAs
Although de novo genes are also regarded as “motherless” genes due to their lack of ancestral protein-coding genes as precursors, we and others have found that at least a proportion of lncRNAs might represent an intermediate stage of their origination, narrowing the gap between non-coding DNA and protein-coding genes. Such an origination process may take advantage of existed genomic contexts. For example, these lncRNA “precursors” usually share the transcriptional context with the nearby protein-coding genes through cis-natural antisense or bi-directional promoters [16,41]. These lncRNAs with stable expression profiles, although not more selectively constrained according to our population genetics study, may then lay the foundation for the emergence of new de novo genes. In addition, the GC-rich sequence property of these lncRNAs further supports stable ORFs of the newly-originated proteins (Fig 3). Overall, these genomic features provide a theoretically favorable foundation for the birth of some functional proteins–a notion well supported by our population genetics data, which revealed that some of these loci already encode human-specific functional proteins (Fig 5).
Although such an origination process is plausible, currently both the definitions of de novo genes and lncRNAs are depending on some arbitrary criteria [9,13,31]. Additional lines of evidence are thus needed to fully support the mechanism through which de novo genes come from the lncRNA pools during the primate evolution. For example, since it is still technically challenging to fully annotate proteomes based on mass-spec studies across tissues, development stages and species, it is inadequate to directly identify human de novo genes on the basis of the presence or absence of peptides across different species. Alternatively, conceptual translation of ORFs between species is still the main strategy in the field to infer the existence of these de novo proteins in different out-group species [9,13]. In this context, although the orthologs of these human de novo genes could be defined as “lncRNAs” in chimpanzee and rhesus macaque by the current criteria, they may actually encode smaller version of these de novo proteins in out-group species.
We thus performed cross-species analyses to test this “functional ORF expansion model”. Briefly, if the functional proteins were indeed absent from out-group species, we would expect similar substitution rates between non-synonymous and synonymous sites when performing comparative genomics analysis. When aligning the truncated forms of the human de novo proteins in non-human primates, we found that the merged dN/dS ratio does not deviate significantly from 1 (dN/dS = 0.90). In line with this finding, our population genetics study on macaque orthologous regions of human de novo genes also indicated that these macaque orthologs may not encode similar functional proteins as in human (Fig 5). Even considering these population genetics evidence, we still could not fully exclude the possibility that some of these so-called “lncRNAs” might actually encode fast-evolving or smaller version of the protein in out-group species. As such, these proteins might be under weak selection, and the signals for selective constraints could not be detected based on the population size of this study. Future mass-spec studies with high sensitivity may aid in clarifying these issues.
“GC-rich inheritance model”: an alternative explanation to the GC-rich feature of coding regions
Protein-coding genes have typically higher GC content than non-coding regions. It has been proposed that the increased GC content in genic regions could be maintained by natural selection, such as the GC preference on the wobble sites potentially shaped by adaptive evolution for the stability of mRNA secondary structure or the efficient protein synthesis [46].
However, here we found that other gene-associated genomic regions are also GC-rich, such as the intronic regions (Fig 3B). Theoretically, considering the models for new gene origination, each protein-coding gene could be traced to an ancient origination event from non-coding DNA. There is thus a formal but as yet unexplored possibility that the biased inheritance from GC-rich lncRNAs could be another major factor underpinning the different extents of GC content between coding regions and genomic background. As we provided an evolutionary and functional connection between protein-coding genes and non-coding DNA regions in the hominoid lineage, we formally tested this “GC-rich inheritance model”. Correspondingly, we found that GC-rich lncRNAs are favorable precursors for new proteins. More importantly, the GC-rich features could be detected in all genomic regions associated with these newly originated de novo genes, even for the wobble sites with established GC preference, as well as their lncRNA precursors, which also resembled those of the well-known protein-coding genes (Figs 3B and S8). Besides being a consequence of adaptive evolution after the acquirements of the ORFs, the GC-rich feature of the protein-coding genes may also inherit from the ancestor lncRNAs, thus complementing previous theory on GC-rich feature for protein-coding genes [46–49].
Materials and Methods
Ethics statement
Rhesus macaque samples were obtained and manipulated from the internationally-accredited (Association for Assessment and Accreditation of Laboratory Animal Care, AAALAC) animal facility of the Institute of Molecular Medicine in Peking University. The present study was approved by the Institutional Animal Care and Use Committee of Peking University.
Identification and characteristics of de novo protein-coding genes in the hominoid lineage
De novo protein-coding genes in the hominoid lineage were identified using a genome-wide pipeline integrating ab initio identifications and meta-analysis of public datasets. On the basis of Ensembl gene annotations (v68), de novo genes were identified using a similar pipeline as we published previously [16]. Briefly, 1) we inferred the locus ages on the basis of the syntenic genomic alignment generated by UCSC, and only human genes assigned with specific locus age were retained; 2) for locus with high-quality alignment (coverage >70% and identity >50%) in the out-group species, the existence of the ORF in multiple out-groups (chimpanzee, orangutan, rhesus macaque, mouse, guinea pig, dog, hedgehog and armadillo) was inferred separately by Exonerate [50], and if the sequence in particular out-group encoded at least one frame-disrupting indel or premature stop codon, with the subsequent maximum continuous ORF shorter than 70% of the human ORF length (a cutoff based on the previous practice in this field [9,13,16]), the ORF was regarded as non-existent in this out-group; 3) we inferred the origination timing of ORFs for these de novo genes by summing up information of their presence or absence in multiple out-group species, along with the phylogenetic tree with the principle of parsimony, and subsequently retained only genes originated in the hominoid lineage; 4) sequence alignments were then performed against all human proteins (BLAST e-value cutoff of 10−6) to ensure these new genes originated through de novo evolution other than gene duplications. Finally, 56 protein-coding genes were identified as candidate de novo genes in the hominoid lineage.
The resulted 56 candidate genes, together with 99 literature-documenting primate-specific de novo genes [9–11,13,16], were then subject to two additional inclusion criteria. First, genome-wide expression filters were introduced to ensure these genes had convincing evidence for transcriptional and translational expression in human. Public RNA-Seq data in 17 human tissues (Human BodyMap 2.0 data from Illumina and data from references [51,52]) were integrated and analyzed to estimate the gene expression level of each gene, according to a standardized pipeline [53]. To distinguish true transcription signals from the background expression, we first estimated the RPKM values for the genomic background represented by 10,000 randomly-selected intergenic regions. The expression levels of intergenic regions were significantly lower than 0.2 RPKM in all tissues (S1A Fig; Monte Carlo p-values ranging from 0.002 to 0.028). Therefore, a more conservative PRKM cutoff of 0.5 was arbitrarily set to confirm the transcriptional expression of these de novo genes in human. Two candidates (ENSG00000205056 and ENSG00000198547) with low RPKM scores were also included due to their reliable experimental evidence for transcriptional expression [9,11]. Peptide evidences from large-scale mass spectrometry studies were then extracted from PRIDE [28], PeptideAtlas [54], ProteomicsDB [55] and Human Proteome Map [29]. A peptide was considered to support the protein expression of a de novo gene only if 1) when performing BLAT similarity searches against all human proteins (Ensembl v68, BLAT settings-t = prot-q = prot-stepSize = 5), its whole sequence exactly match the CDS region of the de novo gene, with the second-best hit in the proteome (if existing) including at least one mismatch; and 2) when performing BLAT similarity searches against the human genome, its whole sequence identically and exclusively match the CDS region of the de novo gene (hg19, BLAT settings-stepSize = 5-stepSize = 5-t = dnax-q = prot). Only genes with 1) RNA-Seq RPKM >0.5 in at least one of the 17 human tissues, and 2) at least one convincing item of peptide evidence in support, were retained (Fig 1A). Second, to verify that these genes are newly-originated rather than old dying genes, we manually checked the corresponding ORF regions in multiple out-group species (chimpanzee, orangutan, rhesus macaque, mouse, guinea pig, dog, hedgehog and armadillo), and only genes with common ancestral disablers shared by multiple out-group species were retained (Fig 1A). Here, a common ancestral disabler refers to a mutation disrupting the ORF in multiple out-group species at the same sequence position [9,11]. In such scenario, the mutation is more likely to be of an ancestral status according to the parsimony principle, thus indicating the gene is newly-originated rather than old dying. Totally, a list of 64 genes was identified to originate recently in the hominoid lineage through de novo evolution (Fig 1B and Tables 1, S1 and S2).
We also studied the characteristics of these de novo genes across primate species in the context of new genomics technologies. According to computational pipelines described previously [9,13,16], mRNA and EST data from UCSC Genome Browser, RNA-Seq data archived in RhesusBase [53,56], as well as single-molecule long-read sequencing data on human transcriptome [57] were downloaded and analyzed to investigate the transcriptional structure of these de novo genes in human. On the basis of public RNA-Seq data in human, chimpanzee and rhesus macaque [16,51,52], comparative transcriptome studies were then performed to compare the transcription level, splicing structure and tissue expression profiles of these de novo genes with their non-coding orthologs in chimpanzee and rhesus macaque, according to a pipeline previously described by us [16]. Specially, an RPKM cutoff of 0.2 was set to distinguish convincing transcription and transcriptional noise as described above (S1B and S1C Fig).
LncRNA identifications in rhesus macaque
Strand-specific, Poly(A)-positive RNA-Seq data in ten tissues (adipose, prefrontal cortex, cerebellum, heart, kidney, liver, lung, muscle, spleen, testis) of the same macaque animal were used to assemble the lncRNAome in rhesus macaque [16,32], following a computational pipeline as described previously [31]. Briefly, RNA-Seq reads of each macaque tissue were aligned separately to the macaque genome (rheMac2) with Tophat (v2.0.6) [58]. Transcriptome assembly was then performed with both Cufflinks (v2.0.2) and Scripture (VPaperR3) [59,60], and redundant transcripts were merged with Cuffcompare (v2.0.2) [60] after boundary correction. To control for false-positives, only long, multi-exonic transcripts (>200 bp) with supportive evidences in ≥2 tissues or by both assemblers were retained [31,61]. To evaluate the performance of this transcriptome assembly, Cuffcompare (v2.0.2) was also introduced to compare the assembled transcripts with multi-exonic protein-coding genes as annotated in RefSeq. Finally, a total of 90,322 multi-exonic transcripts were assembled, which represents transcript structures reconstructing for 95% known multi-exonic protein-coding genes, suggesting the feasibility of this assembly strategy (S3 Fig).
Several stringent criteria, such as proteome annotation - and comparative genomics-based filtering procedures, were incorporated to exclude protein-coding transcripts. Briefly, 1) transcripts with ≥1 splice junction overlapped with known protein-coding genes annotated in either Ensembl or RefSeq were discarded; 2) PhyloCSF was applied to score the coding potential of these candidates (multiple sequence alignments of 9 mammalian genomes,—frames = 3—orf = StopStop3) [62] and only transcripts with PhyloCSFscore <65 were retained, corresponding to a false negative rate of 1% and a false positive rate of 5% on the basis of RefSeq annotation. 3) the nucleotide sequences or 3-frame stop-to-stop translation products were subjected to Blastx, Blastp and HMMER searching against all human proteins or known protein domains (Pfam-A, Pfam-B) [63], and transcripts with significant hits (e-value ≤10−4) were discarded; 4) transcripts with putative ORFs ≥100aa longer were also discarded. The strategy had a good performance in distinguishing lncRNAs from protein-coding transcripts (S3 Fig) and a total of 5,641 lncRNA transcripts were assembled. On the other hand, non-coding genes in human were searched and downloaded from lncRNAdb (http://www.lncrnadb.org/) [35] and the macaque orthologs of these functional human lncRNAs were then retrieved by liftOver.
Characterization of sequence features
For human de novo genes and their orthologs in chimpanzee and rhesus macaque, sequences of different genomic regions were retrieved and the GC contents were calculated by a customized Perl script (https://github.com/Jia-Yu-Chen). Following a previous study [40], we also calculated for each de novo gene the proportion of fragile codons that could become stop codons by single mutation. We further investigated whether the existence of these de novo proteins is beyond anticipation in terms of their theoretical lifespan. Briefly, an ORF would eventually be interrupted if it does not experience any functional constraint, and the rate of ORF interruption under neutrality is largely determined by point mutation rate and insertion/deletion mutation rate. We thus estimated the interruption rate in terms of half-life time of ORF (t1/2) according to the computational simulation method developed by Zhang and Webb [64] (Fig 3E). The half-life time of a given ORF is the time required for an ORF to be interrupted in one-half of 20,000 simulation replicates, with the rates of point and insertion/deletion mutations being set as 1.25 and 0.1 per site per billion years, respectively, as previously defined [64]. Given this half-life time (t1/2), the minimal probability of an ORF remaining intact today under neutrality was then determined by the following equation, by assuming that the lineage-specific gene emerged right after the species divergence from the most recent common ancestor (T) (Fig 3F).

Targeted and whole genome sequencing and polymorphism identification
We firstly profiled the polymorphism data in human populations, by re-analyzing whole genome sequencing data in 67 individuals from different sub-populations and archived with high sequencing coverage by the 1000 Genomes Project (S5 Table). Briefly, deep sequencing reads were mapped to the human genome (hg19) using BWA [65], and the polymorphism sites of each sample were identified and evaluated according to the standard GATK pipeline with UnifiedGenotyper (V2.7–4) [66]. After stringent filtering strategies to remove false-positives in variant calling, 18,186,523 highly reliable single nucleotide variants were identified across the human genome, with 85.4% supported also by the 1000 Genomes Project (Fig 4A).
Accordingly, we profiled the distributions of polymorphic sites in rhesus macaque populations as a reference. For each human de novo gene, we performed targeted capture and ultra-deep sequencing of the macaque orthologous regions (and 1kb flanking regions) in a population of 82 unrelated male animals (S6 Table). Briefly, custom library with >135,000 120-bp DNA oligo probes were designed by Agilent SureSelect XT Target Enrichment System (Agilent Technologies, Inc., Santa Clara, USA), with a 3-folds tiling coverage, to capture the targeted regions in rhesus macaque. Genomic DNA from the macaque animals was isolated from 200–500μl whole blood using the QIAamp DNA Blood Mini Kit (Qiagen, Venlo, Netherlands) and 3μg DNA of each animal was sheared to fragments with a peak at 150–200bp using Covaris S220. Then, the adaptor-ligated libraries were amplified, purified and hybridized with SureSelect Capture Library according to the manufacturer’s instructions. After 16h hybridization at 65°C, the captured targets were pulled down by Dynabeads MyOne Streptavidine T1 (Life Technologies, Ltd., Carlsbad, USA) and amplified for the library preparation, which were then sequenced on Illumina Miseq system with 151-bp paired-end read mode. Totally 222 million 150-bp paired-end reads were generated and uniquely located on the macaque genome (S6 Table). The average effective coverage of the targeted regions reached to 94% in each sample (Fig 4B), and 86% of whole orthologous region (or 95% of CDS regions) were sequenced with coverage of ≥30 in all of the 82 macaque animals (Figs 4C and S6). A total of 10,162 highly-reliable single nucleotide polymorphisms were then identified, according to the standard GATK pipeline with UnifiedGenotyper (V2.7–4) [66].
To evaluate whether allele dropout or other false-positives introduced by target capture may compromise our approach, we further performed whole genome sequencing with 30× coverage in one of these macaque animals (Animal ID: 920653) for evaluation (Fig 4B). Genomic DNA was obtained for the library preparation of whole genome re-sequencing, deep sequencing was performed on a HiSeq 2000 Sequencing System with a 151×2 paired-end read mode, and single nucleotide polymorphisms were identified according to the standard GATK pipeline. Finally, 96.2% polymorphic sites identified in the targeted sequencing were verified by the whole genome sequencing, with 99.5% showing the same genotype (Fig 4D and 4E).
Whole genome sequencing data from 24 macaque animals generated previously in our lab, as well as seven animals published previously [36–38], were also analyzed to profile a genome-wide polymorphism dataset across the macaque genome, according to the pipeline as described above. All deep sequencing data in this study are available at NCBI SRA under accession numbers SRP052932.
Population genetic analyses in human and rhesus macaque
On the basis of the polymorphism data from the population of 31 macaque animals, we measured the nucleotide diversity (π) for the orthologous lncRNA loci of human de novo genes, all lncRNAs, and a list of 89 functional non-coding genes in rhesus macaque. Non-synonymous and synonymous sites of macaque protein-coding genes as annotated by RefSeq were used as benchmarks for the extent of the selective constraints (Fig 2D). Wilcoxon test was performed to test whether the nucleotide diversity between two groups are significantly different, with a p-value cutoff of 0.05 (Fig 2D).
On the basis of the polymorphism data obtained by analyzing the whole genome sequencing data of 67 human individuals and the targeted sequencing data of 82 macaque animals, we estimated the polymorphism levels (θw and π) for different genomic regions (exon, intron, CDS and UTR) of the de novo genes in human and their orthologs in rhesus macaque (Figs 5A and S5). We further performed statistical tests to determine whether the different polymorphism levels between exonic and intronic regions of human de novo genes are statistically significant, with the background estimated by 10,000 times of Monte Carlo simulations, assuming the polymorphic sites were randomly distributed in exonic and intronic regions of human de novo genes. Considering that the average nucleotide diversity in rhesus macaque is higher [67], if the exonic regions are more selectively constrained than intronic regions, we should have greater statistical power to detect the difference. The observation of a comparable nucleotide diversity between macaque exonic and intronic regions then indicates that these macaque orthologs of human de novo genes may not encode similar functional proteins as in human (Figs 5A and S5).
The ratio of the nucleotide diversity between non-synonymous sites to synonymous sites was also determined for each de novo gene, as well as its non-coding ortholog in rhesus macaque (Fig 5B), in which the pseudo-non-synonymous and pseudo-synonymous sites in macaque orthologs were determined by codon-level alignment with human de novo proteins. For each polymorphic site, the derived allele was defined by the EPO pipeline [68,69]. The frequency spectra of derived alleles were then estimated, with 1,000 times of bootstrap performed to estimate the confidence intervals of the proportions of polymorphism sites (Fig 5C–5F). Similar analyses were performed for known protein-coding genes as annotated by RefSeq, as well as human lncRNAs as annotated by GENCODE (v19) as controls.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Jacob F (1977) Evolution and tinkering. Science 196 : 1161–1166. 860134
2. Zhou Q, Zhang G, Zhang Y, Xu S, Zhao R, et al. (2008) On the origin of new genes in Drosophila. Genome Res 18 : 1446–1455. doi: 10.1101/gr.076588.108 18550802
3. Long M, Betran E, Thornton K, Wang W (2003) The origin of new genes: glimpses from the young and old. Nat Rev Genet 4 : 865–875. 14634634
4. Begun DJ, Lindfors HA, Kern AD, Jones CD (2007) Evidence for de novo evolution of testis-expressed genes in the Drosophila yakuba/Drosophila erecta clade. Genetics 176 : 1131–1137. 17435230
5. Begun DJ, Lindfors HA, Thompson ME, Holloway AK (2006) Recently evolved genes identified from Drosophila yakuba and D. erecta accessory gland expressed sequence tags. Genetics 172 : 1675–1681. 16361246
6. Levine MT, Jones CD, Kern AD, Lindfors HA, Begun DJ (2006) Novel genes derived from noncoding DNA in Drosophila melanogaster are frequently X-linked and exhibit testis-biased expression. Proc Natl Acad Sci U S A 103 : 9935–9939. 16777968
7. Cai J, Zhao R, Jiang H, Wang W (2008) De novo origination of a new protein-coding gene in Saccharomyces cerevisiae. Genetics 179 : 487–496. doi: 10.1534/genetics.107.084491 18493065
8. Heinen TJ, Staubach F, Haming D, Tautz D (2009) Emergence of a new gene from an intergenic region. Curr Biol 19 : 1527–1531. doi: 10.1016/j.cub.2009.07.049 19733073
9. Knowles DG, McLysaght A (2009) Recent de novo origin of human protein-coding genes. Genome Res 19 : 1752–1759. doi: 10.1101/gr.095026.109 19726446
10. Toll-Riera M, Bosch N, Bellora N, Castelo R, Armengol L, et al. (2009) Origin of primate orphan genes: a comparative genomics approach. Mol Biol Evol 26 : 603–612. doi: 10.1093/molbev/msn281 19064677
11. Li CY, Zhang Y, Wang Z, Zhang Y, Cao C, et al. (2010) A human-specific de novo protein-coding gene associated with human brain functions. PLoS Comput Biol 6: e1000734. doi: 10.1371/journal.pcbi.1000734 20376170
12. Li D, Dong Y, Jiang Y, Jiang H, Cai J, et al. (2010) A de novo originated gene depresses budding yeast mating pathway and is repressed by the protein encoded by its antisense strand. Cell Res 20 : 408–420. doi: 10.1038/cr.2010.31 20195295
13. Wu DD, Irwin DM, Zhang YP (2011) De novo origin of human protein-coding genes. PLoS Genet 7: e1002379. doi: 10.1371/journal.pgen.1002379 22102831
14. Carvunis AR, Rolland T, Wapinski I, Calderwood MA, Yildirim MA, et al. (2012) Proto-genes and de novo gene birth. Nature 487 : 370–374. doi: 10.1038/nature11184 22722833
15. Sabath N, Wagner A, Karlin D (2012) Evolution of viral proteins originated de novo by overprinting. Mol Biol Evol 29 : 3767–3780. doi: 10.1093/molbev/mss179 22821011
16. Xie C, Zhang YE, Chen JY, Liu CJ, Zhou WZ, et al. (2012) Hominoid-specific de novo protein-coding genes originating from long non-coding RNAs. PLoS Genet 8: e1002942. doi: 10.1371/journal.pgen.1002942 23028352
17. Reinhardt JA, Wanjiru BM, Brant AT, Saelao P, Begun DJ, et al. (2013) De novo ORFs in Drosophila are important to organismal fitness and evolved rapidly from previously non-coding sequences. PLoS Genet 9: e1003860. doi: 10.1371/journal.pgen.1003860 24146629
18. Zhao L, Saelao P, Jones CD, Begun DJ (2014) Origin and spread of de novo genes in Drosophila melanogaster populations. Science 343 : 769–772. doi: 10.1126/science.1248286 24457212
19. Neme R, Tautz D (2014) Evolution: dynamics of de novo gene emergence. Curr Biol 24: R238–240. doi: 10.1016/j.cub.2014.02.016 24650912
20. Palmieri N, Kosiol C, Schlotterer C (2014) The life cycle of Drosophila orphan genes. Elife 3: e01311. doi: 10.7554/eLife.01311 24554240
21. Ruiz-Orera J, Messeguer X, Subirana JA, Alba MM (2014) Long non-coding RNAs as a source of new peptides. Elife 3: e03523. doi: 10.7554/eLife.03523 25233276
22. Chen S, Zhang YE, Long M (2010) New genes in Drosophila quickly become essential. Science 330 : 1682–1685. doi: 10.1126/science.1196380 21164016
23. Charlesworth B (2009) Fundamental concepts in genetics: effective population size and patterns of molecular evolution and variation. Nat Rev Genet 10 : 195–205. doi: 10.1038/nrg2526 19204717
24. Chen HY, Wen JM, Xiao XW, Li DJ, Guo XL, et al. (2010) [Expression of human testis development related gene 1 in testicular cancer detected by tissue microarray]. Zhonghua Nan Ke Xue 16 : 883–886. 21243750
25. Gonzalez D, Else M, Wren D, Usai M, Buhl AM, et al. (2013) CLLU1 expression has prognostic value in chronic lymphocytic leukemia after first-line therapy in younger patients and in those with mutated IGHV genes. Haematologica 98 : 274–278. doi: 10.3324/haematol.2012.070201 22899580
26. Moreaux J, Hose D, Bonnefond A, Reme T, Robert N, et al. (2010) MYEOV is a prognostic factor in multiple myeloma. Exp Hematol 38 : 1189–1198 e1183. doi: 10.1016/j.exphem.2010.09.002 20854874
27. Moyers BA, Zhang J (2015) Phylostratigraphic bias creates spurious patterns of genome evolution. Mol Biol Evol 32 : 258–267. doi: 10.1093/molbev/msu286 25312911
28. Vizcaino JA, Cote RG, Csordas A, Dianes JA, Fabregat A, et al. (2013) The Proteomics Identifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Research 41: D1063–D1069. doi: 10.1093/nar/gks1262 23203882
29. Kim MS, Pinto SM, Getnet D, Nirujogi RS, Manda SS, et al. (2014) A draft map of the human proteome. Nature 509 : 575–+. doi: 10.1038/nature13302 24870542
30. Desiere F, Deutsch EW, King NL, Nesvizhskii AI, Mallick P, et al. (2006) The PeptideAtlas project. Nucleic Acids Research 34: D655–D658. 16381952
31. Pauli A, Valen E, Lin MF, Garber M, Vastenhouw NL, et al. (2012) Systematic identification of long noncoding RNAs expressed during zebrafish embryogenesis. Genome Res 22 : 577–591. doi: 10.1101/gr.133009.111 22110045
32. Chen JY, Peng Z, Zhang R, Yang XZ, Tan BC, et al. (2014) RNA editome in rhesus macaque shaped by purifying selection. PLoS Genet 10: e1004274. doi: 10.1371/journal.pgen.1004274 24722121
33. Gardiner-Garden M, Frommer M (1987) CpG islands in vertebrate genomes. J Mol Biol 196 : 261–282. 3656447
34. Liu Y, Han D, Han Y, Yan Z, Xie B, et al. (2011) Ab initio identification of transcription start sites in the Rhesus macaque genome by histone modification and RNA-Seq. Nucleic Acids Res 39 : 1408–1418. doi: 10.1093/nar/gkq956 20952408
35. Quek XC, Thomson DW, Maag JL, Bartonicek N, Signal B, et al. (2015) lncRNAdb v2.0: expanding the reference database for functional long noncoding RNAs. Nucleic Acids Res 43: D168–173. doi: 10.1093/nar/gku988 25332394
36. Fang X, Zhang Y, Zhang R, Yang L, Li M, et al. (2011) Genome sequence and global sequence variation map with 5.5 million SNPs in Chinese rhesus macaque. Genome Biol 12: R63. doi: 10.1186/gb-2011-12-7-r63 21733155
37. Yan G, Zhang G, Fang X, Zhang Y, Li C, et al. (2011) Genome sequencing and comparison of two nonhuman primate animal models, the cynomolgus and Chinese rhesus macaques. Nat Biotechnol 29 : 1019–1023. doi: 10.1038/nbt.1992 22002653
38. Gokcumen O, Tischler V, Tica J, Zhu Q, Iskow RC, et al. (2013) Primate genome architecture influences structural variation mechanisms and functional consequences. Proc Natl Acad Sci U S A 110 : 15764–15769. doi: 10.1073/pnas.1305904110 24014587
39. Oliver JL, Marin A (1996) A relationship between GC content and coding-sequence length. J Mol Evol 43 : 216–223. 8703087
40. Cusack BP, Arndt PF, Duret L, Roest Crollius H (2011) Preventing dangerous nonsense: selection for robustness to transcriptional error in human genes. PLoS Genet 7: e1002276. doi: 10.1371/journal.pgen.1002276 22022272
41. Wu X, Sharp PA (2013) Divergent transcription: a driving force for new gene origination? Cell 155 : 990–996. doi: 10.1016/j.cell.2013.10.048 24267885
42. Tautz D, Domazet-Loso T (2011) The evolutionary origin of orphan genes. Nat Rev Genet 12 : 692–702. doi: 10.1038/nrg3053 21878963
43. Siepel A (2009) Darwinian alchemy: Human genes from noncoding DNA. Genome Res 19 : 1693–1695. doi: 10.1101/gr.098376.109 19797681
44. Domazet-Loso T, Tautz D (2003) An evolutionary analysis of orphan genes in Drosophila. Genome Res 13 : 2213–2219. 14525923
45. Neme R, Tautz D (2013) Phylogenetic patterns of emergence of new genes support a model of frequent de novo evolution. BMC Genomics 14 : 117. doi: 10.1186/1471-2164-14-117 23433480
46. Chamary JV, Parmley JL, Hurst LD (2006) Hearing silence: non-neutral evolution at synonymous sites in mammals. Nat Rev Genet 7 : 98–108. 16418745
47. Galtier N, Piganeau G, Mouchiroud D, Duret L (2001) GC-content evolution in mammalian genomes: the biased gene conversion hypothesis. Genetics 159 : 907–911. 11693127
48. Singh ND, Davis JC, Petrov DA (2005) Codon bias and noncoding GC content correlate negatively with recombination rate on the Drosophila X chromosome. J Mol Evol 61 : 315–324. 16044248
49. Meunier J, Duret L (2004) Recombination drives the evolution of GC-content in the human genome. Mol Biol Evol 21 : 984–990. 14963104
50. Slater GS, Birney E (2005) Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics 6 : 31. 15713233
51. Brawand D, Soumillon M, Necsulea A, Julien P, Csardi G, et al. (2011) The evolution of gene expression levels in mammalian organs. Nature 478 : 343–348. doi: 10.1038/nature10532 22012392
52. Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, et al. (2008) Alternative isoform regulation in human tissue transcriptomes. Nature 456 : 470–476. doi: 10.1038/nature07509 18978772
53. Zhang SJ, Liu CJ, Yu P, Zhong X, Chen JY, et al. (2014) Evolutionary interrogation of human biology in well-annotated genomic framework of rhesus macaque. Mol Biol Evol 31 : 1309–1324. doi: 10.1093/molbev/msu084 24577841
54. Deutsch EW (2010) The PeptideAtlas Project. Methods Mol Biol 604 : 285–296. doi: 10.1007/978-1-60761-444-9_19 20013378
55. Wilhelm M, Schlegl J, Hahne H, Moghaddas Gholami A, Lieberenz M, et al. (2014) Mass-spectrometry-based draft of the human proteome. Nature 509 : 582–587. doi: 10.1038/nature13319 24870543
56. Zhang SJ, Liu CJ, Shi M, Kong L, Chen JY, et al. (2013) RhesusBase: a knowledgebase for the monkey research community. Nucleic Acids Res 41: D892–905. doi: 10.1093/nar/gks835 22965133
57. Au KF, Sebastiano V, Afshar PT, Durruthy JD, Lee L, et al. (2013) Characterization of the human ESC transcriptome by hybrid sequencing. Proc Natl Acad Sci U S A 110: E4821–4830. doi: 10.1073/pnas.1320101110 24282307
58. Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, et al. (2013) TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14: R36. doi: 10.1186/gb-2013-14-4-r36 23618408
59. Guttman M, Garber M, Levin JZ, Donaghey J, Robinson J, et al. (2010) Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat Biotechnol 28 : 503–510. doi: 10.1038/nbt.1633 20436462
60. Trapnell C, Roberts A, Goff L, Pertea G, Kim D, et al. (2012) Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7 : 562–578. doi: 10.1038/nprot.2012.016 22383036
61. Cabili MN, Trapnell C, Goff L, Koziol M, Tazon-Vega B, et al. (2011) Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev 25 : 1915–1927. doi: 10.1101/gad.17446611 21890647
62. Lin MF, Jungreis I, Kellis M (2011) PhyloCSF: a comparative genomics method to distinguish protein coding and non-coding regions. Bioinformatics 27: i275–282. doi: 10.1093/bioinformatics/btr209 21685081
63. Eddy SR (2011) Accelerated Profile HMM Searches. PLoS Comput Biol 7: e1002195. doi: 10.1371/journal.pcbi.1002195 22039361
64. Zhang J, Webb DM (2003) Evolutionary deterioration of the vomeronasal pheromone transduction pathway in catarrhine primates. Proc Natl Acad Sci U S A 100 : 8337–8341. 12826614
65. Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25 : 1754–1760. doi: 10.1093/bioinformatics/btp324 19451168
66. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, et al. (2010) The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20 : 1297–1303. doi: 10.1101/gr.107524.110 20644199
67. Yuan Q, Zhou Z, Lindell SG, Higley JD, Ferguson B, et al. (2012) The rhesus macaque is three times as diverse but more closely equivalent in damaging coding variation as compared to the human. BMC Genet 13 : 52. doi: 10.1186/1471-2156-13-52 22747632
68. Paten B, Herrero J, Beal K, Fitzgerald S, Birney E (2008) Enredo and Pecan: genome-wide mammalian consistency-based multiple alignment with paralogs. Genome Res 18 : 1814–1828. doi: 10.1101/gr.076554.108 18849524
69. Paten B, Herrero J, Fitzgerald S, Beal K, Flicek P, et al. (2008) Genome-wide nucleotide-level mammalian ancestor reconstruction. Genome Res 18 : 1829–1843. doi: 10.1101/gr.076521.108 18849525
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2015 Číslo 7
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Functional Constraint Profiling of a Viral Protein Reveals Discordance of Evolutionary Conservation and Functionality
- Reversible Oxidation of a Conserved Methionine in the Nuclear Export Sequence Determines Subcellular Distribution and Activity of the Fungal Nitrate Regulator NirA
- Modeling Implicates in Nephropathy: Evidence for Dominant Negative Effects and Epistasis under Anemic Stress
- Nutritional Control of DNA Replication Initiation through the Proteolysis and Regulated Translation of DnaA
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy