
Corp Regulates P53 in via a Negative Feedback Loop
Organisms have exquisitely sensitive mechanisms to detect and respond to DNA damage. If DNA damage in a cell can be repaired, then that cell may resume its normal function. In multi-cellular organisms, if a cell cannot repair DNA damage it usually undergoes programmed cell death. This prevents the proliferation of cells with damaged genomes, which might otherwise differentiate incorrectly or proliferate without limit as cancer. In Drosophila melanogaster we identified corp as a gene that promotes the survival of such cells. Transcription of corp is activated by the P53 tumor suppressor protein, known primarily for its induction of cell death in response to DNA damage. Our experiments show that P53 regulates both pro-death and anti-death genes, and that a competition between these opposing factors determines cell fate. We find that corp functions by down-regulating P53, establishing a negative feedback loop. In vertebrates an identical mode of regulation is known: P53 up-regulates Mdm2, which physically interacts with P53 and is its primary negative regulator. We identified a protein motif on Corp that is shared with Mdm2, and is required for physical interaction between Corp and Drosophila P53. These results reinforce and strengthen the similarities of the P53 pathways and their regulation in vertebrates and in Drosophila.
Published in the journal:
. PLoS Genet 11(7): e32767. doi:10.1371/journal.pgen.1005400
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1005400
Summary
Organisms have exquisitely sensitive mechanisms to detect and respond to DNA damage. If DNA damage in a cell can be repaired, then that cell may resume its normal function. In multi-cellular organisms, if a cell cannot repair DNA damage it usually undergoes programmed cell death. This prevents the proliferation of cells with damaged genomes, which might otherwise differentiate incorrectly or proliferate without limit as cancer. In Drosophila melanogaster we identified corp as a gene that promotes the survival of such cells. Transcription of corp is activated by the P53 tumor suppressor protein, known primarily for its induction of cell death in response to DNA damage. Our experiments show that P53 regulates both pro-death and anti-death genes, and that a competition between these opposing factors determines cell fate. We find that corp functions by down-regulating P53, establishing a negative feedback loop. In vertebrates an identical mode of regulation is known: P53 up-regulates Mdm2, which physically interacts with P53 and is its primary negative regulator. We identified a protein motif on Corp that is shared with Mdm2, and is required for physical interaction between Corp and Drosophila P53. These results reinforce and strengthen the similarities of the P53 pathways and their regulation in vertebrates and in Drosophila.
Introduction
When cells encounter damage to their DNA in the form of DSBs, DNA damage response (DDR) pathways are triggered. The ensuing signaling cascades result in cell cycle arrest, induction of DNA repair genes, and in some cases, apoptosis. It is generally thought that, if damage cannot be repaired, cells will undergo apoptosis rather than continue to divide and propagate a damaged genome. If cells with irreparable damage do survive and proliferate, it can result in widespread genomic instability, creating an early state in the progress towards cancer [1–3]. In Drosophila melanogaster, most cells undergo apoptosis in response to irreparable DNA damage, but a few cells escape, continue to divide and exhibit genomic instability [4–6]. In our current study, we aimed to investigate the genetic mechanisms that allow cells to survive in the presence of irreparably damaged DNA.
One of the key players that controls the fate of a cell following DNA damage is the tumor-suppressor encoded by the p53 gene, which is found to be mutated in most human cancers [7]. In response to DNA damage, the ATM kinase (encoded by tefu in Drosophila) phosphorylates Chk2 (encoded by lok), which in turn phosphorylates and activates P53 [8–10]. Activated P53 is primarily a transcriptional regulator that promotes or inhibits the expression of a large number of target genes that encode a variety of cellular functions such as DNA repair, cell cycle arrest and apoptosis [11–17]. A cell that detects damage and engages the P53 damage response pathway may either repair the damage or experience senescence or death [1–3,18]. Humans that lack one copy of p53 are prone to develop cancers, and p53 knockout mice develop cancers at an increased rate [1–6,19,20]. Similarly, p53-null Drosophila fail to eliminate cells with a DSB in their genome [5,7,20–23].
The Mdm2 gene is a prominent target of P53 in mammals. It encodes a ubiquitin ligase that negatively regulates P53 and promotes its degradation, constituting a negative feedback loop [8–10,24,25]. Mediation of apoptosis by P53 is highly conserved throughout metazoa, including Drosophila melanogaster [11–18,21–23,26]. However, apart from DNA repair genes, targets of p53 that antagonize apoptosis have yet to be reported in flies and no homolog of Mdm2 has been identified.
Here, we report the identification of a gene, companion of reaper (corp), whose overexpression mimics the effects of p53 mutants in the soma and the germline. Our experiments indicate that Corp negatively regulates P53 protein levels. The corp gene has been previously identified as a transcriptional target of P53 [14,15,27], thus Corp acts on P53 in a negative feedback loop. Furthermore, Corp exhibits similarity to Mdm2 in a region essential for the Mdm2-P53 interaction, and we find that this region of Corp mediates a physical interaction with Drosophila P53. These similarities lead us to conclude that corp encodes a functional analog of vertebrate Mdm2 in flies and strengthens the similarities between the regulation and the functions of P53 in Drosophila and mammals.
Results
Corp suppresses tissue ablation resulting from DNA damage
The previously described BARTL (Bar and Telomere Loss) assay [20] was used to screen for insertions of a P-element mis-expression element [28] that modify the eye phenotype resulting from the production of an irreparable DNA DSB. In brief, a combination of eyeless-Gal4 (eyGal4) and UAS-FLP is used to drive FLP recombinase expression in proliferating cells of the eye throughout development [29–31]. These flies also carry a Y chromosome with inverted FRT repeats (DcY(H1), or simply H1). Recombination between FRTs in inverted orientation on sister chromatids produces dicentric chromosomes which break in the subsequent mitotic division, delivering a chromosome with a single broken end to each of the two daughter cells (Fig 1). This results in substantial P53-mediated apoptosis and produces flies with characteristic small and rough eyes (Fig 2B). By introducing an EP transposon insertion, which carries UAS elements that can drive expression of a neighboring gene, the flies’ eyes may become larger or smaller, indicating that the EP element in question modifies the fate of cells in these eyes. We identified one such EP insertion (P{EPgy2}CG1632EY03495) that produced nearly wildtype eyes in the BARTL assay (Fig 2C). This insertion was ideally placed to drive expression of the corp+ gene (CG10965). By qRT-PCR we confirmed that when Gal4 induces the P{EPgy2}CG1632EY03495 element (hereafter referred to as EP-corp+) it drives overexpression of corp+ (S1 Fig). We also constructed a UAS-corp+ transgene, and found that its effect was nearly identical to that produced by EP-corp+ (Fig 2D).


When we tested RNAi-mediated knockdown of corp in the BARTL assay the opposite result was obtained: the eye was completely ablated (Fig 2E; n. b., ey expression extends beyond the eye proper, accounting for, in some cases, nearly complete ablation of the head). A corp mutant was also generated by imprecise excision of a P element located in the 5’ region of the gene. This mutant, corp95B (S2 Fig), is viable in homozygous condition and without obvious phenotype on its own. However, like RNAi-mediated corp knockdown, corp95B completely ablates the eye in the BARTL assay (Fig 2F). This effect can be rescued by the UAS-corp+ transgene (Fig 2G).
To verify that the gene CG1632, in whose intron corp is located and which is transcribed opposite to corp (S2 Fig), is not responsible for these phenotypes we tested RNAi-mediated knockdown of CG1632 in the BARTL assay and found no significant change (Fig 2H). Combined with the observations that a UAS-corp+ construct produces the same phenotype as EP-corp+ and that RNAi against corp has the opposite effect, it is clear that the effects we observe owe to corp and not CG1632.
If EP-corp+ was not induced by Gal4, and eyFLP was instead used to produce dicentric chromosomes in the eye, we found that the EP-corp+ insertion produced slightly larger eyes than the wildtype control, suggesting that the EPgy2 insertion by itself may have slightly elevated corp expression (S3B Fig). This effect, though statistically significant, is small and only eyGal4-mediated corp+ overexpression can generate the wildtype-like eye phenotype in the BARTL assay (Fig 2C and 2D).
To determine whether corp has any influence in the absence of DNA damage we examined wild type or BS flies carrying EP-corp+, induced or uninduced, and flies carrying the corp95B mutant, but without the induction of dicentric chromosomes. There was no change in eye phenotype in any of these cases, indicating that the effects of altered corp+ expression are seen only after DNA damage (S3C–S3G Fig).
EP-corp+-mediated rescue of the eye is not confined to males, or to effects produced by the Y chromosome. We generated XXY females carrying eyGal4, UAS-FLP and the DcY(H1) chromosome and found that EP-corp+ produced almost wildtype eyes, similar to its effect in males (S3H and S3I Fig). Additionally, we found that corp+ overexpression ameliorated the reduction in eye size produced by dicentric induction on chromosome 3 (S3J and S3K Fig). Therefore, the effect of corp+ overexpression is independent of the sex of the fly or the particular chromosome experiencing damage.
Extant corp polymorphisms are functionally indistinguishable
We sequenced the corp genomic regions from five laboratory strains and found two allelic variants of corp that differed by 7 nucleotide changes. Four nucleotide changes were found in the second exon: two are silent mutations (C840T and C891T) and two (T725A and T873G) encode different amino acids (L96H and L145M). These alternate amino acids are also present as polymorphisms in wildtype isolates of D. melanogaster and other Drosophila species (http://www.dpgp.org). There were also three single nucleotide differences (C277T, A398C and A407T) in introns.
The UAS-corp+ transgene that we constructed and tested (as mentioned previously) carries the canonical version, as found in CantonS. The EP-corp+, y w and w1118 strains all carry the variant allele that differs from the reference CantonS strain. Because the overexpression of either allele produces a similar large eye phenotype in the BARTL assay (Fig 2C and 2D), we conclude that both alleles function similarly, and that the two amino acid differences have, at most, minor effects.
corp+ suppresses DNA damage-induced apoptosis in the soma
One mechanism by which Corp could affect eye size in the BARTL assay is through the suppression of apoptosis, thereby allowing survival and proliferation of cells that would otherwise die. To test this we stained wing imaginal discs for apoptotic cells after treatment with ionizing radiation (IR). Untreated discs show similar low rates of apoptosis in both the y w controls and the corp95B mutants, but IR-induced apoptosis was significantly enhanced in corp95B mutants (Fig 3A and 3B). We then examined the effect of corp+ overexpression. An engrailed-Gal4 element [32] was used in combination with EP-corp+ to drive expression in the posterior compartment of wing discs, which was also marked by co-induction of UAS-GFP. Apoptosis was significantly reduced by corp+ overexpression (Fig 3C and 3D). These results show that Corp is a potent negative regulator of apoptosis following DNA damage in the soma.

Overexpression of corp+ restricts the transmission of healed chromosomes through the germline
In the male germline, broken dicentric chromosomes may be healed by de novo telomere addition [33,34]. With DcY(H1) these healed chromosomes (denoted FrY) may be detected in testcrosses to y w females by the loss of the dominant BS marker that lies distal to the inverted FRTs (i.e., by the generation of Bar+ sons). To assess the effect of corp on the transmission of broken-and-healed chromosomes we induced expression of FLP by heat shock (70FLP10) during the first 24 hours of development and used nanosGal4 to drive germ cell-specific overexpression of corp+. Overexpression of corp+ blocked transmission of FrY chromosomes (Table 1).
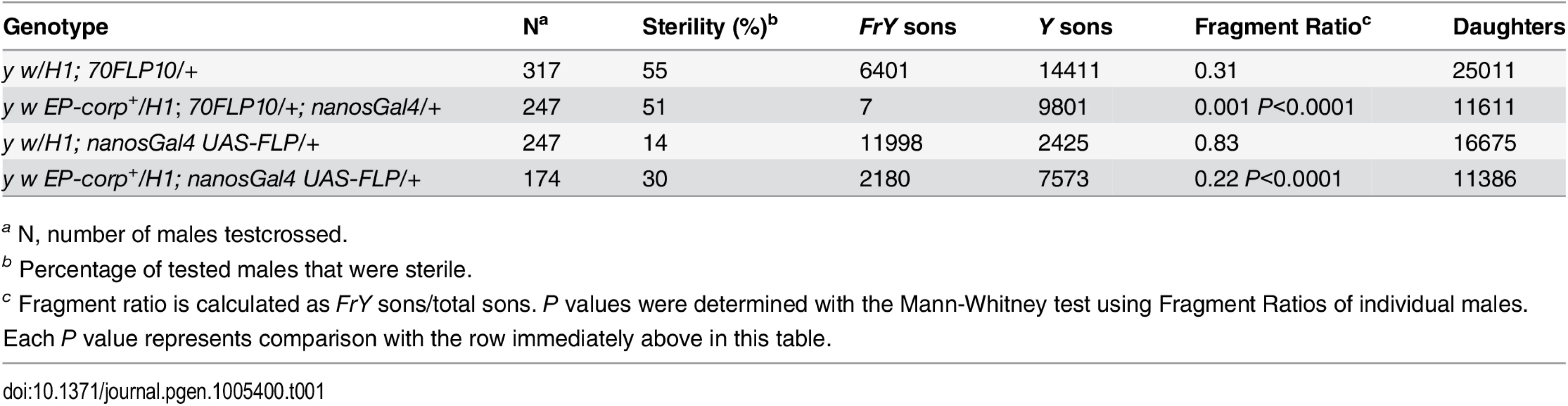
We also drove corp+ and FLP expression specifically in the germline using nanosGal4 (EP-corp+; UAS-FLP nanosGal4) and again observed a large decrease in FrY transmission relative to males with unaltered corp expression (Table 1), confirming that corp+ inhibits transmission of broken-and-healed chromosomes through the male germline.
The relationship between corp and p53
Though it seems surprising that corp+ overexpression produces dissimilar phenotypes in the soma (survival and proliferation of cells with broken chromosomes) vs. the germline (elimination of cells with broken chromosomes), there is precedent: the p535A-1-4 loss of function mutation acts similarly. Homozygous p535A-1-4 flies have almost wildtype eyes in the BARTL assay [20], but strongly reduced transmission of broken-and-healed chromosomes through the male germline [35]. This similarity suggests a functional relationship between corp and p53.
To explore this relationship we generated corp95B; p535A-1-4 double mutants and examined them using the BARTL assay. We found that p53 is epistatic to corp, with the double mutant producing almost wildtype eyes (Fig 4A). In a direct measurement of apoptosis in wing discs following treatment with ionizing radiation (IR) we observed the same epistatic relationship: p535A-1-4 suppressed the elevated apoptosis produced by corp95B, and the double mutant was indistinguishable from the p53 single mutant (Fig 4B and 4C).

In a complementary experiment, we tested the effect of simultaneously overexpressing corp+ and p53+. When GMR-Gal4 drives p53+ overexpression in the developing eye, the adults that eclose have very small eyes owing to an elevated frequency of cell death. We found that if corp+ was simultaneously overexpressed, the eyes became significantly larger. Furthermore, when we combined the corp95B mutant with GMR>p53+, the eyes were much smaller than produced by GMR>p53+ alone (Fig 4D). When apoptosis was directly assayed in eye discs of these genotypes, we observed correlated effects, with corp+ overexpression reducing, and the corp mutant increasing cell death (Fig 4Fa-c and 4G). These results may all be accommodated under the hypothesis that Corp antagonizes P53, either by suppressing its apoptotic effects or by negatively regulating P53 itself.
The level of P53 is inversely correlated with the corp+ expression level
To determine how Corp might affect P53 we examined P53 levels in eye discs by immunostaining. The GMR promoter was used to overexpress p53+ behind the morphogenetic furrow, providing an easily detected level of expression, which is absent in a p535A-1-4 mutant disc. The corp95B mutant eye discs exhibited a significantly higher level of P53 while EP-corp+ overexpression driven by GMR-Gal4 reduced the level of P53 (Fig 5A and 5B). Thus, we conclude that Corp is a negative regulator of P53.

To further verify our results, we knocked down corp in S2 cells by treating with double-stranded RNA (dsRNA) against corp and measured P53 protein levels by Western blot. We found that the quantity of P53 was significantly elevated following corp knockdown (S4 Fig), confirming that Corp promotes P53 downregulation.
To determine whether corp regulates p53 at the transcriptional level, we used qRTPCR to measure p53 mRNA in corp mutant or corp+ overexpressing larvae, with and without irradiation. We found that there are no significant or consistent changes in p53 mRNA levels between these genotypes (Fig 5C). We conclude that Corp regulation of P53 occurs primarily at the level of translation or protein stability.
Effect of Corp on expression of reaper
The pro-apoptotic reaper (rpr) gene is a prominent target of P53 following DNA damage [15,21]. If P53 is negatively regulated by corp then we expect that corp overexpression should also reduce reaper induction following IR. To test this, we measured rpr mRNA levels by qRTPCR in corp+-overexpressing and corp95B mutant larvae, with and without irradiation. We found that, following IR, rpr mRNA levels decrease with corp+ overexpression and increase in corp95B mutants (S5 Fig). Although these results were not significant at the 5% level, they are nonetheless consistent with Corp-mediated downregulation of P53.
Overexpression of corp+ suppresses Hid - and Reaper - mediated cell death
Recently, it was shown that the P53 transcriptional targets hid and rpr act recursively to increase p53 expression and contribute to the apoptotic program [36]. Given that Corp overexpression results in downregulation of P53, we expect that it should also suppress the apoptotic phenotype caused by hid and reaper overexpression by attenuating this positive feedback loop. In order to test this prediction, we overexpressed hid or reaper in the eye under control of the GMR-Gal4 driver. This produced adults with small eyes owing to cell death in the eye discs. When corp+ was simultaneously overexpressed the eyes became significantly larger, confirming that Corp interferes with the hid- and reaper-mediated apoptotic programs (Fig 4E). To confirm that this effect was through inhibition of apoptosis, we directly measured cell death in the eye discs of larvae of the above-mentioned genotypes. We found that rate of apoptosis in GMR-hid and GMR-rpr eye discs was significantly decreased in a corp+ overexpressing background (Fig 4Fd-g and 4H).
If corp+ overexpression rescues the small eye phenotype of GMR-hid flies by attenuating the feedback amplification loop through p53+, we expect similar rescue from a p53 mutant. To test this, we measured eye size of GMR-hid+; p535A-1-4 flies and found that they have significantly larger eyes than GMR-hid+ control eyes (S6 Fig).
These results are fully consistent with the hypothesis that Corp acts via down-regulation of P53. In this particular case, corp+-mediated downregulation of P53 blocks the Hid-P53 amplification loop and thereby reduces apoptosis.
Corp exhibits similarity to the P53-interacting regions of Mdm2
Mdm2 is the major negative regulator of P53 in vertebrates. However, no homolog of Mdm2 has been found in Drosophila. Given that Corp acts in a negative feedback loop on P53, we looked more closely at Corp to see whether any similarities to Mdm2 might be identified. We used the domain analysis tool MEME [37,38] to search for shared protein motifs between four Mdm2 orthologs (H. sapiens, M. musculus, G. gallus and D. rerio) and two Corp orthologs from Drosophila species (D. melanogaster and D. virilis). It identified seven similar motifs, with motifs 4 and 5 shared by Mdm2 and Corp (Fig 6A). Interestingly, motif 4 appears to correspond to the N-terminal region of Mdm2, the primary P53-interacting domain [39,40]. Motif 5 appears to correspond to an additional P53-binding site on Mdm2 [41]. We named the two motifs Corp and Mdm2 Motif-1 (CMM-1) and CMM-2, respectively.

Corp physically interacts with P53 through CMM-1
Motivated by the finding of similarities between Corp and Mdm2 in regions of Mdm2 that bind P53, we asked whether Drosophila Corp and P53 physically interact. We purified GST-DmP53 using a bacterial expression system. C-terminal-tagged Corp-GFP-Flag was then expressed via transient transfection of HeLa or 293 cells. Cell lysates prepared from these cells were incubated with either GST or GST-DmP53. Corp-GFP-Flag was pulled-down specifically by GST-DmP53 but not by GST (Fig 6Bii), indicating that Corp expressed in mammalian cells interacts with DmP53. This result suggests either that Corp can interact directly with DmP53, or that the complex required for their interaction is conserved in mammalian cells. To further probe this, we tested the interaction between GST-DmP53 and in vitro synthesized Corp. We found that GST-DmP53 strongly interacts with in vitro synthesized Corp (Fig 6Biii). Together, these results strongly suggest that Corp can interact directly with DmP53.
To test the role of CMM-1 (amino acids 58–84) and CMM-2 (amino acids 14–54) in mediating the interaction between Corp and DmP53, we generated deletion constructs that lack CMM-1 (Δ58–84), CMM-2 (Δ14–54), or both (Δ14–84) (Fig 6C and 6D). The interaction between DmP53 and Corp mutant was assayed via co-immunoprecipitation (Fig 6C) and GST pull-down (Fig 6D). In both systems, proteins that lack CMM-1 have dramatically diminished affinity to DmP53 as compared to full length Corp or Corp(Δ14–54). This indicates that CMM-1, the motif shared with the N-terminal P53-interacting domain of Mdm2, is required for the physical interaction between Corp and Mdm2.
Discussion
Several studies have identified transcriptional targets of P53 in Drosophila. Some of these play important roles in DNA damage repair or in triggering apoptosis [14–17,42,43]. However, the functions of most P53 target genes have yet to be determined. Our discovery that Corp antagonizes apoptosis by negatively regulating P53 is the first demonstration in Drosophila that a P53-regulated gene (apart from DNA repair genes) is not solely devoted to apoptosis. Our results show that P53 target genes act in competing pathways, defined by the hid- and reaper-mediated pro-apoptotic pathway and the corp-mediated anti-apoptotic pathway (Fig 7). Increased or decreased expression of corp+ shifts the balance in favor of survival or death, respectively.

In vertebrates, the major negative regulator of P53 is Mdm2. It binds to P53 and ubiquitinates it, leading to its degradation, and is responsible for restraining P53 activity in unstressed cells. Furthermore, Mdm2 is also a transcriptional target of P53, and is utilized to turn down the P53 response so that cells that have recovered from the initiating stress, for instance DNA damage, may survive. Though no strict Mdm2 homolog is known in Drosophila, our experiments indicate that Corp provides that function. Similar to Mdm2, the corp gene is a transcriptional target of P53, Corp antagonizes the P53-mediated apoptotic program, P53 levels are inversely correlated with corp+ expression and Corp physically interacts with P53 through a motif similar to the region of Mdm2 that mediates P53-Mdm2 binding. These similarities strongly support the idea that Corp regulates P53 by a direct physical interaction, thus leading us to propose that Corp is the functional analog of mammalian Mdm2 (Fig 7).
There are significant differences between Corp and Mdm2. Mdm2 is an E3 ubiquitin ligase containing a RING domain [44], but Corp shows no evidence of such a domain. Furthermore, in the mouse Mdm2 mutations can cause recessive lethality. These mutants may be rescued by the additional mutation of p53 [45], indicating that lethality results from unrestrained P53 activity. In contrast, the corp95B null mutation is not lethal in flies and exhibits no obvious phenotype in the unstressed condition. Corp appears to function only when DNA damage is detected. In Drosophila the normal level of P53 expression is insufficient to cause lethality in the absence of Corp unless P53 is activated by upstream kinases.
However, these differences between Corp and Mdm2 may not be as significant as they appear. First, recent findings in mice indicate that the constitutive and induced levels of Mdm2 can be functionally separated. When the P53 Response Element was mutated in the promoter of Mdm2, so that Mdm2 was still expressed at a basal level but could no longer be induced to high levels by P53, the resulting mice were viable [46]. Furthermore, when the RING domain of Mdm2 was mutated, so that it no longer functioned as a ubiquitin ligase, but could still interact with its partner Mdmx, the mice were also viable [47]. In both cases the mice were highly sensitive to induced DNA damage, indicating that higher levels of Mdm2 activity are required to recover from DNA damage. Moreover, the latter experiments show that Mdm2 is capable of repressing P53 function without its ubiquitin ligase activity [47]. This may indicate that P53-Mdm2 binding is an ancient mode of regulation, with the ubiquitin ligase activity acquired as a later adaptation. Additionally, recent work has established that Corp physically interacts with the E3 ubiquitin ligase encoded by hyd (hyperplastic discs) and with several proteasomal subunits (S7 Fig), suggesting that Corp, like Mdm2, may participate in proteolytic degradation of P53 [48,49].
There is still room for additional explanations for Corp’s phenotypes. If Corp also targeted downstream components of the apoptotic pathway for degradation it might contribute to the phenotypes we observed. Given the existence of a positive feedback loop between downstream pro-apoptotic genes and p53 [36], Corp might indirectly affect P53 levels by promoting degradation of other components of the apoptotic pathway. However, the physical interaction of Corp and P53 strongly suggests that Corp directly regulates P53, regardless of whether it may also regulate downstream apoptotic components.
Corp is the first reported negative regulator of P53 in Drosophila that is also a transcriptional target of P53. Although Bonus and Rad6 have been identified as negative regulators of P53 in Drosophila [50,51], neither of them are transcriptional targets of P53, and are thus less similar to Mdm2 than is Corp. Recent experimental findings from others [15,36,52,53], and as reported here, indicate that regulation of P53 is complex, with activation by upstream factors and modulation by positive and negative feedback loops. Further investigation of how these pathways are regulated and how they affect these outcomes should greatly improve our understanding of the many functions of P53.
It remains to be understood what benefit might be provided by Corp. If it is normally desirable to eliminate a cell with unrepaired DNA damage to prevent its proliferation, then what purpose could be served by saving such cells? Unlike mammals, where the function of Mdm2 is needed to restrain P53 in normal cells, Corp appears to function only in cells with damaged genomes. However, previous experiments have shown that in wildtype larvae, many cells with damaged genomes are not eliminated by apoptosis immediately, but rather over a period of a few days [5]. Since corp mutants show increased cell death after irradiation, Corp is clearly one factor that restrains the immediate death of cells with damaged genomes. We have often thought it surprising that flies can survive when dicentric chromosomes are formed, and break, in >90% of their cells during development [5,6]. Perhaps if all cells with broken chromosomes immediately succumbed to apoptosis, such flies would not survive. It is easy to imagine that a few remaining survivors, adrift in a sea of dead cells, might not be capable of regenerating a complete imaginal disc. In fact, corp mutants survive poorly after widespread induction of Y chromosome dicentrics (S1 Table). But, if cells with damaged genomes could be eliminated gradually, it might give the surviving cells a suitable matrix to regenerate a disc. Modulating the rate at which cells are eliminated following lethal damage could be the vital function fulfilled by Corp. [54] recently showed that dying cells signal their neighbors to become resistant to damage-induced death. We would not be surprised to find that this pathway acts through Corp.
Materials and Methods
Drosophila stocks
All flies were maintained at 25°C on standard cornmeal food. Construction of the DcY(H1) and Dc3(FrTr61A5)1A chromosomes have been described previously by Kurzhals et al. [20] and p535-A-1-4 by Xie et al. [55]. We obtained the following stocks from the Bloomington, IN (USA) Drosophila stock center: P{UAS-FLP1.D}JD1 (BL 4539), P{Gal4-ey.H}4–8 (BL 5535), P{EPgy2}CG1632EY03495 (BL 15650), P{eyFLP.N}5 (BL5576), M{3xP3-RFP.attP}ZH-86Fb; M{vas-int.B}ZH-102D (BL 23648), P{UAS-2xeGFP}AH2 (BL 6874), nanos-Gal4 [56], P{GMR-p53.Ex}3/TM3, Sb, Ser (BL 8417), GMR-Gal4 [57], P{GMR-hid}G1/CyO (BL 5771), P{GMR-rpr.H}S/TM6B, Tb (BL 5773) and P{Act5C-Gal4}17F01/TM6B, Tb (BL 3954). Two corp-RNAi stocks: v102751 and v16130 and one CG1632-RNAi stock: v106107 were obtained from the Vienna Drosophila Resource Center, Vienna, Austria (VDRC).
The following stocks were obtained from Golic lab collections: heat-shock inducible FLP, P{70FLP}10, P{UAS-GFP} P{Act-Gal4}/CyO and y w; Sp/CyO; nanosGal4 UAS-FLP(95)/TM3, Sb. The engrailed-Gal4 stock was kindly gifted by Mark Metzstein.
Plasmids and transgenic constructions
The coding region of corp from CantonS flies was amplified by PCR with 5’ NotI and 3’ XbaI overhangs (primers used: Fwd-5’CATATTCGCGGCCGCATGGCCGATATCAGGAGCAG3’ and Rev - 5’CCGCGGGTCTAGACTAGATGCGAATCGAGCGCA3’) and cloned into the pUAST-w+-attB vector [58]. Vector plasmid was injected in embryos carrying attP docking sites on chromosome 3 and vasa-ΦC31 integrase on chromosome 4 (BL 23648). w+ flies were selected for establishing stable transgenic stocks.
The corp95B deletion mutation was generated via imprecise excision of the EY03495 P-element insertion (Baylor College of Medicine Genome Disruption Project). The DNA break points were identified by PCR amplification (primer sets used: Fwd 1 : 5’CCAAGCGAACGCATCGCTG3’, Fwd 2 : 5’GAAGAGGTCATCTCCCAAGG3’, Rev1 : 5’CTTAGGAACAATGGTTCAACC3’ and Rev2 : 5’GCAGCCGAGGTATGGAAATC3’ and sequencing of genomic DNA obtained from the homozygous mutant.
DNA sequencing
Sequencing of corp+ from the genomic region in five different genotypes, y w, w1118, EP-corp+, CantonS and v; Sco/Cy; ry, was carried out by the Core Facilities, University of Utah.
Eye photographs
Eye photographs were taken using a Nikon D200 digital camera and processed in Adobe Photoshop.
Quantitative reverse transcriptase PCR
Total RNA was extracted from 12–15 adults or third instar larvae using Trizol Reagent (Sigma Aldrich, MO), treated with DNaseI (Fermentas, PA) and cDNA was synthesized using RevertAid First Strand cDNA synthesis kit (Thermo Scientific, PA) according to manufacturer’s protocol. 1μl of cDNA was used per reaction in triplicates for performing qRT-PCR experiment using Maxima SYBR green/Fluorescein qPCR Master Mix (Fermentas, PA) or PerfeCTa SYBR Green FastMix (Quanta Biosciences, MD) in an iQ-PCR machine (Bio-Rad, CA). Relative quantification of mRNA levels was calculated using the standard curve method. Relative copy numbers of each gene of interest (X) was calculated by normalizing cDNA levels of X over cDNA levels of Ribosomal Protein L32. Primers that were used are: Fwd-corp: 5’ GCAGCCGAGGTATGGAAATC 3’; Rev-corp: 5’ AAGCCGAGGGTCAGAAGG 3’; Fwd-p53 : 5’ GCCGCCTCCTTAATCATGCC 3’; Rev-p53 : 5’ GCCGAGACTGCGACGACTC 3’; Fwd-rpr: 5’ CCAGTTGTGTAATTCCGAACGA 3’;[59] Rev-rpr: 5’ GGATCTGCTGCTCCTTCTGC 3’;[59] Fwd-rpl: 5’ CCGCTTCAAGGGACAGTATC3’; Rev-rpl: 5’ ATCTCGCCGCAGTAAACG 3’.
Irradiation
15–18 wandering third instar larvae were collected in clean 10 mm petri plates and irradiated at 4000 rads using a TORREX120D X-ray generator (Astrophysics Research Corp, CA) or a Mark I irradiator (J. L. Shepherd & Associates, CA). These larvae were returned to fresh food and incubated at 25°C until further experimental treatments.
Eye size measurement
For determining eye sizes, the left eye of each fly was measured along the anterio-posterior axis (A) and the dorso-ventral axis (B), using a digital filar micrometer (Lasico, CA). These two measurements were used to calculate the area of an ellipse (i.e., Π x A/2 x B/2), as the area of the eye, which was then normalized to the mean area of wildtype (w1118 or y w) eyes, and was represented as a fraction of wildtype eye size.
Germline fragment chromosome transmission assay
Flies were allowed to lay eggs and transferred to fresh vials every day. Embryos were collected for 24 hrs, heat-shocked at 38°C for one hour in a circulating water bath and then immediately returned to 25°C. After eclosion, the males were collected and singly mated to 2–3 y w females and their progeny were scored. Alternatively, nosGal4 was used to drive UAS-FLP in the male germline.
Imaginal disc staining and fluorescence microscopy
Wing and eye imaginal discs were dissected from third instar larvae and stained with TUNEL or P53 antibody.
TUNEL staining
TUNEL staining was performed using Apoptag Red In Situ Apoptosis Detection Kit (#S7165, Chemicon International). Briefly, dissected imaginal discs were fixed in 4% paraformaldehyde, rinsed twice in PBTW (0.3% Tween-20 in 1X PBS) for 5 minutes/rinse, post-fixed in 2 : 1 EtOH/1X PBS, rinsed again as before and then incubated with equilibration buffer, TdT enzyme and anti-digoxigenin Rhodamine congujate antibody in subsequent steps, according to manufacturer’s protocol. Finally, the discs were mounted in Vectashield (Vector Laboratories Inc., CA) and photographed in Z projection. The images were exported as 8 bit TIFF files and analyzed in Adobe Photoshop. Fluorescence intensity of TUNEL staining was measured, using ImageJ software, as the total pixel intensity (referred to as Integrated density or InD) within each disc divided by the area of that disc. For wing discs the area of the entire wing disc was considered, or divided into anterior and posterior compartments, as appropriate. For eye discs, where gene expression was driven with GMR, only the region behind the morphogenetic furrow was considered for measuring InD.
P53 immunostaining
Third instar larvae were dissected in 1X PBS and fixed in 4% paraformaldehyde for 15 minutes at room temperature. They were washed in PBTX (0.3% Triton-X in 1X PBS) twice for 30 minutes each, followed by 1 hour blocking in 5% BSA in PBTX. Next, they were incubated overnight at 4°C in primary antibody, p53-7A4 (DSHB, University of Iowa, IA) at 1 : 10 concentration in 5% BSA. The discs were then rinsed twice with PBTX, 30 minutes each and once in blocking buffer for 1 hour and finally incubated with Alexa Fluor 568 goat anti-mouse secondary antibody (Invitrogen, OR) at 1 : 1000 concentration in 5% BSA for two hours. Finally, they were washed twice with PBTX as before, mounted in Vectashield (Vector Laboratories Inc., CA) and photographed in Z projection. All images were taken at 100 ms shutter speed and at neutral density 6. Minimum and maximum intensity range was set to 200–550 for all discs, and images were exported as 8-bit TIFF files. Fluorescence was analyzed in Adobe Photoshop. P53 staining intensity was measured, using ImageJ software, as InD divided by the eye disc area, with only the region behind the morphogenetic furrow considered (Fig 5B). For visual representation (Fig 5A), gamma was set to 0.65 and contrast was increased to 100 to reduce background staining and improve visualization of differences between genotypes.
dsRNA synthesis and purification
To synthesize double stranded RNA for RNA interference experiments with cultured cells, PCR products not more than 700 base pairs, were made of the cDNA of interest flanked by T7 RNA polymerase sites at both ends. After gel purification of the PCR product, it was used as template for in vitro transcription for 6 hours at 37°C in a circulating water bath in 5–6 replicates of 20 μl reaction each for better yield using Ambion Megascript T7 Transcription Kit (Life Technologies), according to manufacturer’s protocol. Then, the reactions were pooled together in a microcentrifuge tube and extracted with phenol-chloroform and chloroform. Finally, dsRNA was precipitated with ispropanol, dissolved in DEPC-treated H2O and quantified in a Nanodrop 1000 spectrophotometer (Thermo Scientific).
Primers used for obtaining PCR products were: Fwd_T7corp: 5’TTAATACGACTCACTATAGGGAGAATGGCCGATATCAGGAGCAG3’; Rev_T7corp: 5’TTAATACGACTCACTATAGGGAGACTAGATGCGAATCGAGCGCA3’; Fwd_T7p53 : 5’TTAATACGACTCACTATAGGGAGAAGATCCAGGCGAACACGCTG3’; Rev_T7p53 : 5’TTAATACGACTCACTATAGGGAGAGGCTTCCGGCACGGACTTG3’; Fwd_T7Pav: 5’TTAATACGACTCACTATAGGGAGAACAACTGCTCTTGGCAGATACC3’; Rev_T7Pav: 5’TTAATACGACTCACTATAGGGAGAAAATCCGTAACGAAACTAACCG3’.
S2 cell culture
S2 cells were cultured at 25°C in Schneider’s Drosophila Medium (Invitrogen) with 10% heat inactivated fetal bovine serum (HyClone) and 1X Antibiotic-Antimycotic (Invitrogen). Cells were passaged into fresh medium every 3–4 days and were discarded after passage 25 (P25).
dsRNA treatment of S2 cells and western blot
The dsRNA treatment protocol was performed as described [60]. Cells were passaged on day 0 at the rate of 2 x 106 cells/ml. On day 1 they were washed and seeded in 24-well plates at 800 μl/well. 15 μg of dsRNA added to each well. The plates were then returned to the 25°C incubator. On day 4–5, cells were re-seeded in 6 well plates at 2 ml/well and retreated with 30 μg of dsRNA. As a control of dsRNA uptake rate, cells were treated with Pavarotti dsRNA, which makes them large and multinucleate[61,62]. On day 6–7, cells were collected, lysed and processed for Western blot.
S2 cells, with or without dsRNA treatment, were irradiated at 4000 rads to observe any elevation/change in P53 levels from unirradiated cells, with or without dsRNA treatment. No significant changes in P53 levels were observed following irradiation, so treated cells were grouped and categorized as (I) no dsRNA control group and (II) + dsRNA experimental group for quantification.
The Western results were pulled from 3 independent experimental sets in the p53-dsRNA treatment experiment and 5 independent experimental sets in the corp-dsRNA treatment experiment. Out of the 3 p53-dsRNA experimental sets, 1 was following irradiation at 4000 rads and allowing 4 hours recovery before cell lysis. Likewise, out of the 5 corp-dsRNA experimental sets, 2 were following irradiation. Relative P53 levels in each experiment were calculated by normalizing total P53 protein level to total β–tubulin level.
Western blotting
S2 cells were collected and lysed in RIPA buffer containing protease inhibitor (Thermo Scientific, IL) to a final concentration of 1X. Protein concentration was measured by BCA assay (Thermo Scientific, IL) and cell lysates were mixed with sample buffer and β–mercaptoethanol to a final concentration of 1X before loading onto a 10% SDS gel at equal concentrations. Western blotting was carried out following standard procedure. Antibodies used: mouse monoclonal anti-Drosophila P53 (# sc-74573, Santa Cruz Biotechnology; as used by Chen et al.[51]) at 1 : 1000 concentration and mouse monoclonal anti-Drosophila β–tubulin (E7, Developmental Studies Hybridoma Bank, University of Iowa, IA) at 1 : 10,000 concentration. After incubation with fluorescent goat anti-mouse secondary antibody (# 926–68020, Li-COR Biosciences) at 1 : 10,000, the membranes were scanned on an infrared Odyssey scanner (LI-COR Biosciences).
GST and GST-DmP53 purification
The open reading frame of DmP53 was cloned into pGEX and transformed to BL21 DE3 cells. The expression was induced by addition of IPTG to a final concentration of 0.1mM in LB/Ampicillin media. Bacterial cells were harvested 4 hours following the induction and resuspended in ice-cold STE buffer (10mM Tris-HCl pH8.0, 150mM NaCl, 1mM EDTA and protease inhibitors) with 1.5% Sarcosyl. Cells were then lysed with sonication and subsequently incubated with STE containing 1% Triton X-100 for 30 minutes. Insoluble proteins were removed by centrifugation at 16,000g for 5 minutes. Supernatant was then incubated overnight with 50% slurry of glutathione-agarose beads at 4°C. The beads were pelleted by centrifuge at 100g and washed 4 more times with 10 ml of ice-cold PBTP (PBS with 0.1% Triton X-100 and protease inhibitors). Washed beads were then resuspended in PBTP with 0.01% sodium azide.
Expression of Corp-GFPFlag in HeLa cells
15ug of pRK5-corp-gfp-flag plasmid DNA was transfected to 2 million HeLa cells with calcium phosphate. Cells were lysed on plate with 1ml RIPA buffer at 48 hours following transfection.
In vitro synthesis of Corp-HA6His
Corp-HA6His was cloned into pCDNA3. In vitro synthesis were carried out using the TnT Coupled Reticulocyte Lysate Systems (Promega, Catalog number L4611) following manufacturer’s instructions.
GST pull-down assays
For in vitro synthesized protein, 1 ug GST or GST-DmP53 bound to Glutathione-agarose beads was incubated with 5 ul of synthesized protein in 500ul Binding buffer (50mM Tris-HCl, pH 8.0; 2mM EDTA; 150mM NaCl; 0.1% NP40; 20uM ZnCl2; 10mM MgCl2; protease inhibitors) containing BSA (0.2ug/ul). Following 1 hour incubation at RT and 1 hour incubation at 4°C, the beads were washed 4 times with Binding buffer. Beads were then pelleted at 100g, re-suspended and boiled in 30 ul sampling buffer, and resolved on SDS-PAGE gel. Following electrophoresis, the gel was fixed in (Isopropenol:dH2O:Acitic acid = 25 : 65 : 10) for 30 minutes and incubated in the Amplify Fluorographic Reagent (GE Healthcare, NAMP100) for 1 hour. The gel was then vacuum dried and processed for autoradiography with an intensify screen at -80°C.
For cellular extract, 1 ug GST or GST-DmP53 bound to Glutathione-agarose beads was first incubated for 30 minutes in 500ul binding buffer with 0.2ug/ul BSA. 500 ul cell lysate was then added and incubated at 4°C for overnight. Following incubation the beads were washed 4 times with 1 ml of RIPA buffer and pelleted by centrifugation at 100g for 5 minutes. Beads were then resuspended in 30ul sampling buffer and resolved on SDS-PAGE gel. Western analysis was performed with anti-Flag M2 antibody (Sigma, F1804).
Graphical methods and statistical analyses
Construction of graphs and calculations of statistical significance were performed using Prism 5.0 (Graphpad). In box-and-whisker plots the ends of whiskers represent 5th and 95th percentiles, top and bottom of the boxes represent 25th and 75th percentile and the horizontal line in the box represents the median, i.e., 50th percentile. The Mann-Whitney test was used in Figs 2, 3, 4, 5 and S1, S3, S6 Figs; Unpaired t-test in S4C and S5 Figs; and, Paired t-test in S4D Fig.
Software
The MEME tool used for searching motif similarity is publicly available software (http://meme.nbcr.net/meme/). Images were quantitatively analyzed using IMAGE J software from National Institutes of Health (http://imagej.nih.gov/ij/index.html) and images were processed using Adobe Photoshop. All line diagrams were composed using Adobe Illustrator.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Zhou B-BS, Elledge SJ (2000) The DNA damage response: putting checkpoints in perspective. Nature 408 : 433–439. 11100718
2. Sancar AA, Lindsey-Boltz LAL, Unsal-Kaçmaz KK, Linn SS (2004) Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Biochemistry 73 : 39–85.
3. Jackson SP, Bartek J (2009) The DNA-damage response in human biology and disease. Nature 461 : 1071–1078. doi: 10.1038/nature08467 19847258
4. Ahmad K, Golic KG (1999) Telomere loss in somatic cells of Drosophila causes cell cycle arrest and apoptosis. Genetics 151 : 1041–1051. 10049921
5. Titen SWA, Golic KG (2008) Telomere Loss Provokes Multiple Pathways to Apoptosis and Produces Genomic Instability in Drosophila melanogaster. Genetics 180 : 1821–1832. doi: 10.1534/genetics.108.093625 18845846
6. Golic KG (1994) Local Transposition of P Elements in Drosophila Melanogaster and Recombination between Duplicated Elements Using a Site-Specific Recombinase. Genetics 137 : 551. 8070665
7. Hollstein M, Sidransky D, Vogelstein B, Harris CC (1991) p53 mutations in human cancers. Science 253 : 49–53. 1905840
8. Hirao A (2000) DNA Damage-Induced Activation of p53 by the Checkpoint Kinase Chk2. Science 287 : 1824–1827. 10710310
9. Matsuoka S, Huang M, Elledge SJ (1998) Linkage of ATM to cell cycle regulation by the Chk2 protein kinase. Science 282 : 1893–1897. 9836640
10. Chaturvedi PP, Eng WKW, Zhu YY, Mattern MRM, Mishra RR, et al. (1999) Mammalian Chk2 is a downstream effector of the ATM-dependent DNA damage checkpoint pathway. Oncogene 18 : 4047–4054. 10435585
11. Chehab NHN, Malikzay AA, Appel MM, Halazonetis TDT (2000) Chk2/hCds1 functions as a DNA damage checkpoint in G(1) by stabilizing p53. Genes & Development 14 : 278–288.
12. Wahl GM, Carr AM (2001) The evolution of diverse biological responses to DNA damage: insights from yeast and p53. Nat Cell Biol 3: E277–E286. 11781586
13. Liu Y (2001) p53 protein at the hub of cellular DNA damage response pathways through sequence-specific and non-sequence-specific DNA binding. Carcinogenesis 22 : 851–860. 11375889
14. Akdemir F, Christich A, Sogame N, Chapo J, Abrams JM (2007) p53 directs focused genomic responses in Drosophila. Oncogene 26 : 5184–5193. 17310982
15. Brodsky MH, Weinert BT, Tsang G, Rong YS, McGinnis NM, et al. (2004) Drosophila melanogaster MNK/Chk2 and p53 Regulate Multiple DNA Repair and Apoptotic Pathways following DNA Damage. Molecular and Cellular Biology 24 : 1219–1231. 14729967
16. Colombani J, Polesello C, Josué F, Tapon N (2006) Dmp53 activates the Hippo pathway to promote cell death in response to DNA damage. Curr Biol 16 : 1453–1458. 16860746
17. Lee JH, Lee E, Park J, Kim E, Kim J, et al. (2003) In vivo p53 function is indispensable for DNA damage-induced apoptotic signaling in Drosophila. FEBS Letters 550 : 5–10. 12935877
18. Vousden KH, Lu X (2002) Live or let die: the cell's response to p53. Nat Rev Cancer 2 : 594–604. 12154352
19. Donehower LAL, Harvey MM, Slagle BLB, McArthur MJM, Montgomery CAC, et al. (1992) Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 356 : 215–221. 1552940
20. Kurzhals RL, Titen SWA, Xie HB, Golic KG (2011) Chk2 and p53 are haploinsufficient with dependent and independent functions to eliminate cells after telomere loss. PLoS Genet 7: e1002103–e1002103. doi: 10.1371/journal.pgen.1002103 21655087
21. Brodsky MH, Nordstrom W, Tsang G, Kwan E, Rubin GM, et al. (2000) Drosophila p53 Binds a Damage Response Element at the reaper Locus. Cell 101 : 103–113. 10778860
22. Ollmann M, Young LM, Di Como CJ, Karim F, Belvin M, et al. (2000) Drosophila p53 is a structural and functional homolog of the tumor suppressor p53. Cell 101 : 91–101. 10778859
23. Jin S, Martinek S, Joo WS, Wortman JR, Mirkovic N, et al. (2000) Identification and characterization of a p53 homologue in Drosophila melanogaster. Proc Natl Acad Sci USA 97 : 7301–7306. 10860994
24. Kubbutat MHG, Jones S, Vousden KH (1997) Regulation of p53 stability by Mdm2. Proc Natl Acad Sci USA 387 : 299–303.
25. Momand J, Zambetti GP, Olson DC, George D, Levine AJ (1992) The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 69 : 1237–1245. 1535557
26. Peters M, DeLuca C, Hirao A, Stambolic V, Potter J, et al. (2002) Chk2 regulates irradiation-induced, p53-mediated apoptosis in Drosophila. proc Natl Acad Sci USA 99 : 11305–11310. 12172011
27. Zhang Y, Lin N, Carroll PM, Chan G, Guan B, et al. (2008) Epigenetic Blocking of an Enhancer Region Controls Irradiation-Induced Proapoptotic Gene Expression in Drosophila Embryos. Developmental Cell 14 : 481–493. doi: 10.1016/j.devcel.2008.01.018 18410726
28. Rørth PP (1996) A modular misexpression screen in Drosophila detecting tissue-specific phenotypes. Proc Natl Acad Sci USA 93 : 12418–12422. 8901596
29. Hauck BB, Gehring WJW, Walldorf UU (1999) Functional analysis of an eye specific enhancer of the eyeless gene in Drosophila. Proc Natl Acad Sci USA 96 : 564–569. 9892673
30. Fan Y, Bergmann A (2008) Distinct Mechanisms of Apoptosis-Induced Compensatory Proliferation in Proliferating and Differentiating Tissues in the Drosophila Eye. Developmental Cell 14 : 399–410. doi: 10.1016/j.devcel.2008.01.003 18331718
31. Brand AH, Perrimon N (1993) Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 118 : 401–415. 8223268
32. Tabata T, Schwartz C, Gustavson E, Ali Z, Kornberg TB (1995) Creating a Drosophila wing de novo, the role of engrailed, and the compartment border hypothesis. Development 121 : 3359–3369. 7588069
33. Ahmad K, Golic KG (1998) The transmission of fragmented chromosomes in Drosophila melanogaster. Genetics 148 : 775–792. 9504924
34. Titen SWA, Golic KG (2010) Healing of euchromatic chromosome breaks by efficient de novo telomere addition in Drosophila melanogaster. Genetics 184 : 309–312. doi: 10.1534/genetics.109.109934 19897748
35. Titen SWA, Lin H-C, Bhandari J, Golic KG (2014) Chk2 and p53 regulate the transmission of healed chromosomes in the Drosophila male germline. PLoS Genet 10: e1004130–e1004130. doi: 10.1371/journal.pgen.1004130 24586185
36. Shlevkov E, Morata G (2012) A dp53/JNK-dependant feedback amplification loop is essential for the apoptotic response to stress in Drosophila. Cell Death Differ 19 : 451–460. doi: 10.1038/cdd.2011.113 21886179
37. Bailey TLT, Elkan CC (1994) Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol 2 : 28–36. 7584402
38. Bailey TLT, Williams NN, Misleh CC, Li WWW (2006) MEME: discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Research 34: W369–W373. 16845028
39. Kussie PH, Gorina S, Marechal V, Elenbaas B, Moreau J, et al. (1996) Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science 274 : 948–953. 8875929
40. Poyurovsky MV, Katz C, Laptenko O, Beckerman R, Lokshin M, et al. (2010) The C terminus of p53 binds the N-terminal domain of MDM2. Nature Structural & Molecular Biology 17 : 982–989.
41. Kulikov R, Winter M, Blattner C (2006) Binding of p53 to the central domain of Mdm2 is regulated by phosphorylation. J Biol Chem 281 : 28575–28583. doi: 10.1074/jbc.M513311200 16870621
42. Sogame N, Kim M, Abrams JM (2003) Drosophila p53 preserves genomic stability by regulating cell death. Proc Natl Acad Sci USA 100 : 4696–4701. 12672954
43. Nordstrom W, Abrams JM (2000) Guardian ancestry: fly p53 and damage-inducible apoptosis. Cell Death Differ 7 : 1035–1038. 11139275
44. Manfredi JJ (2010) The Mdm2-p53 relationship evolves: Mdm2 swings both ways as an oncogene and a tumor suppressor. Genes & Development 24 : 1580–1589.
45. Jones SN, Roe AE, Donehower LA, Bradley A (1995) Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature 378 : 206–208. 7477327
46. Pant V, Xiong S, Jackson JG, Post SM, Abbas HA, et al. (2013) The p53-Mdm2 feedback loop protects against DNA damage by inhibiting p53 activity but is dispensable for p53 stability, development, and longevity. Genes & Development 27 : 1857–1867.
47. Tollini LA, Jin A, Park J, Zhang Y (2014) Regulation of p53 by Mdm2 E3 Ligase Function Is Dispensable in Embryogenesis and Development, but Essential in Response to DNA Damage. Cancer Cell 26 : 235–247. doi: 10.1016/j.ccr.2014.06.006 25117711
48. Guruharsha KG, Rual J-F, Zhai B, Mintseris J, Vaidya P, et al. (2011) A protein complex network of Drosophila melanogaster. Cell 147 : 690–703. doi: 10.1016/j.cell.2011.08.047 22036573
49. Callaghan MJ, Russell AJ, Woollatt E, Sutherland GR, Sutherland RL, et al. (1998) Identification of a human HECT family protein with homology to the Drosophila tumor suppressor gene hyperplastic discs. Oncogene 17 : 3479–3491. 10030672
50. Allton K, Jain AK, Herz H-M, Tsai W-W, Jung SY, et al. (2009) Trim24 targets endogenous p53 for degradation. Proc Natl Acad Sci USA 106 : 11612–11616. doi: 10.1073/pnas.0813177106 19556538
51. Chen S, Wei H-M, Lv W-W, Wang D-L, Sun F-L (2011) E2 ligase dRad6 regulates DMP53 turnover in Drosophila. Journal of Biological Chemistry 286 : 9020–9030. doi: 10.1074/jbc.M110.190314 21205821
52. Zhang W, Durocher D (2010) De novo telomere formation is suppressed by the Mec1-dependent inhibition of Cdc13 accumulation at DNA breaks. Genes & Development 24 : 502–515.
53. Oikemus SR (2004) Drosophila atm/telomere fusion is required for telomeric localization of HP1 and telomere position effect. Genes & Development 18 : 1850–1861.
54. Bilak A, Uyetake L, Su TT (2014) Dying cells protect survivors from radiation-induced cell death in Drosophila. PLoS Genet 10: e1004220. doi: 10.1371/journal.pgen.1004220 24675716
55. Xie HB, Golic KG (2004) Gene Deletions by Ends-In Targeting in Drosophila melanogaster. Genetics 168 : 1477–1489. doi: 10.1534/genetics.104.030882 15579700
56. Rørth P (1998) Gal4 in the Drosophila female germline. Mechanisms of Development 78 : 113–118. 9858703
57. Freeman M (1996) Reiterative use of the EGF receptor triggers differentiation of all cell types in the Drosophila eye. Cell 87 : 651–660. 8929534
58. Bischof JJ, Maeda RKR, Hediger MM, Karch FF, Basler KK (2007) An optimized transgenesis system for Drosophila using germ-line-specific phiC31 integrases. Proc Natl Acad Sci USA 104 : 3312–3317. 17360644
59. Moon N-S, Di Stefano L, Morris EJ, Patel R, White K, et al. (2008) E2F and p53 induce apoptosis independently during Drosophila development but intersect in the context of DNA damage. PLoS Genet 4: e1000153. doi: 10.1371/journal.pgen.1000153 18688282
60. Clemens JC, Worby CA, Simonson-Leff N, Muda M, Maehama T, et al. (2000) Use of double-stranded RNA interference in Drosophila cell lines to dissect signal transduction pathways. Proc Natl Acad Sci USA 97 : 6499–6503. 10823906
61. Somma MP, Fasulo B, Cenci G, Cundari E, Gatti M (2002) Molecular dissection of cytokinesis by RNA interference in Drosophila cultured cells. Mol Biol Cell 13 : 2448–2460. 12134082
62. Goshima G, Vale RD (2003) The roles of microtubule-based motor proteins in mitosis: comprehensive RNAi analysis in the Drosophila S2 cell line. The Journal of Cell Biology 162 : 1003–1016. 12975346
63. Kyte J, Doolittle RF (1982) A simple method for displaying the hydropathic character of a protein. J Mol Biol 157 : 105–132. 7108955
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2015 Číslo 7
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Functional Constraint Profiling of a Viral Protein Reveals Discordance of Evolutionary Conservation and Functionality
- Reversible Oxidation of a Conserved Methionine in the Nuclear Export Sequence Determines Subcellular Distribution and Activity of the Fungal Nitrate Regulator NirA
- Modeling Implicates in Nephropathy: Evidence for Dominant Negative Effects and Epistasis under Anemic Stress
- Nutritional Control of DNA Replication Initiation through the Proteolysis and Regulated Translation of DnaA
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy