
A Genetic Incompatibility Accelerates Adaptation in Yeast
In nature, bacterial populations with high mutation rates can adapt faster to new environments by acquiring beneficial mutations. However, such populations also accumulate harmful mutations that reduce their fitness. We show that the model eukaryote baker’s yeast can use a similar mutator strategy to adapt to new environments. The mutator state that we observed resulted from an incompatibility involving two genes, MLH1 and PMS1, that work together to remove DNA replication errors through a spellchecking mismatch repair mechanism. This incompatibility can occur through mating between baker’s yeast from different genetic backgrounds, yielding mutator offspring containing an MLH1-PMS1 combination not present in either parent. Interestingly, these offspring adapted more rapidly to stress, compared to the parental strains, and did so without an overall loss in fitness. DNA sequencing analyses of baker’s yeast strains from across the globe support the presence of incompatible hybrid yeast strains in nature. These observations provide a powerful model to understand how the segregation of defects in DNA mismatch repair can serve as an effective strategy to enable eukaryotes to adapt to changing environments.
Published in the journal:
. PLoS Genet 11(7): e32767. doi:10.1371/journal.pgen.1005407
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1005407
Summary
In nature, bacterial populations with high mutation rates can adapt faster to new environments by acquiring beneficial mutations. However, such populations also accumulate harmful mutations that reduce their fitness. We show that the model eukaryote baker’s yeast can use a similar mutator strategy to adapt to new environments. The mutator state that we observed resulted from an incompatibility involving two genes, MLH1 and PMS1, that work together to remove DNA replication errors through a spellchecking mismatch repair mechanism. This incompatibility can occur through mating between baker’s yeast from different genetic backgrounds, yielding mutator offspring containing an MLH1-PMS1 combination not present in either parent. Interestingly, these offspring adapted more rapidly to stress, compared to the parental strains, and did so without an overall loss in fitness. DNA sequencing analyses of baker’s yeast strains from across the globe support the presence of incompatible hybrid yeast strains in nature. These observations provide a powerful model to understand how the segregation of defects in DNA mismatch repair can serve as an effective strategy to enable eukaryotes to adapt to changing environments.
Introduction
DNA mismatch repair (MMR) acts primarily during DNA replication in prokaryotes and eukaryotes to correct DNA polymerase misincorporation errors that include base substitutions, frameshift mutations, and insertions/deletions [1–3]. In S. cerevisiae MutS homolog (MSH) heterodimers can track with the replication fork to recognize and bind to DNA mismatches [4,5]. MSH-marked repair sites are recognized primarily by the MutL homolog (MLH) heterodimer Mlh1-Pms1. The resulting ternary complex interacts with downstream excision factors such as Exo1 to remove the newly replicated DNA strand where the misincorporation event had occurred.
Defects in MMR result in the accumulation of deleterious mutations and an overall loss in fitness (e.g. [6]). Interestingly, studies in microbes have shown that the mutation rate per base pair is inversely proportional to genome size, and that changes from the wild-type rate are selected against [7–10]. However, approximately 10% of natural E. coli isolates display a mutator phenotype with 1–3% displaying defects in the MMR pathway [11–12]. The finding that a high mutation rate is typically selected against, but that some bacterial isolates can be observed in populations that are mutators, suggests that mutators may play an important role in adaptive evolution [11, 13–20]. One explanation for this observation is that mutators have an increased likelihood of acquiring the first adaptive mutations within a population. However, such a strategy is not sustainable due to the accumulation of deleterious mutations that ultimately outweigh beneficial mutations. Bacteria appear to solve this problem through horizontal transfer; mismatch repair genes are exchanged between genomes at higher than average rates, which is likely due to the hyper-recombination phenotypes exhibited by MMR-deficient strains [14].
Do eukaryotes also regulate MMR functions to adapt to new selective pressures? Previously Thompson et al. [21] showed that diploid baker’s yeast lacking the MSH2 MMR gene display an adaptive advantage when competed against diploid non-mutators. However, this advantage was not seen in haploids. Previously we hypothesized that MMR function could be modulated in eukaryotes through negative epistatic interactions [22]. This hypothesis was based on experiments in which we mated two S. cerevisiae strains, S288C and SK1, which show 0.7% sequence divergence, and identified one MLH genotype, S288c MLH1-SK1 PMS1, that conferred mutation rates 100-fold higher than wild type in an assay in which mlh1 and pms1 null strains display a 10,000-fold higher rate [22]. The S288c MLH1-SK1 PMS1 combination was defined as ‘incompatible’, while the other three combinations, which did not display a mutator phenotype, were labeled ‘compatible’. A single nucleotide polymorphism (SNP) in PMS1 combined with a single SNP in MLH1 were primarily responsible for the incompatibility [22].
Dobzhansky and Muller proposed a model to explain how hybrid incompatibilities can arise without causing defects within parental strains or species [23–26]. As described previously [22,27], the evolution of the S288c MLH1-SK1 PMS1 MMR incompatibility (Fig 1) fits this model. Mating of S288C and SK1, followed by sporulation and segregation of gene variants within progeny, creates an S288c MLH1-SK1 PMS1 genotype that shows negative epistasis (Fig 1; [27]). Such negative epistasis is similar to the interactions thought to underlie hybrid incompatibility between established [28–32] or incipient species [33].
![A model for how MMR incompatible populations arise in nature [<em class="ref">22</em>,<em class="ref">27</em>].](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/d7317828b386ac0a02d775596687c03a.png)
DNA sequence analyses of natural and laboratory yeast strains indicated that S288c and SK1 strains have mated naturally [22]. This finding suggests that incompatible combinations were likely to have been created in nature but were not maintained due to losses in fitness associated with defects in MMR (e.g. [3,22,27]). We hypothesize that negative epistasis involving MMR gene variants provides a transient advantage critical for adaptive evolution. To test this idea we constructed isogenic compatible and incompatible MLH1-PMS1 strains and subjected them to adaptive evolution in high salt. We found that incompatible populations adapted more rapidly to high salt than compatible strains without displaying an apparent fitness cost. Furthermore, we show that mutations in PMR1 were causative for high salt resistance in incompatible populations. Interestingly, mutations in this same gene, PMR1, subsequently arose in compatible populations though at a slower rate. Together these observations demonstrate an experimentally validated role for genetic incompatibilities in accelerating adaptation to environmental challenges in eukaryotes.
Results
MLH1-PMS1 incompatible strains display a fitness advantage when evolved in high salt
We tested if the negative epistasis phenotype seen in yeast bearing the S288c MLH1-SK1 PMS1 genotype confers an adaptive advantage during stress. This study was initiated by constructing isogenic compatible and incompatible MLH1-PMS1 strains that displayed, prior to adaptation, similar fitness levels in YPD and YPD + 1.2 M NaCl media as measured in growth and competition assays (Materials and Methods; S1 Table and S1 Fig). We assessed the mutator phenotype of compatible and incompatible strains using the lys2-A14 reversion assay. Compared to SK1 MLH1-SK1 PMS1 compatible strains, S288c MLH1-SK1 PMS1 incompatible strains showed increased reversion rates similar to that seen in previously constructed incompatible strains (100 to 120-fold higher than S288c MLH1-S288c PMS1; S2 Table; [22]).
Compatible and incompatible lines were analyzed for adaptation to high salt conditions by growing them in YPD media containing 1.2 M NaCl as described in the Materials and Methods. In YPD media both compatible and incompatible lines completed 8–9 generations per transfer. In YPD + 1.2 M NaCl media both compatible and incompatible lines completed ~5.5 generations after the first transfer. A steady rise in the number of generations completed, from ~6.0 to ~6.8, was seen after Transfers 2 to 20. Cultures obtained after 7 (~50 generations), 10 (~70 generations), and 16 (~120 generations) transfers showed the maximal fitness advantage gained by incompatible lines. Growth rate was determined by measuring the OD600 of cultures every two hours following dilution into YPD + 1.2 M NaCl. Pair-wise competition experiments were performed by mixing equal amounts of cells obtained from randomly chosen incompatible and compatible cultures. The proportion of cells in each culture was determined at T = 0 and T = 24 hrs following mixing (Materials and Methods).
We assessed the growth of isogenic compatible and incompatible lines in YPD and YPD + 1.2 M NaCl. These lines could be distinguished from each other in experimental cultures because they contained different antibiotic resistance markers (KANMX, resistance to G418, and NATMX, resistance to nourseothricin) linked to the MLH1 locus (S1 Table). The markers could be switched between compatible and incompatible strains without an effect on fitness in growth and competition assays, indicating that the antibiotic markers did not confer a selective advantage. As shown in Fig 2, incompatible lines displayed a faster growth rate in YPD + 1.2 M NaCl after Transfer 7 (~50 generations). This advantage was more apparent after Transfer 10 (~70 generations; p < 0.0001, n = 25), but was not seen after Transfer 16 (~120 generations; p >0.05; n = 8).

In addition to direct growth measurements, we measured fitness in competition assays in which cells from randomly chosen compatible and incompatible evolved lines were mixed together at an approximately 1 : 1 ratio. These competitions involved cells adapted in YPD + 1.2 M NaCl that had undergone the same number of transfers. After mixing, the lines were grown in YPD or YPD + 1.2 M NaCl for 24 hrs (seven generations) and the proportion of each type was determined (Materials and Methods). As shown in Table 1 and S2 Fig, neither compatible nor incompatible lines showed a competitive advantage in YPD media. In YPD + 1.2 M NaCl media, neither of the two lines showed a competitive advantage after Transfer 7. However, incompatible lines displayed a competitive advantage in this media after Transfer 10 (p = 0.0031), with an average fitness advantage of 16% over compatible lines (Table 1). This advantage was lost after Transfer 16 (p = 0.39). Together, these data indicate that a MMR incompatibility generated by recombination involving naturally occurring variants in PMS1 and MLH1 can lead to an elevated rate of occurrence of adaptive mutations, and thus accelerate adaptation in a eukaryote.

The fitness advantage seen in incompatible strains depends on mutation supply
How can we explain the temporal rise in fitness advantage seen in incompatible versus compatible lines? The most straightforward explanation is that the supply of mutations in the incompatible lines is higher than in the compatible lines, thus providing a greater likelihood for obtaining beneficial mutations that reach a high enough frequency to be selected and maintained in a population (e.g. [20,21,37]). The mutation supply available is a function of the mutation rate and the population size (N). To test this idea, we lowered the mutation supply by reducing the number of cells (and thus population size) per transfer in YPD-1.2 M NaCl by ten-fold to ~2 x 106 cells per transfer. We then performed cell growth and competition assays. In this experiment we were unable to observe a statistically significant advantage (p> 0.05) in fitness for the incompatible strains even though the number of generations completed, 70 to 75 after Transfer 10, were similar. In this experiment w = 0.99 +/-0.04 (SEM, n = 4) in YPD, and w = 0.96 +/ - 0.02 (SEM, n = 4) in YPD-1.2 M NaCl (see also S3 Fig). These observations thus support the premise that mutation supply is critical to achieve the fitness advantage seen in incompatible strains.
Why was a fitness advantage in high salt seen in incompatible strains at Transfer 10 but not at Transfer 16? One possibility is that compatible populations adapt more slowly due to a lower mutation supply, but eventually obtain beneficial mutations that are selected for and maintained in the population. Alternatively, and/or in conjunction, incompatible populations accumulate a greater number of deleterious mutations and lose fitness over time. As shown in Table 1 and S2 Fig, the fitness of compatible and incompatible cultures was similar in YPD media even after 16 transfers, when the fitness advantage for incompatible lines in YPD + 1.2 M NaCl media was no longer apparent. Together these observations and the mutation supply experiments presented above and S3 Fig indicate that the speed by which compatible and incompatible populations adapt is dependent on the mutation supply rate, which is higher in the incompatible strains.
Mutations in PMR1 were identified in both compatible and incompatible lines grown in YPD + 1.2 M NaCl
Are mutant alleles of the same genes responsible for salt resistance in incompatible and compatible populations? We answered this question by isolating salt-resistant clones (one per line) from independent incompatible and compatible lines. NaCl resistance in evolved strains can be easily phenotyped on YPD + NaCl plates because they grow to larger colony sizes relative to unevolved strains (Fig 3B, left panel). To determine the complexity of the NaCl resistant phenotype, we mated these clones (primarily from transfer 10) to unevolved strains of the opposite mating type (Fig 3) to form diploids. While most (five of eight tested) of the diploid strains were sensitive to NaCl, indicating recessive transmission, three of the eight displayed a semi-dominant phenotype (example in Fig 3B, left panel). Four diploids created by mating evolved and unevolved strains were then sporulated and phenotyped for salt resistance. Interestingly, all four strains displayed primarily a 2 NaClr:2 NaCls segregation phenotype on YPD + NaCl plates, indicating that a single locus in the evolved strain was causative. At least 18 NaClr and 18 NaCls spore clones derived from each of the four matings were pooled separately and subsequently analyzed by whole-genome sequencing using a bulk segregation strategy (Materials and Methods). As shown in Table 2, only one to three mutations were identified in each of the four clones. Interestingly, in all four clones only one locus, PMR1, displayed strong linkage, as measured in sequence read counts, to the NaClr phenotype (p < 10−5 for all linkages to PMR1). As described in further detail below, PMR1 encodes a membrane-bound P-type Ca2+ dependent ATPase involved in transporting Mn2+ and Ca2+ into the Golgi [38]. In subsequent paragraphs we describe a detailed analysis of pmr1 mutants identified in evolved cultures, with the goal of explaining the genetic basis of adaptation to a defined stress, in this case, high salt.


We sequenced in total 37 clones obtained from independent compatible or incompatible lines grown in YPD +1.2 M NaCl. Twelve of these were subjected to whole genome sequencing. For 25 clones Sanger sequencing was performed on the PCR-amplified PMR1 locus. As shown in Table 3 and Fig 4, 21 different mutations in PMR1 were identified in the 37 clones that mapped to the predicted cytoplasmic domain of Pmr1. While most mutations resulted in amino-acid substitutions in the 950 amino acid Pmr1 protein sequence, two involved start codon disruptions, and one was a frameshift mutation predicted to disrupt the reading frame beginning at amino acid 220.

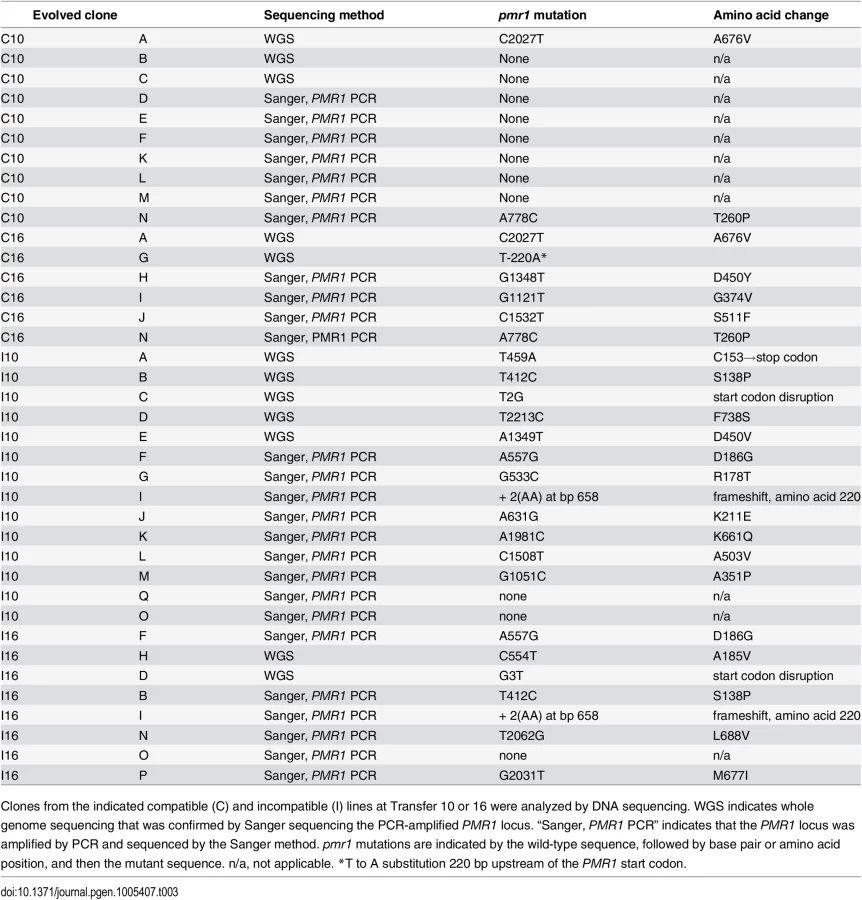
The following observations suggested that pmr1 mutations conferred strong adaptive advantages earlier in incompatible populations due to a higher mutation supply. 1. Almost all of the evolved incompatible clones isolated from evolved lines that completed 10 (twelve of fourteen) or 16 transfers (seven of eight) contained pmr1 mutations. 2. Only two of ten such clones from compatible lines after Transfer 10 contained a pmr1 mutation. 3. For compatible lines at Transfer 16, six of six such clones contained pmr1 mutations (Table 3).
We also sequenced clones isolated from the same line, but at different transfers. For two compatible lines (lines A, N) and three incompatible lines (B, F, I) the same mutation was identified in clones isolated after Transfers 10 and 16 (Table 3). However, for an incompatible line D two different pmr1 mutations were identified in clones isolated after Transfer 10 (pmr1-T2213C) and 16 (pmr1-G3T). Whole genome analysis of these clones showed that two indel mutations were present in both Transfer 10 and 16, suggesting that these mutations reached fixation prior to Transfer 10 (S3 Table). We then sequenced additional clones from line D incompatible line at Transfers 10 and 16. For Transfer 10, the PMR1 gene was sequenced in nine clones; eight of these contained the T2213C mutation and one contained the wild-type sequence. For Transfer 16, all three clones that were sequenced contained the G3T mutation. Together these observations are consistent with the T2213C mutation being adaptive, but before it could fix the G3T mutation appeared in the population and swept to fixation.
Clones obtained from the compatible and incompatible evolved populations showed mutation rates that were similar to those measured in the corresponding unevolved lines (S2 Table). Thus it is not surprising that the total number of mutations detected in the evolved lines were consistent with the genotype of the line (compatible vs incompatible). On average 2.4 mutations were identified per compatible line vs. 8.0 mutations per incompatible line (p < 0.0002). Interestingly, the vast majority of indel mutations (~90%) detected in this study were in homopolymeric runs, with five times as many indels detected per line in incompatible compared to compatible lines. The latter comparison is consistent with the mutation spectra seen in mlh1ts strains grown at the non-permissive temperature [3,6].
Gene replacement analysis shows that pmr1 mutations identified in compatible and incompatible lines are causative for evolved salt tolerance
PMR1 encodes a P-type Ca2+ dependent membrane ATPase involved in protein sorting and calcium homeostasis. It is primarily localized to the Golgi membrane and is involved in transporting Mn2+ and Ca2+ into the Golgi lumen [38]. An uncharacterized pmr1 mutation was identified by Park et al. [39] that conferred increased tolerance to NaCl. The authors proposed that such NaCl tolerance occurred because increased levels of cytosolic calcium in the pmr1 mutant activated calcineurin, a calcium/calmodulin-dependent phosphatase. This activation increased expression of ENA1, a gene encoding a P-type ATPase pump that functions in sodium and lithium efflux to permit salt tolerance [39,40]. In such a model it is not surprising that pmr1 null and hypomorph strains are also both resistant to lithium (Fig 5). However if only this pathway is involved then the pmr1Δ mutation should also confer NaCl tolerance. In fact pmr1Δ confers NaCl hypersensitivity in isogenic cell lines as well as in the S288c yeast knockout collection (Fig 5). We do not have a clear explanation for why the pmr1 alleles identified in this study confer NaCl resistance while the pmr1 null confers hypersensitivity. One possibility, suggested by Park et al. [39] is that factors that act in calcium homeostasis are differentially regulated in the presence or absence of full or partial-length Pmr1, and thus may differentially regulate pumps that function in sodium and lithium efflux.

To further characterize the mutations in PMR1 and their phenotype related to salt resistance, we replaced the wild type PMR1 gene in unevolved strains with constructs containing pmr1 alleles identified in our studies (Fig 5). All assessed alleles (pmr1-T412C, pmr1-T2G, pmr1-A557G, pmr1-C554T) conferred NaClr and LiClr phenotypes in the unevolved strains that were identical to the phenotypes observed in the corresponding evolved strains (Fig 5). These results indicate that the phenotypes identified in evolved lines can be completely explained by mutations in PMR1, supporting the data shown in the bulk segregation experiments (Table 2).
To obtain a better mechanistic explanation for why pmr1 point, frameshift, and initiation codon mutations, but not pmr1Δ conferred NaClr, we transformed two reporter constructs, pKC201 and pMZ11, into wild-type, pmr1Δ, and pmr1 allele strains. pKC201 is a pmr2/ena1::lacZ reporter plasmid used to measure expression levels of the Pmr2/Ena1 ion pump, a major P-type ATPase required for sodium ion flux. pmr2Δ/ena1Δ strains are sensitive to NaCl but strains containing increased copy number or expression of this locus display increased resistance [41–43]. pMZ11 is a UPRE::lacZ reporter used to monitor the unfolded protein response, a signaling pathway that improves endoplasmic reticulum (ER) function during ER stress [44].
As shown in S5 Fig, pmr1Δ and evolved pmr1 strains each displayed constitutive expression of Pmr2/Ena1 at levels that were higher in the absence of NaCl than seen in wild-type. Using the pMZ11 reporter, we found that pmr1 and pmr1Δ strains displayed similar phenotypes with respect to the unfolded protein response (S6 Fig). Together these results suggest that ENA1 overexpression or the induction of the unfolded protein response cannot explain the different NaClr phenotypes seen in pmr1 and pmr1Δ strains. At present we favor the idea that factors acting in calcium homeostasis are differentially regulated in the presence or absence of regulatory sequences during translation of Pmr1 or in the Pmr1 polypeptide, and may differentially regulate pumps that function in sodium and lithium efflux.
It is important to note that not all clones that showed salt resistance contained mutations in PMR1. In fact most NaClr clones obtained from compatible lines that had undergone 10 transfers did not contain pmr1 mutations (S3 Table; S4 Fig). Whole genome sequencing identified mutations in other candidate genes that may be causative. For example, clone C10B isolated from the compatible line at transfer 10 (C10B) contains a mutation in CNB1. Cnb1 is a regulatory subunit of calcineurin that is linked to stress responses ([45]; see below). While cnb1 null mutants show sensitivity to NaCl, mutations in CNB1 were previously identified in lines evolved in NaCl [46]. A clone isolated from a compatible line that had completed 10 transfers (C10C) contained a mutation in GCN2 and a clone isolated from a compatible line that had completed 16 transfers (C16B) contained a mutation in PTK1. Gcn2 is a protein kinase that phosphorylates the alpha-subunit of translation initiation factor eIF2 in response to starvation and Ptk1 is a putative kinase that is involved in polyamine transport [47,48]. Gene replacement approaches will be required to test whether these or other mutations identified in these clones are causative.
Discussion
In this study we showed that S. cerevisiae populations bearing an incompatible Mlh1-Pms1 combination display an adaptive advantage when challenged to adapt to a high salt growth media. The advantage, which was rapid, but transient, was due to the greater mutation supply in incompatible populations. While pmr1 mutations were first seen at high frequency in incompatible lines, they were eventually detected at high frequency in compatible lines. The initial fitness advantage of incompatible lines in which pmr1 mutations had occurred no longer existed when these lines were later competed against compatible lines in which pmr1 mutations had subsequently also arisen. Taken together, these observations suggested that the two loci MMR incompatibility genotype (MLH1, PMS1) generated by recombination among naturally occurring variants at these genes accelerated the appearance of highly beneficial mutations within our yeast populations.
Why did incompatibility accelerate adaptation? The most likely explanation is that the moderate mutator phenotype seen in incompatible strains resulted in a higher rate of mutation (including those that were neutral, deleterious, and beneficial), with strains bearing beneficial mutations selected in our high salt stress condition. Such a model fits observations seen for bacterial populations maintained in stress or selection conditions; in these experiments bacteria displaying high mutation rates were identified at high frequency (e.g. [49]). In one such study E. coli populations displaying high mutation rates, primarily due to MMR defects, showed short-term fitness advantages that were not sustainable [19]. The simplest explanation is that the lack of a long-term fitness advantage was due to the accumulation of deleterious mutations elsewhere in the genomes containing the beneficial mutations [19,50–52]. As indicated in the Introduction, bacterial populations appear to overcome long-term fitness costs associated with high mutation rates by reacquiring functional MMR genes and thus normal mutation rates through horizontal gene transfer [14,19].
Several groups have examined whether a mutator phenotype in eukaryotes confers a fitness advantage when adapting to a stress environment (e.g. [21,37]). Some of the best-known examples involve cancer cells that display mutator phenotypes and/or high rates of genome instability [53]. Such work has suggested that over time mutator populations lose their initial advantage due to fitness costs and clonal interference (e.g. [37]). Fitness costs associated with high mutation rates can occur rapidly; for example, Ma et al. [6] showed that, following 160 generations of growth in non-permissive conditions, a diploid mlh1ts yeast strain accumulated 92 heterozygous mutations, including five in essential genes, and that these mutations account for the poor spore viability (3%) seen in the strain. In addition even a moderate mutator can quickly display a fitness defect. Relevant to this study, Heck et al. [22] showed that the S288c MLH1-SK1 PMS1 incompatible genotype conferred a subtle but significant fitness cost, as measured by decreased spore viability, in diploids grown in rich media for 160 generations. These observations suggest that an adaptive advantage seen in a mutator population is not sustainable. Furthermore, horizontal gene transfer is very rare in yeast [54], indicating that MMR is unlikely to be recovered through such a mechanism.
In our studies the incompatibility seen in S288c MLH1-SK1 PMS1 strains provided an initial adaptive advantage prior to observing a detectable fitness cost. Such an advantage was likely due to incompatible strains displaying a modest mutator phenotype in conjunction with relatively large population sizes of the yeast undergoing serial transfer. How can adapted eukaryotic strains that are mutators escape long-term fitness costs? Sequencing analysis of a 32 kb genomic region provided evidence for recombination between SK1 and S288c strain groups [22]. This observation suggests that an incompatibility could be generated through mating and that adapted incompatible populations can mate back to other strains available in the environment to regain a compatible MMR combination, thus avoiding long-term fitness costs. Several factors are thought to contribute to the likelihood of such a scenario: 1. The frequently of meiotic cycles in wild populations. 2. The number of clonal generations experienced between an outcross. 3. The effects of post-zygotic barriers on the formation of viable progeny. 4. The effect of a stress condition on mating and meiotic cycles.
The ratio between mitotic and meiotic cycles in wild populations of S. cerevisiae is not known, although in S. paradoxus, population genetics approaches have shown that this organism undergoes a sexual cycle approximately once every 1,000 asexual cycles [55]. Magwene et al. [56] used a molecular clock analysis of genomic sequences between yeast strains to estimate the number of clonal generations that two strains would have experienced prior to outcrossing. Their estimates, in conjunction with recombination frequency estimates performed by Ruderfer et al. [57], suggested one outcrossing event per 12,500 to 62,500 generations. Importantly, random mating between spores in natural strains can occur at high rates in the laboratory, and outbreeding was shown to be elevated when spores from different strains were passaged through the fruit fly gut [58,59]. Therefore mating behaviors are likely to be affected by yeast lifestyle conditions that include selection/stress conditions, MMR defects, and population size. Thus it is not difficult to imagine outcrossing restoring MMR compatibility in a large population subjected to strong selection.
Mutations in PMR1 confer a striking adaptive advantage to NaCl tolerance
The majority of mutations that conferred salt tolerance mapped to the PMR1 locus (Table 2 and Fig 5). Previously Anderson et al. [42] identified mutations in other loci that are linked to NaCl tolerance including PMA1, which encodes a proton efflux pump, ENA1, which encodes a sodium efflux pump, and CYC8, which encodes a global transcriptional repressor that regulates ENA1 activity [42]. Possible reasons for why different loci were targeted in the two studies include: 1. The strains used in the two studies were not identical and were likely to have different background mutations. 2. We imposed a stronger selection for NaCl tolerance (1.2 M) than Anderson et al. (1.0 M) [42]. 3. We screened for adaptive advantages at earlier generations (70–100) than Anderson et al. (100–500) [42]. This is of interest because of recent observations made by Lang et al. [60], who studied the appearance of beneficial sterility mutations in haploid S. cerevisiae. In their system they estimated that roughly 100 generations of adaptation were required to generate a threshold level of genetic diversity upon which beneficial mutations could be selected. Thus different target genes might be identified depending on when adaptation is measured. 4. The effective population size per transfer is different in the two studies; we used a 10-fold higher number of cells than Anderson et al. [42]. Such a difference would likely alter the frequency and likelihood that mutations in any one locus would emerge. It is important to note that despite the differences in genes identified between the two studies there is a nice commonality in that NaClr in both studies is likely to involve altered regulation of the Ena1 efflux pump (S5 Fig).
Negative epistasis in MMR genes as a possible adaptation strategy
Epistatic effects involving interacting alleles have been detected for specific fitness measurements between individuals within a population (e.g. [61]). One of the best demonstrations of such effects in yeast was obtained by Brem et al. [62], who crossed two strains of baker’s yeast and then searched for genetic interactions by measuring the levels of all transcripts in a large number of spore progeny. In their analysis they identified statistically significant interactions between locus pairs for 225 transcripts. Based on a population survey of MLH1 and PMS1 alleles, we argued previously that the incompatibility that was identified between MMR genes is similar to epistatic interactions seen in hybrids formed from established or incipient species ([28,29]; see examples in [22]). Support for such an idea is based on the fact that mild reproductive barriers have already been shown to exist between some S. cerevisiae strains [22], and the MMR machinery has been shown to contribute to reproductive isolation when S. cerevisiae strains with sequence divergence are mated [63,64]. The experiments presented in this paper provide an interesting twist to this idea because the incompatibility involving MLH1 and PMS1 might also provide opportunities for adaptive evolution by moderately increasing mutation rates.
Materials and Methods
Media, plasmids and strains
Yeast strains, all isogenic to the FY/S288c background, were grown in YPD (yeast extract, peptone, dextrose), YPD + 1.2 M sodium chloride (NaCl), or YPD + 0.4 M lithium chloride (LiCl) (S1 Table; [65,66]). DNA fragments containing S288c or SK1 derived MLH1 and PMS1 genes (MLH1:KANMX, MLH1::NATMX, and PMS1::HIS3) were introduced into S288c-background strains by gene replacement (S1 Table; [22,67]). S288c derived pmr1::URA3 alleles (URA3 is located 500 bp upstream of PMR1) were also introduced into S288c background strains by gene replacement (S1 Table, pEAA602-606 digested with NotI and XhoI). Integrations were confirmed by PCR amplification of yeast chromosomal DNA, prepared as described by Holm et al. [68] using primers located outside of the ends of the DNA fragments used for integration. Allele integrations were confirmed by sequencing the relevant PCR products using the Sanger method. The sequences of the oligonucleotides used to perform PCR are available upon request. pMZ11 (UPRE::lacZ, ARS-CEN, TRP1, reporter to measure the unfolded protein response) and pKC201 (pmr2::lacZ, 2μ, URA3 reporter to measure PMR2 expression) were generously provided by Jeff Brodsky and Kyle Cunningham, respectively.
Adaptive evolution assays
Single colonies of Saccharomyces cerevisiae were inoculated into 6 ml of YPD and grown for 24 hrs at 30°C in 20 ml glass tubes in a New Brunswick G25 shaker run at 250 RPM. Approximately 2 x 107 of each culture were then transferred into fresh 6 ml YPD or YPD + 1.2 M NaCl (to achieve an initial OD600 of 0.1, Shimadzu UV-1201 spectrophotometer) and then grown for 24 hrs. This procedure was repeated for up to 20 transfers. The number cell generations completed per transfer was determined using the equation log2 (Nt/No), where No = total cell count at 0 hrs and Nt = total cell count at 24 hrs post transfer. A Wilcoxon sign-ranked test was used to compare growth of independent cultures [69].
Competition assays
Incompatible and compatible MLH1-PMS1 strains were created in which the MLH1 gene was marked with KANMX or NATMX (S1 Table). These markers were shown previously to not affect fitness [70,71]. After 7, 10, and 16 transfers in YPD or YPD + 1.2 M NaCl (approximately 50, 70 and 110 generations respectively), incompatible and compatible populations were mixed at a 1 : 1 ratio (1 x 107 cells each inoculated into 5 ml YPD or YPD + 1.2 M NaCl) and grown for an additional 24 hours. The ratio of incompatible and compatible populations was assessed by replica plating YPD plates containing ~ 200 yeast colonies (plated prior to, or after 24 hrs of growth) onto YPD-G418 and YPD-nourseothricin plates [70].
Fitness values (Table 1) were separately determined after competition experiments in which evolved cultures were randomly mixed at a 1 : 1 ratio and grown for an additional 24 hours (estimated to be 7 generations) in YPD or YPD + 1.2 M NaCl. Fitness (w) [34,35] of the incompatible cells relative to the compatible cells was calculated as w = ((pt/qt)/(po/qo))1/t, where po and qo are the number of incompatible and compatible cells, respectively at 0 hrs and pt and qt are the number of incompatible and compatible cells, respectively, at 24 hrs, with t = 7 generations of growth. Fitness differences were analyzed for significance using one-way ANOVA [36].
lys2-A14 reversion assays
lys2-A14 strains (S1 Table; (A)14 inserted into the LYS2 gene) were analyzed for reversion to Lys+ as described previously [27,72]. All strains were inoculated in YPD overnight and plated onto LYS drop out and synthetic complete plates. The 95% confidence intervals were determined as described by Dixon and Massey [73]. Pair-wise Kruskal–Wallis tests were performed between each pair of incompatible and compatible strains to determine the significance of the differences in median reversion rates.
Bulk segregation analysis
Individual NaClr clones isolated from incompatible and compatible strains grown for 10 transfers were phenotyped and then crossed to isogenic, unevolved strains. The resulting diploids were first struck onto YPD + 1.2 M NaCl plates to determine if the NaClr phenotype observed in the evolved haploid strain was dominant or recessive. The diploids were then sporulated using either liquid or solid media containing 1% potassium acetate. Tetrads were dissected and spores clones germinated on YPD were struck onto YPD + 1.2 M NaCl to assess NaCl resistance. The resulting NaClr and NaCls spore clones, at least 18 of each, were pooled in equal cell amounts to create resistant and sensitive bulk pools that were subjected to whole genome sequencing.
Whole genome sequencing
Parental, NaClr evolved compatible and incompatible clones, and the bulk pools described above, were grown in 8 ml cultures. Chromosomal DNA was isolated using Affymetrix Prep-Ease kit and quantified using the Qubit dsDNA HS Assay Kit (Life Technologies). This DNA was then barcoded using Illumina Nextera XT. High throughput sequencing of chromosomal DNA was performed on an Illumina HiSeq 2500 at the Cornell Biotechnology Resource Center.
GATK was used as a platform-independent Java framework. The core system uses the standard sequence alignment program BWA against a reference sequence to create SAM format files [74]. We used the S288c reference sequence in this study (SGD: http://www.yeastgenome.org/) because our strains are isogenic to this background. All differences between our starting unevolved strain and the reference were subtracted. A binary alignment version of the SAM format, called binary alignment/map (BAM), was then compressed and indexed using picard (http://picard.sourceforge.net). Finally, BAM files were analyzed by GATK to optimize the genotyping analysis (http://www.broadinstitute.org/gsa/wiki/index.php/The_Genome_Analysis_Toolkit). SNPs with quality scores of less than 75 were removed from the analysis.
Uniprot was used to predict the topology of Pmr1 (http://www.uniprot.org/).
Beta-galactosidase assays
pKC201 (pmr2::lacZ, 2μ, URA3; [41]) was transformed into evolved strains to determine if mutations in PMR1 activated ENA1/PMR2 expression. Transformants were grown overnight in uracil dropout media in the presence or absence of 1.2 M NaCl and then analyzed in liquid assays for beta-galactosidase activity (permeabilized yeast cell assay, [51]). pMZ11 (UPRE::lacZ, ARS-CEN, TRP1; [44]) was transformed into evolved strains to determine if mutations in PMR1 activated the unfolded protein response pathway. Transformants were grown for the indicated times in tryptophan dropout media with or without 1.2 M NaCl and then analyzed in liquid assays for beta-galactosidase activity. DTT was included at 5 mM to serve as a positive control for the unfolded protein response.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Kunkel TA. Erie DA (2005) DNA mismatch repair. Annu Rev Biochem 74 : 681–710. 15952900
2. Nishant KT, Wei W, Mancera E, Argueso JL, Schlattl A, et al. (2010) The baker's yeast diploid genome is remarkably stable in vegetative growth and meiosis. PLoS Genet 6: e1001109. doi: 10.1371/journal.pgen.1001109 20838597
3. Zanders S, Ma X, Roychoudhury A, Hernandez RD, Demogines A, Barker B et al. (2010) Detection of heterozygous mutations in the genome of mismatch repair defective diploid yeast using a Bayesian approach. Genetics 186 : 493–503. doi: 10.1534/genetics.110.120105 20660644
4. Hombauer H, Campbell CS, Smith CE, Desai A, Kolodner RD (2011) Visualization of eukaryotic DNA mismatch repair reveals distinct recognition and repair intermediates. Cell 147 : 1040–1053. doi: 10.1016/j.cell.2011.10.025 22118461
5. Jiricny J (2013) Postreplicative mismatch repair. Cold Spring Harb Perspect Biol 5: a012633. doi: 10.1101/cshperspect.a012633 23545421
6. Ma X, Rogacheva MV, Nishant KT, Zanders S, Bustamante CD, Alani E. (2012) Mutation hot spots in yeast caused by long-range clustering of homopolymeric sequences. Cell Rep 1 : 36–42. doi: 10.1016/j.celrep.2011.10.003 22832106
7. Lynch M. (2010) Evolution of the mutation rate. Trends in Genetics 26 : 345–352. doi: 10.1016/j.tig.2010.05.003 20594608
8. Drake JW (1991) A constant rate of spontaneous mutation in DNA-based microbes. Proc Natl Acad Sci USA 88 : 7160–7164. 1831267
9. Drake JW, Charlesworth B, Charlesworth D, Crow JF (1998) Rates of Spontaneous Mutation. Genetics 148 : 1667–1686. 9560386
10. Zeyl C, DeVisser JA (2001) Estimates of the rate and distribution of fitness effects of spontaneous mutation in Saccharomyces cerevisiae. Genetics 157 : 53–61. 11139491
11. LeClerc JE, Li B, Payne WL, Cebula TA (1996) High mutation frequencies among Escherichia coli and Salmonella pathogens. Science 274 : 1208–1211. 8895473
12. Matic I, Radman M, Taddei F, Picard B, Doit C, et al. (1997) Highly variable mutation rates in commensal and pathogenic Escherichia coli. Science 277 : 1833–1834. 9324769
13. Boe L, Danielsen M, Knudsen S, Petersen JB, Maymann J, et al. (2000) The frequency of mutators in populations of Escherichia coli. Mutat Res 448 : 47–55. 10751622
14. Denamur E., Lecointre G, Darlu P, Tenaillon O, Acquaviva C et al. (2000) Evolutionary implications of the frequent horizontal transfer of mismatch repair genes. Cell 103 : 711–721. 11114328
15. Sniegowski PD, Dombrowski PG, Fingerman E (2002) Saccharomyces cerevisiae and Saccharomyces paradoxus coexist in a natural woodland site in North America and display different levels of reproductive isolation from European conspecifics. FEMS Yeast Res 1 : 299–306. 12702333
16. Taddei F, Radman M, Maynard-Smith J, Toupance B, Gouyon PH, et al. (1997) Role of mutator alleles in adaptive evolution. Nature 387 : 700–702. 9192893
17. Tanaka MM, Bergstrom CT, Levin BR (2003) The evolution of mutator genes in bacterial populations: the roles of environmental change and timing. Genetics 164 : 843–854. 12871898
18. Townsend JP, Nielsen KM, Fisher DS, Hartl DL (2003) Horizontal acquisition of divergent chromosomal DNA in bacteria: effects of mutator phenotypes. Genetics 164 : 13–21. 12750317
19. Giraud A, Matic I, Tenaillon O, Clara A, Radman M, et al. (2001) Costs and benefits of high mutation rates: adaptive evolution of bacteria in the mouse gut. Science 291 : 2606–2608. 11283373
20. Chao L, Cox EC (1983) Competition between high and low mutating strains of Escherichia coli. Evolution 37 : 125–134.
21. Thompson DA, Desai MM, Murray AW. (2006) Ploidy controls the success of mutators and nature of mutations during budding yeast evolution. Curr Biol 16 : 1581–1590. 16920619
22. Heck JA, Argueso JL, Gemici Z, Reeves RG, Bernard A, et al. (2006) Negative epistasis between natural variants of the Saccharomyces cerevisiae MLH1 and PMS1 genes results in a defect in mismatch repair. Proc Natl Acad Sci USA 103 : 3256–3261. 16492773
23. Muller HJ, Pontecorvo G (1940) Recombinants between Drosophila Species the F1 Hybrids of which are sterile. Nature 146 : 199–200.
24. Muller HJ (1939) Reversibility in evolution considered from the standpoint of genetics. Biol Rev Camb Philos Soc 14 : 261–280.
25. Orr HA (1995) The population genetics of speciation: the evolution of hybrid incompatibilities. Genetics 139 : 1805–1813. 7789779
26. Dobzhansky T (1936) Studies on Hybrid Sterility. II. Localization of Sterility Factors in Drosophila Pseudoobscura Hybrids. Genetics 21 : 113–135. 17246786
27. Demogines A, Wong A, Aquadro C, Alani E (2008) Incompatibilities involving yeast mismatch repair genes: a role for genetic modifiers and implications for disease penetrance and variation in genomic mutation rates. PLoS Genetics 4: e1000103. doi: 10.1371/journal.pgen.1000103 18566663
28. Wu CI, Ting CT (2004) Genes and speciation. Nat Rev Genet 5 : 114–122. 14735122
29. Coyne JA, Orr HA (2004) Speciation, Sinauer Associates, Sunderland, MA.
30. Ting CT, Tsaur SC, Wu ML, Wu CI (1998) A rapidly evolving homeobox at the site of a hybrid sterility gene. Science 282 : 1501–1504. 9822383
31. Barbash DA, Siino DF, Tarone AM, Roote J (2003) A rapidly evolving MYB-related protein causes species isolation in Drosophila. Proc Natl Acad Sci USA 100 : 5302–5307. 12695567
32. Wittbrodt J, Adam D, Malitschek B, Maueler W, Raulf F et al. (1989) Novel putative receptor tyrosine kinase encoded by the melanoma-inducing Tu locus in Xiphophorus. Nature 341 : 415–421. 2797166
33. Rawson RD, Burton RS (2002) Functional coadaptation between cytochrome c and cytochrome c oxidase within allopatric populations of a marine copepod. Proc Natl Acad Sci USA 99 : 12955–12958. 12271133
34. Hartl D, Clark A (2007) Principles of Population Genetics. 4rth Edition. Sinauer Associates.
35. Dykhuizen D, Hartl DL (1980) Selective neutrality of 6GPD allozymes in E. coli and the effects of genetic background. Genetics 96 : 801–817. 7021316
36. McDonald JH (2014) Handbook of Biological Statistics (3rd ed) p 145–156. Sparky House Publishing, Baltimore, Maryland.
37. Raynes Y, Gazzara MR, Sniegowski PD (2012) Contrasting dynamics of a mutator allele in asexual populations of differing size. Evolution 66 : 2329–2334. doi: 10.1111/j.1558-5646.2011.01577.x 22759305
38. Antebi A, and Fink GR (1992) The yeast Ca2+-ATPase homologue, PMR1, is required for normal Golgi function and localizes in a novel Golgi-like distribution. Mol Biol Cell 3 : 633–654. 1379856
39. Park SY, Seo SB, Lee SJ, Na JG, Kim YJ (2001). Mutation in PMR1, a Ca(2+)-ATPase in Golgi, confers salt tolerance in Saccharomyces cerevisiae by inducing expression of PMR2, an Na(+)-ATPase in plasma membrane. J Biol Chem 276 : 28694–28699. 11387321
40. Zhao J, Lin W, Ma X, Lu Q, Ma X, Bian G, Jiang L (2010) The protein kinase Hal5p is the high-copy suppressor of lithium-sensitive mutations of genes involved in the sporulation and meiosis as well as the ergosterol biosynthesis in Saccharomyces cerevisiae. Genomics 95 : 290–298. doi: 10.1016/j.ygeno.2010.02.010 20206679
41. Cunningham KW, Fink GR (1996) Calcineurin inhibits VCX1-dependent H+/Ca2+ exchange and induces Ca2+ ATPases in Saccharomyces cerevisiae. Mol Cell Biol 16 : 2226–2237. 8628289
42. Anderson JB, Funt J, Thompson DA, Prabhu S, Socha A et al. (2010) Determinants of divergent adaptation and Dobzhansky-Muller interaction in experimental yeast populations. Curr Biol 20 : 1383–1388. doi: 10.1016/j.cub.2010.06.022 20637622
43. Lenassi M, Gostinčar C, Jackman S, Turk M, Sadowski I, Nislow C, et al. (2013) Whole genome duplication and enrichment of metal cation transporters revealed by de novo genome sequencing of extremely halotolerant black yeast Hortaea werneckii. PLoS One 8: e71328. doi: 10.1371/journal.pone.0071328 23977017
44. Zhou M, Schekman R (1999) The engagement of Sec61p in the ER dislocation process. Mol Cell 4 : 925–934. 10635318
45. Cyert MS and Thorner J (1992) Regulatory subunit (CNB1 gene product) of yeast Ca2+/calmodulin-dependent phosphoprotein phosphatases is required for adaptation to pheromone. Mol Cell Biol 12 : 3460–3469. 1321337
46. Kohn LM, Anderson JB (2014) The underlying structure of adaptation under strong selection in 12 experimental yeast populations. Eukaryot Cell 13 : 1200–1206. doi: 10.1128/EC.00122-14 25016004
47. Hinnebusch AG and Natarajan K (2002) Gcn4p, a master regulator of gene expression, is controlled at multiple levels by diverse signals of starvation and stress. Eukaryot Cell 1 : 22–32. 12455968
48. Kakinuma Y, Maruyama T, Nozaki T, Wada Y, Ohsumi Y, Igarashi K (1995) Cloning of the gene encoding a putative serine threonine protein kinase which enhances spermine uptake in Saccharomyces cerevisiae. Biochem Biophys Res Commun 216 : 985–992. 7488221
49. Sniegowski PD, Gerrish PJ, Lenski RE (1997) Evolution of high mutation rates in experimental populations of E. coli. Nature 387 : 703–705. 9192894
50. Funchain P, Yeung A, Stewart JL, Lin R, Slupska MM, Miller JH (2000) The consequences of growth of a mutator strain of Escherichia coli as measured by loss of function among multiple gene targets and loss of fitness. Genetics 154 : 959–970. 10757746
51. Woods RJ, Barrick JE, Cooper TF, Shrestha U, Kauth MR, Lenski RE. (2011) Second-order selection for evolvability in a large Escherichia coli population. Science 331 : 1433–1436. doi: 10.1126/science.1198914 21415350
52. Elena SF, Lenski RE (2003) Evolution experiments with microorganisms: the dynamics and genetic bases of adaptation. Nat Rev Genet 4 : 457–469. 12776215
53. Sprouffske K, Merlo LM, Gerrish PJ, Maley CC, Sniegowski PD (2012) Cancer in light of experimental evolution. Curr Biol 22: R762–771. doi: 10.1016/j.cub.2012.06.065 22975007
54. Liti G, Louis EJ (2005) Yeast evolution and comparative genomics. Annu Rev Microbiol 59 : 135–153. 15877535
55. Tsai IJ, Bensasson D, Burt A, Koufopanou V (2008) Population genomics of the wild yeast Saccharomyces paradoxus: Quantifying the life cycle. Proc Natl Acad Sci USA 105 : 4957–4962. doi: 10.1073/pnas.0707314105 18344325
56. Magwene PM, Kayıkçı Ö, Granek JA, Reininga JM, Scholl Z, Murray D (2011) Outcrossing, mitotic recombination, and life-history trade-offs shape genome evolution in Saccharomyces cerevisiae. Proc Natl Acad Sci USA 108 : 1987–1992. doi: 10.1073/pnas.1012544108 21245305
57. Ruderfer DM, Pratt SC, Seidel HS, Kruglyak L (2006) Population genomic analysis of outcrossing and recombination in yeast. Nat Genet 38 : 1077–1081. 16892060
58. Murphy HA, Zeyl CW (2010) Yeast Sex: Surprisingly high rates of outcrossing between asci. PLoS One 5: e10461. doi: 10.1371/journal.pone.0010461 20463964
59. Reuter M, Bell G, Greig D (2007) Increased outbreeding in yeast in response to dispersal by an insect vector. Curr Biol 17: R81–83. 17276903
60. Lang GI, Botstein D, Desai MM (2011) Genetic variation and the fate of beneficial mutations in asexual populations. Genetics 188 : 647–661. doi: 10.1534/genetics.111.128942 21546542
61. Corbett-Detig RB, Zhou J, Clark AG, Hartl DL, Ayroles JF (2013) Genetic incompatibilities are widespread within species. Nature 504 : 135–137. doi: 10.1038/nature12678 24196712
62. Brem RB, Storey JD, Whittle J, Kruglyak L (2005) Genetic interactions between polymorphisms that affect gene expression in yeast. Nature 436 : 701–703. 16079846
63. Hunter N, Chambers SR, Louis EJ, Borts RH (1996) The mismatch repair system contributes to meiotic sterility in an interspecific yeast hybrid. EMBO J 15 : 1726–1733. 8612597
64. Naumov GI, Naumova ES, Lantto RA, Louis EJ, Korhola M (1992) Genetic homology between Saccharomyces cerevisiae and its sibling species S. paradoxus and S. bayanus: electrophoretic karyotypes. Yeast 8 : 599–612. 1441740
65. Winston F, Dollard C, Ricupero-Hovasse SL (1995) Construction of a set of convenient Saccharomyces cerevisiae strains that are isogenic to S288C. Yeast 11 : 53–55. 7762301
66. Rose MD, Winston F, Hieter P (1990) Methods in yeast genetics: A Laboratory Course Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
67. Gietz RD, Schiestl RH (2007) Large-scale high-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat Protoc 2 : 38–41. 17401336
68. Holm C, Meeks-Wagner DW, Fangman WL, Botstein D (1986) A rapid, efficient method for isolating DNA from yeast. Gene 42 : 169–173. 3015730
69. Rosner B, Glynn RJ, Lee M-LT (2006) The Wilcoxon signed rank test for paired comparisons of clustered data. Biometrics 62 : 185–192. 16542245
70. Goldstein AL, McCusker JH (1999) Three new dominant drug resistance cassettes for gene disruption in Saccharomyces cerevisiae. Yeast 15 : 1541–1553. 10514571
71. Baganz F, Hayes A, Marren D, Gardner DC, Oliver SG (1997) Suitability of replacement markers for functional analysis studies in Saccharomyces cerevisiae. Yeast 13 : 1563–1573. 9509575
72. Tran HT, Keen JD, Kricker M, Resnick MA, Gordenin DA (1997) Hypermutability of homonucleotide runs in mismatch repair and DNA polymerase proofreading yeast mutants. Mol Cell Biol 17 : 2859–2865. 9111358
73. Dixon WJ, Massey FJ (1969) Introduction to Statistical Analysis. New York: McGraw-Hill.
74. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, et al. (2010) The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20 : 1297–1303. doi: 10.1101/gr.107524.110 20644199
75. Wanat JJ, Singh N, Alani E (2007) The effect of genetic background on the function of Saccharomyces cerevisiae mlh1 alleles that correspond to HNPCC missense mutations. Hum Mol Genet 16 : 445–452. 17210669
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2015 Číslo 7
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Functional Constraint Profiling of a Viral Protein Reveals Discordance of Evolutionary Conservation and Functionality
- Reversible Oxidation of a Conserved Methionine in the Nuclear Export Sequence Determines Subcellular Distribution and Activity of the Fungal Nitrate Regulator NirA
- Modeling Implicates in Nephropathy: Evidence for Dominant Negative Effects and Epistasis under Anemic Stress
- Nutritional Control of DNA Replication Initiation through the Proteolysis and Regulated Translation of DnaA
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy