
Genetic and Functional Studies Implicate Synaptic Overgrowth and Ring Gland cAMP/PKA Signaling Defects in the Neurofibromatosis-1 Growth Deficiency
Neurofibromatosis type 1 (NF1), a genetic disease that affects 1 in 3,000, is caused by loss of a large evolutionary conserved protein that serves as a GTPase Activating Protein (GAP) for Ras. Among Drosophila melanogaster Nf1 (dNf1) null mutant phenotypes, learning/memory deficits and reduced overall growth resemble human NF1 symptoms. These and other dNf1 defects are relatively insensitive to manipulations that reduce Ras signaling strength but are suppressed by increasing signaling through the 3′-5′ cyclic adenosine monophosphate (cAMP) dependent Protein Kinase A (PKA) pathway, or phenocopied by inhibiting this pathway. However, whether dNf1 affects cAMP/PKA signaling directly or indirectly remains controversial. To shed light on this issue we screened 486 1st and 2nd chromosome deficiencies that uncover >80% of annotated genes for dominant modifiers of the dNf1 pupal size defect, identifying responsible genes in crosses with mutant alleles or by tissue-specific RNA interference (RNAi) knockdown. Validating the screen, identified suppressors include the previously implicated dAlk tyrosine kinase, its activating ligand jelly belly (jeb), two other genes involved in Ras/ERK signal transduction and several involved in cAMP/PKA signaling. Novel modifiers that implicate synaptic defects in the dNf1 growth deficiency include the intersectin-related synaptic scaffold protein Dap160 and the cholecystokinin receptor-related CCKLR-17D1 drosulfakinin receptor. Providing mechanistic clues, we show that dAlk, jeb and CCKLR-17D1 are among mutants that also suppress a recently identified dNf1 neuromuscular junction (NMJ) overgrowth phenotype and that manipulations that increase cAMP/PKA signaling in adipokinetic hormone (AKH)-producing cells at the base of the neuroendocrine ring gland restore the dNf1 growth deficiency. Finally, supporting our previous contention that ALK might be a therapeutic target in NF1, we report that human ALK is expressed in cells that give rise to NF1 tumors and that NF1 regulated ALK/RAS/ERK signaling appears conserved in man.
Published in the journal:
. PLoS Genet 9(11): e32767. doi:10.1371/journal.pgen.1003958
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003958
Summary
Neurofibromatosis type 1 (NF1), a genetic disease that affects 1 in 3,000, is caused by loss of a large evolutionary conserved protein that serves as a GTPase Activating Protein (GAP) for Ras. Among Drosophila melanogaster Nf1 (dNf1) null mutant phenotypes, learning/memory deficits and reduced overall growth resemble human NF1 symptoms. These and other dNf1 defects are relatively insensitive to manipulations that reduce Ras signaling strength but are suppressed by increasing signaling through the 3′-5′ cyclic adenosine monophosphate (cAMP) dependent Protein Kinase A (PKA) pathway, or phenocopied by inhibiting this pathway. However, whether dNf1 affects cAMP/PKA signaling directly or indirectly remains controversial. To shed light on this issue we screened 486 1st and 2nd chromosome deficiencies that uncover >80% of annotated genes for dominant modifiers of the dNf1 pupal size defect, identifying responsible genes in crosses with mutant alleles or by tissue-specific RNA interference (RNAi) knockdown. Validating the screen, identified suppressors include the previously implicated dAlk tyrosine kinase, its activating ligand jelly belly (jeb), two other genes involved in Ras/ERK signal transduction and several involved in cAMP/PKA signaling. Novel modifiers that implicate synaptic defects in the dNf1 growth deficiency include the intersectin-related synaptic scaffold protein Dap160 and the cholecystokinin receptor-related CCKLR-17D1 drosulfakinin receptor. Providing mechanistic clues, we show that dAlk, jeb and CCKLR-17D1 are among mutants that also suppress a recently identified dNf1 neuromuscular junction (NMJ) overgrowth phenotype and that manipulations that increase cAMP/PKA signaling in adipokinetic hormone (AKH)-producing cells at the base of the neuroendocrine ring gland restore the dNf1 growth deficiency. Finally, supporting our previous contention that ALK might be a therapeutic target in NF1, we report that human ALK is expressed in cells that give rise to NF1 tumors and that NF1 regulated ALK/RAS/ERK signaling appears conserved in man.
Introduction
RASopathies, caused by mutations that activate Ras/ERK signaling, are a group of related disorders with features that include facial dysmorphism, skeletal, skin and cardiac defects, cognitive deficits, reduced growth and an increased cancer risk [1]. Neurofibromatosis type 1 (NF1; OMIM 162200), caused by loss of a RasGAP, and Noonan syndrome, caused by mutations that alter Ras/ERK pathway proteins SOS1, KRAS, NRAS, RAF1, BRAF, CBL, PTPN11, or SHOC2, are the most common members of this group, affecting 1 in 3,000, or as many as 1 in 1,000 live births, respectively [2], [3]. The genetics of these disorders provides a strong argument that excess Ras/ERK signaling underlies common RASopathy symptoms, and much effort remains focused on attenuating Ras/ERK signaling as a strategy for therapeutic intervention. However, whether life-long pharmacological inhibition of Ras/ERK signaling is a viable strategy to treat the full range of often non-life-threatening, but nonetheless serious symptoms of these chronic disorders, remains an open question. This motivates our work to better understand the molecular and cellular pathways responsible for NF1 symptom development, in the hope this will identify more specific therapeutic targets.
We have been interested in using Drosophila melanogaster as a model to investigate NF1 functions in vivo, following our identification of a conserved dNf1 ortholog predicting a protein that is 60% identical to human neurofibromin over its entire 2802 amino acid length [4]. Like human neurofibromin, the Drosophila protein functions as a GAP for conventional (dRas1) and R-Ras-like (dRas2) GTPases [4], [5]. This functional conservation made it all the more surprising when both initially identified dNf1 homozygous null mutant phenotypes, a postembryonic growth deficiency and a neuropeptide-elicited NMJ electrophysiological defect, appeared insensitive to genetic manipulations that attenuate Ras signaling strength, but were suppressed by increasing signaling through the cAMP-dependent PKA pathway [4], [6]. The genetic link between dNf1 and cAMP/PKA led to further studies, which demonstrated that similar to many children with NF1 [7], and Nf1+/− mice [8], dNf1−/− flies exhibit specific learning and memory deficits [9]. Biochemical studies with fly brain extracts further revealed that loss of dNf1 is associated with reduced GTP-γS-stimulated but not basal adenylyl cyclase (AC) activity [9], and with defects in both classical and unconventional AC pathways [10]. Arguing that the cAMP related function of NF1 is evolutionary conserved, GTP-γS-stimulated AC activity and cAMP levels were also reduced in E12.5 Nf1−/− mouse brain [11], and defects in cAMP generation appear to explain the unique sensitivity to Nf1 heterozygosity of murine central nervous system neurons [12]. Arguing that NF1 may regulate cAMP signaling at least in part in a cell autonomous manner, reduced cAMP levels and AC activity were also found in NF1 deficient human astrocytes [13]. Thus, while there is little doubt that aberrant AC signaling is an evolutionary conserved NF1 phenotype, we and others have reached conflicting conclusions about the underlying mechanism.
Based on Drosophila phenotypic rescue studies with human NF1 transgenes, others reported that neurofibromin has physically separable functions as a negative regulator of Ras and a positive mediator of AC/PKA signaling. This conclusion followed from findings that NF1-GAP activity was not required to rescue dNf1 size [10] or learning [14] phenotypes, whereas a transgene encoding a C-terminal part of human neurofibromin that did not include the GAP catalytic domain did suppress both defects. In obvious conflict, in similar experiments with dNf1 transgenes, we found that neuronal expression of a functional NF1-GAP catalytic segment was necessary and sufficient to suppress the systemic growth defect, and that other protein segments had no effect. Moreover, the dNf1 growth defect was also suppressed by neuronal expression of the Drosophila p120RasGAP ortholog, and although we extended earlier findings by showing that heterozygous loss of dRas1 or dRas2, or of a comprehensive set of Ras effector proteins did not modify the growth defect, these mutations also did not reduce the elevated phospho-ERK level in the dNf1 central nervous system (CNS). However, some Ras/ERK pathway double mutants did suppress both defects, leading us to conclude that excess neuronal Ras/ERK signaling is the proximal cause of the non-cell-autonomous dNf1 growth defect [5]. Further supporting this notion, recent work implicated the neuronal dAlk tyrosine kinase receptor and its activating ligand jelly belly (jeb) as rate-limiting activators of dNf1 regulated Ras/ERK pathways responsible for both systemic growth and olfactory learning defects [15].
The above evidence underlies our hypothesis that loss of dNf1 increases neuronal dAlk/Ras/ERK activity, which in turn causes reduced cAMP/PKA signaling, which may or may not be cell-autonomous. Obviously, identifying additional components of dNf1-regulated growth controlling pathways followed by functional analysis might help to test this hypothesis. Here we report results of a dNf1 growth deficiency modifier screen, which identified components of tyrosine kinase/Ras/ERK and neuropeptide/cAMP/PKA pathways in addition to genes involved in synaptic morphogenesis and functioning. Further analysis showed that the requirement for dNf1 and cAMP/PKA in Drosophila growth regulation involves different tissues, with dNf1 required broadly in larval neurons, and cAMP/PKA signaling specifically in AKH-producing cells and perhaps in other parts of the neuroendocrine ring gland. These results, and the recent discovery of a novel dNf1 synaptic overgrowth phenotype [16] that is also suppressed by several genes identified in our screen, set the stage for further work to more precisely define how loss of dNf1 causes Ras/ERK and other signaling defects, the ultimate consequence of which is reduced systemic growth.
Results
Loss of dNf1 Does Not Phenocopy Starvation or Alter Developmental Timing
Animals use elaborate hormonal mechanisms to coordinate nutrient availability and feeding with changes in metabolism and overall growth. Since starvation or crowding during the larval phase of the Drosophila life cycle reduces systemic growth [17], we first examined whether the small size of dNf1 mutants reflected reduced feeding. Arguing against this hypothesis, wild-type and dNf1 larvae ingested similar amounts of dye-stained food throughout their development (Figure 1A). Unlike a pumpless (ppl) mutant [18], dNf1 larvae also showed no tendency to move away from a food source (Figure 1B). Analysis of the expression of the starvation-inducible Pepck and Lip3 genes [18] provided further evidence that loss of dNf1 does not phenocopy starvation (Figure 1C).

Mechanisms that control Drosophila growth have been the topic of intense study and much has been learned about how an interplay between insulin-like peptide (ILP) controlled growth rate and ecdysone controlled growth duration determines overall growth (see [19] and [20] for reviews). Arguing against an important role for ecdysone or other factors that control the length of the larval growth period, no differences in the expression of canonical ecdysone-regulated genes was found (results not shown) and no difference in developmental timing between wild-type and dNf1 mutants was detected (Figure 1D and S1). Rather, a reduced growth rate throughout larval development results in an approximately 25% weight reduction of dNf1 pupae relative to isogenic controls (Figure 1E and S1).
Drosophila ILPs control systemic growth, metabolism, longevity, and female fecundity [21]–[24]. Among the eight Drosophila ILP genes, Ilp2, Ilp3 and Ilp5 are co-expressed in bilateral clusters of seven insulin-producing neurosecretory cells (IPCs) in the larval brain [21]. Ablation of these cells causes a severe reduction in overall size, which is rescued by inducing the expression of a hsp70-Ilp2 transgene [22], [23]. However, several results argue against a role for ILPs in the dNf1 growth defect. Firstly, two hypomorphic insulin receptor alleles, InR05545 and InR327, did not affect dNf1 pupal size (Figure 1F). Secondly, qRT-PCR analysis of RNA extracted from wandering wild-type and dNf1 third instar larvae detected no major differences in the expression of Ilp1 (not shown), Ilp2, Ilp3, Ilp5, Ilp6 and Ilp7 in fed larvae. Among the three IPC expressed ILP genes, the expression of Ilp3 and Ilp5 is reduced in response to starvation [21]. Starved wild-type and dNf1 larvae showed a similar reduction in Ilp5 expression, whereas Ilp3 showed a less pronounced response (Figure 1G). Thirdly, while certain insulin receptor or insulin receptor substrate (chico) mutants have an up to 85% increased life span [25], [26], the lifespan of dNf1 mutants and isogenic controls was comparable (Figure 1H). We note that others previously reported a reduced life span for the originally identified dNf1 p-element alleles, generated in a different genetic background [27]. Finally, we previously showed that Ilp2-GAL4 driven UAS-dNf1 expression in IPCs did not rescue the dNf1 size defect [5]. Although daily heat shocking of hsp70-ilp2 carrying larvae increased the size of dNf1 pupae, indicating that mutants do not lack the ability to respond to insulin, similar induction of this transgene, as previously noted [21], also substantially increased the size of wild-type controls (Figure 1I). Thus, reduced insulin signaling does not provide an obvious explanation for the slower dNf1 growth rate, prompting us to perform a screen to identify other genes involved in dNf1-mediated systemic growth control.
Screen for Dominant Modifiers of dNf1 Systemic Growth Phenotype
While most dNf1 defects are poorly suited for use in modifier screens, the postembryonic growth defect is robust and readily quantified during the pupal stage [4]. However, using this phenotype in a screen is complicated by the fact that organismal size is sexually dimorphic (females are larger than males) and affected by population density, feeding, environmental factors and genetic background differences. With these confounding factors in mind, we used the crossing schemes outlined in Figure 2 to test collections of isogenic 1st and 2nd chromosome deficiencies for dNf1E2 pupal size modifier effects or synthetic lethal interactions. For each of 139 1st and 347 2nd chromosome deficiencies from the Exelixis [28], DrosDel [29] or Bloomington Stock Center (BSC) collections, we generated Df(1)/+; Nf1E2/Nf1E2 (Figure 2A) or Df(2)/+; Nf1E2/Nf1E2 (Figure 2B) stocks, respectively. Notably, our work identified only few synthetic lethal interactions, and in all cases tested the synthetic lethality has been specific to the chromosome carrying the Nf1E2 allele, and not observed when the same deficiency was tested in Nf1E2/Nf1E1 null trans-heterozygotes [5]. To guard against size differences caused by inadvertent differences in population density or environmental conditions, each deficiency was scored at least twice using an initial rough caliper measurement of pupae attached to the side of culture vials. For each candidate modifying deficiency thus identified, microscopy combined with image analysis was used to determine the precise head-to-tail length of at least 40 pupae, which were then allowed to individually eclose in order to establish their sex. Several controls were next performed to eliminate non-specific modifiers or artifactual results. First, for all suppressors the continued presence of the Nf1E2 nonsense mutation was confirmed by a PCR assay (Figure S2). Secondly, as a critical specificity control, all modifying deficiencies were analyzed in a wild-type background to eliminate those that affect pupal size irrespective of dNf1 genotype. Further analysis of some of these non-specific modifiers demonstrated that loss of Act57B dominantly increases pupal size, whereas heterozygous loss of the glutamate transporter Eaat1 has the opposite effect. Thirdly, because pupal size is a function of larval growth rate and duration, modifying deficiencies were monitored for obvious changes in developmental timing. Table 1 shows the number of screened chromosome 1, 2L and 2R deficiencies, the fraction of genes uncovered and the number of dNf1 and wild-type pupal size modifying deficiencies and loci identified. Figure 2C shows the magnitude of the pupal size modification of typical enhancers and suppressors. The number of modifying deficiencies exceeds the number of identified loci, because many modifying deficiencies uncover overlapping genomic segments (Figure 3). Not unexpectedly, individual modifying deficiencies increase or decrease dNf1 pupal size to different extents (Figure 4).




Some large non-modifying deficiencies identified in our screen completely overlapped with smaller modifying ones. In such cases, stocks were re-ordered and reanalyzed. If these tests replicated the original results, genetic complementation analysis or PCR amplification using transposon and flanking sequence-specific primers was used to confirm the mapping of the deficiencies in question. This procedure identified several mismapped or mislabeled deficiencies, most of which have since been withdrawn by stock centers. Any suspect or recessive modifying deficiency, or any deficiency that uncovers genes with non-specific size phenotypes, such as Minute loci [30], [31], were eliminated from further analysis. Table S1 lists these deficiencies and the reason for their exclusion.
During work to identify genes responsible for observed effects, we prioritized genes uncovered by suppressing deficiencies over those uncovered by enhancers. We also prioritized modifying loci uncovered by more than one deficiency, strong modifiers over weak ones, and genes uncovered by smaller deficiencies over those uncovered by larger ones, reasoning that effects of smaller deficiencies are more likely due to the loss of single genes. Validating the screen, suppressing Df(2R)Exel7144 uncovers dAlk and partially overlapping suppressing Df(2R)BSC199 and Df(2R)BSC699 each uncover the gene for its activating ligand, jeb, both previously identified as dominant suppressors of dNf1 size, learning, and neuronal ERK over-activation phenotypes [15]. Other uncovered candidate modifiers, such as PKA catalytic and regulatory subunit genes, were tested in crosses with loss-of-function alleles and/or by tissue-specific knockdown using at least two independent UAS-RNAi transgenes, most of which were obtained from the Vienna Drosophila Stock Center (VDRC) [32]. For deficiencies that lacked obvious candidate modifiers, we used the UAS-RNAi approach to more broadly screen uncovered genes. Figure S3 shows examples of modifiers identified by this latter approach. Although the nutrient sensing fat body and other tissues outside of the CNS play important roles in Drosophila growth control [33], [34], candidate modifiers have only been tested by RNAi knockdown in neurons or glial cells. We focused on these cell types, because neuronal UAS-dNf1 expression sufficed to suppress the growth phenotype [5].
The dNf1 pupal size modifiers identified to date can be classified into three non-exclusive categories, the first of which consists of the previously implicated dAlk/jeb receptor/ligand pair and two not previously implicated other genes involved in Ras-mediated signal transduction. Another expected category includes genes involved in cAMP/PKA signaling, including the previously reported dnc cAMP phosphodiesterase suppressor [35], and the newly identified PKA catalytic subunit gene, PKA-C1, which acts as an enhancer. This group also includes the CCKLR-17D1 drosulfakinin receptor, recently implicated as a cAMP-coupled promoter of synaptic growth [36], which is particularly interesting given the recent identification of a dNf1 larval NMJ overgrowth phenotype [16]. Finally, our screen also identified multiple genes whose roles in dNf1 growth control had not been anticipated and whose functional relevance remains to be established. Several genes in this group are predominantly expressed in brain or have known neuronal functions, including genes coding for the aforementioned CCKLR-17D1 receptor, the synaptic scaffold protein Dap160, the neuronal RNA binding protein elav, the neuronal Na,K ATPase interacting protein NKAIN [37], and the larval brain and alimentary channel expressed amino acid transporter NAAT1 [38]. Other genes in this group include CKIIbeta2, encoding a casein kinase regulatory subunit, the endosomal trafficking proteins deep-orange and carnation, the Notch modifier heparan sulfate 3-O sulfotransferase Hs3st-B [39], and the ubiquitin E3 ligases HERC2, which acts as a suppressor, and CUL3, which has the opposite effect. Table 2 lists deficiencies that modify dNf1 but not wild-type pupal size, limited to those for which the responsible gene has been identified. Table S2 identifies all analyzed deficiencies, indicates which modified dNf1 pupal size (providing female pupal sizes as a gauge of modification strength), which also altered wild-type pupal size, and which deficiencies altered developmental timing.

dNf1 Pupal Size Modifiers Involved in Jeb/dAlk/Ras/ERK Signaling
We previously reported that the dAlk receptor tyrosine kinase [40] acts as a rate-limiting activator of neuronal Ras/ERK pathways responsible for dNf1 size and learning defects [15]. Therefore, the fact that the dAlk and jeb genes are uncovered by one and two suppressing deficiencies, respectively (Table 2), validates our screen. Others recently reported that Jeb/dAlk signaling allows brain growth to be spared at the expense of other tissues in nutrient restricted Drosophila, and identified a glial cell niche around neuroblasts as the source of Jeb under these conditions [41]. To determine whether glial cells also produce Jeb involved in overall growth control under normal conditions, we used glial and neuronal Gal4 drivers to test the effect of tissue-specific jeb and dAlk knockdown. Arguing that neurons are the main source of Jeb involved in systemic growth control under non-starvation conditions, jeb knockdown with the Ras2-Gal4, C23-Gal4, and n-syb-Gal4 neuronal drivers [5] increased dNf1E2 pupal size (Figure 5A), whereas the Nrv2-Gal4, Eaat1-Gal4 and Gli-Gal4 glial drivers had no effect (data not shown). The only glial driver that gave rise to partial rescue was the pan-glial repo-Gal4 line, although this effect was not enhanced by co-expressing UAS-Dcr2. Control experiments showed that any driver used in these and other experiments had no effect on pupal size in the absence of UAS transgenes or vice-versa, that UAS transgenes had no effect in the absence of Gal4 drivers (Figure 5A and data not shown). Finally, extending previous findings and further confirming a role for jeb as a dominant dNf1 size defect suppressor, the jebweli loss-of-function allele [42] dominantly increased dNf1 pupal size (Figure 5B)

Previously, heterozygous mutations affecting RAF/MEK/ERK kinase cascade components Draf (pole hole; phl), Dsor1/dMEK, or ERK/rolled (rl), did not modify dNf1 size [5]. In agreement, two phl-uncovering deficiencies, Df(1)ED6574 and Df(1)ED11354, did not score as modifiers (Table S2). No rl uncovering deficiencies were analyzed, but Df(1)Exel9049, which is among the stronger suppressors identified, deletes Dsor1 and only two other genes, the neurogenic gene almondex (amx), and CG17754, predicting a BTB and Kelch domain protein. Arguing that reduced Ras/ERK signaling upon loss of Dsor1 combined with abnormal neuronal differentiation due to loss of amx may synergistically cause the observed strong effect, Ras2-Gal4 driven UAS-RNAi transgenes targeting either gene, while causing pupal lethality at 25°C, increased dNf1 pupal size at lower temperatures (Figure 5C). Moreover, suppression of the dNf1 pupal size defect was also observed upon individual heterozygous loss of either Dsor1 or amx, although at least with the tested alleles, combined loss of both genes did not have a more pronounced effect (Figure 5B). Previously, we did not observe suppression of the dNf1E2 pupal size defect in crosses with the Dsor1S-1221 allele [5]. A potential explanation may be that Dsor1LH110 is a null mutant [43], whereas the molecular nature of Dsor1S-1221 is undetermined. Genetic background differences between these Dsor1 alleles are another potential explanation for the discrepant results.
Multiple screens aimed at identifying genes involved in Drosophila tyrosine kinase/Ras signaling have been performed [44]–[52]. Among the genes identified, several are uncovered by 1st and 2nd chromosome deficiencies that do not modify dNf1 size. Suppressing Df(2R)BSC161 uncovers 27 genes including connector enhancer of KSR (cnk), a scaffold protein that functions as a bimodal (both positive and negative) regulator of RAS/MAPK signaling [53], [54]. Supporting a role for cnk as a dNf1 modifier, the cnkXE-385 and cnkE-2083 alleles acted as dominant suppressors (Figure 5B), and suppression was also observed upon RNAi-mediated Cnk knockdown using Ras2-Gal4 or P(GawB)C23-Gal4 neuronal drivers (Figure 5C). However, Df(2R)BSC154, which uncovers cnk and only nine other genes, did not score as a modifier (Table S2).
dNf1 Size Modifiers Involved in cAMP/PKA Signaling
The dNf1 growth defect is suppressed by heat shock-induced expression of a constitutively active murine PKA catalytic subunit transgene, called PKA* [4], or by loss of the dunce (dnc) cAMP phosphodiesterase [35]. Further validating our screen, two dnc uncovering deficiencies and another that removes the region immediately upstream of the dnc coding region, all scored as suppressors (Table 2). Moreover, the Pka-R2 gene, encoding a cAMP binding regulatory PKA subunit, whose dissociation from the catalytic subunit activates the latter, is uncovered by two additional suppressing deficiencies, whereas a deficiency that uncovers the major Pka-C1 catalytic subunit gene scored as an enhancer (Table 2). Df(1)ED7261, which uncovers the rutabaga (rut) adenylyl cyclase, did not score as a modifier (not shown). Confirmation of dnc and Pka-C1 as the genes responsible for the observed effects was obtained in crosses with three dnc and three Pka-C1 loss-of-function alleles (Table 2). Pka-R2 remains an attractive candidate suppressor, but expression Pka-R2RNAi transgenes in neurons had no effect and its role as a dNf1 modifier remains unconfirmed (results not shown).
Novel dNf1 Modifiers
Recently, the cAMP-coupled CCKLR-17D1 drosulfakinin receptor, but not its closely related CCKLR-17D3 paralog, was identified as a positive regulator of synaptic growth [36]. The CCKLR-17D1 gene is uncovered by three suppressing deficiencies, including Df(1)Exel9051, which uncovers only three other genes. The closely linked CCKLR-17D3 paralog is not uncovered by Df(1)Exel9051, and while Ras2-Gal4 or P(GawB)C23-Gal4 driven neuronal CCKLR-17D1 RNAi expression strongly suppressed the dNf1 pupal size defect, similar suppression of CCKLR-17D3 had no effect (Figure 6A).
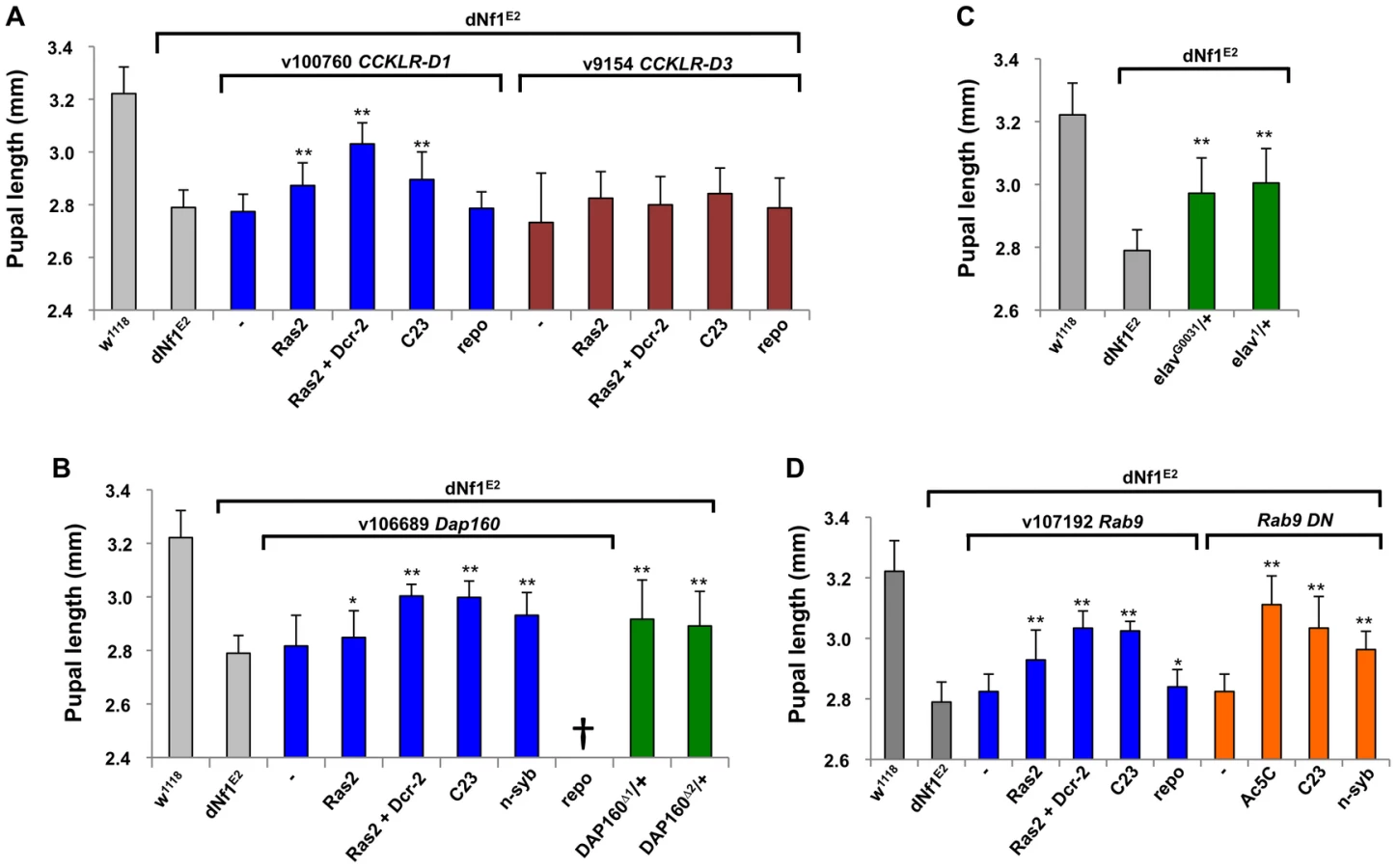
Beyond CCKLR-17D1, several dNf1 size modifiers are expressed in brain and/or have neuronal functions. Among these, dynamin-associated protein 160 (Dap160) is an intersectin-related scaffold implicated in synaptic vesicle exocytosis and neuroblast proliferation [55]–[58]. Dap160 is uncovered by suppressing deficiencies Df(2L)Exel6047 and Df(2L)BSC302, whose region of overlap encompasses ten genes. We note that Df(2L)Exel6047 also uncovers the Drosophila Ret tyrosine kinase gene, the human ortholog of which is the receptor for glial-derived neurotrophic factor. Ret initially appeared an especially attractive candidate suppressor, because activating RET and inactivating NF1 mutations can both lead to human pheochromocytoma [59], and because Drosophila Ret is expressed in larval brain neurons that resemble neuroendocrine cells [60]. However, among multiple lines of evidence that argue against a role for Ret in the dNf1 growth defect, UAS-dNf1 re-expression directed by a newly generated Ret-Gal4 driver that recapitulates the endogenous larval brain Ret expression pattern (Figure S4B), or RNAi-mediated Ret inhibition, did not modify dNf1 pupal size, nor did expression of a UAS-Ret K805A kinase dead transgene. Moreover, Ret-Gal4 driven expression of UAS-Ret transgenes carrying the activating C695R mutation, which mimics a mutation found in multiple endocrine neoplasia type 2 did not phenocopy the dNf1 reduced growth phenotype, although the same transgene did produce the previously described rough eye phenotype when driven by GMR-Gal4 [60]; Figure S4C]. Further arguing against a role in dNf1 growth control, Ret is uncovered by non-modifying Df(2L)BSC312. By contrast, Dap160 loss-of-function alleles (Dap160Δ1 and Dap160Δ2; [56]), or Dap160 RNAi expression driven by three neuronal Gal4 drivers, suppressed the dNf1 pupal size defect, identifying it as the responsible modifier (Figure 6B).
The gene for the neuronal RNA binding protein elav is uncovered by suppressing Df(1)Exel6221 and Df(1)ED6396 whose region of overlap includes just three other genes. Identifying elav as the responsible modifier, elav1 and elavG0031 alleles strongly suppressed (Figure 6C). Rab9 is a modifier uncovered by suppressing deficiency Df(2L)Exel8041. Neuronal but not glial Rab9RNAi expression increases dNf1 pupal size, and the same result is seen upon neuronal expression of a Rab9 dominant negative [61] mutant (Figure 6D).
NAAT1, coding for a larval gut and brain expressed amino acid transporter with a unique affinity for D-amino acids [38], is uncovered by suppressing Df(1)Exel6290 and Df(1)BSC533 whose region of overlap includes only four other genes. Identifying NAAT1 as the responsible suppressor, three neuronal Gal4 lines driving the expression of three NAAT1 targeting RNAi transgenes suppressed the dNf1 size defect, whereas Repo-Gal4 driven glial expression had no effect (Figure 7A and Table 2).

Mammalian E3 ubiquitin ligase HERC2 controls the ubiquitin-dependent assembly of DNA repair proteins on damaged chromosomes [62]. Drosophila HERC2 is uncovered by suppressing deficiency Df(1)Exel6254, which also uncovers the syx16, coding for syntaxin 16. No HERC2 alleles exist, but Ras2-Gal4 driven expression of a UAS-HERC2RNAi transgene (v105374) strongly suppressed the dNf1 pupal size defect (Figure 7A), whereas similar knockdown of Syx16 had no statistically significant effect (not shown). The gene for another E3 ligase component, Cul-3, is uncovered by three enhancing deficiencies, and a Cul-3 loss-of-function allele or Ras2-Gal4 driven expression of a Cul-3 RNAi transgene both enhanced the dNf1 size defect, identifying it as the responsible gene (Table 2).
Suppressing Df(1)Exel9068 uncovers only four genes, including one encoding the TORC2 complex subunit Rictor. However, systematic Ras2-Gal4 driven RNAi knockdown of Df(1)Exel9068 uncovered genes identified Hs3st-B, encoding one of two Drosophila heparan sulfate 3-O sulfotransferases, as a potent dNf1 size defect suppressor (Figure 7A), whereas knockdown of Rictor had no effect (not shown). Others previously identified Hs3st-B as a positive regulator of Notch signaling [39]. However, the heparan sulfate proteoglycan substrates of Hs3st-B bind various growth factors and other ligands and have been implicated in a variety of biological processes. Exactly why loss of Hs3st-B suppresses the dNf1 growth defect remains to be determined.
Two functionally related dNf1 growth defect suppressors carnation (car/Vps33A) and deep-orange (dor/Vps18), encode subunits of the Class C Vacuolar Protein Sorting (VPS) complex, required for the delivery of endosomal vesicles to lysosomes [63]; Figure 7B]. The Vps16A gene encodes a third member of this complex [64], but whether Vps16A located on the 3rd chromosome also acts as a dNf1 suppressor, or whether pharmacological inhibition of lysosomal degradation affects dNf1 pupal size are questions that remain to be answered.
B4/Susi is a coiled-coil protein without obvious orthologs outside of insects. It functions as a negative regulator of Drosophila class I phosphatidylinositol-3 kinase Pi3K92E/Dp110 by binding to its Pi3K21B/dP60 regulatory subunit. Homozygous B4 mutants have an increased body size [65], which may explain why Ras2-Gal4-driven RNAi-mediated suppression of B4, uncovered by suppressing deficiency Df(2L)BSC147, increased dNf1 pupal size (not shown). However, whether B4 is the responsible dominant modifier is doubtful, given that it is also uncovered by Df(2L)BSC692, a non-modifying deficiency. Moreover, we previously found that heterozygous loss of Pi3K21B, or neuronal expression of a dominant negative Pi3K92E transgene, did not modify dNf1 pupal size [5]. Beyond B4, dNf1 size modifying deficiencies uncovered no genes involved in the canonical growth regulating pathways mediated by insulin and ecdysone. Indeed, several such genes were uncovered by non-modifying deficiencies. Among these genes, fat body expressed insulin-like growth factor Ilp6, which regulates larval growth in the post-feeding phase [66], [67], is uncovered by two non-modifying deficiencies. A single non-modifying deficiency, Df(2L)BSC206, uncovers both the chico and pten genes, whose products antagonistically control insulin-stimulated Pi3K92E/Dp110 activity, leading to changes in body, organ, and cell size [68], [69]. Among subunits of the cell growth regulating mTORC1 complex, raptor is uncovered by three and Tor by one non-modifying deficiency. Among genes implicated in ecdysone signaling, the ecdysone co-receptor ultraspiracle and the ecdysone-induced growth regulating DHR4 nuclear receptor [70] are each uncovered by non-modifying deficiencies, and two such deficiencies uncover Ptth, coding for prothoracicotropic hormone, which provides developmental timing cues by stimulating the production of ecdysone [71], [72]. These results reinforce our conclusion that the canonical growth regulating pathways involving insulin and ecdysone play no obvious roles in dNf1 growth control.
Manipulating cAMP/PKA Signaling in the Ring Gland Affects dNf1 Systemic Growth Non-Cell-Autonomously
Several results argue that defects in Ras/ERK and cAMP/PKA signaling responsible for the dNf1 growth defect involve non-overlapping cell populations. Firstly, heat shock-induced hsp70-PKA*, or Ras2-Gal4 induced attenuated UAS-PKA* transgene (see below) expression rescued the dNf1 pupal size defect, but failed to reduce the elevated larval brain phospho-ERK level (Figure 8A). Moreover, several neuronal RNAi drivers that increase dNf1 pupal size when driving UAS-dNf1 [5], failed to modify this phenotype when driving dncRNAi transgenes, even in the presence of the UAS-Dcr-2 RNAi enhancer (Table 3). This prompted us to investigate whether genetic manipulation of cAMP/PKA signaling in cells other than dNf1 requiring neurons was more effective.


To manipulate cAMP/PKA signaling tissue-specifically we used three UAS-dncRNAi transgenes. We also generated a series of attenuated UAS-PKA* transgenes using vectors with modified Gal4-inducible promoters harboring just 2, 3 or 4 Gal4-binding UAS elements (Figure 8B and C). We made the latter transgenes because a UAS-PKA* expression using the five UAS element containing standard UAS-T vector is lethal in combination with most Gal4 drivers [73]. As reported previously [74], driving UAS-dNf1 ubiquitously with Act5C-Gal4, or broadly in neurons with elav-Gal4, Ras2-Gal4, c23-Gal4, or 386Y-Gal4 restored dNf1 pupal size, whereas driving the same transgene with more restricted neuronal or non-neuronal drivers had no effect (Figure 8D and Table 3). By contrast, driving the expression of UAS-dncRNAi or attenuated UAS-PKA* transgenes with the same set of broadly expressed neuronal drivers was ineffective (Tables 3 and S5). We note that expression of the 2×UAS-PKA* and 3×-UAS-PKA* transgenes was generally well tolerated, whereas the 4×UAS-PKA* and the 5×UAS-PKA* transgenes exhibited increasing levels of lethality (Tables 3 and S5). Arguing that rescue of the dNf1 growth defect by manipulating cAMP/PKA signaling or dNf1 expression involves different cells, strong pupal size rescue was observed by increasing cAMP/PKA signaling in adipokinetic hormone-producing cells at the base of the neuroendocrine ring gland using the Akh-Gal4 driver (Figure 8D). Rescue was also observed with the Feb36-Gal4 and Aug21-Gal4 ring gland drivers (Figure 8D), which give rise to expression in the corpora allata, the source of juvenile hormone, but not with the P0206-Gal4 or Mai60-Gal4 drivers, which express predominantly in the prothoracic gland (Table 3). The tissue specificity of all Gal4 drivers used in this and other experiments was verified by microscopic observation of dissected UAS-GFP expressing larvae (Table S4 and Figures 8E–H and S5).
dAlk, Jeb, Cnk and CCKLR-17D1 Suppress a dNf1 NMJ Architectural Defect
During larval development, significant expansion of the NMJ arbor must occur, reflecting the steady muscle growth that takes place during larval life. As the NMJ grows, additional branches and boutons are added to the initial synaptic arbor that forms during late embryonic stages upon motor axon contact with its target muscle. As a result, at the wandering third instar stage, wild-type NMJs contain a highly stereotyped, segment specific number of synaptic boutons [75]. Recently, it was reported that dNf1 functions presynaptically to constrain NMJ synaptic growth and neurotransmission [16]. In dNf1 null mutant wandering third instar larvae, while the distribution of major presynaptic proteins is unaffected, increased overall size and synaptic bouton number is apparent at multiple NMJs, supporting a specific role for dNf1 in restricting NMJ expansion [16]. Several dNf1 suppressors that emerged in the current screen have also been linked to synapse morphogenesis, including CCKLR-17D1, which functions as a promoter of NMJ growth [36]. As our screen identified CCKLR-17D1 as a dominant dNf1 size defect suppressor, we wanted to confirm the dNf1 NMJ phenotype and test whether CCKLR-17D1 and other suppressors affected this defect.
By quantifying bouton number at the NMJ on muscles 6 and 7, we confirmed that dNf1 mutants have a significant increase in mean bouton number (Figure 9A and B). In addition, this analysis confirmed previously published phenotypes for dAlk, jeb and CCKLR-17D1 [36], [76]. Importantly, the dNf1 synaptic overgrowth phenotype is dominantly suppressed by CCKLR-17D1, dAlk, jeb, and cnk alleles (Figure 9B), arguing that all four genes are epistatic to dNf1. As a control we analyzed an allele of spitz (spi), which encodes an EGF-like growth factor and is uncovered by suppressing Df(2L)Exel8041. However, spi shows no genetic interaction with dNf1, as loss of spi modified neither the pupal size nor the NMJ overgrowth phenotypes (Figure 9B and data not shown).

Human ALK Is Expressed in Schwann Cells and May Serve as a Therapeutic Target in NF1
The identification of dAlk as a suppressor of all hitherto analyzed dNf1 defects prompted us to explore whether human ALK represents a therapeutic target in NF1. Given our hypothesis that NF1 negatively regulates ALK stimulated Ras/ERK signaling, in order to play such a role, ALK and NF1 must be co-expressed in cells that give rise to symptoms. We previously found that dNf1 and dAlk expression overlaps extensively in Drosophila larval and adult CNS [15], and the expression of orthologs of both genes also overlaps in the murine CNS [77], [78]. While overlapping CNS expression is compatible with a role for ALK in NF1-associated cognitive dysfunction, a causative role in another hallmark NF1 symptom, peripheral nerve-associated tumors, is less obvious. Among the near universal symptoms on NF1, benign neurofibromas consist of Schwann cells, perineurial fibroblasts, infiltrating mast cells, and nerve elements, with the Schwann cells sustaining the second NF1 hit [79]. To test whether increased ALK signaling in the absence of NF1 might play a role in the development of neurofibromas, we used reverse transcription/PCR to detect the presence or absence of ALK mRNA in neurofibroma-derived NF1−/− Schwann cells and NF1+/− fibroblasts, using RNAs kindly provided by Drs. Eric Legius and Eline Beert. In these experiments, two different primer sets readily detected ALK mRNA in NF1−/− Schwann cells, but not in NF1+/− fibroblasts derived from the same tumors (Figure S6).
To test whether functional interactions between NF1 and ALK exist in human cells, we used the SK-SY5Y and Kelly neuroblastoma cells, both of which harbor constitutively active F1174L ALK alleles, and both of which are highly sensitive to pharmacological ALK inhibition [80]. Compatible with a role for NF1 as a negative regulator of mitogenic ALK/RAS signals, qRT-PCR verified NF1 knockdown with two shRNA retroviral vectors increased the resistance of both lines to ALK inhibitors NVP-TAE684 and Crizotinib (Figures 10A, 10C and S7). Compatible with a model in which NF1 negatively regulates ALK/RAS signaling, NF1 knockdown resulted in elevated ERK and AKT activation (Figures 10B). Moreover, expression of activated KRAS, BRAF, or MEK transgenes, but not of other Ras effector transgenes, in SH-SY5Y cells conferred similar resistance to ALK inhibition (Figure S8).

Discussion
The work reported here was motivated by the fact that human NF1 is a characteristically variable disease, the severity of which is controlled at least in part by symptom-specific modifier genes [81]. Thus, a genetic analysis in Drosophila might not only reveal molecular pathways controlled by the highly conserved (50% identical) dNf1 protein, but also provide clues to the identity of human modifiers, which by virtue of their rate-limiting roles in symptom development might serve as therapeutic targets. The current work was also motivated by the fact that, for reasons that remain poorly understood, most dNf1 null mutant phenotypes are rescued by increasing, or phenocopied by decreasing, cAMP/PKA signaling. The identification of genetic modifiers of a cAMP/PKA sensitive defect might reveal how loss of dNf1 affects cAMP/PKA signaling, and help to resolve the long-standing controversy as to whether dNf1 affects cAMP/PKA signaling directly, independent of its role as a Ras regulator [10], [27], or indirectly, secondary to a Ras signaling defect [5], [15].
While recognizing that none of the thus far identified dNf1 phenotypes are ideally suited for use in modifier screens, we selected the pupal size defect as the phenotype to analyze in our screen for three main reasons. First, pupariation occurs at the end of the larval growth period, and pupal size is readily assessed by inspecting pupae attached to the side of culture vials, making this phenotype amenable to a large-scale screen. Second, the growth defect is among several cAMP/PKA sensitive dNf1 phenotypes. Finally, reduced growth is also a symptom of human NF1 and other RASopathies [1], [82]. However, while compelling reasons support the selection of this phenotype, confounding factors include that Drosophila size is a sexually dimorphic phenotype affected by population density, feeding, environmental conditions such as temperature, and genetic background differences. Moreover, while heterozygous dNf1 mutants are marginally smaller than wild-type pupae [5], the more robust size phenotype (∼15% reduction in linear dimensions, ∼25% reduction in weight) used in our screen is only observed upon homozygous loss of dNf1. Thus, our screen was not designed to find modifiers that act on the dNf1 protein itself, like the recently identified SPRED proteins [83]. Finally, organism size is a function of growth rate and duration, both of which are regulated by hormonal cascades that involve cross-talk between the larval brain, the neuroendocrine ring gland, the fat body and other tissues [19], [84]. Thus, a screen for modifiers of dNf1-regulated growth may uncover genes involved in various aspects of systemic growth control.
Early attempts to identify dNf1 pupal size modifiers were abandoned when >95% of large X-ray induced 2nd chromosome deficiencies were found to be lethal in a dNf1 background (Glenn Cowley, Iswar Hariharan and A.B., unpublished), or when a pilot chemical mutagenesis screen found the reliable mapping of identified enhancer or suppressor mutations to be impracticable (Suzanne Brill, Iswar Hariharan and A.B., unpublished). Both aborted screens informed the current effort, which used precisely defined small deficiencies, isogenic crossing schemes and experimental protocols that guarded against population density differences. In total we analyzed 486 1st and 2nd chromosome deficiencies that together uncover well over 80% of chromosome 1, 2L and 2R genes (Table 1). Among the screened deficiencies, 132 (27.2%) significantly modified dNf1 pupal size (p<0.01; two-tailed Student's t-test). While this is a large number, 20 deficiencies were subsequently eliminated because they also affect wild-type size. Several modifying deficiencies also uncover overlapping genomic segments, further reducing the number of dNf1 modifying loci to 76. During follow-up studies aimed at identifying responsible genes, we prioritized genes uncovered by suppressing deficiencies over those uncovered by enhancing ones, modifiers uncovered by overlapping deficiencies over those uncovered by single deletions, modifiers uncovered by small deficiencies over those uncovered by larger ones and stronger modifiers over weaker ones. We also limited ourselves to genes that function in the nervous system, based on the consideration that dNf1 re-expression in larval neurons is sufficient to suppress the growth defect [5].
We previously reported that dNf1 growth and learning defects are phenocopied by increasing neuronal Jeb/dAlk/ERK signaling, and suppressed by genetic or pharmacological attenuation of this pathway [15]. Validating our screen, deficiencies that uncover jeb and dAlk were identified as dominant dNf1 size defect suppressors. Others recently reported that Jeb/dAlk signaling allows brain growth to be spared at the expense of other tissues in nutrient restricted Drosophila and identified a glial cell niche around neuroblasts as the source of Jeb under these conditions [41]. However, Jeb involved in systemic growth appears of mainly neuronal origin, as RNAi-mediated jeb knockdown in neurons increased dNf1 pupal size, whereas only one of four tested glial drivers produced partial rescue (Figure 5A).
The identification of cAMP/PKA pathway modifiers dnc, PKA-C1 and tentatively PKA-R2 further validates our screen. Arguing that increased PKA activity doesn't suppress dNf1 defects by attenuating Ras/Raf/MEK/ERK signaling, hsp70-PKA* transgene expression, using a daily heat shock regimen that suppresses the dNf1 size defect [4], does not reduce the elevated dNf1 larval brain phospho-ERK level, and neither does Ras2-Gal4 driven neuronal UAS-PKA* expression (Figure 8D). Providing further mechanistic clues, our results demonstrate that dNf1 and cAMP/PKA both affect systemic growth non-cell-autonomously, but not necessarily in the same cells. Thus, we previously showed that only relatively broadly expressed neuronal Gal4 drivers restored mutant growth when driving UAS-dNf1, whereas multiple drivers expressed in specific subsets of neurons, including several expressed in the ring gland, lacked the ability to restore dNf1 growth [5]. By contrast, using UAS-dncRNAi or a series of newly generated attenuated UAS-PKA* transgenes that avoid the toxicity associated with high level PKA expression [73], we now show that manipulating cAMP/PKA signaling with broadly expressed neuronal Gal drivers does not affect the dNf1 size phenotype, whereas the same transgenes induced with three ring gland drivers did suppress. Intriguingly, the most potent rescue was observed when UAS-dncRNAi or attenuated UAS-PKA* transgenes were driven in AKH-producing cells at the base of the ring gland, whereas weaker rescue was also observed with two ring gland drivers that show overlapping expression in the juvenile hormone producing corpora allata. This suggests that the dNf1 growth deficiency involves a defect in processes controlled by one or both of these neuroendocrine hormones.
As might be expected of a screen that used systemic growth as a read-out, our work identified a diverse set of potential modifiers. Notably, however, among a non-exhaustive set of 18 1st or 2nd chromosome genes implicated in various aspects of Drosophila body, organ, and/or cell size control (dAlk, B4, chico, hpo, Hr4, Ilp6, jeb, Mer, mir-8, Pi3K21B, Pten, Ptth, SNF1A, sNPF, step, Tor, ush and yki; see Table S3 for details), only dAlk and jeb scored as dominant dNf1 pupal size modifiers, whereas the remaining 16 genes were uncovered by non-modifying deficiencies, or in the case of Ptth, by two deficiencies that altered developmental timing (Table S2). Further explaining this lack of overlap, the previously implicated PI3 kinase regulator B4 act in a recessive manner and several of the above listed genes function outside of the CNS. Our screen excluded such genes, because dNf1 controls growth non-cell-autonomously by regulating neuronal Ras [5]. As previously noted, a special case is provided by insulin pathway components chico and Pten, which affect growth antagonistically. Both genes map within 5 kb of each other on the 2nd chromosome and are uncovered by the same non-modifying deficiency.
Two newly identified dNf1 growth defect suppressors, Dap160 and CCKLR-17D1, affect synaptic architecture or functioning [36], [56], [57]. Because dNf1 was recently reported to function downstream of focal adhesion kinase to restrain NMJ synaptic growth and neurotransmission [16], and because the cholecystokinin receptor related CCKLR-17D1 drosulfakinin receptor stimulates NMJ growth [36], we analyzed whether this and three Ras signaling related dNf1 size defect suppressors also affected NMJ architecture. Our results confirm that dNf1 mutants exhibit synaptic overgrowth, and show that loss of CCKLR-17D1 suppresses this defect. Importantly, loss of jeb, dAlk, or cnk similarly suppresses both size and synaptic overgrowth defects, suggesting that both phenotypes may be related.
The results presented here further support our previous conclusion that excess neuronal Jeb/dAlk/Ras/MEK/ERK signaling is the root cause of the cAMP/PKA sensitive dNf1 systemic growth defect. What happens downstream of this primary defect remains less clear, although our demonstration that increasing cAMP/PKA signaling in AKH-producing cells and other parts of the neuroendocrine ring gland suppresses the size defect provides an important new clue, not only about pathways involved in the dNf1 growth defect, but also about the likely non-cell-autonomous cause of similar growth defects of PKA-C1 or dCreb2 mutants [85], [86]. Other questions that remain to be fully answered concern the role of the NMJ architectural defect in the dNf1 growth deficiency and the role of Jeb/dAlk signaling in the NMJ defect. We note in this respect that that C. elegans ALK ortholog, T10H9.2, has been implicated in synapse formation [87], and that recent work suggests a role for trans-synaptic Jeb/dAlk signaling in the control of neurotransmission and synaptic morphology [88]. However, while the dNf1 growth defect is due to excess dAlk signaling in neurons, NMJ synapse formation has been suggested to involve the release of presynaptic Jeb activating postsynaptic dAlk [88]. Further work will have to establish whether the suppression of the dNf1 NMJ overgrowth phenotype by jeb, dAlk and cnk involves cell autonomous roles for these genes at synapses, or non-cell-autonomous functions elsewhere in the CNS. Further work is also required to reveal the functional significance and the sites of action of other novel modifiers identified in our screen.
From a clinical perspective, perhaps the most relevant questions raised by our work are whether NF1 regulated ALK/RAS/ERK signaling is evolutionarily conserved and whether excessive ALK/RAS/ERK signaling contributes to human NF1 symptoms. Much indirect evidence hints at a positive answer to both questions. First, the expression of ALK and NF1 largely overlaps in the murine nervous system [77], [78], same as it does in Drosophila [15]. Second, ALK functions as an oncogene and NF1 as a tumor suppressor in neuroblastoma [89]–[94]. Third, midkine, a ligand that activates mammalian ALK [95], is produced by NF1−/− Schwann cells, present at elevated levels in NF1 patient skin and serum, and acts as a mitogen for NF1 tumor cell lines [96]–[98]. We add to this evidence by showing that shRNA-mediated NF1 knockdown renders two oncogenic ALK-driven human neuroblastoma cell lines resistant to pharmacological ALK inhibition, and by confirming that ALK mRNA is expressed in neurofibroma-derived NF1−/− human Schwann cells. These findings make a strong case that ALK should be explored as a therapeutic target in NF1, and that loss of NF1 expression should be considered as a potential mechanism in cases of acquired resistance to ALK inhibition [99].
Materials and Methods
Fly Stocks and Experiments
The dNf1E1 and dNf1E2 alleles have been described [5]. Exelixis, DrosDel and BSC deficiencies were obtained from the Bloomington Stock Center. Transgenic RNAi lines were obtained from the Vienna Drosophila Research Center (VDRC) and the TRiP Collection at Harvard Medical School. Eaat1SM1 and Eaat1SM2 were provided by D. van Meyel, dALK8 and jebweli by R. Palmer, cnkXE-385 and cnkE-2083 by M. Therrien, and carΔ146 by H. Kramer, ppl06913 by M. Pankratz, hs-Ilp2 transgenic line by E. Rulifson and UAS-Rab9 DN by R. Hiesinger. Flies were maintained on agar-oatmeal-molasses medium at 25°C, unless otherwise indicated.
To assess feeding, larvae at various stages of development were placed on blue food dye-stained yeast paste, removed after 20 min, washed and photographed. To analyze wandering behavior, 100 larvae (age 40–44 hr after egg deposition (AED)) were placed on an agar plate with a central blob of yeast paste, and their position after 24 hr was documented. To assess the expression of starvation-sensitive genes, larvae at 72 h AED were placed in vials with water for 16 hr, after which RNA was prepared and subjected to blot analysis. To determine developmental timing, L1 larvae were collected 24 hr AED using a 2 hr egg collection and reared at 140 animals per vial. The number of larvae that pupariated was scored at hourly intervals. To determine the larval weight, L1 larvae were collected 24 hr AED using a 2 hr egg collection. Larvae were reared at 140 larvae per vial and groups of 10 larvae were weighed at 8 hr intervals. Longevity was assessed by maintaining adult flies under standard conditions and counting the number of dead flies at regular intervals. In each of these assays, genotypes were tested in duplicate. To induce hs-Ilp2 transgene expression, culture vials were placed in a circulating water bath at 37°C for 10 min once or twice a day with an 8 hr interval.
Insulin-Like Protein mRNA Quantification
The 7500 Fast Real-Time PCR System from Applied Biosystems was used to determine Ilp mRNA levels in RNA prepared from dissected larval brains or from whole wandering stage 3rd instar larvae. Results were normalized to RpL32. The following primers were used: IIp2-Forward, GGCCAGCTCCACAGTGAAGT,Ilp2-Reverse, TCGCTGTCGGCACCGGGCAT, Ilp3-Forward, CCAGGCCACCATGAAGTTGT. Ilp3-Reverse, TTGAAGTTCACGGGGTCCAA, Ilp5-Forward, TCCGCCCAGGCCGCAAACTC, Ilp5-Reverse, TAATCGAATAGGCCCAAGGT, Ilp6-Forward, CGATGTATTTCCCAACAGTTTCG, Ilp6-Reverse, AAATCGGTTACGTTCTGCAAGTC, Ilp7-Forward, CAAAAAGAGGACGGGCAATG, Ilp7-Reverse, GCCATCAGGTTCCGTGGTT. Expression of the distantly related Ilp8 and the midgut-expressed Ilp4 genes [21] was not analyzed.
Genetic Screening, Validation, and Statistical Analysis
The crossing schemes in Figure 2 were used to generate dNf1E2 mutants carrying 1st and 2nd chromosome deficiencies. To avoid crowding, cultures were maintained at 100–200 pupae per culture vial. Initial scoring used calipers set at the length of dNf1 female pupae, ignoring dNf1 heterozygotes recognizable by the presence of the TM6B balancer. Next, the length of individual pupae carrying candidate modifying deficiencies was measured by determining their head-to-tail length using a microscope fitted with NIS-Elements AR 3.0 imaging software. Measured pupae were then placed in 96-well plates (Falcon) to determine their gender and, if necessary, the genotype of eclosed flies. At least 40 pupae were measured for each genotype, and only measurements of female pupae were used to calculate mean values and standard deviations. Statistical significance was assessed with a two-tailed Student's t-test. Throughout this report, single or double asterisks denote p-values<0.05 or <0.01 respectively.
To identify responsible modifiers we used specific alleles or UAS-RNAi knockdown. Alleles and UAS-RNAi lines on the 1st and 2nd chromosomes were crossed into the dNf1E2 background. UAS-RNAi lines on the 3rd chromosome were recombined with dNf1E2. UAS-RNAi lines in the dNf1E2 background were crossed to Gal4 drivers in the same background. The few deficiencies that gave rise to synthetic lethal interactions were backcrossed with dNf1E1 flies to produce Df/+; dNf1E2/dNf1E1 progeny.
To test whether genetic suppression reflected the inadvertent introduction of a wild-type dNf1 allele, we used fly DNA prepared using DNAzol (Molecular Research Inc.) in a PCR assay with AGTCACATTAATTGATCCTG and GAGATCGTTGATAAAGAAGT primers. The second primer introduces a penultimate single nucleotide change, which together with the E2 mutation results in the introduction of an RsaI restriction site. RsaI digestion of the PCR product gives rise to 370 and 61 bp fragments for the wild-type allele, and 348, 61 and 22 bp fragments for the dNf1E2 allele. Digests were run on 8% acrylamide gels using both wild-type (w1118) and dNf1E2 controls.
Construction of Akh-Gal4 and Attenuated UAS-PKA* Transgenes
The Akh promoter region was amplified with Akh-FORWARD (AGATCTAATCTCCTGAATGCCGCAGCG) and Akh-REVERSE (AGATCTATGCTGGTCCACTTCGATTC) primers. The resulting PCR fragment was subcloned into the BamHI site of a GAL4 coding region containing pCaSpeR derivative. The final construct was sequenced to ensure correct orientation of the Akh promoter before being used generate transgenic flies by standard protocols.
To reduce the toxicity associated with high-level PKA expression, we generated modified pUAS-T vectors containing 1, 2, 3 or 4, rather than 5 Gal4-binding sites. The primers used to generate these vectors were: 1×UAS-FOR: AACTGCAGAGCGGAGTACTGTCCTCCGAGCGGAGACTCTAG; 2×UAS-FOR: AACTGCAGCGGAGTACTGTCCTCCGAGCGGAGTACTGTCCTCCG; 3×UAS-FOR: AACTGCAGCGGAGTACTGTCCTCCGAGCGGAGTACTGTCCTCCGAGCGGAGTACTGTCCTCCG, and UAS-REV: CTAGAGGTACCCTCGAGCGCGGCCGCAAGAT. An initial PCR was performed using the 1×UAS-FOR and UAS-REV primers with the standard pUAS-T vector as a template. The resulting amplified fragment was TA subcloned into pCR2.1 to make pCR2.1-1×UAS. The 2×UAS-FOR and UAS-REV primers were then used with pCR2.1-UAS(1×) as a template to generate a UAS(2×) clone, which was subcloned to produce pCR2.1-UAS(2×). Similarly, 3×UAS-FOR and UAS-REV primers in a PCR reaction with pCR2.1-UAS(2×) as template generated pCR2.1-UAS(3×) and pCR2.1-UAS(4×)). The pCR2.1-UAS clones were sequenced, their inserts excised with PstI and subcloned into PstI-digested p-UAST. Correct insert orientation was verified by sequence analysis, after which the mutationally activated murine PKA* coding region [100] was subcloned into the modified vectors using XbaI and NotI.
Immunofluorescence and Analysis of NMJ Morphology
Wandering third instar larvae were dissected in Ca2+-free saline and fixed in 4% paraformaldehyde for 25 min at room temperature. Following fixation, larval pelts were washed three times in phosphate-buffered saline (PBS) and then blocked for one hour in PBT (PBS+0.1% Triton-X 100)+5% normal goat serum. Larvae were incubated in primary antibody solution for three hours at room temperature. Anti-HRP 568 (1∶1000, Invitrogen) was used to visualize neurons and Alexa Fluor 488 phalloidin (1∶500, Invitrogen) was used to visualize F-actin in the musculature. Images were collected using a Yokogawa CSU-X1 spinning-disk confocal microscope with the Spectral Applied Research (Richmond Hill, ON, Canada) Borealis modification on a Nikon (Melville, NY) Ti-E inverted microscope using a 60× Plan Apo (1.4 NA) objective. The microscope was equipped with a Prior (Rockland, MA) Proscan II motorized stage. Larval samples were excited with 488-nm (for phalloidin) and 561-nm (for HRP) 100-mW solid-state lasers from a Spectral Applied Research LMM-5 laser merge module and was selected and controlled with an acousto-optical tunable filter. Emission was collected with a Semrock (Rochester, NY) quad pass (405/491/561/642 nm) dichroic mirror and 525/50 nm (for phalloidin) and 620/60 nm (for HRP) Chroma (Bellows Falls, VT) emission filters. Images were acquired using a Hamamatsu ORCA-ER-cooled CCD camera. Hardware was controlled with MetaMorph (version 7.7.9) software (Molecular Devices, Sunnyvale, CA.). Five individual animals were imaged for subsequent morphological analysis. Motor nerve terminals of muscles 6 and 7 were imaged in abdominal segments A2 and A3 and Z-stacks (0.25 µM between images) and were captured from the top to bottom of each NMJ. Morphological analysis of the NMJ was performed using NIH Image J and was assessed by quantifying the number of synaptic boutons per square micron. The number of synaptic boutons was counted as previously described [16], [101] and muscle area covered by the NMJ was quantified by tracing a polygon connecting each terminal branch point [102].
Human NF1 Experiments
The retroviral RNAi vectors targeting human NF1 and expression constructs of active alleles of RAS effectors were as described previously [94]. Crizotinib (S1068) and NVP-TAE648 (S1108) were purchased from Selleck Chemicals. Antibody against NF1 was from Bethyl Laboratories (A300-140A); antibodies against pAKT(S473) and ATK1/2 were from Cell Signalling; antibodies against p-ERK (E-4), ERK1 (C-16), ERK2 (C-14) and CDK4 (C-22) were from Santa Cruz Biotechnology; A mixture of ERK1 and ERK2 antibodies was used for detection of total ERK from human cell lines. Antibody against mouse PKAα-cat (A-2) SC-28315 was from Santa Cruz Biotechnology, β-Tubulin E7 from Developmental Studies Hybridoma Bank.
SH-SY5Y, Kelly and Phoenix cells were cultured in DMEM with 8% heat-inactivated fetal bovine serum, penicillin and streptomycin at 5% CO2. Subclones of each cell line expressing the murine ecotropic receptor were generated and used for all experiments shown. Phoenix cells were used to produce retroviral supernatants as described at http://www.stanford.edu/group/nolan/retroviral_systems/phx.html.
To measure cell proliferation, single cell suspensions were seeded into 6-well plates (1–2×104 cells/well) and cultured both in the absence and presence of ALK inhibitors. At the indicated endpoints, cells were fixed, stained with crystal violet and photographed. All knockdown and overexpression experiments were done by retroviral infection as described previously [103].
The 7500 Fast Real-Time PCR System from Applied Biosystems was used to determine mRNA levels. NF1 mRNA expression levels were normalized to expression of GAPDH. The following primers sequences were used in the SYBR Green master mix (Roche): GAPDH-Forward, AAGGTGAAGGTCGGAGTCAA; GAPDH-Reverse, AATGAAGGGGTCATTGATGG; NF1-Forward, TGTCAGTGCATAACCTCTTGC; NF1-Reverse, AGTGCCATCACTCTTTTCTGAAG. ALK mRNA levels in neurofibroma-derived NF1−/− Schwann cells and NF1+/− fibroblasts were analyzed using the following two primer sets: ALK-N-Forward, GGAGTGCAGCTTTGACTTCC; ALK-N-Reverse, TGGAGTCAGCTGAGGTGTTG; ALK-C-Forward, GCAACATCAGCCTGAAGACA; ALK-C-Reverse, GCCTGTTGAGAGACCAGGAG.
Supporting Information
Zdroje
1. ZenkerM (2011) Clinical manifestations of mutations in RAS and related intracellular signal transduction factors. Current opinion in pediatrics 23 : 443–451.
2. EvansDG, HowardE, GiblinC, ClancyT, SpencerH, et al. (2010) Birth incidence and prevalence of tumor-prone syndromes: estimates from a UK family genetic register service. American journal of medical genetics Part A 152A: 327–332.
3. AllansonJE (2007) Noonan syndrome. American journal of medical genetics Part C, Seminars in medical genetics 145C: 274–279.
4. TheI, HanniganGE, CowleyGS, ReginaldS, ZhongY, et al. (1997) Rescue of a Drosophila NF1 mutant phenotype by protein kinase A. Science 276 : 791–794.
5. WalkerJA, TchoudakovaAV, McKenneyPT, BrillS, WuD, et al. (2006) Reduced growth of Drosophila neurofibromatosis 1 mutants reflects a non-cell-autonomous requirement for GTPase-Activating Protein activity in larval neurons. Genes & development 20 : 3311–3323.
6. GuoHF, TheI, HannanF, BernardsA, ZhongY (1997) Requirement of Drosophila NF1 for activation of adenylyl cyclase by PACAP38-like neuropeptides. Science 276 : 795–798.
7. HymanSL, ShoresA, NorthKN (2005) The nature and frequency of cognitive deficits in children with neurofibromatosis type 1. Neurology 65 : 1037–1044.
8. SilvaAJ, FranklandPW, MarowitzZ, FriedmanE, LaszloGS, et al. (1997) A mouse model for the learning and memory deficits associated with neurofibromatosis type I. Nature genetics 15 : 281–284.
9. GuoHF, TongJ, HannanF, LuoL, ZhongY (2000) A neurofibromatosis-1-regulated pathway is required for learning in Drosophila. Nature 403 : 895–898.
10. HannanF, HoI, TongJJ, ZhuY, NurnbergP, et al. (2006) Effect of neurofibromatosis type I mutations on a novel pathway for adenylyl cyclase activation requiring neurofibromin and Ras. Human molecular genetics 15 : 1087–1098.
11. TongJ, HannanF, ZhuY, BernardsA, ZhongY (2002) Neurofibromin regulates G protein-stimulated adenylyl cyclase activity. Nature neuroscience 5 : 95–96.
12. BrownJA, GianinoSM, GutmannDH (2010) Defective cAMP generation underlies the sensitivity of CNS neurons to neurofibromatosis-1 heterozygosity. The Journal of neuroscience: the official journal of the Society for Neuroscience 30 : 5579–5589.
13. DasguptaB, DuganLL, GutmannDH (2003) The neurofibromatosis 1 gene product neurofibromin regulates pituitary adenylate cyclase-activating polypeptide-mediated signaling in astrocytes. The Journal of neuroscience: the official journal of the Society for Neuroscience 23 : 8949–8954.
14. HoIS, HannanF, GuoHF, HakkerI, ZhongY (2007) Distinct functional domains of neurofibromatosis type 1 regulate immediate versus long-term memory formation. The Journal of neuroscience: the official journal of the Society for Neuroscience 27 : 6852–6857.
15. GouziJY, MoressisA, WalkerJA, ApostolopoulouAA, PalmerRH, et al. (2011) The receptor tyrosine kinase Alk controls neurofibromin functions in Drosophila growth and learning. PLoS genetics 7: e1002281.
16. TsaiPI, WangM, KaoHH, ChengYJ, WalkerJA, et al. (2012) Neurofibromin Mediates FAK Signaling in Confining Synapse Growth at Drosophila Neuromuscular Junctions. The Journal of neuroscience: the official journal of the Society for Neuroscience 32 : 16971–16981.
17. Ashburner M, Thompson JN (1978) The laboratory culture of Drosophila. In: Ashburner M, Wright TRF, editors. The Genetics and Biology of Drosophila. London: Academic Press. pp. 1–109.
18. ZinkeI, KirchnerC, ChaoLC, TetzlaffMT, PankratzMJ (1999) Suppression of food intake and growth by amino acids in Drosophila: the role of pumpless, a fat body expressed gene with homology to vertebrate glycine cleavage system. Development 126 : 5275–5284.
19. MirthCK, ShingletonAW (2012) Integrating body and organ size in Drosophila: recent advances and outstanding problems. Frontiers in endocrinology 3 : 49.
20. AndersenDS, ColombaniJ, LeopoldP (2013) Coordination of organ growth: principles and outstanding questions from the world of insects. Trends in cell biology 23 (7) 336–44.
21. BrogioloW, StockerH, IkeyaT, RintelenF, FernandezR, et al. (2001) An evolutionarily conserved function of the Drosophila insulin receptor and insulin-like peptides in growth control. Current biology: CB 11 : 213–221.
22. IkeyaT, GalicM, BelawatP, NairzK, HafenE (2002) Nutrient-dependent expression of insulin-like peptides from neuroendocrine cells in the CNS contributes to growth regulation in Drosophila. Current biology: CB 12 : 1293–1300.
23. RulifsonEJ, KimSK, NusseR (2002) Ablation of insulin-producing neurons in flies: growth and diabetic phenotypes. Science 296 : 1118–1120.
24. HwangboDS, GershmanB, TuMP, PalmerM, TatarM (2004) Drosophila dFOXO controls lifespan and regulates insulin signalling in brain and fat body. Nature 429 : 562–566.
25. ClancyDJ, GemsD, HarshmanLG, OldhamS, StockerH, et al. (2001) Extension of life-span by loss of CHICO, a Drosophila insulin receptor substrate protein. Science 292 : 104–106.
26. TatarM, KopelmanA, EpsteinD, TuMP, YinCM, et al. (2001) A mutant Drosophila insulin receptor homolog that extends life-span and impairs neuroendocrine function. Science 292 : 107–110.
27. TongJJ, SchrinerSE, McClearyD, DayBJ, WallaceDC (2007) Life extension through neurofibromin mitochondrial regulation and antioxidant therapy for neurofibromatosis-1 in Drosophila melanogaster. Nature genetics 39 : 476–485.
28. Artavanis-TsakonasS (2004) Accessing the Exelixis collection. Nature genetics 36 : 207.
29. RyderE, AshburnerM, Bautista-LlacerR, DrummondJ, WebsterJ, et al. (2007) The DrosDel deletion collection: a Drosophila genomewide chromosomal deficiency resource. Genetics 177 : 615–629.
30. SchultzJ (1929) The Minute Reaction in the Development of DROSOPHILA MELANOGASTER. Genetics 14 : 366–419.
31. MarygoldSJ, RooteJ, ReuterG, LambertssonA, AshburnerM, et al. (2007) The ribosomal protein genes and Minute loci of Drosophila melanogaster. Genome biology 8: R216.
32. DietzlG, ChenD, SchnorrerF, SuKC, BarinovaY, et al. (2007) A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature 448 : 151–156.
33. GutierrezE, WigginsD, FieldingB, GouldAP (2007) Specialized hepatocyte-like cells regulate Drosophila lipid metabolism. Nature 445 : 275–280.
34. TennessenJM, ThummelCS (2011) Coordinating growth and maturation - insights from Drosophila. Current biology: CB 21: R750–757.
35. WilliamsJA, SuHS, BernardsA, FieldJ, SehgalA (2001) A circadian output in Drosophila mediated by neurofibromatosis-1 and Ras/MAPK. Science 293 : 2251–2256.
36. ChenX, GanetzkyB (2012) A neuropeptide signaling pathway regulates synaptic growth in Drosophila. The Journal of cell biology 196 : 529–543.
37. GorokhovaS, BibertS, GeeringK, HeintzN (2007) A novel family of transmembrane proteins interacting with beta subunits of the Na,K-ATPase. Human molecular genetics 16 : 2394–2410.
38. MillerMM, PopovaLB, MeleshkevitchEA, TranPV, BoudkoDY (2008) The invertebrate B(0) system transporter, D. melanogaster NAT1, has unique d-amino acid affinity and mediates gut and brain functions. Insect biochemistry and molecular biology 38 : 923–931.
39. KamimuraK, RhodesJM, UedaR, McNeelyM, ShuklaD, et al. (2004) Regulation of Notch signaling by Drosophila heparan sulfate 3-O sulfotransferase. The Journal of cell biology 166 : 1069–1079.
40. LorenCE, ScullyA, GrabbeC, EdeenPT, ThomasJ, et al. (2001) Identification and characterization of DAlk: a novel Drosophila melanogaster RTK which drives ERK activation in vivo. Genes to cells: devoted to molecular & cellular mechanisms 6 : 531–544.
41. ChengLY, BaileyAP, LeeversSJ, RaganTJ, DriscollPC, et al. (2011) Anaplastic lymphoma kinase spares organ growth during nutrient restriction in Drosophila. Cell 146 : 435–447.
42. StuteC, SchimmelpfengK, Renkawitz-PohlR, PalmerRH, HolzA (2004) Myoblast determination in the somatic and visceral mesoderm depends on Notch signalling as well as on milliways(mili(Alk)) as receptor for Jeb signalling. Development 131 : 743–754.
43. LuX, MelnickMB, HsuJC, PerrimonN (1994) Genetic and molecular analyses of mutations involved in Drosophila raf signal transduction. The EMBO journal 13 : 2592–2599.
44. SimonMA, BowtellDD, DodsonGS, LavertyTR, RubinGM (1991) Ras1 and a putative guanine nucleotide exchange factor perform crucial steps in signaling by the sevenless protein tyrosine kinase. Cell 67 : 701–716.
45. DicksonBJ, van der StratenA, DominguezM, HafenE (1996) Mutations Modulating Raf signaling in Drosophila eye development. Genetics 142 : 163–171.
46. KarimFD, ChangHC, TherrienM, WassarmanDA, LavertyT, et al. (1996) A screen for genes that function downstream of Ras1 during Drosophila eye development. Genetics 143 : 315–329.
47. MaixnerA, HeckerTP, PhanQN, WassarmanDA (1998) A screen for mutations that prevent lethality caused by expression of activated sevenless and Ras1 in the Drosophila embryo. Developmental genetics 23 : 347–361.
48. HuangAM, RubinGM (2000) A misexpression screen identifies genes that can modulate RAS1 pathway signaling in Drosophila melanogaster. Genetics 156 : 1219–1230.
49. RebayI, ChenF, HsiaoF, KolodziejPA, KuangBH, et al. (2000) A genetic screen for novel components of the Ras/Mitogen-activated protein kinase signaling pathway that interact with the yan gene of Drosophila identifies split ends, a new RNA recognition motif-containing protein. Genetics 154 : 695–712.
50. ZhuMY, WilsonR, LeptinM (2005) A screen for genes that influence fibroblast growth factor signal transduction in Drosophila. Genetics 170 : 767–777.
51. FriedmanA, PerrimonN (2006) A functional RNAi screen for regulators of receptor tyrosine kinase and ERK signalling. Nature 444 : 230–234.
52. FriedmanAA, TuckerG, SinghR, YanD, VinayagamA, et al. (2011) Proteomic and functional genomic landscape of receptor tyrosine kinase and ras to extracellular signal-regulated kinase signaling. Science signaling 4: rs10.
53. TherrienM, WongAM, RubinGM (1998) CNK, a RAF-binding multidomain protein required for RAS signaling. Cell 95 : 343–353.
54. DouziechM, RoyF, LabergeG, LefrancoisM, ArmengodAV, et al. (2003) Bimodal regulation of RAF by CNK in Drosophila. The EMBO journal 22 : 5068–5078.
55. RoosJ, KellyRB (1998) Dap160, a neural-specific Eps15 homology and multiple SH3 domain-containing protein that interacts with Drosophila dynamin. The Journal of biological chemistry 273 : 19108–19119.
56. KohTW, VerstrekenP, BellenHJ (2004) Dap160/intersectin acts as a stabilizing scaffold required for synaptic development and vesicle endocytosis. Neuron 43 : 193–205.
57. MarieB, SweeneyST, PoskanzerKE, RoosJ, KellyRB, et al. (2004) Dap160/intersectin scaffolds the periactive zone to achieve high-fidelity endocytosis and normal synaptic growth. Neuron 43 : 207–219.
58. ChabuC, DoeCQ (2008) Dap160/intersectin binds and activates aPKC to regulate cell polarity and cell cycle progression. Development 135 : 2739–2746.
59. OpocherG, ContonP, SchiaviF, MacinoB, ManteroF (2005) Pheochromocytoma in von Hippel-Lindau disease and neurofibromatosis type 1. Familial cancer 4 : 13–16.
60. ReadRD, GoodfellowPJ, MardisER, NovakN, ArmstrongJR, et al. (2005) A Drosophila model of multiple endocrine neoplasia type 2. Genetics 171 : 1057–1081.
61. ChanCC, ScogginS, WangD, CherryS, DemboT, et al. (2011) Systematic discovery of Rab GTPases with synaptic functions in Drosophila. Current biology: CB 21 : 1704–1715.
62. Bekker-JensenS, Rendtlew DanielsenJ, FuggerK, GromovaI, NerstedtA, et al. (2010) HERC2 coordinates ubiquitin-dependent assembly of DNA repair factors on damaged chromosomes. Nature cell biology 12 : 80–86 sup pp 81–12.
63. SevrioukovEA, HeJP, MoghrabiN, SunioA, KramerH (1999) A role for the deep orange and carnation eye color genes in lysosomal delivery in Drosophila. Molecular cell 4 : 479–486.
64. PulipparacharuvilS, AkbarMA, RayS, SevrioukovEA, HabermanAS, et al. (2005) Drosophila Vps16A is required for trafficking to lysosomes and biogenesis of pigment granules. Journal of cell science 118 : 3663–3673.
65. WittwerF, JaquenoudM, BrogioloW, ZarskeM, WustemannP, et al. (2005) Susi, a negative regulator of Drosophila PI3-kinase. Developmental cell 8 : 817–827.
66. OkamotoN, YamanakaN, YagiY, NishidaY, KataokaH, et al. (2009) A fat body-derived IGF-like peptide regulates postfeeding growth in Drosophila. Developmental cell 17 : 885–891.
67. SlaidinaM, DelanoueR, GronkeS, PartridgeL, LeopoldP (2009) A Drosophila insulin-like peptide promotes growth during nonfeeding states. Developmental cell 17 : 874–884.
68. BohniR, Riesgo-EscovarJ, OldhamS, BrogioloW, StockerH, et al. (1999) Autonomous control of cell and organ size by CHICO, a Drosophila homolog of vertebrate IRS1-4. Cell 97 : 865–875.
69. GoberdhanDC, ParicioN, GoodmanEC, MlodzikM, WilsonC (1999) Drosophila tumor suppressor PTEN controls cell size and number by antagonizing the Chico/PI3-kinase signaling pathway. Genes & development 13 : 3244–3258.
70. King-JonesK, CharlesJP, LamG, ThummelCS (2005) The ecdysone-induced DHR4 orphan nuclear receptor coordinates growth and maturation in Drosophila. Cell 121 : 773–784.
71. MirthC, TrumanJW, RiddifordLM (2005) The role of the prothoracic gland in determining critical weight for metamorphosis in Drosophila melanogaster. Current biology: CB 15 : 1796–1807.
72. McBrayerZ, OnoH, ShimellM, ParvyJP, BecksteadRB, et al. (2007) Prothoracicotropic hormone regulates developmental timing and body size in Drosophila. Developmental cell 13 : 857–871.
73. KigerJAJr, EklundJL, YoungerSH, O'KaneCJ (1999) Transgenic inhibitors identify two roles for protein kinase A in Drosophila development. Genetics 152 : 281–290.
74. WalkerJA, GouziJY, HuangS, MaherR, XiaH, et al. (2013) A Genetic Screen For Genes Involved In The Drosophila Neurofibromatosis-1 Growth Defect Implicates ALK And Other Potential Therapeutic Targets. PLoS genetics In Press.
75. KeshishianH, BroadieK, ChibaA, BateM (1996) The drosophila neuromuscular junction: a model system for studying synaptic development and function. Annual review of neuroscience 19 : 545–575.
76. RohrboughJ, BroadieK (2010) Anterograde Jelly belly ligand to Alk receptor signaling at developing synapses is regulated by Mind the gap. Development 137 : 3523–3533.
77. DastonMM, RatnerN (1992) Neurofibromin, a predominantly neuronal GTPase activating protein in the adult, is ubiquitously expressed during development. Developmental dynamics: an official publication of the American Association of Anatomists 195 : 216–226.
78. IwaharaT, FujimotoJ, WenD, CupplesR, BucayN, et al. (1997) Molecular characterization of ALK, a receptor tyrosine kinase expressed specifically in the nervous system. Oncogene 14 : 439–449.
79. SerraE, RosenbaumT, WinnerU, AledoR, ArsE, et al. (2000) Schwann cells harbor the somatic NF1 mutation in neurofibromas: evidence of two different Schwann cell subpopulations. Human molecular genetics 9 : 3055–3064.
80. McDermottU, IafrateAJ, GrayNS, ShiodaT, ClassonM, et al. (2008) Genomic alterations of anaplastic lymphoma kinase may sensitize tumors to anaplastic lymphoma kinase inhibitors. Cancer research 68 : 3389–3395.
81. EastonDF, PonderMA, HusonSM, PonderBA (1993) An analysis of variation in expression of neurofibromatosis (NF) type 1 (NF1): evidence for modifying genes. American journal of human genetics 53 : 305–313.
82. SoucyEA, Van OppenD, NejedlyNL, GaoF, GutmannDH, et al. (2012) Height Assessments in Children With Neurofibromatosis Type 1. Journal of child neurology 28 (3) 303–7.
83. StoweIB, MercadoEL, StoweTR, BellEL, Oses-PrietoJA, et al. (2012) A shared molecular mechanism underlies the human rasopathies Legius syndrome and Neurofibromatosis-1. Genes & development 26 : 1421–1426.
84. GrewalSS (2012) Controlling animal growth and body size - does fruit fly physiology point the way? F1000 biology reports 4 : 12.
85. LaneME, KalderonD (1993) Genetic investigation of cAMP-dependent protein kinase function in Drosophila development. Genes & development 7 : 1229–1243.
86. BelvinMP, ZhouH, YinJC (1999) The Drosophila dCREB2 gene affects the circadian clock. Neuron 22 : 777–787.
87. LiaoEH, HungW, AbramsB, ZhenM (2004) An SCF-like ubiquitin ligase complex that controls presynaptic differentiation. Nature 430 : 345–350.
88. RohrboughJ, KentKS, BroadieK, WeissJB (2012) Jelly belly trans-synaptic signaling to anaplastic lymphoma kinase regulates neurotransmission strength and synapse architecture. Developmental neurobiology 73 (3) 189–208.
89. TheI, MurthyAE, HanniganGE, JacobyLB, MenonAG, et al. (1993) Neurofibromatosis type 1 gene mutations in neuroblastoma. Nature genetics 3 : 62–66.
90. GeorgeRE, SandaT, HannaM, FrohlingS, LutherW2nd, et al. (2008) Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature 455 : 975–978.
91. ChenY, TakitaJ, ChoiYL, KatoM, OhiraM, et al. (2008) Oncogenic mutations of ALK kinase in neuroblastoma. Nature 455 : 971–974.
92. Janoueix-LeroseyI, LequinD, BrugieresL, RibeiroA, de PontualL, et al. (2008) Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature 455 : 967–970.
93. MosseYP, LaudenslagerM, LongoL, ColeKA, WoodA, et al. (2008) Identification of ALK as a major familial neuroblastoma predisposition gene. Nature 455 : 930–935.
94. HolzelM, HuangS, KosterJ, OraI, LakemanA, et al. (2010) NF1 is a tumor suppressor in neuroblastoma that determines retinoic acid response and disease outcome. Cell 142 : 218–229.
95. StoicaGE, KuoA, PowersC, BowdenET, SaleEB, et al. (2002) Midkine binds to anaplastic lymphoma kinase (ALK) and acts as a growth factor for different cell types. The Journal of biological chemistry 277 : 35990–35998.
96. MashourGA, WangHL, Cabal-ManzanoR, WellsteinA, MartuzaRL, et al. (1999) Aberrant cutaneous expression of the angiogenic factor midkine is associated with neurofibromatosis type-1. The Journal of investigative dermatology 113 : 398–402.
97. MashourGA, RatnerN, KhanGA, WangHL, MartuzaRL, et al. (2001) The angiogenic factor midkine is aberrantly expressed in NF1-deficient Schwann cells and is a mitogen for neurofibroma-derived cells. Oncogene 20 : 97–105.
98. MashourGA, DrieverPH, HartmannM, DrisselSN, ZhangT, et al. (2004) Circulating growth factor levels are associated with tumorigenesis in neurofibromatosis type 1. Clinical cancer research: an official journal of the American Association for Cancer Research 10 : 5677–5683.
99. KatayamaR, ShawAT, KhanTM, Mino-KenudsonM, SolomonBJ, et al. (2012) Mechanisms of acquired crizotinib resistance in ALK-rearranged lung Cancers. Science translational medicine 4 : 120ra117.
100. LiW, OhlmeyerJT, LaneME, KalderonD (1995) Function of protein kinase A in hedgehog signal transduction and Drosophila imaginal disc development. Cell 80 : 553–562.
101. JohnsonKG, TenneyAP, GhoseA, DuckworthAM, HigashiME, et al. (2006) The HSPGs Syndecan and Dallylike bind the receptor phosphatase LAR and exert distinct effects on synaptic development. Neuron 49 : 517–531.
102. LoyaCM, LuCS, Van VactorD, FulgaTA (2009) Transgenic microRNA inhibition with spatiotemporal specificity in intact organisms. Nature methods 6 : 897–903.
103. HuangS, LaoukiliJ, EppingMT, KosterJ, HolzelM, et al. (2009) ZNF423 is critically required for retinoic acid-induced differentiation and is a marker of neuroblastoma outcome. Cancer cell 15 : 328–340.
104. RewitzKF, YamanakaN, GilbertLI, O'ConnorMB (2009) The insect neuropeptide PTTH activates receptor tyrosine kinase torso to initiate metamorphosis. Science 326 : 1403–1405.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2013 Číslo 11
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- and Are Required for Growth under Iron-Limiting Conditions
- Genetic and Functional Studies Implicate Synaptic Overgrowth and Ring Gland cAMP/PKA Signaling Defects in the Neurofibromatosis-1 Growth Deficiency
- The Light Skin Allele of in South Asians and Europeans Shares Identity by Descent
- RNA∶DNA Hybrids Initiate Quasi-Palindrome-Associated Mutations in Highly Transcribed Yeast DNA
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy