
Prion Formation and Polyglutamine Aggregation Are Controlled by Two Classes of Genes
Prions are self-perpetuating aggregated proteins that are not limited to mammalian systems but also exist in lower eukaryotes including yeast. While much work has focused around chaperones involved in prion maintenance, including Hsp104, little is known about factors involved in the appearance of prions. De novo appearance of the [PSI+] prion, which is the aggregated form of the Sup35 protein, is dramatically enhanced by transient overexpression of SUP35 in the presence of the prion form of the Rnq1 protein, [PIN+]. When fused to GFP and overexpressed in [ps−] [PIN+] cells, Sup35 forms fluorescent rings, and cells with these rings bud off [PSI+] daughters. We investigated the effects of over 400 gene deletions on this de novo induction of [PSI+]. Two classes of gene deletions were identified. Class I deletions (bug1Δ, bem1Δ, arf1Δ, and hog1Δ) reduced the efficiency of [PSI+] induction, but formed rings normally. Class II deletions (las17Δ, vps5Δ, and sac6Δ) inhibited both [PSI+] induction and ring formation. Furthermore, class II deletions reduced, while class I deletions enhanced, toxicity associated with the expanded glutamine repeats of the huntingtin protein exon 1 that causes Huntington's disease. This suggests that prion formation and polyglutamine aggregation involve a multi-phase process that can be inhibited at different steps.
Published in the journal:
. PLoS Genet 7(5): e32767. doi:10.1371/journal.pgen.1001386
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1001386
Summary
Prions are self-perpetuating aggregated proteins that are not limited to mammalian systems but also exist in lower eukaryotes including yeast. While much work has focused around chaperones involved in prion maintenance, including Hsp104, little is known about factors involved in the appearance of prions. De novo appearance of the [PSI+] prion, which is the aggregated form of the Sup35 protein, is dramatically enhanced by transient overexpression of SUP35 in the presence of the prion form of the Rnq1 protein, [PIN+]. When fused to GFP and overexpressed in [ps−] [PIN+] cells, Sup35 forms fluorescent rings, and cells with these rings bud off [PSI+] daughters. We investigated the effects of over 400 gene deletions on this de novo induction of [PSI+]. Two classes of gene deletions were identified. Class I deletions (bug1Δ, bem1Δ, arf1Δ, and hog1Δ) reduced the efficiency of [PSI+] induction, but formed rings normally. Class II deletions (las17Δ, vps5Δ, and sac6Δ) inhibited both [PSI+] induction and ring formation. Furthermore, class II deletions reduced, while class I deletions enhanced, toxicity associated with the expanded glutamine repeats of the huntingtin protein exon 1 that causes Huntington's disease. This suggests that prion formation and polyglutamine aggregation involve a multi-phase process that can be inhibited at different steps.
Introduction
Prions are associated with transmissible spongiform encephalopathies, a family of neurological diseases that include Creutzfeldt-Jakob disease in humans, Scrapie in sheep, and the well-publicized “Mad Cow” disease. Transmission of prions occurs when a normally folded protein is converted to an alternate conformation that has the ability to further convert additional molecules of the normal protein into the misfolded infectious form.
Prions also exist in Saccharomyces cerevisiae. The well characterized cytoplasmically transferred elements [URE3], [PSI+], and [PIN+] (see reviews [1]–[4]), as well as several other recently characterized elements [5]–[9], have been identified as yeast prions.
Prions occur spontaneously in laboratory strains, although at very low frequency [10]–[12]. De novo appearance of prions can be facilitated by overexpression of either the whole prion protein or distinct regions that are required for propagation, called prion domains [13]–[19]. Most known yeast prion domains are glutamine (Q) and asparagine (N) rich [3], [5]–[9], [20]. In the case of de novo appearance of the prion form of the Sup35 translational termination factor, [PSI+], overexpression of Sup35 or its prion domain (Sup35PD) dramatically increases the appearance of [PSI+]. However, this increase requires either Q/N-rich domains that are simultaneously overexpressed [21], [22] or the presence of another Q/N rich prion, like the prion form of the Rnq1 protein, [PIN+] (also called [RNQ+]; [22]–[24]).
Similar to prion strains found in mammals (reviewed in [25]), yeast prions have also been shown to exist in different conformations called “variants” [14], [26]–[30]. The introduction of in vitro generated Sup35 amyloid fibers into yeast not only infects the cells with the prion, proof of the “protein-only” hypothesis, but also demonstrates that distinct forms of the in vitro made amyloid cause the appearance of distinct variants that are heritable [31]–[32].
To further understand how prions are formed and maintained, recent studies have focused on specific host factors that affect the propagation and appearance of yeast prions. Chaperones, which are normally involved in proper protein folding, play a role in prion maintenance and appearance. Hsp104 and Sis1 break prion aggregates into smaller pieces that efficiently segregate into daughter cells, a requirement for prion transmission [33]–[41]. Deletion of the N-terminal activation domain of Hsf1, a heat shock transcription factor, prevents [PSI+] formation, while deletion of the Hsf1 C-terminal region promotes [PSI+] appearance [42]. Furthermore, disruption of the non-essential human Hsp110 ortholog, SSE1, or overexpression of HSP82 and HSC82 that encode members of the Hsp90 family of chaperones, dramatically reduces, but does not eliminate, the induction of [PSI+] caused by the overexpression of Sup35PD [43].
Factors that affect [PSI+] induction are not limited to chaperones. Deletion of actin cytoskeletal genes, such as SLA1 or SLA2, reduces [PSI+] induction [44], suggesting that the actin cytoskeleton may play a role in prion appearance. Deletion of the ubiquitin-conjugating enzyme, Ubc4, enhances the de novo appearance of [PSI+] [45], and exposure to environmental stress can also alter the frequency of [PSI+] appearance [46].
Here, we identify deletions of several genes (bug1Δ, bem1Δ, arf1Δ, hog1Δ, las17Δ, vps5Δ, and sac6Δ) that reduce the efficiency with which overexpression of Sup35PD can induce the de novo appearance of [PSI+]. Deletion of LAS17, VPS5, or SAC6, which are associated with endocytosis and the actin cytoskeleton, not only inhibit [PSI+] induction, but also suppress the toxicity and aggregation associated with the expanded glutamine repeats of the huntingtin protein exon 1 that causes Huntington's disease.
Results
Deletions of BUG1, BEM1, ARF1, HOG1, LAS17, VPS5, and SAC6 show low or no induction of [PSI+] but maintain propagation of [PSI+] and [PIN+]
Our goal was to identify genes that influence the induction of the [PSI+] prion. Previous work approached this problem by making use of the observation that overexpression of Sup35PD-GFP in [PSI+] [PIN+] cells causes toxicity due to excessive sequestration of essential Sup35 into large aggregates [46], [47]. When Sup35PD-GFP is highly overexpressed in [psi−] [PIN+] cells, the frequent induction of [PSI+] results in an intermediate level of toxicity, because only cells that have switched to the [PSI+] state are sick [23], [46]. Therefore, genes whose deletions enhance or reduce the toxicity associated with overexpression seemed likely to increase or decrease [PSI+] induction frequency, respectively [46]. We tested 238 deletion strains that enhanced toxicity, 151 that reduced toxicity, and nine other strains studied in Tyedmers et al. [46] (Table S1; Tyedmers and Lindquist, unpublished) for their effects on [PSI+] induction.
First, to distinguish the inability to induce [PSI+] from the inability to propagate [PSI+] we tested if the 398 deletions could maintain weak and strong variants of [PSI+] in a propagating culture after cytoduction with [PSI+]. A plasmid encoding a copper inducible Sup35PD-GFP fusion was plasmiduced into the deletion strains simultaneously with the cytoduction of weak or strong [PSI+]. After over 50 generations of growth, we scored for maintenance of [PSI+] by overexpressing Sup35PD-GFP and examining cells for the presence of fluorescent aggregates. All deletion strains cytoduced with either weak or strong [PSI+] contained fluorescent aggregates, indicative of [PSI+], except for hsp104Δ, which had the characteristic diffuse fluorescence of [psi−] cells (data not shown). Additionally, we have previously shown that [PIN+] is maintained in all strains of the deletion library except rnq1Δ and hsp104Δ [48].
Next, we used a standard nonsense suppression assay for [PSI+] to screen the 398 yeast deletion library strains, in the BY4741 background, for their effects on induction of stable propagating [PSI+]. To do this, the [PIN+] prion, the Sup35PD-GFP plasmid, and a plasmid containing a ura3–14 nonsense allele to score for [PSI+] cells [49] were simultaneously cytoduced into the 398 deletion strains as described previously [48]. Following overexpression of Sup35PD-GFP to induce [PSI+], cells were plated on–Ura where suppression of the plasmid borne ura3–14 nonsense allele allowed [PSI+], but not [psi−], cells to grow.
Six novel deletion strains: bem1Δ, def1Δ, scp160Δ, rpp1aΔ, spt4Δ, and pre9Δ, as well as the expected rnq1Δ and hsp104Δ deletions, failed to grow on -Ura (Table 1). Previous work has shown that in the presence of the SUP35 R2E2 allele, which increases the appearance of [PSI+] without SUP35 overexpression, bem1Δ and pre9Δ decreased [PSI+] appearance [46]. In addition, 29 other deletions showed either low or extremely low induction of [PSI+] (Figure 1; Table 1). While strains carrying deletions of SPT4 or YML010C-B failed to express Sup35PD-GFP, all other deletion strains expressed Sup35PD-GFP at similar levels (data not shown).
![Scoring for [<i>PSI</i><sup>+</sup>] induction in deletion strains.](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/2b829341f4c7ca707974371088cf4838.png)
![Yeast deletion library strains that show no, very low, or low induction of [<i>PSI</i><em class="ref">+</em>].](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/9c27ddf79ebe480b374327ed8f2b2a28.png)
Deletion strains that decrease the efficiency of [PSI+] induction (Table 1) were from both enhanced and reduced toxicity groups. This makes sense if we consider that toxic side effects of the deletions could overlay the positive effect on growth rate due to the reduced [PSI+] induction. Furthermore, in some deletions, even the overexpression of YFP alone (without the Sup35 prion domain) causes strong toxicity that must be, therefore, completely unrelated to prion induction frequency [46].
To eliminate the effects of secondary mutations known to have accumulated in library strains [50], [51], multiple independent deletions of nineteen of the best candidates that showed no or reduced induction of [PSI+] (Table 1) were re-engineered in a wildtype 74-D694 [PIN+] strain. This 74-D694 genetic background contains a [PSI+] suppressible ade1–14 allele that provides the ability to directly score for [PSI+] by examining growth on -Ade. Of the 19 re-engineered deletions, only the six deletions (bre1Δ, bug1Δ, bem1Δ, arf1Δ, pre9Δ, and hog1Δ) that reproducibly reduced the frequency of [PSI+] induction, relative to the wildtype induction frequency of approximately 7.5 X 10−3 (Figure 2), were pursued further. Since a slow growth phenotype complicates the scoring for [PSI+], deletions that significantly inhibited growth in the 74D-694 background, like lst7Δ and swa2Δ (data not shown), were eliminated from further analysis.
![Nine deletions show reduced [<i>PSI</i><sup>+</sup>] appearance.](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/163f695473652e80c306321e76cc4c40.png)
In addition to the above deletions, we made a deletion of LAS17, which was previously shown to inhibit the aggregation of the polyglutamine 103Q repeat [52]. We also made deletions of VPS5 and SAC6, because they, like Las17 and other proteins shown to affect the appearance of [PSI+], are associated with endocytosis and the actin cytoskeleton. Even though prion propagation is unaffected in these three deletion strains because they were all able to maintain [PSI+] over many generations after cytoduction (data not shown), vps5Δ caused a significant decrease in prion induction, and las17Δ and sac6Δ strains completely failed to induce [PSI+] (Figure 2).
To eliminate deletions that reduced [PSI+] induction by altering the levels of Sup35, Sup35PD-GFP, or chaperones, we examined these levels in all strains. None of the deletions caused significant changes in Hsp104, Ssa1, Sis1, Sse1, or Ssb1/2 (data not shown). Since Sup35PD-GFP levels were reduced in bre1Δ strains and Sup35 endogenous levels were decreased in pre9Δ strains (Figure S1), these deletions were dropped from further study.
To eliminate the possibility that the reduced [PSI+] induction was due to the loss of [PIN+] during the construction of any of the deletion strains, we showed that the maintenance of [PIN+] was unaffected by the deletions. Each independently constructed deletion was crossed to a [pin-] strain carrying a plasmid with the CUP1 controlled RNQ1-GFP fusion. When grown on medium containing copper, induction of the resulting diploid containing the RNQ-GFP construct caused the appearance of punctate dots indicative of [PIN+] (data not shown). Furthermore, we showed that [PIN+] was maintained in deletion strains by the presence of [PIN+] characteristic SDS-resistant oligomeric species after 24 hours of Sup35PD-GFP overexpression (Figure S2). Differences in the migration of Rnq1 SDS-resistant oligomers have been shown to be associated with different [PIN+] variants [53]. We observed similar migration of Rnq1 oligomeric species in the deletion strains, suggesting that the [PIN+] prion variants were not altered during construction of the strain or by Sup35PD-GFP overexpression. These results and other properties of the deletions investigated further below are summarized in Table 2.
![Properties of deletions that inhibit <i>de novo</i> induction of [<i>PSI</i><sup>+</sup>].](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/003cb919cc465633d86a6f73eed50019.png)
Reduced [PSI+] formation is not always associated with reduced ring formation
Transient overexpression of Sup35PD-GFP in [psi−] [PIN+] cells leads to the appearance of cytoplasmic fluorescent rings and lines [54]. Daughter cells derived from micromanipulated cells that contain such rings or lines, where overexpression of Sup35PD-GFP is turned off but where endogenous SUP35 is tagged with GFP, always contained fluorescent punta indicative of [PSI+] [55]. In contrast, mother cells without ring or line aggregates always gave rise to daughter cells with diffuse fluorescence, indicative of [psi−] [44], [54]–[56]. Thus the appearance of rings/lines, while not necessarily a direct intermediate in the formation of [PSI+], is nonetheless a hallmark of the potential appearance of induced [PSI+]. No such rings have ever been observed during spontaneous appearance of [PSI+] in the absence of Sup35PD overexpression (unpublished).
To determine the deletions that inhibit the induction of [PSI+] and also prevent ring formation, we compared the number of cells that contained rings after 24 hours of expressing Sup35PD-GFP in the seven deletion strains. Ring containing cells in four of the deletions, bug1Δ, bem1Δ, arf1Δ, and hog1Δ, appeared at levels similar to wildtype (30%, Figure 3A). The rnq1Δ strains, which are [pin−], always displayed diffuse fluorescence (Figure 3A), as expected, since [PIN+] has been previously shown to be required for ring formation [54].
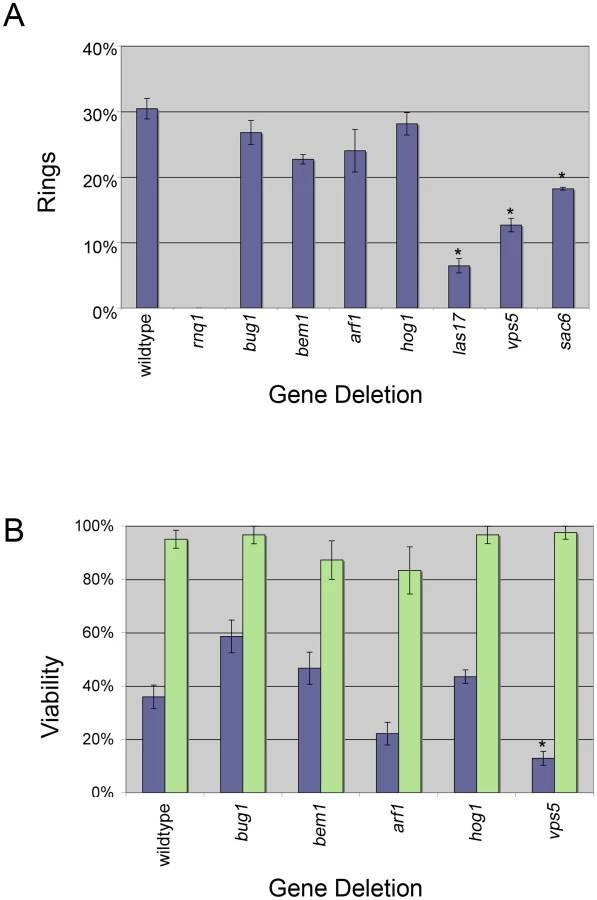
Interestingly, when measured after 24 hrs of Sup35PD-GFP induction, the three deletions associated with endocytosis and organization of the actin cytoskeleton, sac6Δ, las17Δ, and vps5Δ, all showed a significant reduction in cells containing rings. Since by 25 hrs of induction, wildtype, sac6Δ, and vps5Δ cultures were in stationary phase, where ring formation peaks [54], the inhibition of ring formation in sac6Δ and vps5Δ was not the result of a failure of these cultures to reach stationary phase (Figure S3). Also, since las17Δ cells reached saturation phase only after 36 hours, we also measured ring formation in las17Δ cultures following 50 hrs of induction, well after the culture reached stationary phase. These cultures still showed a 50% reduction in ring formation relative to the wildtype cultures (data not shown).
Ring-associated toxicity
Ring formation is associated with [PSI+] appearance [54]. However, it has been shown that cells that contain rings have a higher rate of cell death than those that have diffuse fluorescence [44], [47], [54]. Therefore, we asked if the deletions might be more toxic to ring bearing cells, which would result in a decrease in [PSI+] induction. To test this, we micromanipulated individual ring containing cells from selected deletion strains and assessed whether they were viable. Similar to previous findings [47], only 36% of wildtype ring containing cells were viable, whereas 95% of wildtype cells with diffuse cytoplasmic fluorescence were viable (Figure 3B). In general, the viability of ring containing wildtype, bem1Δ, and hog1Δ cells appeared to be similar (Figure 3B) and therefore cannot explain the strong reduction of de novo induction of [PSI+]. Ring containing arf1Δ cells had reduced viability (21%), which could account for the decrease in [PSI+] induction (Figure 2). In contrast, bug1Δ ring cells showed an increase in viability (59%), but apparently these cells did not efficiently give rise to [PSI+] cells. In the absence of rings, there appeared to be no effect on the viability of any of the deletion strains (Figure 3B, green bars).
Since isolating ring cells in strains with low ring formation was difficult, we focused on one example and found that of the small population of cells in vps5Δ strains that formed rings, a majority were inviable (Figure 3B). This suggests that rings in the absence of VPS5 may be harmful to the cell and likely explains the decreased percentage of rings observed in Figure 3A.
Deletion strains affect aggregation and toxicity of polyglutamine
We next asked if our deletion strains also affect the [PIN+] dependent aggregation of the polyglutamine (polyQ) expanded repeat found in the mutant huntingtin protein associated with Huntington's disease. Since las17Δ strains, carrying a galactose inducible expanded polyQ repeat (103Q) fused to GFP, were previously shown to delay 103Q-GFP aggregate formation compared to wildtype strains [52], we tested our other deletions for similar aggregation patterns. When wildtype [PIN+] cells expressed 103Q-GFP for approximately one to two hours, 82% of cells displayed strong fluorescent puncta, while [pin-] cells showed mostly cells with diffuse fluorescence (73%) and a minor population that contained a few faint fluorescent foci on a diffuse background (27%; Figure 4A and 4B). Similar analyses of [PIN+] strains with bug1Δ, bem1Δ, arf1Δ, and hog1Δ deletions resulted in a reduced number of cells that contained strong fluorescent puncta (Figure 4B, green bars) and an increased number of cells that had faint fluorescent foci with a diffuse background (Figure 4B, purple bars). As in the previous study [52], [PIN+] las17Δ strains had barely any strong fluorescent foci. Examination of [PIN+] vps5Δ and [PIN+] sac6Δ strains also showed a strong reduction in cells having bright foci and a similar distribution of cells with faint foci vs. no foci as seen in wildtype [pin-] strains.

We next examined the effect of these deletions on toxicity associated with expression of the expanded polyglutamine protein in the presence of the [PIN+] prion [57]. Our controls verified earlier findings [57] that wildtype [PIN+] cells carrying a galactose inducible 103Q-GFP plasmid show toxicity compared to cells carrying the non-toxic 25Q-GFP plasmid. As expected, polyglutamine expressing cells that are either [pin-] or rnq1Δ did not exhibit toxicity, whereas wildtype [PIN+] cells were sick (Figure 4C). Since previous studies have correlated the presence of polyQ aggregates with cell toxicity [57], the lack of strong polyQ fluorescent foci in the class of deletions that also reduced ring formation after Sup35PD-GFP overexpression (las17Δ, vps5Δ, and sac6Δ; Figure 4A and B) nicely explained the observed decrease in polyQ toxicity. Interestingly, the class of deletions that reduced [PSI+] induction, but not ring formation (bug1Δ, bem1Δ, arf1Δ, and hog1Δ), increased the frequency of cells containing faint polyQ-GFP fluorescent foci on diffuse backgrounds and clearly enhanced 103Q toxicity.
Discussion
We scored 398 gene deletions, previously identified by their ability to increase or decrease toxicity caused by overexpression of Sup35PD-GFP [46], for effects on the induction of [PSI+] and established effects for four of the deletions (see Table 2 summarizing all results). Since there is not always a strong correlation between the toxicity associated with SUP35 overexpression and the induction of [PSI+], we tested all of the enhanced and reduced toxicity candidates found in the previous study [46] for [PSI+] induction. We observed that induction of [PSI+] was inhibited by deletions that either reduced (arf1Δ and bem1Δ) or enhanced (bug1Δ and hog1Δ) toxicity. Possibly the rings or other less visible aggregates have an altered property in the presence of bug1Δ or hog1Δ that inhibits detectable [PSI+] appearance and causes toxicity. It has been shown that rings cause toxicity by titrating Sup35 and/or Sup45 (the essential release factor that binds to Sup35) into aggregates and away from the ribosome [46]. The toxicity associated with bug1Δ and hog1Δ during [PSI+] induction could be an enhancement of this effect or could be by the formation of a different type of toxic [PSI+] intermediate.
We also examined three deletions (las17Δ, vps5Δ, and sac6Δ), which were chosen for their association with endocytosis and the actin cytoskeleton, and found that they reduce [PSI+] induction. While las17Δ and vps5Δ were not included in the library originally scored for SUP35PD-GFP toxicity, the sac6Δ BY4741 library strain was not associated with changes in toxicity [46]. We found that sac6Δ inhibits [PSI+] induction in both the deletion library (BY4741; data not shown) and the 74-D694 (Figure 2) backgrounds. This suggests that other non-essential deletions that inhibit [PSI+] induction were likely missed in the toxicity screen.
All seven of our deletion strains reduce [PSI+] induction caused by overexpression of Sup35PD-GFP. While a deletion of BEM1 was previously shown to reduce the spontaneous appearance of [PSI+] associated with the SUP35 R2E2 expanded repeat [46], it is unknown whether our deletions affect the spontaneous appearance of [PSI+] without overexpression or mutated alleles (e.g. R2E2) of Sup35. Since the spontaneous appearance of [PSI+] is very infrequent and Mendelian suppressors with the phenotype of [PSI+] [58] appear at a higher rate than [PSI+], scoring for mutations that lower the appearance of [PSI+] is challenging.
Prion and polyglutamine formation and maturation
Time lapse examination of individually micromanipulated [psi−] [PIN+] cells, containing an endogenously tagged Sup35-GFP fusion and transiently overexpressing Sup35PD-GFP from a plasmid, previously showed that a fluorescent ring initially forms at the cell periphery and then internalizes around the vacuole. Later, such cells with rings give rise to [PSI+] daughter cells with fluorescent foci (Figure 5A) [44], [55].

In this paper, we identified two classes of gene deletions that reduce the de novo induction of stable [PSI+] but differ in effects on ring formation. Class I deletions (bug1Δ, bem1Δ, arf1Δ, and hog1Δ) form Sup35PD-GFP rings at approximately wildtype levels (Figure 3). Class II deletions (las17Δ, vps5Δ, and sac6Δ) have a significantly reduced number of cells with Sup35PD-GFP rings (Figure 3).
The existence of these two deletion classes suggests that the prion formation pathway can be inhibited at different steps. In the case of class I deletions, problems in peripheral and internal ring formation were not detected (data not shown), suggesting that these genes are important for ring containing cells to transmit heritable [PSI+] aggregates to daughter cells (Figure 5A). In the case of class II deletions, peripheral rings form infrequently, suggesting that these genes are important in the initial formation of the ring even in the presence of [PIN+] (Figure 5A). While rings contained in vps5Δ (class II) cells have reduced viability, it is unlikely that ring formation is so toxic that cells die before the ring appears because there is no corresponding increase in toxicity in non-ring containing cells (Figure 3B).
The formation of polyglutamine aggregates, upon overexpression of Q103:GFP in a [PIN+] strain, is not associated with ring formation. Instead, large bright aggregates are observed within one to two hours of induction [52]. All of our deletions affected polyglutamine aggregation, but the type of effect differed based on the class of the deletion. Class I deletions (bem1Δ, bug1Δ, arf1Δ, and hog1Δ) caused a decreased level of bright fluorescent aggregates and an increase in diffuse cells with faint foci (Figure 4A and 4B). Interestingly, these deletions enhanced the toxicity associated with 103Q-GFP (Figure 4C). In the presence of the class II deletions (las17Δ, vps5, and sac6Δ) 103Q-GFP usually remained diffuse, and very few cells had bright aggregates (Figure 4B). Additionally, all class II deletions suppressed polyglutamine toxicity (Figure 4C).
The formation of large huntingtin aggregates in mammals was initially thought to be the cause of huntingtin-mediated cell death (reviewed in [59]), but emerging evidence suggests that these large aggregates are neuroprotective ([60], reviewed in [61]) and toxicity is due to a soluble pool of oligomeric conformers [62]-[63]. Also in yeast, small multiple foci of 103Q appear to be more toxic than a large aggregate [64], [65]. Thus, we propose that class I genes are important for the formation of large protective aggregates but not small toxic oligomers (Figure 4B), and that when a class I gene product is absent, the reduction of large protective aggregates permits the increased propagation of deadly soluble oligomers.
In contrast, in cells with class II deletions, 103Q-GFP toxic oligomers or aggregates may form only rarely. While the [PSI+] and polyglutamine formation pathways are not directly comparable, we propose that class II genes affect the initial steps of polyglutamine formation, and the formation of large protective aggregates (promoted by class I genes) is a downstream event (Figure 5B).
Actin and prion formation
Although endocytosis is relatively unaffected in bem1Δ, bug1Δ, arf1Δ, hog1Δ, las17Δ, vps5, and sac6Δ strains, six out of seven of our deletion mutants display fragmented vacuoles (Figure S4) [66], [67]. Possibly, vacuole fragmentation may affect the perivacuolar deposition site for aggregated proteins, called IPOD [68], which has been suggested to play a role in prion formation [55], [56]. Some of the deletion library strains we screened induced [PSI+] normally but had fragmented vacuoles (Table SI, asterisk; [66]), suggesting that intact vacuoles are not necessarily a requirement for efficient prion appearance. Vacuole fragmentation has been shown to be associated with microtubular defects [69] as well as fluctuations in the soluble actin pool [67]. We showed that treatment with the microtubule disrupting drugs Nocodazole (Figure S4B and S4C) and Thiabendazole (data not shown) at concentrations that did not inhibit Sup35PD-GFP induction do not affect [PSI+] appearance. Conversely, the actin disrupting drug Latrunculin A [70] and act1-R117A alleles [44] do inhibit [PSI+] induction. Furthermore, many of the deletions identified in this study are involved with actin (Table 2), suggesting that actin organization plays a critical role in the aggregation of prion and polyglutamine proteins.
Possibly, proper actin organization on the cell periphery is required for the initial formation of the [PSI+] ring or polyglutamine oligomers, where as actin organization elsewhere in the cell could be required for downstream events such as the formation of a propagating [PSI+] conformer or the formation of large protective polyglutamine aggregates. Actin could possibly be involved in facilitating the addition of monomer to newly formed aggregates. In assaying for [PSI+], ring cells containing functional monomer not yet integrated into an aggregate would appear to be [psi−].
Our data suggests that prion and polyglutamine formation involves a multi-step process that is dependent upon actin organization. Interestingly, six of the seven proteins identified here have mammalian homologues (Table 2), suggesting that similar mechanisms may be involved in aggregation and oligomerization of QN-rich proteins in higher eukaryotes. Further elucidation of how actin nucleation contributes to prion induction will not only shed light on how toxic oligomeric species are formed, but also could provide clues to the molecular mechanisms underlying many human aggregating neurodegenerative diseases.
Methods
Plasmids and strains
In this work, 398 yeast deletion library strains (parent strain BY4741: MATa ura3Δ his3Δ1 leu2Δ met15Δ; Open Biosystems, Huntsville, AL; Table S1) previously obtained from an earlier toxicity screen ([46]; Tyedmers and Lindquist unpublished) were scored for effects on [PSI+] induction in a wildtype Sup35 background. The “kar1 plasmid donor” strain (GF667; MATα CEN1–16::pGal1-CEN1–16-URA3Kl kar1Δ15 lys2 rad5-535 leu2-3,112 can1-100 his3-11,15 trp1-1 cyhR) was used to introduce plasmids and prions into the deletion library strains via cytoduction, as described in Manogaran et al. [48]. A plasmid containing a copper inducible prion and middle domain of Sup35 (Sup35PD) fused to green fluorescent protein (Sup35PD-GFP; p1181: CEN2 HIS3 ori ARS AmpR pCup1-Sup35PD-GFP) was used to induce [PSI+] in deletion strain derivates of BY4741, while a LEU2 version of the plasmid (p1182) was used in 74-D694 (L1749; [33]; MATa ade1–14 leu2–3,115 his3Δ200 ura3–52 trp1–289 high [PIN+]). To score for [PSI+] in the BY4741 strains, a plasmid containing the [PSI+] suppressible ura3–14 allele ([49]; p1513; CEN2 LEU2 ura3–14 ori ARS AmpR) was used. A tester strain (L2174; MATα leu2 ura2 his3 [pin-]) transformed with a copper inducible RNQ1 fused to GFP (p1186; CEN LEU2 ori ARS AmpR pCUP1-RNQ1:GFP) was used to confirm the presence of [PIN+] in 74D-694 deletion strains (see below). Plasmids p1572 and p1838 [52], which contain a fusion of GFP to the galactose inducible 25Q or 103Q repeats in exon 1 of the huntingtin gene, respectively, were used to examine polyglutamine aggregation and toxicity. These fusion constructs do not contain the proline-rich region of the huntingtin exon 1 [52].
Cultivation procedures
Yeast strains were cultivated using standard media and growth protocols [71] and grown at 30oC except when indicated. Complex media contained 2% dextrose (YPD), and synthetic complete media contained the required amino acids and 2% dextrose (SD) or 2% galactose (SGal).
Curing of BY4741 deletion strains
Pre-existing prions were cured by growing strains on media containing low levels of guanidine hydrochloride (GuHCl), which cures through the inactivation of Hsp104 [72], [73]. Strains were spotted onto YPD plates containing 5mM GuHCl and repeated two to three additional times to ensure that prions were cured.
Preparation of the kar1 plasmid donor and introduction of plasmids and prions into deletion strains
Prions and plasmids were introduced into the kar1 plasmid donor strain. To make a [PSI+] kar1 plasmid donor strain to test for [PSI+] maintenance in the deletion strains, the kar1 plasmid donor strain was crossed to either a weak [PSI+] (L1759) or strong [PSI+] (L1763) strain containing Sup35PD-GFP. kar1 plasmiductants containing [PSI+] were chosen as described in Manogaran et al. [48], and confirmed to contain [PSI+] by the formation of Sup35PD-GFP aggregates after 16 hours. The [PSI+] kar1 plasmid donor was mated to the BY4741 deletion strains, deletion strain cytoductants were confirmed by testing for auxotrophic markers, and the presence of [PSI+] Sup35PD-GFP aggregate formation was examined as described above [54].
To test for the induction of [PSI+], prions and plasmids were introduced into the kar1 plasmid donor strains by crossing to either a [PIN+] (L1749 high [PIN+]) or [pin-] strain (L2910) containing the Sup35PD-GFP and ura3–14 plasmids. [PIN+] kar1 plasmid donor strains were confirmed as above and tested for the presence of [PIN+] by the formation of Sup35PD-GFP fluorescent rings after 24 hours of induction on copper [54]. Cytoduction of plasmids and prions into the BY4741 library deletion strains has been described previously [48]. To ensure reproducibility, cytoductions were performed in duplicate.
Induction of [PSI+] in cytoduced deletion strains
Cytoduced BY4741 deletion strains, containing [PIN+] and both Sup35PD-GFP (HIS3) and ura3–14 (LEU2) plasmids, were spotted onto plasmid selective SD-His-Leu plates plus 50 µM copper sulfate and grown for approximately two days. Strains were resuspended in 300 µL of sterile water and either spotted onto SD-Leu (grown two days) to assess growth, or SD-Leu-Ura (grown at room temperature for five to seven days) to score for [PSI+] induction. Induction experiments on all 398 yeast deletion library strains were repeated six times to ensure reproducibility. Spots that exhibited no or reduced growth on SD-Leu-Ura, compared to controls, were chosen as candidate genes that affect [PSI+] appearance (Table 1).
Re-engineering of candidate strains
Genetic recombination was used to replace candidate genes (Table 1) with HIS3 in [PIN+] 74-D694. Primers (Table S2), adjacent to sequences flanking the 5′ or 3′ ends of the candidate gene, were used to PCR amplify the HIS3 gene. PCR products were transformed and His+ transformants were confirmed for insertion of the HIS3 gene in the correct locus. Two to three independent knockout lines (Table S3) were obtained for each deletion, except for pre9Δ. To confirm the presence of [PIN+], deletion strains were mated to a [pin-] tester strain containing a RNQ1-GFP plasmid. Diploids were checked for the formation of fluorescent Rnq1-GFP aggregates after induction overnight. To test whether las17Δ, vps5Δ, or sac6Δ maintain [PSI+], the deletions were cytoduced with [PSI+] and checked for growth on –Ade.
Scoring the induction frequency of [PSI+]
Re-engineered deletion strains were transformed with the Sup35PD-GFP (LEU2) plasmid and grown in SD-Leu plus copper sulfate liquid media for 24 hours. After induction, approximately 10,000 cells were plated on SD–Ade, and a 50-fold dilution of cells was plated on SD+12. Colony counts were obtained from at least one transformant from each independent knock out (see Table S3) on SD+12 vs. SD-Ade. Colony counts from at least three transformants were used to determine the induction frequency.
Sup35PD-GFP ring formation in re-engineered deletion strains
After 24 hours of induction, cells were examined for the formation of GFP fluorescent rings [54] using a Zeiss Axioskop2 deconvolution workstation equipped with either a X40 Plan-Neofluar or X100 Plan-Apochromat objective lens (Zeiss). Approximately 300 cells were counted from at least one transformant from each independent knock out, for a total of three transformants.
Determining cell viability of ring-containing cells
Ring containing cells were simultaneously visualized and micromanipulated onto 2% sterile Noble Agar slabs. Slabs were transferred onto YPD media and grown for one to two days.
Aggregation and toxicity of polyglutamine in re-engineered deletion strains
Strains containing the galactose inducible 25Q or 103Q GFP fusion plasmids were grown overnight in SD-Ura and then washed in water approximately four times to remove residual glucose. To score for aggregation, washed cells were grown in liquid Gal-Ura for one to two hours with shaking and then examined for GFP aggregates. To score for toxicity, log phase uninduced washed cells were serially diluted 20-fold and spotted onto SD-Ura or SGal-Ura.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. LiebmanSW
DerkatchIL
1999
The yeast [PSI+] prion: making sense of nonsense.
J Biol Chem
274
1181
1184
2. UptainSM
LindquistSL
2002
Prions as protein-based genetic elements.
Ann Rev Microbiol
56
703
741
3. WicknerRB
LiebmanSW
SaupeSJ
2004
Prions of yeast and filamentous fungi: [URE3], [PSI+], [PIN+] and [Het-s].
Prion Biology and Disease, 2nd edtion
PrusinerSB
Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press
305
372
4. DerkatchIL
LiebmanSW
2007
Prion-prion interactions.
Prion
1
161
169
5. DuZ
ParkKW
YuH
FanQ
LiL
2008
Newly identified prion linked to the chromatin-remodeling factor Swi1 in Saccharomyces cerevisiae.
Nat Genet
40
460
465
6. PatelBK
Gavin-SmythJ
LiebmanSW
2009
The yeast global transcriptional co-repressor protein Cyc8 can propagate as a prion.
Nat Cell Biol
11
344
349
7. NemecekJ
NakayashikiT
WicknerRB
2009
A prion of yeast metacaspase homolog (Mca1p) detected by a genetic screen.
Proc Natl Acad Sci U S A
106
1892
1896
8. AlbertiS
HalfmannR
KingO
KapilaA
LindquistS
2009
A systematic survey identifies prions and illuminates sequence features of prionogenic proteins.
Cell
137
146
158
9. RogozaT
GoginashviliA
RodionovaS
IvanovM
ViktorovskayaO
2010
Non-Mendelian determinant [ISP+] in yeast is a nuclear-residing prion form of the global transcriptional regulator Sfp1.
Proc Natl Acad Sci U S A
107
10573
10577
10. AigleM
LacrouteF
1975
Genetic aspects of [URE3], a non-mendelian cytoplasmically-inherited mutation in yeast.
Mol Gen Genet
136
327
335
11. CoxBS
1965
ψ, a cytoplasmic suppressor of super-suppressors in yeast.
Heredity
20
505
521
12. LundPM
Cox
B
1981
Reversion analysis of [psi−] mutations in Saccharomyces cerevisiae.
Genet. Res
37
173
182
13. WicknerRB
1994
[URE3] as an altered Ure2 protein: evidence for a prion analog in Saccharomyces cerevisiae.
Science
264
566
569
14. DerkatchIL
ChernoffYO
KushnirovVV
Inge-VechtomovSG
LiebmanSW
1996
Genesis and variability of [PSI+] prion factors in Saccharomyces cerevisiae.
Genetics
144
1375
1386
15. Ter-AvanesyanMD
DagkesamanskayaAR
KushnirovVV
SmirnovVN
1994
The SUP35 omnipotent suppressor gene is involved in the maintenance of the non-Mendelian determinant [PSI+] in the yeast Saccharomyces cerevisiae.
Genetics
137
671
676
16. MasisonDC
WicknerRB
1995
Prion-inducing domain of yeast Ure2p and protease resistance of Ure2p in prion-containing cells.
Science
270
93
95
17. SondheimerN
LindquistSL
2000
Rnq1: an epigenetic modifier of protein function in yeast.
Mol Cell
5
163
172
18. VitrenkoYA
GrachevaEO
RichmondJE
LiebmanSW
2006
Visualization of aggregation of the Rnq1 prion domain and cross-seeding interactions with Sup35NM.
J BIol Chem
282
1779
1787
19. TanejaV
MaddeleinM
TalarekN
SaupeSJ
LiebmanSW
2007
A non Q/N-rich prion domain of a foreign prion, [Het-s], can propagate as a prion in yeast.
Mol Cell
27
67
77
20. DePaceAH
SantosoA
HillnerP
WeissmanJS
1998
A critical role for amino-terminal glutamine/asparagine repeats in the formation and propagation of a yeast prion.
Cell
93
1241
1252
21. OsherovichLZ
WeissmanJS
2001
Multiple Gln/Asn-rich prion domains confer susceptibility to induction of the yeast [PSI+] prion.
Cell
106
183
194
22. DerkatchIL
BradleyME
HongJY
LiebmanSW
2001
Prions affect the appearance of other prions: the story of [PIN+].
Cell
93
171
182
23. DerkatchIL
BradleyME
ZhouP
ChernoffYO
LiebmanSW
1997
Genetic and environmental factors affecting the de novo appearance of the [PSI+] prion in Saccharomyces cerevisiae.
Genetics
147
507
519
24. DerkatchIL
BradleyME
MasseSV
ZadorskySP
PolozkovGV
2000
Dependence and independence of [PSI+] and [PIN+]: a two-prion system in yeast?
EMBO J
19
1942
1952
25. BruceME
2003
TSE strain variation.
Br Med Bull
66
99
108
26. ZhouP
DerkatchIL
PatinoM
UptainS
LindquistS
1999
The yeast non-Mendelian factor [ETA+] is a variant of [PSI+], a prion-like form of release factor eRF3.
EMBO J
18
1182
1191
27. SchlumpbergerM
PrusinerSB
HerskowitzI
2001
Induction of distinct [URE3] yeast prion strains.
Mol Cell Biol
21
7035
7046
28. BradleyME
EdskesHK
HongJY
WicknerRB
LiebmanSW
2002
Interactions among prions and prion “strains” in yeast.
Proc Natl Acad Sci U S A
99
16392
16399
29. ToyamaBH
KellyMJ
GrossJD
WeissmanJS
2007
The structural basis of yeast prion strain variants.
Nature
449
233
237
30. TessierPM
LindquistS
2007
Prion recognition elements govern nucleation, strain specificity and species barriers.
Nature
447
556
561
31. KingCY
Diaz-AvalosR
2004
Protein-only transmission of three yeast prion strains.
Nature
428
319
323
32. TanakaM
ChienP
NaberN
CookeR
WeissmanJS
2004
Conformational variations in an infectious protein determines prion strain differences.
Nature
428
323
328
33. ChernoffYO
LindquistSL
OnoB
Inge-VechtomovSG
LiebmanSW
1995
Role of the chaperone protein hsp104 in propagation of the yeast prion-like factor [PSI+].
Science
268
880
884
34. PaushkinSV
KushnirovVV
SmirnovVN
Ter-AvanesyanMD
1996
Propagation of the yeast prion-like [PSI+] determinant is mediated by oligomerization of the Sup35-encoded polypeptide chain release factor.
EMBO J
15
3127
3134
35. MoriyamaH
EdskesHK
WicknerRB
2000
[URE3] prion propagation in Saccharomyces cerevisiae: Requirement of chaperone Hsp104 and curing by overexpressed chaperone Ydj1p.
Mol Cell Biol
20
8912
8922
36. Satpute-KrishnanP
LangsethSX
SerioTR
2007
Hsp104-dependent remodeling of prion complexes mediates protein-only inheritance.
PLoS Biol
2
e24
doi:10.1371/journal.pbio.0050024
37. WegrzynRD
BapatK
NewnamGP
ZinkAD
ChernoffYO
2001
Mechanism of prion loss after Hsp104 inactivation in yeast.
Mol Biol Cell
21
4656
4669
38. KryndushkinDS
AlexandrovIM
Ter-AvanesyanMD
KushnirovVV
2003
Yeast [PSI+] prion aggregates are formed by small Sup35 polymers fragmented by Hsp104.
J Biol Chem
278
49636
49643
39. LopezN
AronR
CraigEA
2003
Specificity of class II Hsp40 Sis1 in maintenance of yeast prion [RNQ+].
Mol Biol Cell
14
1172
1181
40. ShorterJ
LindquistSL
2004
Hsp104 catalyzes formation and elimination of self-replicating Sup35 prion conformers.
Science
304
1793
1797
41. DerdowskiA
SindiSS
KlaipsCL
DiSalvoS
SerioTR
2010
A size threshold limits prion transmission and establishes phenotypic diversity.
Science
330
680
3
42. ParkK
HahnJ
QingF
ThieleDJ
LiL
2006
De novo appearance and “strain” formation of yeast prion [PSI+] are regulated by the Heat-Shock Transcription Factor.
Genetics
173
35
47
43. FanQ
ParkK
DuZ
MoranoKA
LiL
2007
The role of Sse1 in the de novo formation and variant determination of the [PSI+] prion.
Genetics
177
1583
1593
44. GanusovaEE
OzolinsLN
BhagatS
NewnamGP
WegrzynRD
2006
Modulation of prion formation, aggregation, and toxicity by the actin cytoskeleton in yeast.
Mol Cell Biol
26
617
629
45. AllenKD
ChernovaTA
TennantEP
WilkinsonKD
ChernoffYO
2007
Effects of the ubiquitin system alterations on the formation and loss of a yeast prion.
J Biol Chem
282
3004
3013
46. TyedmersJ
MadariagaML
LindquistSL
2008
Prion switching in response to environmental stress.
PLoS Biol
6
e294
doi:10.1371/journal.pbio.0060294
47. VishveshwaraN
BradleyME
LiebmanSW
2009
Sequestration of essential proteins causes prion associated toxicity in yeast.
Mol Microbiol
73
1101
1114
48. ManogaranAL
FajardoVM
ReidRJ
RothsteinR
LiebmanSW
2010
Most, but not all, yeast strains in the deletion library contain the [PIN+] prion.
Yeast
27
159
166
49. ManogaranAL
KirklandKT
LiebmanSW
2006
An engineered nonsense URA3 allele provides a versatile system to detect the presence, absence and appearance of the [PSI+] prion in Saccharomyces cerevisiae.
Yeast
23
141
147
50. LehnerKR
StoneMM
FarberRA
PetesTD
2007
Ninety-six haploid yeast strains with individual disruptions of open reading frames between YOR097C and YOR192C constructed for the Saccharomyces genome deletion project, have an additional mutation in the mismatch repair gene MSH3.
Genetics
177
1951
1953
51. ReidRJ
SunjevaricI
VothWP
CicconeS
DuW
2008
Chromosome-scale genetic mapping using a set of 16 conditionally stable Saccharomyces cerevisiae chromosomes.
Genetics
180
1799
1808
52. MeriinAB
ZhangX
AlexandrovAM
SalnikovaAB
Ter-AvanesianMD
2007
Endocytosis machinery is involved in aggregation of proteins with expanded polyglutamine domains.
FASEB
21
1915
1925
53. BagriantsevS
LiebmanSW
2004
Specificity of prion assembly in vivo. [PSI+] and [PIN+] form separate structures in yeast.
J Biol Chem
279
51042
51048
54. ZhouP
DerkatchIL
LiebmanSW
2001
The relationship between visible intracellular aggregates that appear after overexpression of Sup35 and the yeast prion-like elements [PSI+] and [PIN+].
Mol Microbiol
39
37
46
55. MathurV
TanejaV
SunY
LiebmanSW
2010
Analyzing the birth and propagation of two distinct prions in yeast.
Mol Biol Cell
21
1449
1461
56. TyedmersJ
TreuschS
DongJ
McCafferyJM
BevisB
2010
Prion induction involves an ancient system for the sequestration of aggregated proteins and heritable changes in prion fragmentation.
Proc Natl Acad Sci USA
107
8633
8638
57. MeriinAB
ZhangX
HeX
NewnamGP
ChernoffYO
2002
Huntington toxicity in yeast model depends on polyglutamine aggregation mediated by a prion-like protein Rnq1.
J Cell Biol
157
997
1004
58. BradleyME
BagriantsevS
VishveshwaraN
LiebmanSW
2003
Guanidine reduces stop codon read-through caused by missense mutations in SUP35 or SUP45.
Yeast
20
625
632
59. TruantR
AtwalRS
DesmondC
MunsieL
TranT
2008
Huntington's disease: revisiting the aggregation hypothesis in polyglutamine neurodegenerative diseases.
FEBS J
275
4252
62
60. ArrasateM
MitraS
SchweitzerES
SegalMR
FinkbeinerS
2004
Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death.
Nature
431
805
10
61. TreuschS
CyrDM
LindquistSL
2009
Amyloid deposits: protection against toxic protein species?
Cell Cycle
8
1668
74
62. KayedR
HeadE
ThompsonJL
McIntireTM
MiltonSC
2004
Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis.
Science
300
486
9
63. LesneS
KohMT
KotilinekL
KayedR
GlabeCG
2006
A specific amyloid-beta protein assembly in the brain impairs memory.
Nature
440
352
357
64. DehayB
BertolottiA
2006
Critical role of the proline-rich region in Huntingtin for aggregation and cytotoxicity in yeast.
J Biol Chem
35608
35615
65. WangY
MeriinAB
ZaarurN
RomanovaNV
ChernoffYO
2009
Abnormal proteins can form aggresome in yeast: aggresome-targeting signals and components of the machinery. FASEB J.
23
451
463
66. SeeleyES
KatoM
MargolisN
WicknerW
EitzenG
2002
Genomic analysis of homotypic vacuole fusion.
Mol Biol Cell
13
782
794
67. EitzenG
WangL
ThorngrenN
WicknerW
2002
Remodeling of organelle-bound actin is required for yeast vacuole fusion.
J Cell Biol
158
669
679
68. KaganovichD
KopitoR
FrydmanJ
2008
Misfolded proteins partition between two distinct quality control compartments.
Nature
454
1088
1095
69. GuthrieBA
WicknerW
1988
Yeast vacuoles fragment when microtubules are disrupted.
J Cell Biol
107
115
120
70. Bailleul-WinslettPA
NewnamGP
WegrzynRD
ChernoffYO
2002
An antiprion effect of the anticytoskeletal drug latrunculin A in yeast.
Gene Expr
9
145
156
71. ShermanF
FinkGR
HIcksJB
1986
Methods in Yeast Genetics.
Plainview NY
Cold Spring Harbor Laboratory Press
72. TuiteMF
MundyCR
CoxBS
1981
Agents that cause a high frequency of genetic change from [PSI+] to [psi−] in Saccharomyces cerevisiae.
Genetics
98
691
711
73. JungG
MasisonDC
2001
Guanidine hydrochloride inhibits Hsp104 activity in vivo: a possible explanation for its effect in curing yeast prions.
Cur Microbiol
43
7
10
74. BehniaR
BarrFA
FlanaganJJ
BarloweC
MunroS
2007
The yeast orthologue of GRASP65 forms a complex with a coiled-coil protein that contributes to ER to golgi traffic.
J Cell Biol
176
255
261
75. IrazoquiJE
GladfelterAS
LewDJ
2003
Scaffold-mediated symmetry breaking by Cdc42p.
Nat Cell Biol
5
1062
1079
76. HanBK
BogomolnayaLM
TottenJM
BlankHM
DangottLJ
2005
Bem1p, a scaffold signaling protein, mediates cyclin-dependent control of vacuolar homeostasis in Saccharomyces cerevisiae.
Genes Dev
19
2606
2618
77. FuciniRV
NavarreteA
VadakkanC
LacomisL
Erdjument-BromageH
2000
Activated ADP-ribosylation factor assembles distinct pools of actin on golgi membranes.
J Biol Chem
275
18824
18829
78. KahnRA
KernFG
ClarkJ
GelmannEP
RulkaC
1991
Human ADP-ribosylation factors. A functionally conserved family of GTP-binding proteins.
J Biol Chem
266
2606
14
79. MotizukiM
YokotaS
TsurugiK
2008
Effect of low pH on organization of the actin cytoskeleton in Saccharomyces cerevisiae.
Biochim Biophys Acta
1780
179
184
80. HanJ
LeeJD
BibbsL
UlevitchRJ
1994
A MAP kinase targeted by endotoxin and hyperosmolarity in mammalian cells.
Science
265
808
811
81. LiR
1997
Bee1, a yeast protein with homology to Wiscott-Aldrich syndrome protein, is critical for the assembly of cortical actin cytoskeleton.
J Cell Biol
136
649
658
82. NothwehrSF
Hindes
1997
The yeast VPS5/GRD2 gene encodes a sorting nexin-1-like protein required for localizing membrane proteins to the late golgi.
J Cell Sci
110
1063
1072
83. AdamsAE
ShenW
LinCS
LeavittJ
MatsudairaP
1995
Isoform-specific complementation of yeast sac6 null mutation by human fimbrin.
Mol Cell Biol
15
69
75
84. BagriantsevSN
GrachevaEO
RichmondJE
LiebmanSW
2008
Variant-specific [PSI+] infection is transmitted by Sup35 polymers within [PSI+] aggregates with heterogeneous protein composition.
Mol Biol Cell
19
2433
2443
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2011 Číslo 5
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Nodal-Dependent Mesendoderm Specification Requires the Combinatorial Activities of FoxH1 and Eomesodermin
- SHINE Transcription Factors Act Redundantly to Pattern the Archetypal Surface of Arabidopsis Flower Organs
- Association of Genetic Variants in Complement Factor H and Factor H-Related Genes with Systemic Lupus Erythematosus Susceptibility
- STAT Is an Essential Activator of the Zygotic Genome in the Early Embryo
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy