
Embryonálne tumory s mnohovrstvovými rozetami – vzácne tumory centrálneho nervového systému v detskom veku
Embryonal Tumors with Multilayer Rosettes – Rare Central Nervous System Tumors in Infants
Introduction:
The most recent findings show a histopathological, genetic and clinical uniformity in cases of tumors called embryonal tumors with multilayer rosettes. This group is composed of medulloepithelioma, ependymoblastoma and embryonal tumor with abundant neuropil and true rosettes. Amplification of locus 19q13.42, which includes C19MC cluster containing genes for microRNA, and also LIN28A positivity are present in all three entities. Dysregulation of epigenetic modifiers is very important in pathogenesis of the disease. These tumors manifest in little children (median less than 3 years of age); overall survival is 5–10%.
Case report:
Almost three year-old boy diagnosed with brainstem tumor: meduloepithelioma, WHO grade IV confirmed by histological investigation. He presented with dysarthria, bulbar syndrome, central lesion of the facial nerve, quadriparesis with right-side dominancy. He received three induction cycles of chemotherapy from March to May 2014 (according to protocol COG ACNS0334). Only partial improvement of his clinical state was reached. Signs of an intracranial hypertension appeared resulting in VP shunt insertion; impairment of consciousness developed after the induction cycles and before any other treatment could be initiated. He underwent radiotherapy due to vital indication. After application of two fractions (boost in the center of the tumor), the patient became quickly comatose. Spinal cord metastasis was demarked by MRI scan (in the level of 3rd cervical vertebra). A bilateral infiltration in pulmonary parenchyma, according to a radiologist metastasis-wise, was detected by CT scan (histologisation of infiltration was not implemented). The patient died in August 2014 – six months after manifestation of first symptoms.
Conclusion:
We reported our first documented case of a patient with tumor from embryonal tumors with multilayer rosettes group in Slovakia. Nowadays, there is no effective treatment of these tumors. Research of molecules targeting to epigenetic modifiers would be one of the possible promises for future therapy.
Key words:
medulloepithelioma – ependymoblastoma – embryonal tumors with multilayer rosettes – microRNA – 19q13.42 – C19MC – LIN28
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Submitted:
26. 5. 2015
Accepted:
24. 6. 2015
Autoři:
M. Pleško 1; K. Husáková 1; E. Kaiserová 1; M. Tichý 2; J. Zámečník 3
Působiště autorů:
Klinika detskej hematológie a onkológie LF UK a DFNsP Bratislava, Slovenská republika
1; Neurochirurgická klinika dětí a dospělých 2. LF UK a FN v Motole, Praha
2; Laboratoř neuropatologie, Ústav patologie a molekulárni medicíny, 2. LF UK a FN v Motole, Praha
3
Vyšlo v časopise:
Klin Onkol 2015; 28(4): 288-292
Kategorie:
Kazuistika
doi:
https://doi.org/10.14735/amko2015288
Souhrn
Úvod:
Najnovšie poznatky dokázali histopatologickú, genetickú, aj klinickú uniformitu v prípade tumorov označovaných ako embryonálne tumory s mnohovrstvovými rozetami, kam patrí meduloepitelióm, ependymoblastóm a embryonálny tumor s abundatným neuropilom a pravými rozetami. Spoločným znakom je pozitivita LIN28A a amplifikácia lokusu 19q13.42, ktorý zahrňuje C19MC klaster obsahujúci gény pre mikroRNA. V patogenéze ochorenia hrá významnú úlohu dysregulácia epigenetických modifikátorov. Tieto tumory sú pozorované u najmenších detí (medián veku pod 3 roky), miera celkového prežívania je menej ako 5 – 10 %.
Kazuistika:
Temer trojročný chlapec s histologicky dokázaným tumorom mozgového kmeňa: meduloepiteliom WHO grade IV. Prišiel s príznakmi dyzartrie, bulbárneho syndrómu, centrálnej lézie n.V a kvadruparézy s pravostrannou prevahou. Od marca do konca mája 2014 dostal tri cykly indukčnej chemoterapie (protokol COG ACNS0334). Dosiahlo sa iba prechodné zlepšenie klinického stavu. V prestávke liečby sa objavili príznaky intrakraniálnej hypertenzie s potrebou zavedenia ventrikulo ‑ peritoneálnej drenáže, vznikla porucha vedomia v zmysle soporu. Z vitálnej indikácie dostal rádioterapiu. Po podaní dvoch frakcií – boost na ložisko tumoru – pacient upadol do kómy, na MRI sa demarkovala metastáza v mieche v úrovni C3. CT vyšetrením sa zistilo obojstranné ložiskové postihnutie v pľúcnom parenchýme, podľa popisu rádiológa mali charakter metastáz (histologizácia ložísk nerealizovaná). Pacient zomrel v auguste 2014, šesť mesiacov od prvých príznakov ochorenia.
Záver:
Referovaním tejto kazuistiky sme dokumentovali prvý na Slovensku zaznamenaný prípad tumoru zo skupiny embryonálnych tumorov s mnohovrstvovými rozetami. V súčasnosti neexistuje účinná liečba týchto tumorov. Prísľubom do budúcna je výskum molekúl zacielených na epigenetické modifikátory.
Klíčové slová:
meduloepitelióm – ependymoblastóm – embryonálne tumory s mnohovrstvovými rozetami – mikroRNA – 19q13.42 – C19MC – LIN28
Úvod
Embryonálne nádory sú najčastejšie malignity centrálneho nervového systému (CNS) u detí [1]. Podľa WHO klasifikácie z roku 2007 sa rozdeľujú do troch skupín – meduloblastóm, atypický teratoidný/ rabdoidný tumor (AT/ RT) a primitívny neuroektodermálny tumor (PNET). Skupina PNET sa ďalej člení do týchto piatich podskupín – PNET CNS, neuroblastóm CNS, ganglioneuroblastóm CNS, meduloepitelióm (MEPL), ependymoblastóm (EBL). V skupine PNET CNS je zaradený aj veľmi vzácny a agresívny typ tumoru – embryonálny tumor s abundantným neuropilom a pravými rozetami (embryonal tumour with abundant neuropil and true rosettes – ETANTR) [2]. Prvých deväť prípadov ETANTR popísali v roku 2000 Eberhart et al, pričom poukázali na unikátny imunohistochemický nález, ako aj histologickú a ultraštrukturálnu skladbu tumoru [3]. Ďalší výskum ukázal, že ETANTR spolu s MEPL a EBL vykazujú zhodu v základných histopatologických črtách (predovšetkým prítomnosť mnohovrstvových roziet), ako aj v mimoriadne nepriaznivom klinickom priebehu ochorenia [4]. Všetky tri entity sa vyskytujú u detí v najnižšej vekovej kategórii (medián pod 3 roky), s miernou prevahou dievčat nad chlapcami [5]. Odpoveď na chemoterapiu a rádioterapiu je zlá, celkové prežívanie (overall survival – OS) je menej ako 5 – 10 %, medián prežívania je 6 – 9 mesiacov [6 – 8].
V roku 2009 Pfister et al a o niečo neskôr aj Li et al prvýkrát popísali zjednocujúci genetický marker – amplifikáciu C19MC klastra na lokuse 19q13.42, ktorý kóduje gény pre mikroRNA (miRNA) [9,10]. V roku 2012 Korshunov et al identifikovali ďalší vysoko špecifický imunohistochemický marker – LIN28A [11]. Na základe rozširujúcich sa poznatkov ešte v roku 2010 navrhli Paulus a Kleihues zlúčenie troch popisovaných tumorov do samostatnej skupiny, ktorú nazvali embryonálne tumory s mnohovrstvovými rozetami (ETMR) [12]. Presná incidencia ETMR nie je známa. Khatua et al vo svojej epidemiologickej štúdii z roku 2012 uviedli menej ako 75 hlásených prípadov [13]. V rámci práce publikovanej v roku 2014 Korshunov et al urobili molekulárnu analýzu 97 dostupných vzoriek tumorov zo skupiny ETMR [14].
Biológia malígnej transformácie ETMR
Malígna transformácia pri ETMR je determinovaná predovšetkým epigenetickými mechanizmami, a to metylovaním a deacetyláciou vybraných úsekov jadrového chromatínu, čo vedie k dysregulácii expresie kľúčových génov bunkového cyklu. Metylácia DNA je kritický moment vo vývoji ochorenia. Reakcia je katalyzovaná troma enzymaticky aktívnymi DNA ‑ metyltransferázami (DNMT) – DNMT1, DNMT3A a DNMT3B [15]. Za normálnych okolností je najviac exprimovaná DNMT1. Následkom amplifikácie C19MC dochádza k epigenetickej remodelácii jadrového chromatínu, čo vedie k oslabeniu transkripcie niektorých nádorových supresorových génov, vrátane p53 [16,17]. Podrobnosti tohto procesu zobrazuje obr. 1.

Gén LIN28 kóduje proteín – LIN28A, ktorý podnecuje delenie buniek zvýšením transkripcie produktov protoonkogénov. Normálne tkanivá CNS, ako aj iné tkanivá organizmu, neexprimujú LIN28A [11]. Molekula je však prepisovaná počas včasnej embryogenézy a jej tvorba je utlmená po diferenciácii tkanív plodu. Okrem ETMR je nadexpresia LIN28A pozorovaná v klinicky nepriaznivo sa vyvíjajúcich prípadoch rakoviny ovárií, ezofágu, kolorekta, ale aj pri neuroblastómoch [18,19]. Strata regulačného systému spôsobená nadexpresiou LIN28A má za následok nadmernú expresiu protoonkogénov (podrobnejší popis na obr. 2), čo spoločne s vyblokovaním nádorových supresorových génov amplifikáciou C19MC vedie k malígnej transformácii buniek CNS v prípadoch ETMR.

Ďalšou skúmanou cestou malignizácie v prípadoch ETMR je dysregulácia signálnej cesty PI3K ‑ mTOR, overuje sa tiež úloha WNT signálnej dráhy [9,20].
Kazuistika
Predstavujeme kazuistiku chlapca, ktorému bolo v čase diagnózy 2 a 3/ 4 roka. Dieťa bolo narodené z prvej fyziologickej gravidity, pôrod bol vedený sekciou pre panvovú polohu. Apgarovej skóre bolo 9/ 10/ 10, pôrodná hmotnosť 2 400 g, pôrodná dĺžka 52 cm. Popôrodná adaptácia, psychomotorický vývoj aj chorobnosť boli v norme. V rodinnej anamnéze je zaujímavý údaj o otcovom bratovi, ktorý ako trojročný exitoval pre tumor CNS (liečený iba rádioterapiou). Jednoznačný hereditárny súvis nie je dokázateľný, nakoľko histológia tohto tumoru nie je k dispozícii.
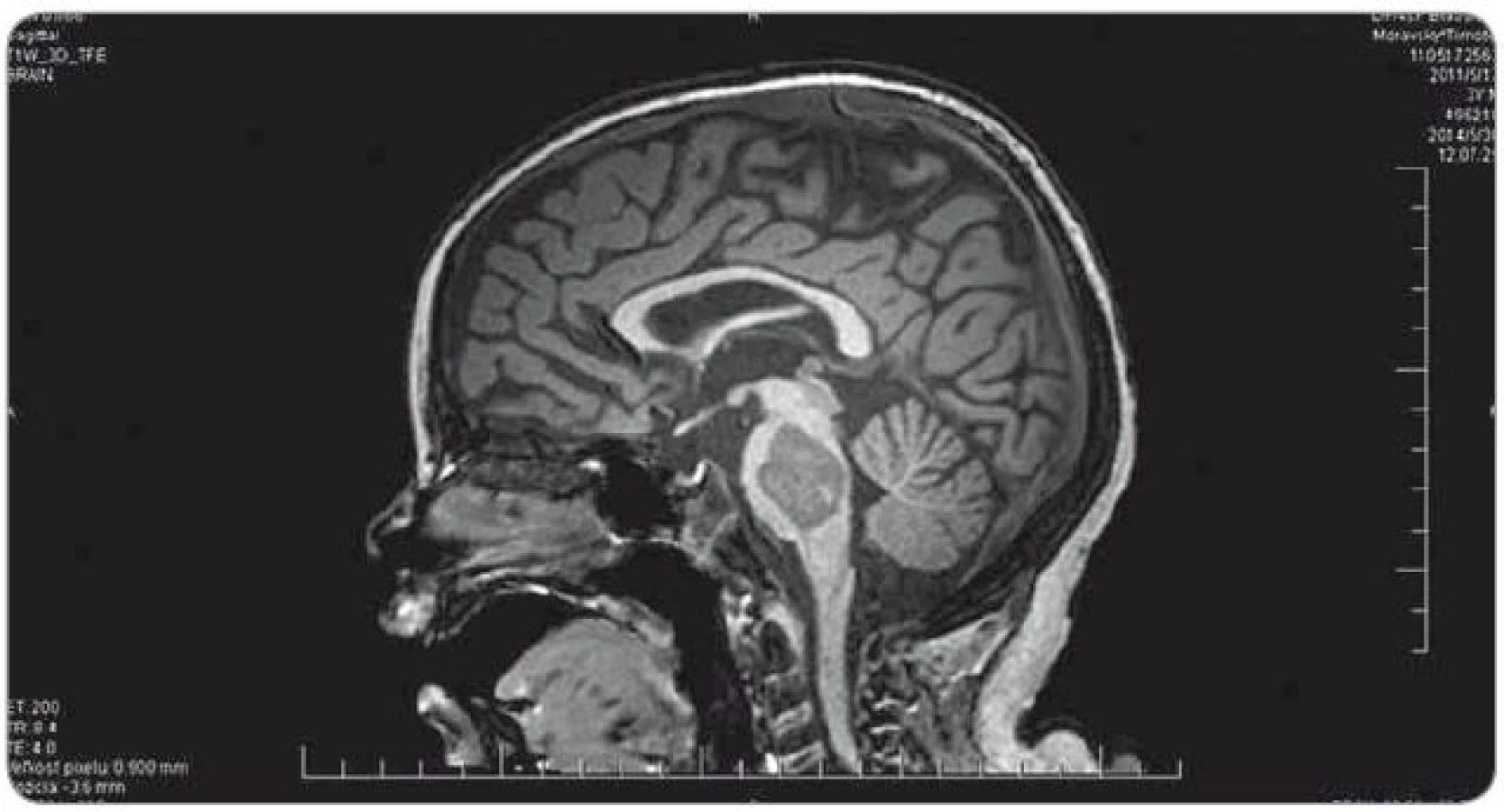
Ťažkosti pacienta vznikli začiatkom februára 2014. Prejavili sa ako vertigo,poruchy rovnováhy, ataxia, únava, zmeny správania. Objektívne sa postupne rozvinula dyzartria, bulbárny syndróm, centrálna lézia n. V, kvadruparéza až plégia vpravo. Na iniciálnom MRI mozgu bol nález intrakraniálnej tumoróznej expanzie v ponse a mezencefale vľavo, bez hydrocefalu, veľkosť ložiska bola 3,2 × 2,6 × 3,6 cm. Na predoperačnom MRI bola popisovaná mierna progresia veľkosti na 3,7 × 2,9 × 3,6 cm. Parciálna extirpácia v zmysle rozšírenej biopsie bola vykonaná na Neurochirurgickej klinike detí a dospelých vo FN v Motole, Praha. Bolo ponechané významné reziduum o veľkosti 3,3 × 2,8 × 3,2 cm. Histologicky sa zistil nezrelý embryonálny tumor zo skupiny PNET, mozgová varianta – meduloepitelióm WHO grade IV (diagnostika vykonaná v Neuropatologickom laboratóriu Ústavu patológie a molekulárnej medicíny, 2. LF UK v Prahe). V marci 2014 bol pacient prijatý na Kliniku detskej hematológie a onkológie v Detskej fakultnej nemocnici s poliklinikou v Bratislave. MRI spinálneho kanála vylúčilo metastatickú infiltráciu, vyšetrením likvoru a kostnej drene sa nenašli malígne bunky. Dostal chemoterapiu podľa protokolu COG ACNS0334 (jedná sa o protokol kooperatívnej skupiny Children‘s Oncology Group (COG) pre liečbu supratentoriálneho PNET a vysokorizikového meduloblastómu u detí mladších ako 36 mesiacov). Podali sme tri indukčné cykly (úvodná 24 - hodinová infúzia vysoko dávkovaného metotrexátu, potom pokračovanie radov cytostatík – etopozid, cyklofosfamid, cisplatina, vinkristín). Postupne sa zlepšil subjektívny aj objektívny klinický stav, reč bola artikulovanejšia, vymizli známky bulbárneho syndrómu, pacient postupne začínal opäť chodiť, intenzívne rehabilitoval. Po skončení indukčnej chemoterapie bolo dňa 30. 5. 2014 urobené kontrolné MRI mozgu (obr. 3). Nález bol v porovnaní s pooperačným MRI popísaný ako stacionárny (rozmery tumoru: 3,3 × 2,8 × 3,2 cm). V prestávke liečby, pred plánovanými konsolidačnými cyklami formou myeloablatívnej megachemoterapie s autológnym prevodom periférnych krvotvorných kmeňových buniek, nastalo promptné zhoršenie klinického stavu. Objavila sa nauzea, zvracanie, točenie hlavy, opätovne bola akcetovaná porucha reči, prehĺtania, chôdze aj rovnováhy, pridala sa porucha vedomia manifestovaná somnoleciou až soporom. Na kontrolných CT snímkach mozgu sa zaznamenalo postupné rozširovanie komorového systému. Pacientovi bol zavedený VP ‑ shunt, dostával antiedémovú liečbu, vrátane kortikoidov, s minimálnym efektom. Pre zhoršenie klinického stavu sme sa rozhodli z vitálnej indikácie predsunúť rádioterapiu pred konsolidačnú chemoterapiu. Naplánovalo sa ožiarenie 30,6 Gy boost na reziduum tumoru a 23,4 Gy na kraniospinálnu os do celkovej dávky 54,0 Gy s konkomitantným podávaním temozolomidu. Po podaní dvoch frakcií na tumorózne ložisko pacient upadol do kómy. Na MRI z 11. 7. 2014 (obr. 4) sa popisovalo temer deväťnásobné zväčšenie objemu nádoru, s obrazom počínajúcej descendentnej herniácie a demarkovalo sa nové ložisko – metastáza v oblasti C3 miechy. Nález v mozgovom kmeni sme hodnotili ako quo ad vitam infaustný. Pacient ďalej dostával symptomatickú liečbu. Pre zvyšujúcu sa potrebu oxygenoterapie a úsilné dýchanie sa 30. 7. 2014 urobilo CT vyšetrenie hrudníka s nálezom početného obojstranného ložiskového postihnutia pľúcneho parenchýmu, podľa popisu rádiológa charakteru metastáz (obr. 5). Histológiu pľúcnych ložísk sme nerealizovali. Preterminálne vznikli u pacienta centrálne horúčky, decerebračné kŕče, poruchy dýchania a srdcovej akcie. Zomrel 2. 8. 2014, šesť mesiacov od iniciálnych príznakov ochorenia.


Diskusia a záver
Referovaním tejto kazuistiky chceme prispieť k zvýšeniu povedomia o extrémne vzácnom type nádorového ochorenia u detí, upozorniť na jeho mimoriadne nepriaznivú prognózu a neexistenciu účinnej liečby. V deviatich prípadoch opísaných Eberhartom et al prežil iba jeden detský pacient, u ktorého bol tumor resekovaný úplne. Jeden pacient zomrel na recidívu tumoru po subtotálnej resekcii, iba sedem mesiacov od prvých príznakov. Ostatní zomreli v rozmedzí 5 – 14 mesiacov od objavenia sa iniciálnych ťažkostí [3]. Aj ďalšie literárne údaje zo zahraničia dokumentujú rovnako nepriaznivo sa vyvíjajúce prípady nádorov zo skupiny ETMR [5]. V súčasnosti sa skúma niekoľko cieľov možnej terapie:
- DNMT3B – štúdie na bunkových líniách malígnych gliómov dokázali zníženie DNA metylácie a reaktiváciu p53 pri použití cytidínových analógov: 5-azacytidín (5 - AzaC) a 5 - aza ‑ 2’ - deoxycitidín [21].
- HDAC – zvažuje sa použitie inhibítora histón deacetylázy (HDAC) – vorinostatu. Jeho synergický účinok s inhibítormi DNMT je známy [22].
- Kyselina valproová (valproát) – pôsobí ako inhibítor histón deacetylázy (HDAC), v kombinácii s 5 - azacitidínom má synergický antikancerózny účinok in vitro aj in vivo [24].
- mTOR a WNT – ďalšie štúdie budú zamerané aj na použitie mTOR inhibítorov (everolimus, temsirolimus, siroli-mus) a inhibítorov WNT signálnej dráhy, ktorá reguluje kľúčové oblasti bunkovej diferenciácie a migrácie počas embryogenézy [9,23].
Doterajšie výsledky výskumov na bunkových líniách dávajú prísľub, že zacielenie terapie na epigenetické modifikátory môže priniesť požadovaný nový terapeutický prístup v liečbe tohto obzvlášť rezistentného nádorového ochorenia. Najväčšou výzvou do budúcna je realizácia predklinických štúdií zameraných na vyhodnotenie účinnosti a toxicity potencionálnych liečiv. Potreba týchto štúdií a ďalšieho bádania, je vzhľadom na charakter ochorenia, viac než urgentná [24,25].
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
Obdržané: 26. 5. 2015
Prijaté: 24. 6. 2015
MUDr. Marek Pleško
Klinika detskej hematológie a onkológie
LF UK a DFNsP Bratislava
Limbová 1
833 40 Bratislava
Slovenská republika
e-mail: marekplesko@gmail.com
Zdroje
1. Kaiserová E. Nádory centrálneho nervového systému. In: Ondruš D et al (eds). Všeobecná a špeciálna onkológia. 1. vyd. Bratislava: Polygrafické stredisko UK 2006 : 278 – 280.
2. Louis DN, Ohgaki H, Wiestler OD et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol 2007; 114(2): 97 – 109.
3. Eberhart CG, Brat DJ, Cohen KJ et al. Pediatric neuroblastic tumors containing abundant neuropil and true rosettes. Pediatr Dev Pathol 2000; 3(4): 346 – 352.
4. Ryzhova MV, Kushel YV, Zheludkova OG et al. Medulloepithelioma, ependymoblastoma and embryonal tumor with multilayered rosettes: Are the Same Disease Entity? N N Burdenko J Neurosurgery 2013; 77(6): 45 – 48.
5. Ferri Niguez B, Martinez ‑ Lage JF, Almagro MJ et al. Embryonal tumor with neuropil and true rosettes (ETANTR): a new distinctive of pediatric PNET: a case‑based update. Childs Nerv Syst 2010; 26(8): 1003 – 1008. doi: 10.1007/ s00381 ‑ 010 ‑ 1179 ‑ x.
6. Adamek D, Sofowora KD, Cwiklinska M et al. Embryonal tumor with abudant nuropil and true rosettes: an autopsy case‑based update and review of the literature. Childs Nerv Syst 2013; 29(5): 849 – 854. doi: 10.1007/ s00381 ‑ 013 ‑ 2037 ‑ 4.
7. Gessi M, Giangaspero F, Lauriola L et al. Embryonal tumors with abundant neuropil and true rosettes: a distinctive CNS primitive neuroectodermal tumor. Am J Surg Pathol 2009; 33(2): 211 – 217. doi: 10.1097/ PAS.0b013e318186235b.
8. Woehrer A, Slavc i, Peyrl A et al. Embryonal tumor with abundant neuropil ant true rosettes (ETANTR) with loss of morphological but retained genetic key features during progression. Acta Neuropathol 2011; 122(6): 787 – 790. doi: 10.1007/ s00401 ‑ 011 ‑ 0903 ‑ 2.
9. Li M, Lee KF, Lu Y et al. Frequent amplification of a chr19q13.41 microRNA polycistron in aggressive primitive neuroectodermal brain tumors. Cancer Cell 2009; 16(6): 533 – 546. doi: 10.1016/ j.ccr.2009.10.025.
10. Pfister S, Remke M, Castoldi M et al. Novel genomic amplification targeting the microRNA cluster at 19q13.42 in a pediatric embryonal tumor with abundant neuropil and true rosettes. Acta Neuropathol 2009; 117(4): 457 – 464. doi: 10.1007/ s00401 ‑ 008 ‑ 0467 ‑ y.
11. Korshunov A, Ryzhova M, Jones TW et al. LIN28Aimmunoreactivity is a potent diagnostic marker of embryonal tumor with multilayered rosettes (ETMR). Acta Neuropathol 2012; 124(6): 875 – 881. doi: 10.1007/ s00401 ‑ 012 ‑ 1068 ‑ 3.
12. Paulus W, Kleihues P. Genetic profiling of CNS tumors extends histological classification. Acta Neuropathol 2010; 120(2): 269 – 270. doi: 10.1007/ s00401 ‑ 010 ‑ 0710 ‑ 1.
13. Khatua S, Brown R, Pearlman M et al. ETANTR – tumor with undefined biology. Neuro‑Oncology 2012; 14 : 143 – 148.
14. Korshunov A, Sturm D, Ryzhova M et al. Embryonal tumor with abundant neuropil and true rosettes (ETANTR), ependymoblastoma, and medulloepithelioma share molecular similarity and comprise a single clinicopathological entity. Acta Neuropathol 2014; 128(2): 279 – 289. doi: 10.1007/ s00401 ‑ 013 ‑ 1228 ‑ 0.
15. Robertson KD. DNA metylation, methyltransferases, and cancer. Oncogene 2001; 20(24): 3139 – 3155.
16. Ioshikhes IP, Zhang MQ. Large ‑ scale human promoter mapping using CpG islands. Nat Genet 2000; 26(1): 61 – 63.
17. Suganuma T, Workman JL. Crosstalk among histone modifications. Cell 2008; 135(4): 604 – 607. doi: 10.1016/ j.cell.2008.10.036.
18. Hamano R, Miyata H, Yamaski M et al. High expression of Lin28 is associated with tumour aggressiveness and poor prognosis of patients in oesophagus cancer. Br J Cancer 2012; 106(8): 1415 – 1423. doi: 10.1038/ bjc.2012.90.
19. Molenaar JJ, Domingo ‑ Fernandez R, Ebus ME et al. LIN28B induces neuroblastoma and enhances MYCN levels via let ‑ 7 suppression. Nat Genet 2012; 44(11): 1199 – 1206. doi: 10.1038/ ng.2436.
20. Spence T, Perotti C, Sin‑Chan P et al. A novel C19MC amplified cell line links LIN28/ let7 to mTOR signaling in embryonal tumor with multilayered rosettes. Neuro Oncol 2014; 16(1): 62 – 71. doi: 10.1093/ neuonc/ not162.
21. Huang J, Chen K, Huang J et al. Regulation of the leucocyte chemoattractant receptor FPR in glioblastoma cells by cell differentiation. Carcinogenesis 2009; 30(2): 348 – 355. doi: 10.1093/ carcin/ bgn266.
22. Spence T, Sin‑Chan P, Picard D et al. CNS ‑ PNETS with C19MC amplification and/ or LIN28 expression comprise a distinct histogenetic diagnostic and therapeutic entity. Acta Neuropathol 2014; 128(2): 291 – 303. doi: 10.1007/ s00401 ‑ 014 ‑ 1291 ‑ 1.
23. Komiya Y, Habas R. Wnt signal transduction pathway. Organogenesis 2008; 4(2): 68 – 75.
24. Braiteh F, Soriano AO, Garcia ‑ Manero G et al. Phase I study of epigenetic modulation with 5 - azacytidine and valproic acid in patients with advanced cancers. Clin Cancer Res 2008; 14(19): 6296 – 6301. doi: 10.1158/ 1078 ‑ 0432.CCR ‑ 08 ‑ 1247.
25. Sin‑Cahn P, Huang A. DNMTs as potential therapeutic targets in high‑risk pediatric embryonal brain tumors. Expert Opin Ther Targets 2014; 18(10): 1103 – 1107. doi: 10.1517/ 14728222.2014.938052.
Štítky
Dětská onkologie Chirurgie všeobecná OnkologieČlánek vyšel v časopise
Klinická onkologie

2015 Číslo 4
- Diosmin a hesperidin: Co ukazuje farmakokinetika?
- Zkušenosti s axitinibem v léčbě metastatického renálního karcinomu
- Evaluace skóre m7-FLIPI u pacientů s folikulárním lymfomem odhalila slibný prediktivní biomarker k určení vhodné chemoterapie
- Neuropatie výrazně zpomaluje proces hojení u povrchových ran
- Zeolit-jodový komplex pomáhá v péči o infikované rány
Nejčtenější v tomto čísle
- Extraoseální Ewingův sarkom, primární postižení děložního čípku – kazuistika
- Embryonálne tumory s mnohovrstvovými rozetami – vzácne tumory centrálneho nervového systému v detskom veku
- Domácí parenterální výživa v onkologii
- Potenciál volné cirkulující DNA v diagnostice nádorových onemocnění
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy