
Systémový lupus erythematosus – súčasný pohľad na genetickú determináciu, imunopatogenézu a liečbu
Systemic Lupus Erythematosus – A Contemporary View of its Genetic Determination, Immunopathogenesis and Therapy
Systemic lupus erythematosus (SLE) is an organ non-specific autoimmune disorder, with multiple immunopathogenic mechanisms being implicated in its development. The most conspicuous feature of the disease is an exaggerated synthesis of various types of autoantibodies, followed by the formation of immune complexes that deposit in tissues and elicit an inflammatory response. Apart from antibodies, dendritic cells, T cells and cytokines are substantially involved in the pathogenesis of SLE and class I interferons seem to play a crucial role. SLE is a genetically determined disease. HLA system and complement system genes, apoptosis regulating genes and IgG Fc-gamma receptor genes are among the multiple genes implicated in SLE. The role of hormones, both estrogen and progesterone, in SLE activity has been reported. Some monoclonal antibodies have recently proved effective in the treatment of SLE.
Key words:
autoantibodies, HLA complex – complement – interferon signature – monoclonal antibodies – SLE – T cells.
Autoři:
M. Buc 1; J. Rovenský 2
Působiště autorů:
Imunologický ústav Lekárskej fakulty UK, Bratislava
1; Národný ústav reumatických chorôb, Piešťany
2
Vyšlo v časopise:
Epidemiol. Mikrobiol. Imunol. 58, 2009, č. 1, s. 3-14
Souhrn
Systémový lupus erythematosus (SLE) patrí k orgánovo nešpecifickým autoimunitným chorobám, na ktorého rozvoji sa podieľajú početné imunopatogenetické mechanizmy. V popredí je nadmerná tvorba autoprotilátok a tvorba imunokomplexov s následným ukladaním do tkanív a indukciou zápalového procesu. Na jeho patogenéze sa okrem protilátok podstatnou mierou podieľajú aj dendritové bunky, T-lymfocyty a cytokíny, výrazný je najmä vplyv interferónov prvého typu. Ide o geneticky podmienenú chorobu; do mozaiky génov patria gény HLA-komplexu, gény komplementového systému, gény regulujúce apoptotický proces, determinujúce Fc-receptory pre IgG a iné. Chorobu výrazne modifikujú hormóny, okrem estrogénov aj progesterón. V liečbe SLE sa v ostatnej dobe začínajú presadzovať viaceré monoklonové protilátky.
Kľúčové slová:
autoprotilátky – HLA-komplex – interferónový podpis – komplement – monoklonové protilátky – SLE – T-lymfocyty.
1. Genetická podmienenosť vývoja SLE
Podobne ako ostatné autoimunitné choroby, aj SLE sa vyskytuje častejšie v rodinách. Konkordantnosť pre jednovaječné dvojčatá sa pohybuje okolo 34 %, pre dvojvaječné dvojčatá medzi 2–5 %. Silnú rodinnú predispozíciu najlepšie odráža rizikový index λs1, ktorý pri SLE dosahuje hodnotu 29,0. Napriek vysokej konkordantnosti choroby u jednovaječných dvojčiat, predsa len táto nedosahuje hodnotu 100 %, čo poukazuje na dôležitosť faktorov vonkajšieho prostredia pri vývoji choroby. Hoci SLE sa vyskytuje vo všetkých populáciách, jeho prevalencia je v odlišných etnických skupinách rozdielna. Černosi sú na SLE vnímavejší ako belosi (u nich je prevalencia 1 : 20002; je pritom zaujímavé, že africkí černosi nemajú tak vysokú prevalenciu SLE ako americkí, čo opäť poukazuje na superimponovanie faktorov prostredia nad genetickú predispozíciu k chorobe [7, 16, 24].
Genetická determinácia SLE je komplexná. Jej pochopenie značne uľahčila existencia myší, ktorých samičia polovica spontánne vyvíja chorobu, ktorá sa veľmi podobá SLE. Ide o myši prvej generácie (F1), ktoré vznikli krížením inbrédnych čiernych (black) a bielych (white) myší, pôvodom z Nového Zélandu [(NZB x NZW)F1]. Rodičovské kmene NZB trpeli miernou formou autoimunitnej hemolytickej anémie, myši rodičovského kmeňa NZW tvorili zase anti-DNA protilátky. Avšak až kombinácia genetického vybavenia pochádzajúceho z oboch rodičov v F1-generácii podmienila plné prepuknutie choroby [9].
Podobná situácia existuje aj pri iných dvoch inbrédnych kmeňoch myší – MLR lpr/lpr a MLR gld/gld. myši MLR lpr/lpr majú defektný gén pre molekulu Fas, myši gld/gld zase mutáciu v géne pre ligand Fas-molekuly, t.j. FasL. Ide o molekuly, ktoré regulujú apoptotický proces, ktorým sa odstraňujú aktivované bunky (najmä lymfocyty CD8+, CD4+, B-lymfocyty a makrofágy) po splnení ich efektorovej funkcie a to práve preto, aby nespôsobili autoimunitné poškodenie organizmu. Ich prežívanie pre poruchu apoptotických mechanizmov im umožňuje pokračovať v svojej efektorovej aktivite, ktorá sa prejaví napr. pretrvávajúcou produkciou (auto)protilátok, s následným vývojom imunopatologického poškodenia. Dôležitý je však poznatok, že genotyp lpr/lpr, resp. gld/gld indukuje imunopatologické zmeny iba pri myšiach MLR. Mutácie génov Fas, resp. FasL pri myšiach iných inbrédnych kmeňov k týmto zmenám neviedlo [31]. Tieto výsledky naznačujú, že gény Fas, resp. FasL sú jedni z viacerých, ktoré determinujú vývoj choroby a že mutácie génov Fas, resp. FasL iba urýchľujú začatý proces. Takýchto „imunopatologických“ génov sa pri myšiach doteraz našlo 11 a možno predpokladať, že sa objavia aj iné [9, 31]. Za zmienku snáď ešte stoja myši, ktorým sa cielene znefunkční gén (myši „knock out“) pre sérový amyloid P (SAP). Tieto tiež spontánne vyvinú chorobu podobnú SLE s početnými autoprotilátkami a ťažkú glomerulonefritídu [19]. Fyziologická úloha SAP je viazať apoptotické telieska a tak sa podieľať na ich ľahšom odstraňovaní.
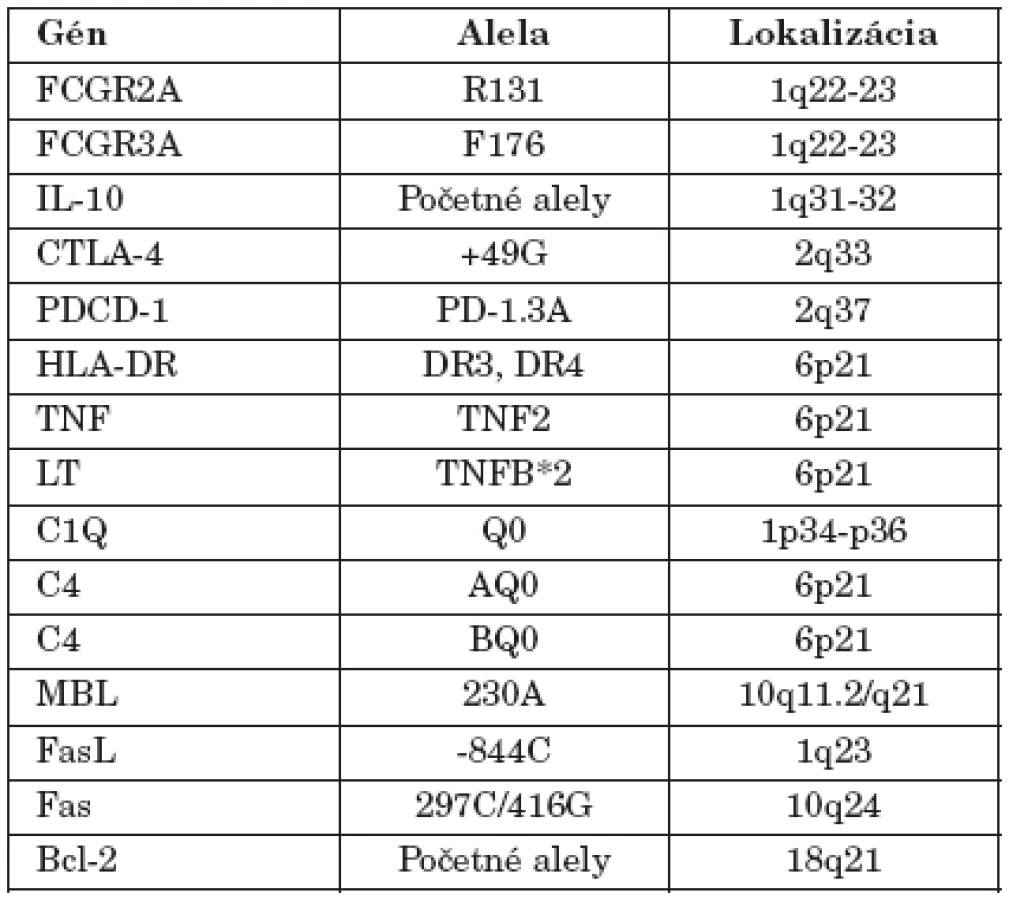
Podobná situácia je aj u človeka, aj keď genetická predispozícia k chorobe sa tu zisťuje omnoho ťažšie. Tak isto sa potvrdilo, že aj u niektorých chorých s SLE dochádza k mutáciám v génoch Fas (CD95), resp. FasL (CD95L) [40]. Z ďalších doteraz známych génov sa na genetickej determinácii podieľajú gény HLA-komplexu, gény pre niektoré zložky komplementového systému resp. ich receptory, pre receptory pre Fc-fragment IgG a receptory pre niektoré cytokíny. Penetrancia každého z uvedených prispievajúcich génov je pomerne malá, čo je príčinou toho, že SLE je tak komplexne determinovaná choroba [24, 33].
Štandardne genetickú predispozíciu k autoimunitným chorobám a teda aj SLE, ovplyvňujú gény HLA-komplexu, aj keď možno konštatovať, že pri SLE táto asociácia nie je, na rozdiel od iných chorôb, najvýraznejšia, čo naznačuje, že podiel HLA-oblasti na rozvoji choroby sa rovná podielu iných génov. S predispozíciou k SLE v kaukazoidnej populácii sa spájajú dva haplotypy – HLA-DRB1*1501 (determinuje molekulu DR15)3, -DQB1*0602 (kóduje DQ6) a HLA-DRB1*0301 (determinuje DR3), -DQB1*0201 (kóduje DQ2). Alely DRB1*0301, -DQB1*0201 sú súčasťou rozšíreného HLA-haplotypu, tak typického pre kaukazoidnú populáciu – HLA-A1, -B8, -DR3, v ktorom sa navyše nachádzajú aj ďalšie SLE-predisponujúce alely, tie, ktoré patria komplementovému, resp. cytokínovému systému, (C4A*Q0, resp. TNF2), takže kompletný predisponujúci haplotyp s relatívnym rizikom 2,6 v heterozygótnom, resp. 6,8 v homozygótnom stave, má nasledovnú skladbu génov: HLA-A*01, -Cw*07, B*08, C4AQ*0, C4B*1, TNF2, DRB1*0301, DQB1*0201 [7, 41].
Najvýznamnejšia asociácia SLE je však s génmi komplementového systému, v poradí C1q > C4 > C2. Jedinci, ktorým chýba C1q sú veľmi náchylní na vývoj SLE, až 90 % osôb, ktorým chýba gén pre C1q na oboch chromozómoch (1p34.1-p36.3), vyvinie SLE. Príčina tejto asociácie je úloha C1q pri likvidácii apoptotických buniek. Jeho chýbanie je však veľmi zriedkavé, doteraz sa celosvetovo identifikovalo približne iba 40 chorých [10, 11].
Druhá významná asociácia SLE s génmi komplementového systému sa týka alely C4AQ*0. C4-zložku komplementového systému kódujú dva gény – C4A a C4B, ktoré sa nachádzajú v HLA-genetickej oblasti, medzi lokusmi HLA-DR a HLA-B (obr. 1). Oba lokusy sú polymorfné, striedajú sa na nich viaceré alely. Alela C4AQ*0, resp. C4BQ*0, označuje skutočnosť, že nekóduje nijaký proteín. Jednotlivá nulová alela sa vyskytuje asi u 30 % ľudí4 pričom C4AQ*0-homozygótnych jedincov je asi 4 % a C4BQ*0-homozygotov približne 1 %; SLE vyvinie až 75 % C4AQ*0 osôb. Proteíny (izotypy) C4A a C4B sú funkčne rovnaké, i keď mierne rozdiely medzi nimi existujú: C4A-izotyp sa viaže na proteíny, napr. na imunokomplexy, lepšie ako izotyp C4B, čo vysvetľuje významnosť asociácie SLE s C4AQ*0 – deficiencia C4A-izotypu vedie k menej efektívnej zábrane tvorby imunokomplexov, ktoré majú pri tejto chorobe tak významnú úlohu v patogenetických procesoch. Alela C4AQ*0 je súčasťou vyššie uvedeného charakteristického HLA-haplotypu, ktorý sa spája aj so zníženou expresiou Fas-molekuly (CD95) na lymfocytoch SLE-pacientov, čo prispieva k neúčinnej eliminácii autoreaktívnych buniek apoptotickým procesom [10, 11].

Chýbanie C2-zložky komplementového systému tiež predisponuje k SLE. Deficiencia C2 je v kaukazoidnej populácii pomerne častá, vyskytuje sa približne u 1 z 10 000 jedincov, ale len asi 20 % C2-deficientnych osôb vyvinie SLE. C2-deficiencia sa v kaukazoidnej populácii v absolútnej väčšine spája s haplotypom HLA-A25, -B18, -DR2 (DRB1*1501), -DQ6 (DQB1*0602) [11]. Význam zložiek komplementu v rozvoji SLE podčiarkuje aj jeho indukcia pri myšiach, ktorým chýba CR1 (CD35) (receptor pre C3b, C4b, iC3b a iC4b) [11].
Nemenej významná je aj asociácia s nízkoafinitnými Fc-receptormi pre IgG. Gény pre jednotlivé receptory sa nachádzajú v 10 kb oblasti na dlhom ramene 1. chromozómu (1q22 –23), v poradí FCGR2A, FCGR3A, FCGR3B a FCGR2B, pričom prvé dva sú asociované s SLE [41, 45].
FCGR2A (FcγRIIa) sa nachádza na makrofágoch, neutrofiloch, eozinofiloch, Langerhansových bunkách. Jeho karboxylový koniec obsahuje ITAM (immunoreceptor tyrosine-based activating motif), ktorý po väzbe ligandu na receptor, prenáša signál a aktivuje procesy fagocytózy. Sú známe dve alely FcγRIIa, jedna determinuje v pozícii 131 (glyko)proteínového reťazca histidín (H131) a druhá arginín (R131). Receptor R131 viaže IgG2 horšie ako receptor H131 a práve táto alela sa spája s SLE, riziko vývoja SLE je 1,3. Nedávno sa dokázalo, že R131 (ale nie H131) vysokoafinitne viaže C-reaktívny proteín (CRP). Pretože CRP sa konštantne nachádza v renálnych imunokomplexových depozitoch, podobne ako IgG2, je pravdepodobné, že R131 môže prispievať k riziku proliferačnej lupusovej nefritídy. Homozygoti R131 navyše majú zvýšené riziko na infekcie baktériami druhu Streptococcus pneumoniae, ktoré vyvolávajú zápaly pľúc [8, 45].
FCGR3A (FcRIIIa = CD16) sa exprimuje v membránach NK-buniek, monocytov a makrofágov a viaže aj IgG1 aj IgG3. Existujú tiež dve alely – alela determinujúca v pozícii 158 reťazca fenylalanín (F) a alela, ktorá v tejto pozícii kóduje valín (V). Jedinci homozygotní pre F-alelu viažu IgG1 a IgG3 menej účinne (nízkoafinitný receptor) ako jedinci s genotypom VV (vysokoafinitný receptor), čo ich opäť predisponuje k vývoju lupusovej nefritídy [8, 45].
Aj polymorfizmus promótorovej oblasti génu pre faktor nekrotizujúci nádory (TNF – tumour necrosis factor) sa tiež spája s rizikom vývoja SLE. Alela, ktorá má v pozícii –308 adenín (TNF2), namiesto bežného guanínu (TNF1), determinuje vyššiu produkciu TNF; homozygoti pre TNF2 produkujú až päťnásobne viac TNF.
Existujú údaje aj o iných génoch, ktoré sa podieľajú na predispozícii k SLE. doteraz sa ich identifikovalo vyše 40 (tab. 1) a možno ich zoradiť do troch skupín: gény determinujúce odstraňovanie apoptotických teliesok (napr. FcR, SAP, CRP, C1q), regulujúce apoptotický proces (napr. TNF, Fas, FasL, Bim, Bcl-x) a napokon gény, ktoré ovplyvňujú aktiváciu a expanziu lymfocytov (napr. HLA, IL-10, CD19, CD22, CD45, PD1, TACI) [20, 24, 30, 33].
2. Hormonálne faktory pri vývoji SLE
Okrem genetických faktorov na vývoji SLE sa výrazne podpisujú aj hormóny. Pomer žien k mužom trpiacich na SLE je 9 : 1, pričom najviac sú postihnuté ženy v reprodukčnom veku. O vplyve estrogénov na vývoj SLE svedčia aj údaje o vyššej frekvencii vývoja SLE u postmenopauzových žien berúcich estrogénové prípravky. Podobne, liečba chorých žien so ľahšou a strednou formou SLE s dehydro-epiandrosterónom (DHEA), adrenálnym steroidom s miernou androgénnou aktivitou, mala priaznivý efekt [5, 17, 44]. Ženy trpiace na SLE majú totiž v svojej plazme zvýšenú hladinu enzýmu aromatózy, ktorý znižuje hladinu disponibilných androgénov tým, že ich konverguje na estrogény [43].
Tehotenstvo tiež nie je nečastou udalosťou u SLE-žien. Choroba sa u niektorých nich zhorší, resp. ak bola predtým v pokojnom štádiu, môže znovu prepuknúť. Tehotenstvo sa od tehotenstva u zdravých žien líši nižším počtom prežívajúcich plodov, vyšším počtom predčasných pôrodov alebo oneskoreným vnútromaternicovým vývinom plodu [17, 44].
Experimenty na myšiach tiež dokazujú, že estrogény zvyšujú vnímavosť k chorobe. Naopak, ovariektómia myši pred vývojom choroby chráni. Príčinný vzťah medzi estrogénmi a imunitným systémom je daný tým, že estrogény podporujú syntézu cytokínov (IL-4, IL-6, IL-10), ktoré stimulujú B-lymfocyty, aby vo zvýšenej miere produkovali protilátky a potláčajú bunkovú imunitu [13].
Okrem estrogénov priebeh SLE môže ovplyvňovať aj prolaktín. Dôkaz o vplyve tohto hormónu sa získal na myšiach [(NZB x NZW)F1]; ak sa im transplantovala ďalšia hypofýza, koncentrácie prolaktínu sa zvýšili 3 až 18 násobne, nastal vzostup anti-dsDNA protilátok a zvýšila sa mortalita [29]. U časti chorých s SLE (20–30 %) sa tiež častejšie deteguje hyperprolaktémia a hladina prolaktínu u niektorých z nich korelovala s aktivitou choroby [21, 22].
3. Poruchy imunity
SLE sa všeobecne považuje za chorobu, pri ktorej za vývoj klinických príznakov zodpovedajú priamo alebo nepriamo protilátky. V skutočnosti pri SLE ide o komplexnejšiu poruchu v imunitnom systéme. Časť autoprotilátok vzniká určite na základe nešpecifickej aktivácie viacerých klonov B-lymfocytov bez účasti T-lymfocytov; dôkazom môžu byť zvýšené hladiny imunoglobulínov [15, 27]. Početné autoprotilátky pri SLE sú však antigénovo špecifické, patria do triedy IgG. Ďalej, pri analýze genetickej determinácie tvorby autoprotilátok pri myšiach lpr/lpr, sa dokázali normálne procesy preskupovania imunoglobulínových génov, somatické mutácie, afinitná maturácia, priešmyk z IgM na IgG, t.j. ako pri fyziologickej odpovedi na antigén [13]. Znamená to, že na indukciu ich produkcie sú potrebné T-lymfocyty a celý autoimunitný proces zakladá tá istá základná trojica buniek ako je tomu pri fyziologickej imunitnej odpovedi na exogénny antigén proteínovej povahy, t.j. bunka prezentujúca antigén (APC), T-lymfocyt a B-lymfocyt. APC prostredníctvom svojich HLA-molekúl druhej triedy prezentuje antigén, resp. peptid, ktorý z neho pochádza, T-lymfocyt na jednej strane rozpoznáva prezentovaný peptid, aktivuje sa a na strane druhej kooperuje s B-lymfocytom, ktorý po získaní patričných signálov sa diferencuje na plazmatickú bunku, produkujúcu špecifické (auto)protilátky [8, 18].
Pri analýze cytokínového profilu sa zistilo, že pri rozvoji SLE sa zúčastňujú interferóny prvej triedy, najmä IFN-α. Jeho hladina je plazme chorých s SLE zvýšená a pacienti, ktorí sa liečia IFN α2 (chronická hepatitída B, C, chronická myeloidná leukémia, malígny melanóm, renálny karcinóm a iné) niekedy ako komplikáciu liečby vyvinú SLE. Navyše pri štúdiu expresie génov mikročipovou metódou sa zistilo, že u SLE chorých sú nápadne aktivované gény, ktorých expresiu podmieňujú práve interferóny; naopak ich expresia sa potlačí, ak sa pacienti liečia vysokými dávkami glukokortikoidov, ktoré syntézu IFN-α inhibujú; hovorí sa o interferónovom podpise. IFN-α podporuje diferenciáciu monocytov do dendritových buniek, ktoré sú najúčinnejším aktivátorom pomocných T-lymfocytov (TH) v primárnej imunitnej odpovedi na antigén. Aktivované T-lymfocyty následne stimulujú autoreaktívne B-lymfocyty, aby produkovali protilátky, najmä tie, ktoré sú namierené proti DNA a nukleoproteínom. Následné zvýšené hladiny cirkulujúcich imunokomplexov pôsobia ako faktor indukujúci IFN-α, čím sa bludný kruh uzatvára [3, 4, 6]. Okrem IFN-α sa v plazme chorých vyskytujú aj zvýšené hladiny receptora pre IL-2, čo svedčí o aktivácii T-lymfocytov a zvýšené hladiny molekúl ICAM-1 a CD40L, ktoré korelujú so závažnosťou choroby [23].
Základnou otázkou je, ktoré autoantigény sú prezentované a prečo dochádza k poruche periférnej tolerancie, keďže u zdravých ľudí existujú viaceré mechanizmy, ktoré autoreaktívne T-lymfocyty „držia na uzde“. niektoré autoantigény sú známe, píše sa o nich nižšie, v časti o autoprotilátkach. Čo je však na samom začiatku aktivácie týchto T-lymfocytov, rozpoznávajúcich, ale nereagujúcich na autoantigény do autoagresívneho stavu? Najčastejšie sa za spustenie tohto „circulus vitiosus“ obviňujú mikroorganizmy a známy fenomén molekulového mimikri. Vskutku existujú epidemiologické údaje, že chorí s SLE prekonali častejšie infekcie spôsobené vírusom Epsteina a Barrovej (EBV); prirodzene, okrem EBV imunopatologické procesy môže spustiť aj iný mikroorganizmus. V súčasnosti sa však viac zo spustenia autoimunitných procesov obviňuje porucha odstraňovania apoptotických teliesok, ktoré vznikajú pri fyziologickej obnove vlastných tkanív, imunitnej odpovedi a zápalových procesoch. Tieto namiesto makrofágov pohlcujú dendritové bunky, potentné induktory primárnej odpovede. Tieto pohltený materiál spracujú a prezentujú T-lymfocytom, ktoré po prekročení určitého prahu sa aktivujú; v ďalšom kroku tieto kooperujú s B-lymfocytmi a stimulujú ich do produkcie autoprotilátok so všetkými následnými patologickými dôsledkami (obr. 2) [25].
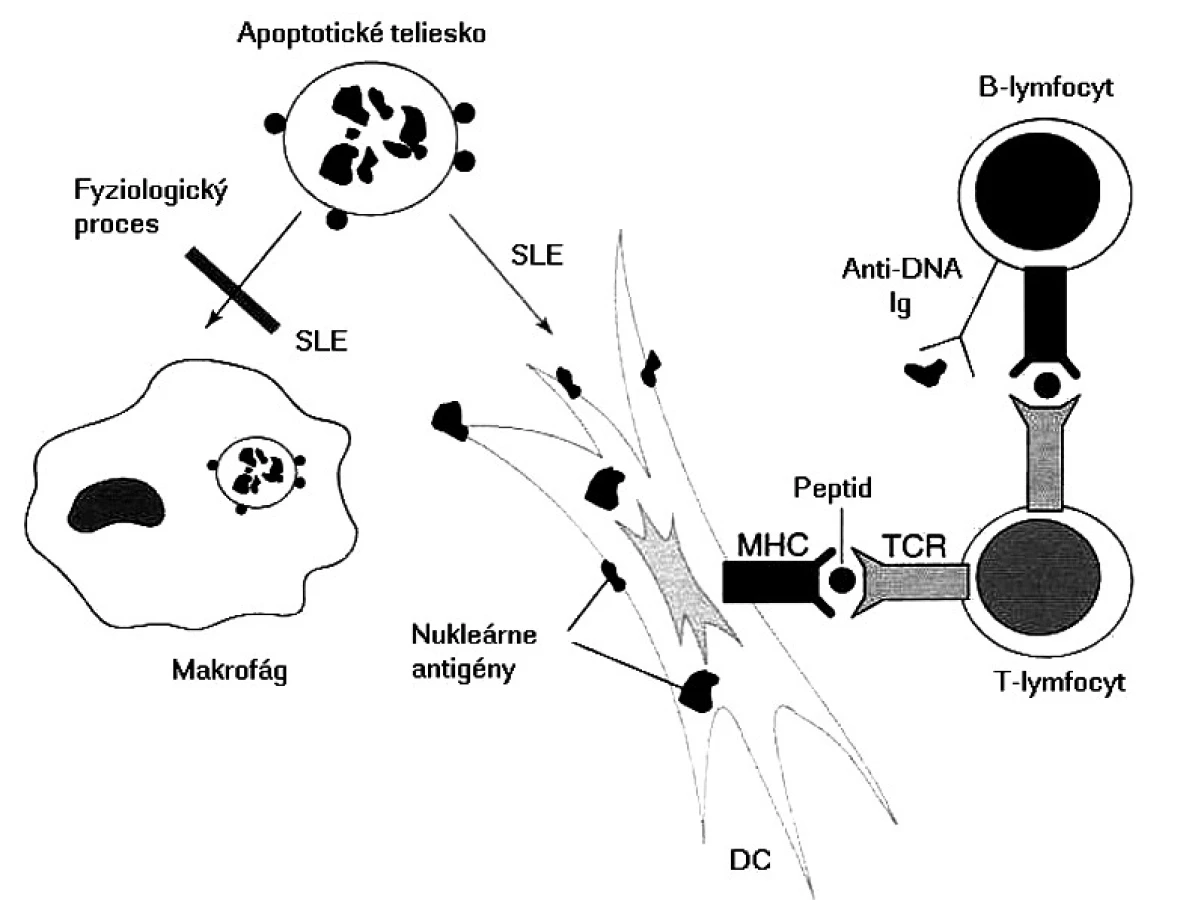
Najnovšie výskumy poukazujú na možné prepojenie prirodzenej a adaptívnej imunity. Zistilo sa, že k aktivácii B-lymfocytov a následnej tvorbe autoprotilátok dochádza vtedy, keď B-lymfocyty sa súčasne aktivujú cez dva typy receptorov – imunoglobulínové (BCR – Bcell receptor) špecificky rozpoznávajúce antigény a Toll-like receptory (TLR), ktoré rozpoznávajú vzorkové antigény mikroorganizmov (PAMPs pathogen-associated molecular patterns). Experimentálne práce dokázali, že anti-IgG protilátky (= reumatoidný faktor) vznikali stimuláciou B-lymfocytov imunokomplexmi, ktoré obsahujú DNA. BCR, ktorý rozpoznáva imunoglobulínovú časť imunokomplexu, sprostredkoval prvý signál a TLR9, ktorý špecificky rozpoznáva DNA časť imunokomplexu, sprostredkoval druhý signál; možno, že aj u chorých s SLE, početné protilátky vznikajú aj týmto mechanizmom [26, 32].
Pre SLE je charakteristická nielen polyklonová hyperaktivita B-lymfocytov, ale aj výrazná porucha ich homeostázy. Nápadná je najmä redukcia CD27- buniek (ide o naivné B-lymfocyty), kým počty CD27+ buniek (pamäťové B-lymfocyty) sú zvýšené5. Obzvlášť je zvýšený počet buniek exprimujúcich tento diferenciačný antigén vo veľkej miere (CD27high); ďalej exprimujú CD38, CD95, ale nie CD20, resp. CD5 a len veľmi málo CD19. Úspešná imunosupresívna terapia vedie k ich dramatickému zníženiu, čo má praktický dopad na diagnózu a terapiu SLE. Cytometrické monitorovanie B-lymfocytovej populácie, využijúc znak CD27 v spojení s CD19, poskytuje takto diagnostický parameter aktivity choroby [32].
V diagnostike možno využiť aj sledovanie hladín IL-10. U SLE-pacientov sa vo zvýšenej miere tvorí IL-10, ktoré hladiny odrážajú klinický priebeh choroby a korelujú s titrom anti-dsDNA protilátok. IL-10 syntetizujú monocyty stimulované imunokomplexmi a podporuje proliferáciu a diferenciáciu B-lymfocytov [24, 27].
Našli sa početné poruchy aj na úrovni T-lymfocytov. Predovšetkým pomocné T-lymfocyty sú aktivované, v svojich membránach majú napr. zvýšenú expresiu kostimulačných molekúl CD40, ktoré po interakcii so svojim ligandom CD40L (CD154) na B-lymfocytoch ich stimulujú, aby produkovali viac autoprotilátok. Príčina aktivovaného stavu pomocných T-lymfocytov spočíva v zmene štruktúry ich antigénového receptoru. Kým T-lymfocyty zdravých osôb exprimujú TCR v asociácii s 5 reťazcami diferenciačného antigénu CD3 (α, δ, ε a homodimér ζ, prípadne heterodimér η, ζ), tak T-lymfocyty SLE-chorých majú namiesto ζ-homodiméru, FcR-homodimér, ktorý za fyziologických okolností tvorí súčasť vysokoafinitného Fc-receptora pre IgE (FcεRI). Biologická funkcia CD3ζ-homodiméru spočíva v prenose signálu do vnútra bunky, ktorý zabezpečuje s ním asociovaná kináza ZAP-70 (obr. 3). Podobnú funkciu pri FcεRI zabezpečuje γ-homodimér, s ktorým sa spája Syk-kináza [8]. Zásadný rozdiel medzi reťazcami CD3ζ a FcRγ je v ich účinnosti: prenos signálov komplexu FcRγ-Syk je až 100-krát vyšší ako prenos signálov komplexu CD3ζ-ZAP70. Táto výmena reťazcov spôsobuje, že prah aktivácie T-lymfocytov u SLE-chorých je podstatne nižší ako pri T-lymfocytoch zdravých osôb. Príčinou výmeny CD3ζ-homodiméru za FcRγ-homodimér je nedostatočná syntéza CD3ζ-reťazca v dôsledku nedostatočnej glykozylácie transkripčného faktora Elf-1, čo mu zabraňuje viazať sa na promótorovú oblasť CD3ζ-génu. Znížená syntéza CD3ζ-reťazca je pravdepodobne aj príčinou zníženého počtu T-lymfocytov, ktorý sa u chorých s SLE často pozoruje [42).

Druhá výrazná porucha na úrovni pomocných T-lymfocytov chorých spočíva v nedostatočnej tvorbe IL-2, z čoho vyplýva následná znížená aktivácia aj cytotoxických T-lymfocytov aj NK-buniek; o.i. aj tým sa vysvetľuje zvýšená vnímavosť SLE-chorých na infekcie. Príčina tohto stavu je v nedostatočnej, resp. chybnej tvorbe transkripčného faktora NFκB. Za fyziologických okolností sa skladá z heterodiméru p50 a p65, pri SLE p65 nahrádza p50, čím vzniká homodimér p50 p50, ktorý na IL-2 promótor pôsobí skôr represívne ako aktivačne. Príčinou je pravdepodobná zvýšená degradácia p65-reťazca kaspázou 8, pretože tento proteín je pre ňu jedným zo substrátov a pretože pri SLE je zvýšená spontánna apoptóza [38].
Napokon pozoroval sa aj znížený počet prirodzených regulačných T-lymfocytov v periférnej krvi SLE-chorých, čo určite tiež prispieva k celkovému hyperaktívnemu stavu imunitného systému pri tejto chorobe [12, 28].
Celkovo súčasnú predstavu o imunopatogenéze SLE zachytáva obr. 4.

3. Charakter autoprotilátok
SLE charakterizujú početné autoprotilátky, ktoré sú namierené proti viac ako 100 rôznym autoantigénom. Typické je, že sa v plazme chorého objavujú už mnoho rokov pred prepuknutím klinických príznakov, pred určením diagnózy [16, 24, 35, 36].
Antinukleárne protilátky (ANA) sa detegujú nepriamou imunofluorescenciou, pričom možno rozlíšiť 3 základné obrazy (homogénny, membránový a škvrnitý) – tab. 2. Detekcia ANA nie je veľmi špecifická, ale zato vysoko senzitívna metóda. Negatívny výsledok skoro vylučuje diagnózu SLE (95 % chorých je pozitívnych na ANA), kým vysoký titer protilátok je s veľkou pravdepodobnosťou naznačuje, že ide o SLE. Treba však podotknúť, že samotný dôkaz ANA diagnózu SLE nepotvrdzuje, lebo tieto protilátky možno dokázať aj pri iných systémových autoimunitných chorobách a pri chronických infekciách.

Protilátky proti DNA sú pri SLE najdôležitejšie, pretože ich v svojej plazme má až 2/3 chorých. Môžu reagovať s jednovláknovou DNA (ssDNA – single-stranded DNA) alebo s dvojvláknovou DNA (dsDNA – double-stranded DNA). Hoci protilátky anti-ssDNA sa vyskytujú aj pri iných chorobách ako SLE, protilátky anti-dsDNA sa nachádzajú takmer výlučne iba pri SLE (u 40–60 % chorých).
Svojráznosťou pri SLE je nález tzv. LE-buniek. LE-bunka je polymorfonukleárny leukocyt, ktorý pohltil uvoľnený jadrový materiál iného leukocytu. Za LE-fenomén zodpovedajú protilátky proti nukleozómu. Tieto, spolu so zložkami komplementu, opsonizujú uvoľnenú DNA, ktorú napokon pohltí fagocyt. LE-bunka má veľké bledé fagocytované jadro, okolo ktorého sa nachádza malé množstvo cytoplazmy. Pôvodné jadro fagocytujúceho leukocytu je zatlačené k bunkovej membráne.
Viac ako 65 % chorých s SLE má v svojej plazme protilátky proti histónom, a to proti všetkým 5 typom (H1, H2A, H2B, H3 a H4). Okrem nich sú známe aj protilátky proti nehistónovým proteínom, ktoré sú súčasťou aparátu, ktorý sa podieľa na úprave heterogénnej RNA; úprava spočíva vo vyštepovaní intrónov a spájaní exónov do funkčného reťazca mrna. Anglický výraz pre vyštepovanie je „splice“, a preto sa tento aparát v anglickej literatúre označuje ako „splicesome“, slovensky „vyštepovač“. Vyštepovač je komplex ribonukleovej kyseliny bohatej na uridín (existuje 5 rozdielnych typov, označujú sa U1, U2, U4-6, U5 alebo U6), ktorá sa spája so skupinou proteínov (70, A, B/B1, C, D, E, F, G; všeobecne, spolu s antigénmi Ro a La /pozri ďalej/, sa označujú aj ako ENA – extrahovateľné nukleové antigény) (obr. 5). Pri SLE vznikajú protilátky proti týmto proteínom a označujú sa ako anti-Sm a anti-U1-RNP. Kým protilátky anti-Sm sú namierené proti proteínom asociovaných s RNA U1, U2, U4-6 alebo U5, tak anti-U1-RNP len proti proteínu, ktorý sa spája s U1-RNA.

Ďalšie malé RNA (označované ako hY1 – hY5) kooperujú s polymerázou RNA-III pri transkripcii mRNA. Nachádzajú sa v cytoplazme, niektoré aj v jadre a spájajú sa s proteínmi SS-A/Ro (Mr 60 000, alebo 52 000 ) a SS-B/La (Mr 48 000) (obr. 6), buď len s jedným z nich, alebo s oboma. Protilátky špecifické pre tieto proteíny sa vyskytujú aj u chorých so Sjögrenovým syndrómom.

Protilátky anti-Sm sa vyskytujú asi u jednej tretiny chorých a sú pre SLE špecifické, pri iných chorobách sa nenašli; ich detekcia má preto diagnostickú hodnotu.
Protilátky anti-U1-RNP nie sú pre SLE charakteristické, pretože sa vyskytujú aj pri iných chorobách spojivového tkanivá. Chorí s týmito protilátkami typicky nevytvárajú anti-dsDNA protilátky, priebeh choroby je mierny, obličky bývajú postihnuté iba výnimočne.
Protilátky anti-SSA/Ro sa vyskytujú aj bez protilátok anti-SSB/La, ale nie naopak. Vyskytujú sa u tretiny chorých s SLE a dvoch tretín chorých trpiacich na Sjögrenov syndróm [36]. Protilátky anti-Ro sa častejšie detegujú u tých chorých, ktorí nemajú ANA. Deti, ktoré sa narodia matkám s protilátkami anti-Ro môžu trpieť poruchami frekvencie srdca, leukopéniou a dermatopatiami. U matiek s protilátkami namierenými proti Ro-antigénu s Mr 52 000 sa dokázala signifikantná asociácia medzi prítomnosťou týchto protilátok s kompletným srdcovým atrio-ventrikulovým blokom ich detí [14].
R. 2004 sa objavila informácia, že myši, ktorým sa znefunkčnil gén pre Ro (Ro-/-) vyvinú chorobu podobnú SLE u ľudí [14, 36]. Ide o prvý prípad choroby, kedy chýbanie molekuly, ktorá má určitú fyziologickú funkciu, spôsobí vývoj autoimunity. Akú úlohu môže mať Ro-proteín? Výsledky naznačujú, že sa podieľa na správnej funkcii 5S ribozómovej RNA (5S rRNA), pretože zle poskladaná 5S rRNA sa spája s Ro-proteínom, ktorý ju takto označí, ako molekulu, ktorá je nefunkčná a ktorú treba odstrániť. Pri chýbaní Ro-proteínu nedochádza k identifikácii a následnému odstraňovaniu apoptotických teliesok obsahujúcich pozmenenú 5S rRNA. Táto porucha sa takto pripája k mechanizmom porúch odstraňovania apoptotických teliesok pri SLE, čo sa ukazuje ako jeden z najdôležitejších mechanizmov imunopatogenézy choroby. Hlavné miesto fyziologického pôsobenia Ro-proteínu bude pravdepodobne v koži, kde sa zúčastňuje na odstraňovaní apoptotických teliesok vznikajúcich po UV-žiarení. Anti-Ro protilátky u SLE-chorých môžu s týmto procesom interferovať, čo by vysvetľovalo ich zvýšenú fotosenzitivitu [14, 36].
Protilátky anti-SSB/La sa vyskytujú u tretiny chorých s SLE a približne polovice chorých trpiacich na Sjögrenov syndróm.
Primárny antifosfolipidový syndróm (PAS) existuje ako samostatná klinická jednotka, alebo je súčasťou inej autoimunitnej choroby, najčastejšie SLE. Napokon sa u niektorých chorých vytvárajú protilátky proti trombocytom, resp. jednotlivým faktorom zrážania krvi, čo spôsobuje ich náchylnosť na krvácavé stavy. Protilátky proti erytrocytom zodpovedajú za hemolytickú anémiu, ktorá sa môže u niektorých chorých tiež vyvinúť [16].
4. Klinické príznaky podmienené patogenetickým pôsobením protilátok
Hlavným patogenetickým faktorom pri vývoji klinických príznakov SLE sú protilátky. Pestrosť klinických príznakov je daná práve heterogénnosťou produkovaných protilátok a ich imunopatologického pôsobenia (tab. 3). Ako sa uvádza vyššie, autoimunitná trombocytopénia pri SLE je výsledkom pôsobenia antitrombocytových protilátok. Podobne autoimunitná hemolytická anémia je následkom deštrukcie červených krviniek IgG-autoprotilátkami. Protilátky proti neurónom môžu byť nejakým spôsobom zodpovedné za niektoré neurologické zmeny (poruchy citlivosti, rôzne parézy), imunokomplexy usadené v plexus chorioideus a vaskulitídy mozgových ciev môžu zase zodpovedať za psychické zmeny (od poruchy správania sa až po ťažké psychotické stavy), ktoré sa pozorujú u niektorých SLE-chorých (neuropsychický lupus). Protilátky SSA/Ro sa spájajú s fotosenzibilitou a vývojom dermatologických príznakov (pozri vyššie) [16, 24].

Najvážnejšou komplikáciou pri SLE je lupusová glomerulonefritída. Existuje množstvo dôkazov, že ju podmieňuje ukladanie cirkulujúcich imunokomplexov do steny glomerulov. Objavuje sa najmä u chorých, ktorý majú vysokoafinitný Fc-receptor pre IgG determinovaný alelou FcRIIIaV [2, 45].
Komplexy antigénu s protilátkou (imunokomplexy) vznikajú v organizme pri každom stretnutí protilátky so solubilným antigénom, ktoré sa vo väčšine prípadov vychytávajú a odstraňujú bunkami mononukleárno-fagocytového systému [8]. Za určitých okolností však tvorba imunokomplexov môže mať imunopatologické dôsledky. Závisí to predovšetkým od relatívnych pomerov medzi antigénom a protilátkou (absolútne množstvo určuje intenzitu reakcie). Pri nadbytku protilátok vytvorené imunokomplexy rýchlo precipitujú, zostávajú lokalizované v mieste prieniku antigénu do organizmu a degradujú sa vo fagocytoch. Naproti tomu pri nadbytku antigénu, ako je tomu aj pri SLE, sa utvárajú malé a stredné imunokomplexy, ktoré majú tendenciu k depozícii do tkanív a orgánov s ich následným patologickým poškodením. Imunokomplexy sa ukladajú najmä do stien artérií, glomerulov a synovie kĺbov, vyvolávajúc tak vaskulitídy, glomerulonefritídy a artritídy. Preferencia ukladania sa imunokomplexov do glomerulov a synovie vyplýva zo skutočnosti, že na týchto miestach pod značným hydrostatickým tlakom dochádza cez stenu kapilár k filtrácii plazmy, aby sa vytvoril moč, resp. synoviová tekutina. Rozsah depozície imunokomplexov závisí aj od celkovej schopnosti organizmu odstraňovať ich z cirkulácie. Zbavovanie sa imunokomplexov závisí jednak od funkčného stavu mononukleárno - -fagocytového systému a jednak od väzby proteínov komplementového systému na imunokomplexy, ktoré zvyšujú ich odstraňovanie. Pri poruche fagocytózy sa pozoruje o. i. aj perzistencia imunokomplexov a ich depozícia do tkanív. Podobne je tomu pri deficiencii niektorých zložiek komplementového systému, ako sa o tom píše vyššie [7].
Vytvorené imunokomplexy vyvolávajú v organizme celý rad zápalových procesov. Môžu reagovať s komplementom, kedy počas kaskádovitých aktivačných reakcií vznikajú fragmenty C3a a C5a, ktoré majú anafylaktoidné a chemotaktické vlastnosti. Spôsobia jednak uvoľnenie histamínu z mastocytov s následným zvýšením cievnej permeability a jednak priťahujú neutrofilné leukocyty. Tieto sa snažia pohlcovať imunokomplexy6, ale pre ich pevné uchytenie v tkanive sa fagocytom túto ich funkciu darí plniť len ťažko. V dôsledku toho fagocyty začnú uvoľňovať proteolytické enzýmy a intermediálne produkty kyslíka, ktoré poškodzujú tkanivo (obr. 7). Za vhodných podmienok môže nastať aj agregácia trombocytov, čo spôsobí uvoľnenie ďalších vazoaktívnych látok; môžu vznikať aj mikrotromby, ktoré zapríčinia lokálnu ischémiu. Morfologicky sa imunokomplexové poškodenie tkaniva prejaví nekrózou, ktorá často obsahuje fibrín, a preto sa aj označuje ako fibrinoidná nekróza. Typický je aj celulárny infiltrát, ktorý pozostáva hlavne z neutrofilov. V niektorých prípadoch by za lupusovú nefritídu nemuseli zodpovedať imunokomplexy, ale DNA uvoľnená z poškodených buniek. Táto prostredníctvom svojich pozitívne nabitých častíc sa dokáže viazať na bazálnu membránu glomerulov. Následne by sa proti tomuto komplexu vytvárali protilátky, aktivoval komplementový systém a indukoval zápal, tak ako sa to opisuje vyššie [8, 24].

Sumárne možno konštatovať, že SLE je choroba, na ktorej indukcii sa u geneticky vnímavých jedincov a pod výrazným vplyvom hormónov podieľajú imunitné mechanizmy, ktoré potom aj v nasledovnom vývoji zodpovedajú na vznik patologických zmien a klinických príznakov.
5. Terapia
Charakteristickou črtou SLE, že v jeho chronickom, dlhodobom priebehu sa striedajú obdobia plne rozvinutej choroby s obdobiami ich relatívneho ústupu – remisiami. Liečebný postup musí preto byť taký, aby zahrňoval lieky, ktorými lekár zvládne aj akútnu formu choroby, ktorá v niektorých situáciách môže ohroziť dokonca život pacienta a aj taký, ktorý v chronickej fáze bude brániť progresii choroby. Liečba SLE je prísne individuálna a vychádza z klinických a laboratórnych analýz. V liečbe SLE sa využívajú viaceré lieky. Nesteroidné antiflogistiká (NSA), vrátane salicylátov sa veľmi často používajú na zvládnutie svalových a kĺbových ako aj nešpecifických (horúčka či mierne zápaly seróznych blán) prejavov. Glukokortikoidy sú doteraz najúspešnejším prostriedkom na zvládnutie akútnych prejavov a u niektorých chorých s hyperakútnym priebehom aj liekom, ktorý im zachráni život. Antimalariká (chlorochin a hydrochlorochin) sú nezastupiteľné pri všetkých kožných formách, najmä pri subaktútnom kožnom LE a podávajú sa aj chorým s postihnutými kĺbmi, od atralgií až po stredne ťažkú formu polyartritídy či sprievodnú myozitídu. Azathioprin sa považuje za alternatívny liek k cyklofosfamidu; považuje sa za menej účinný, ale bezpečnejší liek. Cyklofosfamid sa najviac osvedčil pri liečbe lupusovej nefritídy. Metotrexát sa obyčajne podáva tým chorým, ktorým sa predtým aplikovali antimalariká; ide prevažne o chorých s postihnutím kĺbov, kožnými prejavmi, serozitídou alebo s horúčkami bez vážneho postihnutia orgánov. Cyklosporín A sa najviac osvedčil pri liečbe lupusovej nefritídy. Danazol sa používa na zvládnutie lupusovej trombocytopénie alebo niekedy aj pri liečbe diskoidnej formy LE. Na odstránenie imunokomplexov sa vyžíva plazmaferéza; aj keď sa po tomto zákroku ich koncentrácia výrazne zníži, dochádza v relatívne krátkom časovom úseku k návratu do pôvodného stavu. Odporúča sa preto kombinovať plazmaferézu s následným jednorázovým podaním cyklofosfamidu s cieľom utlmiť syntézu autoprotilátok [16, 24, 35].
S príchodom monoklonových protilátok a ich využitím v terapii, sa začali klinické skúšky aj pri SLE s tými protilátkami, ktoré sú namierené proti diferenciačným antigénom B-lymfocytov. Ide o rituximab, humanizovanú monoklonovú protilátku (MoPr) anti-CD20. Vyžíva sa predovšetkým v liečbe non-Hodgkinových lymfómov, ale keďže SLE charakterizuje polyklonová aktivácia B-lymfocytov, usúdilo sa, že rituximab by mohol byť úspešný aj v liečbe tejto choroby; prvé výsledky sú priaznivé. Podáva sa obyčajne v kombinácii s metotrexátom. Najnovšie sa liečby zavádzajú humanizované MoPr anti-CD22 (epratuzumab) a MoPr anti-BAFF (belimumab), ktoré blokujú aktiváciu B-lymfocytov prostredníctvom neutralizácie rastového faktora B-lymfocytov syntetizovaného aktivovanými makrofágmi, BAFF (B-cell-activating factor). Rituximab a epratuzumab pôsobia na B-lymfocyty rozdielne, čo umožní ich vzájomnú kombináciu [2, 34, 37].
Skúsenosti s liečbou rituximabom ukázali, že účinnosť liečby závisí aj od genotypu jedinca chorí, ktorí boli homozygótni pre F-alelu FcRIIIa-receptora si vyžadovali približne 10-násobne vyššie dávky ako jedinci s genotypom VV alebo VF [2]. Ďalej sa ukázalo, že pacientov s SLE nemožno liečiť MoPr anti-TNF. Dôvodom je, že TNF je v určitej protiváhe k IFN-I a zníženie jeho hladín by situáciu ešte ďalej zhoršili; napokon aj skúsenosti s liečbou týmito MoPr pri RA ukazujú, že u niektorých pacientov sa vytvoria anti-dsDNA protilátky, ba dokonca sa objavia aj klinické prejavy SLE [4].
V liečbe SLE, najmä pri chronickej lupusovej nefritíde, sa osvedčili aj ACE-inhibítory, ktoré redukujú syntézu cytokínov, najmä IL-4 a TGF - [39].
Práca vyšla v rámci riešenia a s podporou grantu MŠ SR VEGA: 1/3437/06
Vysvětlivky:
1Index s sa vypočíta, keď riziko choroby súrodencov sa vydelí rizikom choroby v populácii; číslo 1,0 vyjadruje hodnotu pre choroby, ktoré nemajú rodinnú záťaž.
2Je zaujímavé, že pri sclerosis multiplex je tomu naopak.
3DR15 je podtyp molekuly DR2 (druhý podtyp je DR16)
4Platí pre kaukazoidnú populáciu.
5CD27 patrí do rodiny TNF-receptorov. CD27 je signalizačná molekula, po interakcii so svojim ligandom CD70, prítomným v membránach T-lymfocytov, prenáša kľúčový signál na dozrievanie B-lymfocytov do buniek, ktoré sú už schopné produkovať protilátky.
6Neutrofily viažu imunokomplexy prostredníctvom svojho receptora CR1, ktorý je špecifický pre fragment C3b komplementového systému.
Do redakce došlo 8.8.2008
Prof. MUDr. Milan Buc, DrSc.
Imunologický ústav LFUK
Odborárske nám. 14
811 08 Bratislava 1
Slovenská republika
e-mail: milan.buc@fmed.uniba.sk
Zdroje
1 . Abraham, L. J., Kroeger, K. M. Impact of the -308 TNF promoter polymorphism on the transcriptional regulation of the TNF gene: relevance to disease. J Leukoc Biol, 1999, 66, 562-566.
2. Anolik, J. H., Campbell, D., Felgar, R. E., Young F. et al. The relationship of FcgammaRIIIa genotype to degree of B cell depletion by rituximab in the treatment of systemic lupus erythematosus. Arthritis Rheumatism, 2003, 48, 455-459.
3. Baechler, E. C., Gregersen, P. K., Behrens, T. W. The emerging role of interferon in human systemic lupus erythematosus. Curr Opin Immunol, 2004, 416, 801-807.
4. Banchereau, J., Pascual, V. Type I interferon in systemic lupus erythematosus and other autoimmune diseases. Immunity, 2006, 25, 383-392.
5. Bijlsma, J. W., Straub, R. H., Masi, A. T., Lahita RG, Cutolo, M. Neuroendocrine immune mechanisms in rheumatic diseases. Trends Immunol, 2002, 23, 59-61.
6. Blanco, P., Palucka, A. K., Gill, M., Pascual, V., Banchereau. J. Induction of dendritic cell differentiation by IFN-alpha in systemic lupus erythematosus. Science, 2001, 294, 1540-1543.
7. Buc, M. Autoimunita a autoimunitné choroby. Bratislava: Veda, 2005, 491 s.
8. Buc, M. Imunológia. Bratislava: Veda, 2001, 434 s.
9. Carlsten, H., Holmdahl, R., Tarkowski, A. Analysis of the genetic encoding of oestradiol suppression of delayed-type hypersensitivity in (NZB x NZW) F1 mice. Immunol, 1991, 73, 186-190.
10. Carroll, M. C. A protective role for innate immunity in systemic lupus erythematosus. Nat Rev Immunol, 2004, 4, 825-831.
11. Crawford, K., Alper, C. A. Genetics of the complement system. Rev Immunogenet, 2000, 2, 323-338.
12. Crispin, J. C., Vargas, M. I., Alcocer-Varela, J. Immunoregulatory T cells in autoimmunity. Autoimmun Rev, 2004, 3, 45-51.
13. Dhaher, Y. Y., Greenstein, B. D., Khamashta, M. A., Hughes, G. R. Effects of oestradiol and the oestrogen antagonist Ici 182,780 on the delayed type hypersensitivity (DTH) index and on serum levels of IgM and IgG in ovariectomised Balb/C and MRL/Mp-Lpr/Lpr mice, a model of systemic lupus erythematosus (SLE). Autoimmunity, 2001, 33, 237-243.
14. Dorner, T., Chaoui, R., Feist, E., Goldner, B. et al. Significantly increased maternal and fetal IgG autoantibody levels to 52 kD Ro (SS-A) and La(SS-B) in complete congenital heart block. J Autoimmun, 1995, 8, 675-684.
15. Dorner, T., Lipsky, P. E. Immunoglobulin variable-region gene usage in systemic autoimmune diseases. Arthritis and rheumatism, 2001, 44, 2715-2727.
16. Dostál, C. Systémový lupus erytematodes. In: Rovenský, J. Klinická reumatológia. Martin: Osveta, 2000, 275-301.
17. Dostál, C. Systémový lupus erytematosus a gravidita. In: Rovenský, J. Reumatologia v teórii a praxi. Martin: Osveta, 1998, 309-316.
18. Doyle, H. A., Yan, J., Liang, B., Mamula, M. J. Lupus autoantigens: their origins, forms, and presentation. Immunol Res, 2001, 24, 131-147.
19. Gillmore, J. D., Hutchinson, W. L., Herbert, J., Bybee, A. et al. Autoimmunity and glomerulonephritis in mice with targeted deletion of the serum amyloid P component gene: SAP deficiency or strain combination? Immunol, 2004, 112, 255-264.
20. Harley, J. B., Moser, K. L., Gaffney, P. M., Behrens, T. W. The genetics of human systemic lupus erythematosus. Curr Opin Immunol, 1998, 10, 690-696.
21. Jara, L. J., Benitez, G., Medina, G. Prolactin, dendritic cells, and systemic lupus erythematosus. Autoimmun Rev, 2008, 7, 251-255.
22. Jara, L. J., Vera-Lastra, O., Miranda, J. M., Alcala M., Alvarez-Nemegyei, J. Prolactin in human systemic lupus erythematosus. Lupus 2001, 10, 748‑756.
23. Katsiari, C. G., Tsokos, G. C. Transcriptional repression of interleukin-2 in human systemic lupus erythematosus. Autoimmun Reviews, 2006, 5, 118-121.
24. Kotzin, B. L., West, S. G. Systemic lupus erythematosus. In Rich R. R. Clinical Immunology Principles and practice. Edinburg, New York, Philadelphia, St. Louis, Sydney, Toronto: Mosby, 2001, 60.61-60.24.
25. Larsson, M., Fonteneau, J. F., Bhardwaj, N. Dendritic cells resurrect antigens from dead cells. Trends Immunol, 2001, 22, 141-148.
26. Leadbetter, E. A., Rifkin, I. R., Hohlbaum, A. M., Beaudette, B. C. et al. Chromatin-IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nature, 2002, 416, 603-607.
27. Lipsky, P. E. Systemic lupus erythematosus: an autoimmune disease of B cell hyperactivity. Nat Immunol, 2001, 2, 764-766.
28. Maloy, K. J., Powrie, F. Regulatory T cells in the control of immune pathology. Nat Immunol 2001, 2, 816-822.
29. McMurray, R., Keisler, D., Kanucke, K., Izui, S., Walker, S. E. Prolactin influences autoimmune disease activity in the female B/W mouse. J Immunol, 1991, 147, 3780-3787.
30. Nath, S. K., Kilpatrick, J., Harley, J. B. Genetics of human systemic lupus erythematosus: the emerging picture. Curr Opin Immunol, 2004, 16, 794-800.
31. Nose, M., Nishihara, M., Kamogawa, J., Terada, M., Nakatsuru, S. Genetic basis of autoimmune disease in MRL/lpr mice: dissection of the complex pathological manifestations and their susceptibility loci. Rev Immunogenet, 2000, 2, 154-164.
32. Odendahl, M., Jacobi, A., Hansen, A., Feist, E. et al. Disturbed peripheral B lymphocyte homeostasis in systemic lupus erythematosus. J Immunol, 2000, 165, 5970-5979.
33. Raman, K., Mohan, C. Genetic underpinnings of autoimmunity lessons from studies in arthritis, diabetes, lupus and multiple sclerosis. Curr Opin Immunol, 2003, 15, 651-659.
34. Ramanujam, M., Davidson, A. BAFF blockade for systemic lupus erythematosus: will the promise be fulfilled? Immunol Rev, 2008, 223, 156-174.
35. Rovenský, J. Klinická reumatológia. Martin: Osveta, 2000, 1048 s.
36. Scofield, R. H. Genetic knock out of 60kD Ro (or SSA), a common lupus autoantigen, induces lupus. Trends Immunol, 2004, 25, 1-3.
37. Shaw, T., Quan, J., Totoritis, M. C. B cell therapy for rheumatoid arthritis: the rituximab (anti-CD20) experience. Ann Rheum Dis, 2003, 62, Suppl. 2, 55-59.
38. Tenbrock, K., Juang, Y. T., Kyttaris, V. C., Tsokos, G. C. Altered signal transduction in SLE T cells. Rheumatol, 2007, 46, 1525-1530.
39. Teplitsky, V., Shoenfeld, Y., Tanay, A. The renin-angiotensin system in lupus: physiology, genes and practice, in animals and humans. Lupus, 2006, 15, 319-325.
40. Thome, M., Tschopp, J. Regulation of lymphocyte proliferation and death by FLIP. Nat Rev Immunol, 2001, 1, 50-58.
41. Tsao, B. P. The genetics of human systemic lupus erythematosus. Trends Immunol, 2003, 24, 595-602.
42. Tsokos, G. C., Nambiar, M. P., Tenbrock, K., Jung, Y. T. Rewiring the T-cell: signaling defects and novel prospects for the treatment of SLE. Trends Immunol, 2003, 24, 259-263.
43. Vidaver, R. Molecular and clinical evidence of the role of estrogen in lupus. Trends Immunol, 2002, 23, 229-230.
44. Whitacre, C. C. Sex differences in autoimmune disease. Nat Immunol, 2001, 2, 777-780.
45. Woof, J. M., Burton, D. R. Human antibody-Fc receptor interactions illuminated by crystal structures. Nat Rev Immunol, 2004, 4, 89-99.
Štítky
Hygiena a epidemiologie Infekční lékařství MikrobiologieČlánek vyšel v časopise
Epidemiologie, mikrobiologie, imunologie

2009 Číslo 1
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Systémový lupus erythematosus – súčasný pohľad na genetickú determináciu, imunopatogenézu a liečbu
- Výskyt a charakteristika salmonel ve vybraných lokalitách České republiky – porovnání epidemiologických a laboratorních dat
- Srovnání citlivosti spor Bacillus subtilis a spor českých kmenů Clostridium difficile vůči dezinfekčním prostředkům
- Molekulárno-biologická a fenotypová charakterizácia humánnych izolátov Salmonella enterica serovar Paratyphi B dT+, alebo Salmonella Java
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy