
Nutriční terapie u pacientů s dědičnými poruchami metabolismu
Nutritional therapy in patients with inherited metabolic disorders
Most of inherited metabolic disorders are rare, but a number of these disorders increases and so does a number of diagnosed patients due to a newborn screening and a development of new analytic methods. Inherited metabolic disorders are mostly serious. Nutrition management has a significant role in a therapy of a part of these disorders, especially in so called small molecules disorders. It involves a regulated intake of a substrate, which metabolism is limited in a patient due to an enzymatic or a protein deficiency or functional disruption. Supplementation of lacking metabolic products and essential nutrients is also a part of a nutrition management. The goals of a nutrition management are to reach and maintain a normal level of all metabolites and a normal growth and development of a patient.
This article reviews the basic principles of a nutrition management of most often occuring inherited metabolic disorders, which are managible by a diet.
KEY WORDS:
nutrition therapy, inherited metabolic disorders, low protein diet, low fat diet, antihypoglycemic regiment, lactose-free diet, low fructose diet, low cholesterol diet, low plant sterol diet, ketogenic diet, low purine diet
Autoři:
M. Floriánková; Š. Bláhová; M. Pencová; T. Honzík; P. Ješina
Působiště autorů:
Klinika dětského a dorostového lékařství 1. LF UK a VFN, Praha
Vyšlo v časopise:
Čes-slov Pediat 2018; 73 (6): 395-407.
Kategorie:
Sympozium: dědičné poruchy metabolismu
Souhrn
Dědičné poruchy metabolismu převážně patří mezi vzácná a většinou závažná onemocnění. Díky novorozeneckému screeningu a rozvoji analytických metod stoupá počet objevených onemocnění i počet diagnostikovaných pacientů. Především u tzv. nemocí malých molekul má významnou roli v léčbě nutriční management, spočívající v regulaci přívodu látek, jejichž metabolismus je u pacienta limitován poruchou funkce některého z enzymů či proteinů. K nutriční terapii patří rovněž dodání chybějících produktů metabolismu a doplnění stravy o preparáty s obsahem potřebných nutrientů. Cílem nutriční terapie je celoživotní normalizace hladin jednotlivých metabolitů a dosažení normálního růstu a vývoje nemocného dítěte.
Následující sdělení přináší přehled hlavních zásad nutričního managementu u nejčastěji se vyskytujících dietou ovlivnitelných dědičných metabolických poruch.
KLÍČOVÁ SLOVA:
nutriční terapie, dědičné poruchy metabolismu, nízkobílkovinná dieta, nízkotuková dieta, antihypoglykemický režim, bezlaktózová dieta, nízkofruktózová dieta, nízkocholesterolová dieta, nízkofytosterolová dieta, ketogenní dieta, nízkopurinová dieta
ÚVOD
Dědičné poruchy metabolismu (DPM) zahrnují více než 1000 závažných onemocnění, z nichž většina se vyskytuje vzácně, v souhrnu však postihují až 1 % populace [1]. Tato onemocnění jsou způsobena mutací v nukleární či mitochondriální DNA, která má za následek nedostatečnou tvorbu nebo poruchu funkce jednoho či více enzymů, jejich kofaktorů nebo jiných proteinů podílejících se na transportu látek a regulaci. Porucha funkce je zapříčiněna změnou složení nebo strukturálního uspořádání. V důsledku poruchy se určité látky hromadí, zatímco jiných je nedostatek. Hromadící se substráty či jejich metabolity mohou působit toxicky. Dědičnost onemocnění může být autosomálně recesivní (AR) nebo autosomálně dominantní (AD), méně často X-vázaná nebo mitochondriální.
První klinické projevy DPM se mohou objevit v kterémkoli věku v závislosti na typu defektu nebo zachování aktivity postiženého enzymu či proteinu. Klinické projevy mohou být ovlivněny i faktory vnějšího prostředí. Ke zhoršení klinického obrazu dochází při katabolismu – při zvracení, průjmech, hladovění, zvýšené námaze nebo při infekčních onemocněních s horečkami (kdy kromě zvýšení nutričních nároků organismu dochází i k dalšímu snížení funkce proteinů změnou jejich sekundární a terciární struktury vlivem teploty a vyplavení proteáz).
Léčitelná je pouze část DPM, většinou se jedná o léčbu celoživotní. Spočívá ve snižování tkáňových a plazmatických koncentrací hromadících se metabolitů (jejich sníženým přívodem nebo zvýšením jejich exkrece). Koncentrace chybějících produktů je zvyšována buď jejich dodáním, nebo zvýšením aktivity enzymu suplementací specifických kofaktorů [2]. Specifické léčebné postupy zahrnují enzymovou substituční terapii, substrát redukující léčbu anebo transplantace orgánů či buněk. Cílem léčby je dosažení a udržení fyziologických koncentrací metabolitů a zároveň zachování normálního růstu a vývoje. Nutriční terapie se jako pilíř léčby uplatňuje jen asi u 100 DPM, především u tzv. nemocí malých molekul, kdy je narušena funkce enzymu v metabolické dráze látky, přiváděné stravou [3]. Nutriční terapie zahrnuje přesně monitorovaný přívod kritického substrátu nebo jeho prekurzorů s tím, že jeho množství je individuálně přizpůsobováno pacientově toleranci, kterou ověřujeme opakovanou kontrolou laboratorních parametrů a celkového klinického stavu. Nedílnou součástí nutriční terapie je i doplňování nezbytných živin, které jsou díky dietní restrikci ve stravě nedostatečně zastoupeny. Podle individuální potřeby probíhá edukace pacienta nebo jeho zákonného zástupce v nutričním režimu.
LÉČBA
U nutričně ovlivnitelných DPM je základem léčby dieta s restrikcí kritické látky a podávání potravin pro zvláštní lékařské účely (PZLÚ), které obsahují směs esenciálních i neesenciálních živin s omezením nebo vyloučením kritické sloučeniny a zajišťují dostatek všech nutrientů pro správný růst a vývoj. Celková dávka bílkoviny obsažené v přirozené stravě a v PZLÚ má být taková, aby pokryla denní doporučený referenční příjem (DRI) bílkovin na kg hmotnosti pro zdravou populaci daného věku [4]. U některých onemocnění se doporučují dávky vyšší než DRI. Stejně tak i dávky ostatních živin a energie mají odpovídat zdravé populaci, případně jsou navýšeny podle individuálních potřeb pacienta a specifik konkrétního onemocnění. Energetický příjem je nastaven tak, aby nedocházelo ke katabolismu, ale zároveň jej regulujeme ve snaze předejít nadváze [2]. U neprospívajících nebo naopak obézních dětí je nutno upravit doporučené dávky energie a živin vzhledem k ideální hmotnosti [5]. U některých metabolických vad je dieta dále doplněna modulárními dietetiky (maltodextriny, MCT oleji) pro dosažení žádoucího energetického příjmu nebo pro udržení stabilní glykémie.
Při akutním onemocnění nebo netoleranci stravy, při dekompenzacích po zvýšené fyzické námaze nebo z jiných příčin je často nutná hospitalizace s podáváním parenterální výživy a korekcí vnitřního prostředí. Parenterální výživa je podávána až do obnovení plné tolerance perorální nebo enterální výživy. U pacientů s DPM se častěji vyskytují potíže s perorálním příjmem stravy, ať už z důvodu rozvoje centrálního nechutenství nebo jiné příčiny. Při krátkodoběji trvajících obtížích se zavádí nazogastrická či nazojejunální sonda, při déletrvajících obtížích PEG, PEJ nebo chirurgická gastrostomie s fundoplikací. Získání a udržení dobré compliance je pro pacientovu prognózu zásadní. V terapii se uplatňuje multidisciplinární tým. Nutriční péče zahrnuje i pravidelné edukace pacienta nebo ošetřující osoby a dlouhodobé monitorování nutričního příjmu.
PORUCHY METABOLISMU AMINOKYSELIN
1. Fenylketonurie (PKU)
Fenylketonurie je první dědičnou poruchou metabolismu, léčenou speciálním nutričním režimem (poprvé popsáno v roce 1953), a současně také první nemocí vůbec, vyhledávanou novorozeneckým screeningem (ve světě od r. 1960, v ČR od r. 1975) [1, 6]. Fenylketonurie má AR dědičnost a rozlišujeme 3 formy: klasickou PKU, hyperfenyl-alaninémii (HPA) a maligní PKU. Klasická PKU i HPA jsou způsobeny poruchou jaterního enzymu fenylalaninhydroxylázy (PAH), který katalyzuje přeměnu fenylalaninu (Phe) na tyrosin (Tyr). U klasické PKU je snížená aktivita PAH <1–2 % normy, neléčení pacienti mají hladinu Phe v krvi >600 µmol/l (norma 30–120 µmol/l). U HPA (někdy označované jako non-PKU) jsou hladiny Phe 120 až 600 µmol/l [1, 2]. Maligní forma PKU je přítomna u 1–2 % pacientů s poruchou metabolismu Phe a je způsobena poruchou syntézy tetrahydrobiopterinu (BH4, kofaktor PAH) nebo poruchou recyklace dihydropteridinreduktázy (DHPR), která je nezbytná k obnově BH4. U gravidních žen s PKU při nedodržování diety hrozí rozvoj fenylalaninové embryopatie s projevy intrauterinní růstové retardace, s vrozenými srdečními vadami a mikrocefalií s rozvojem mentální retardace [2, 6, 7]. Koncentrace Phe je v krvi plodu dvojnásobná oproti hladině Phe u matky.
V krvi a tkáních pacientů s PKU se hromadí Phe, který se nemůže dostatečně metabolizovat na tyrosin a místo toho dochází k přeměně Phe na ketolátky – kyseliny fenylpyrohroznovou, fenyloctovou a fenylmléčnou. Vysoká hladina Phe snižuje funkci 3-hydroxy-3-metylglutaryl-CoA (HMG-CoA) dehydrogenázy a přispívá k hypomyelinizaci CNS a k tvorbě lézí v bílé hmotě mozkové. Hladina tyrosinu je u pacientů s neléčenou PKU naopak nízká, jeho hladina v CNS je ještě nižší než v séru díky soutěžení Tyr s nadbytečným Phe o vazby při transportu přes hematoencefalickou bariéru. Dochází k nedostatečné tvorbě melaninu a dopaminergních neurotransmiterů.
Kyseliny fenylpyrohroznová, fenylmléčná a fenyloctová způsobují typický zápach moči a potu „po myšině“, kyselina fenyloctová také dermatitidu. Nedostatek Tyr a následně melaninu vede k rozvoji hypopigmentace (světlé vlasy, světlá pleť i duhovky). Pokud není brzy po narození zahájena dieta s restrikcí fenylalaninu nebo v případě maligní PKU substituce BH4, dochází k nevratnému poškození CNS, zpomalení psychomotorického vývoje a mikrocefalizaci. Pacienti s neléčenou fenylketonurií mají těžkou mentální retardaci (DQ/IQ <50 bodů), u dětí je častá hyperaktivita, záchvaty vzteku, poruchy koncentrace a paměti, poruchy růstu a chůze, spasticita, u 25 % pacientů se rozvíjí epilepsie [1]. V dospělém věku se mohou rozvinout záchvaty úzkosti, deprese nebo obsese [6, 7, 8].
Léčba:
Základem léčby PKU je dieta s nízkým obsahem Phe, která je doplněná o podávání PZLÚ se směsí aminokyselin bez Phe, obohacené o zvýšenou dávku tyrosinu a mikronutrienty [7, 8]. Povolené množství Phe v dietě se mění s věkem a hmotností a reguluje se podle hladin Phe v krvi tak, aby se hladina Phe v krvi udržovala v doporučeném rozmezí (tab. 1) [8]. Orientačně se dá říci, že pacienti s klasickou PKU tolerují jen zhruba čtvrtinu (cca 10–15 mg/kg/den) běžného příjmu Phe ve stravě. Frekvence monitorování hladiny Phe je individuální a závisí na věku a kompenzaci [1]. Hladinu Phe je možné stanovit i vyšetřením vzorku suché krevní kapky, odebrané 2–3 hodiny po hlavním jídle [7].
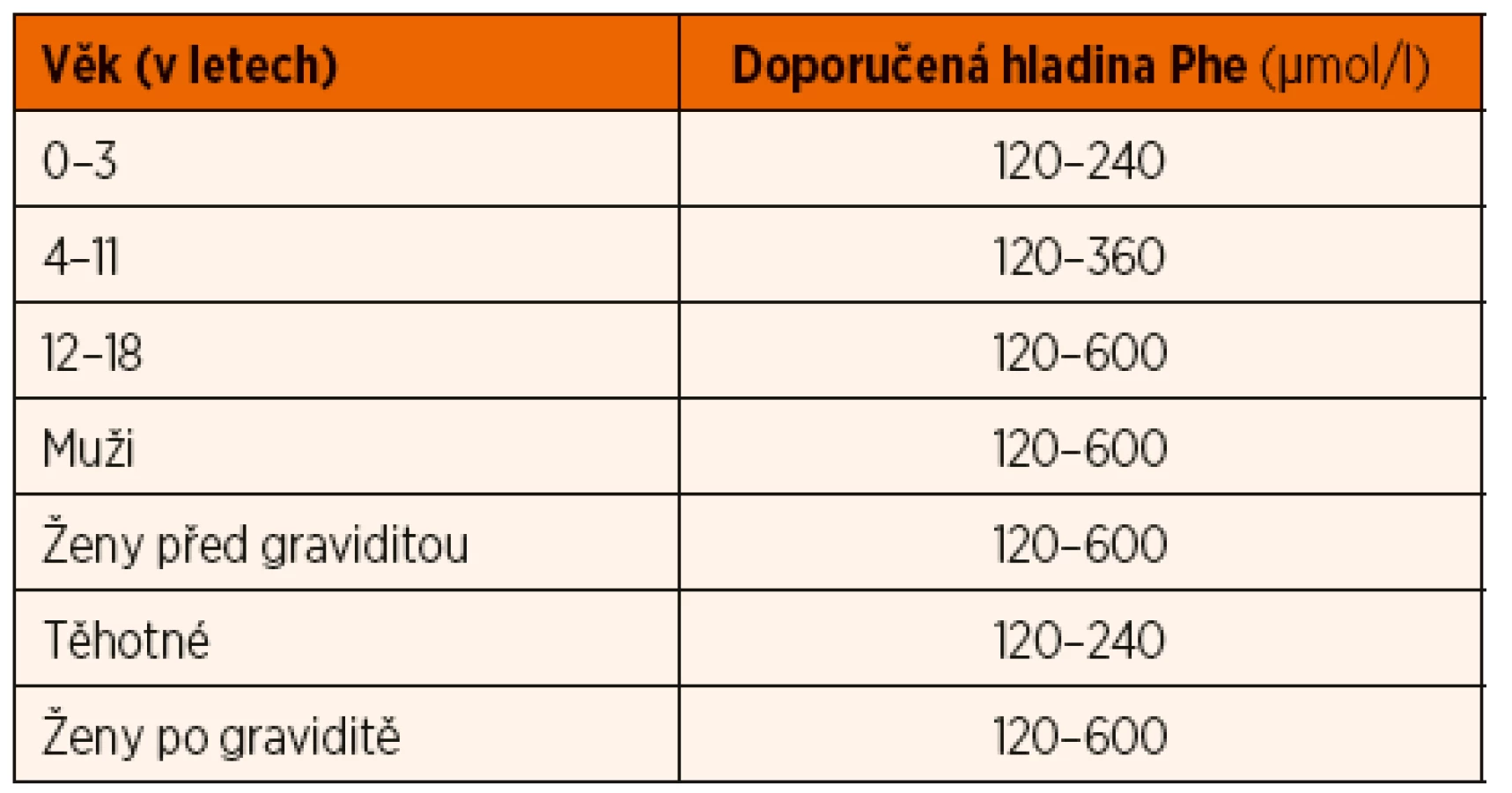
*Doporučené hladiny platí pro Phe, měřený v kapilární krvi z prstu
metodou tandemové hmotnostní spektrometrie. Hladina Phe, měřená
v séru/plazmě pomocí automatického analyzátoru aminokyselin,
je o cca 20 % vyšší.
V prvních měsících života je dieta založena na pravidelných dávkách mateřského mléka nebo počáteční kojenecké formule, doplněných PZLÚ. V případě potřeby (hlad, nedostatečné prospívání) je možno dávku PZLÚ navýšit, dávka přirozené stravy se upravuje podle kontrolních hladin Phe. Obsah Phe v přirozené stravě se počítá podle speciálních tabulek (ukázka tab. 2), nebo přepočtem z obsahu bílkovin uvedeným výrobcem (1 g bílkoviny = 40 mg Phe). Celková dávka bílkovin z přirozené stravy a PZLÚ by měla být dle guidelines z r. 2016 u dětí s PKU do 4 let o 50 % vyšší a u dětí nad 4 roky o 20–40 % vyšší než DRI u zdravých jedinců ve stejné věkové kategorii [6]. Energetický příjem a příjem všech nutrientů kromě Phe, Tyr a přirozené bílkoviny má být stejný jako u zdravé populace stejného věku [7]. První nemléčné příkrmy se přidávají podle prospívání dítěte po ukončení 4.–6. měsíce. Každá potravina je zaváděna samostatně. Jídelníček má být co nejpestřejší, jeho základem jsou ovoce, zelenina a speciální nízkobílkovinné potraviny (potraviny s uměle sníženým obsahem bílkovin, nahrazující běžné pečivo, mouky, cukrovinky, vejce, masné nebo mléčné výrobky). Obvykle zcela vyloučeny jsou maso, masné výrobky, ryby, luštěniny, běžné obiloviny, vejce, semena a ořechy. V malém množství je možno zařazovat mléčné výrobky a rostlinné náhrady mléka.

Postupně se přechází ze stejných dávek Phe v každém jídle na přirozené rozdělení, kdy hlavní jídla obsahují cca 25–30 % denní dávky Phe a svačiny a druhá večeře do 10 %. Důležité je vyčerpat celou doporučenou denní dávku Phe (Phe je esenciální aminokyselina, potřebná pro proteosyntézu). U některých pacientů s mírnou HPA je možno postupně přejít z diety s nízkým obsahem fenylalaninu na běžnou dietu s pravidelnou monitorací hladin Phe.
Zvláštní dietní režim je nutný u dívek s PKU v reprodukčním věku. Jedinou prevencí fenylalaninové embryopatie u těhotných žen s PKU je plánovaná gravidita, pečlivá prekoncepční příprava a přísná restrikce Phe v dietě s paralelním snížením jeho sérových hladin <240 µmol/l již před otěhotněním.
Při akutních infekčních onemocněních obvykle dochází k vzestupu hladiny Phe v krvi díky katabolismu. Pro minimalizaci dekompenzace je potřebná neprodlená léčba akutního onemocnění, udržení perorálního příjmu k zabránění katabolismu (podáváním sacharidových potravin s nízkým obsahem Phe, případně přidáním sacharidů do orálních rehydratačních roztoků, pokud potrava není tolerována). Při déletrvající intoleranci p.o. příjmu může být nutná parenterální výživa [6, 7].
U pacientů s maligní PKU zahrnuje léčebný režim navíc podávání BH4 ve formě sapropterin-dihydrochloridu a prekurzory dopaminu a serotoninu.
Vzhledem k tomu, že dieta při fenylketonurii není plno-hodnotná, je potřeba monitorovat hladiny vitaminů, minerálních látek a stopových prvků. Riziko nedostatku je zejména u železa, vitaminu D, B12, kyseliny listové, zinku, selenu [8]. Protože u pacientů s PKU se častěji rozvíjí osteopenie a osteoporóza, je zapotřebí u nich dbát na dostatečný příjem vápníku ve formě PZLÚ a doplňků stravy a pravidelně monitorovat stav kostní hmoty. Některé studie považují za nezbytnou také suplementaci esenciálních mastných kyselin, především DHA (kyselina dokosahexanová) [7].
Další terapeutickou možností PKU je suplementace neutrálních aminokyselin (LNAA) – tryptofanu, valinu, leucinu, isoleucinu a tyrosinu, bez fenylalaninu. Tyto aminokyseliny soutěží s fenylalaninem o stejné vazby při transportu přes střevní a hematoencefalickou bariéru a mohou snižovat hladinu Phe v krvi a mozkomíšním moku. Novinkou ve fázi výzkumu je podávání glykomakropeptidu (GMP). Jde o proteinový izolát z pšenice, který přirozeně neobsahuje Phe, díky kontaminaci se v něm však malé množství Phe vyskytuje. Vzhledem k tomu, že se jedná o intaktní protein, jeho nutriční hodnota je vyšší než u aminokyselinového preparátu. GMP se používá k obohacení nízkofenylalaninových potravin bílkovinou [2, 7].
2. Klasická homocystinurie
Homocystinurie je AR dědičné onemocnění způsobené mutací genu CBS, kódující enzym cystathionin-β-syntázu (CBS), který katalyzuje přeměnu homocysteinu na cystathionin [1]. Lze říct, že CBS řídí tok síry od methioninu k cysteinu, který je mimo jiné prekurzorem gluta-thionu, taurinu a sírovodíku. U klasické formy rozlišujeme pyridoxin (vit. B6) responzivní, pyridoxin-rezistentní a pyridoxin částečně responzivní formu.
K projevům homocystinurie patří oční projevy, které zahrnují ektopii čoček, těžkou myopii, glaukom, atrofii optického nervu, kataraktu nebo odchlípení sítnice. Pacienti mívají marfanoidní habitus, skoliózu, osteoporózu, asi 50 % pacientů má arachnodaktylii, častá jsou genua valga, pectus carinatum nebo pectus excavatum. Poměrně časté jsou tromboembolické příhody. U některých pacientů může být přítomen opožděný vývoj a poruchy chování či učení. Mírný deficit CBS se může manifestovat až v dospělosti tromboembolickými příhodami. Tito pacienti mají obvykle normální IQ a nemívají jiné komplikace. Pyridoxin-responzivní formy mají mírnější projevy a pozdější manifestaci než pyridoxin-rezistentní. Pacienti mají obvykle významně lepší mentální kapacitu [9].
Onemocnění je v ČR od roku 2016 součástí novorozeneckého screeningu. Pro onemocnění je charakteristický laboratorní nález zvýšené hladiny celkového homocysteinu (tHcy) a methioninu (Met) a zároveň snížené hladiny cysteinu a cystathioninu v krvi a zvýšená hladina homocystinu v moči. Diagnózu lze potvrdit na enzymatické úrovni průkazem snížené aktivity enzymu CBS a na molekulárně-genetické úrovni nálezem mutace v genu CBS [1].
Léčba:
U časně diagnostikovaných pacientů lze léčbou předejít komplikacím onemocnění, dosáhnout normálního růstu a vývoje. U později diagnostikovaných pacientů s již závažným klinickým nálezem je hlavním cílem léčby předejít tromboembolickým komplikacím, i když byl popsán i vliv léčby na zlepšení intelektu a chování [9]. U pacientů s pyridoxin-responzivní formou se doporučuje perorálně podávat denně pyridoxin. Jsou popsány případy pacientů s extrémní responzibilitou na pyridoxin a normalizací nálezu již při nízkých dávkách kolem 10 mg vitamínu B6. Obvykle se ale podává mnohonásobně vyšší dávka, která by však neměla překročit 500 mg denně. Na této dávce se u plně responzivní formy snižují hladiny Met a Hcy v krvi a pacienti nepotřebují dietu s nízkým obsahem Met [9]. Doporučuje se i podávání nízkých dávek folátu. U pacientů s pyridoxin-rezistentní formou homocystinurie nedochází při podávání vitaminu B6 k významnému poklesu hladin Hcy a je u nich nutné nastavit dietu s nízkým obsahem methioninu, aby došlo k žádoucímu poklesu hladin Hcy v krvi <80–100 µmol/l. U pacientů s částečně responzivní formou dochází při terapii pyridoxinem k poklesu hladin Met a Hcy, ale k dostatečné kompenzaci, za kterou je považována hladina Hcy <50 µmol/l, je obvykle nutná kombinace pyridoxinu a diety s nízkým obsahem methioninu, která však není tolik přísná jako u pyridoxin non responzivní formy [1, 9]. Dovolený příjem methioninu u pyridoxin-rezistentní formy je velmi nízký, zpravidla nepřesahující 10 mg/kg//den, u pacientů s částečnou responzivitou je tolerován příjem vyšší, kolem 20 mg/kg/den, s maximem do cca 600 mg/den. Příjem Met se monitoruje podle tabulek s přehledem potravin s analyzovaným obsahem Met nebo přepočtem obsahu bílkovin na mg Met (1 g bílkovin obsahuje průměrně 25 mg Met). Obsah methioninu ve vybraných potravinách ukazuje tabulka 3. Dieta s restrikcí Met (často až na čtvrtinové dávky běžného příjmu methio-ninu) musí být doplněna o PZLÚ bez Met, obohaceným o L-cystin, vitaminy, minerální látky, stopové prvky a další nutrienty. Skladba jídelníčku je velmi podobná dietě při fenylketonurii, stejně tak rozdělení Met do jednotlivých jídel je podobné doporučenému rozdělení Phe u PKU. Energetický příjem a příjem mikronutrientů při homocystinurii má splňovat DRI pro zdravou populaci, specifické doporučení pro pacienty s CBS deficitem není stanoveno [9].

Pokud se nedaří dosáhnout optimální kompenzace dietou nebo dietou v kombinaci s léčbou pyridoxinem, podáváme betain (N,N,N-trimethylglycin), který poskytuje metylovou skupinu k přeměně Hcy na Met, čímž snižuje hladinu Hcy [9]. Hladinu Met je nutné pravidelně monitorovat, zejména v dětství mnohem častěji. Z nutričních parametrů mají být u pacientů na dietě monitorovány hladiny zinku, mědi a selenu, vit. D, vit. B12, kyseliny listové, plazmatické hladiny aminokyselin, albuminu a ferritinu [9].
Při akutním onemocnění se léčba homocystinurie upravuje podle aktuální hladiny homocysteinu a methioninu, doporučuje se zabránit dehydrataci a katabolismu a předcházet, pokud možno, imobilizaci kvůli zvýšenému riziku tromboembolických komplikací. Při plánovaném chirurgickém výkonu je snaha nejprve optimalizovat nutriční stav pacienta.
3. Organické acidurie
Organické acidurie jsou skupinou onemocnění s mnoha společnými biochemickými a klinickými projevy. Rozlišujeme u nich těžkou novorozeneckou formu, akutní intermitentní formu s pozdním začátkem a chronickou progresivní formu [10]. Těžká novorozenecká forma se rozvíjí v hodinách až týdnech po porodu a projevuje se především toxickou encefalopatií s ketózou nebo ketoacidózou. Dítě odmítá stravu, je apatické, dehydratované, hyper - nebo hypotonické, stav progreduje do kómatu. Akutní intermitentní forma s pozdním začátkem se projevuje v batolecím věku nebo později, k dekompenzacím dochází při febrilním infektu nebo po vysokém příjmu bílkovin, mezi atakami je pacient bez potíží. Ataky se projevují kómatem, letargií s ataxií, Reye-like syndromem, těžkými hematologickými změnami [10]. Do klinického obrazu chronické progresivní formy patří dlouhodobá anorexie, časté zvracení, neprospívání, osteoporóza, hypotonie, psychomotorická retardace, křeče. Mezi komplikace organických acidurií patří například edém mozku, chronická renální insuficience, akutní nebo chronická pankreatitida či kardiomyopatie [10]. Základem léčby organických acidurií je nízkobílkovinná dieta se suplementací směsi vitaminů, minerálních látek a stopových prvků a aminokyselin bez limitujícího substrátu. Je nutno zajistit dostatečný kalorický příjem a zabránit delšímu hladovění.
Nemoc javorového sirupu – leucinóza (MSUD)
Leucinóza je porucha metabolismu větvených aminokyselin valinu (Val), leucinu (Leu) a isoleucinu (Ile) a od nich odvozených α-oxokyselin následkem mutace v jednom ze 4 genů pro mitochondriální enzymový komplex dehydrogenázy větvených α-oxokyselin. Díky enzymovému deficitu mají pacienti zvýšené hladiny větvených aminokyselin a α-oxokyselin, porucha zasahuje i do glykolýzy a způsobuje vzestup hladiny laktátu. Jde o AR dědičné onemocnění se vzácným výskytem [1]. Rozlišujeme 4 formy leucinózy: klasickou, intermediární, intermitentní a thiamin-responzivní.
Leucinóza je součástí novorozeneckého screeningu. Diagnózu potvrzuje nález zvýšených hladin Val, Leu, Ile a alloisoleucinu v séru a nález zvýšených hladin 2-oxokyselin v moči [1]. Leucinózu lze diagnostikovat také změřením aktivity dekarboxylázy při dekarboxylaci kyseliny 2-oxoisokapronové a molekulárně-genetickým vyšetřením. Na rozdíl od methylmalonové či propionové acidurie není u leucinózy přítomna výrazná hyperamonémie ani těžká dehydratace a metabolická acidóza, krevní obraz bývá normální [10].
Léčba:
Pokud je klasická leucinóza léčena až po rozvoji neurologických příznaků, mohou mít pacienti trvalé poškození CNS s těžkou psychomotorickou retardací. Příjem přirozené bílkoviny je omezen na 0,3–1,2 g bílkovin/kg/den podle závažnosti enzymatického defektu. Je nutný dostatečný kalorický příjem (DRI dle věku). Hladina Leu by měla být pod 200 µmol/l u dětí do 5 let a do 300 µmol/l u pacientů nad 5 let [11]. Dieta je doplněna preparátem se směsí aminokyselin bez Val, Leu a Ile, obohacena o vitaminy, minerální látky a stopové prvky. Někdy je nutno doplňovat Val a Ile, protože jejich nedostatek vede ke zvýšení hladiny Leu a jejich obsah v přirozené bílkovině je nižší [2]. U thiamin-responzivní leucinózy se podává 10–1000 mg vit. B1 denně, doporučuje se podávat B1 u všech forem leucinózy kromě klasické [11]. V době akutního onemocnění se doporučuje snížit na 24–48 hodin příjem přirozené bílkoviny na 50–100 % pacientovy denní dávky a nahradit chybějící bílkovinu PZLÚ bez Val, Leu a Ile [11]. Při akutní dekompenzaci je nutná hospitalizace pacienta s co nejrychlejším snížením hladin neurotoxických metabolitů a zabránění katabolismu pomocí infuzí glukózy s ionty, inzulinu a tukových emulzí, případně hemodiafiltrací. Doporučuje se zvýšit energetický příjem na 150 % DRI [11]. Krajním řešením při opakovaných dekompenzacích pacienta je transplantace jater.
V těhotenství je nutno navýšit příjem celkové bílkoviny i energie v souladu s DRI pro zdravou populaci, s obsahem přirozené bílkoviny podle tolerance pacienta. V těhotenství a laktaci může být nutné suplementovat některé vitaminy, minerální látky a L-karnitin. Pozornost je nutné věnovat prevenci katabolismu při těhotenské nevolnosti, v době porodu a 2–6 týdnů po porodu [11].
4. Poruchy cyklu močoviny
V cyklu močoviny dochází k přeměně toxického amoniaku na netoxickou močovinu, která se bezpečně vyloučí močí. Metabolická dráha je tvořena 6 enzymy: N-acetylglutamát syntázou (NAGS), karbamoylfosfát syntázou I (CPS I), ornitintranskarbamylázou (OTC), argininosukcinát syntázou (ASS), argininosukcinát lyázou (ASL) a arginázou (ARG) [2, 11]. Nejčastější poruchou cyklu močoviny je deficit OTC, při kterém je narušená přeměna ornitinu a karbamoylfosfátu na citrulin. Dědičnost OTC deficitu je vázaná na X chromosom. Ostatní poruchy cyklu močoviny jsou AR dědičné, nejvzácněji se vyskytují deficity NAGS a ARG [4].
Klinické projevy všech poruch cyklu močoviny kromě deficitu arginázy jsou podobné. V důsledku hyperamonémie a hromadění glutaminu v astrocytech dochází k poškozování mozku, letargii, kómatu a stav může končit úmrtím. Projevem deficitu ARG je pouze mírný vzestup amoniaku v plazmě a neurologické obtíže [4]. První projevy poruch cyklu močoviny se objevují tím dříve, čím nižší je zbytková aktivita postiženého enzymu. U časných forem začínají obvykle 24–48 hodin po porodu odmítáním pití, zvracením, letargií, zvýšenou dráždivostí, hypotermií s progresí do kómatu. Symptomy jsou někdy zaměňovány s novorozeneckou sepsí. Pacienti, kteří přežijí první ataku hyperamonémie, mohou mít neurologické potíže a psychomotorickou retardaci. U pozdní formy s vyšší zbytkovou aktivitou enzymu (nebo u přenašeček deficitu OTC) se mohou projevy vyskytnout až v období zvýšené zátěže organismu, například při zvýšené konzumaci bílkovin, infektu nebo operaci. Projevy jsou méně specifické – změny chování, nechutenství, apatie, tonicko-klonické křeče [4].
Citrulinémie typu I (ASS) a argininémie jsou od roku 2016 součástí novorozeneckého screeningu, u ostatních poruch cyklu močoviny je význam screeningu omezen tím, že se obvykle projeví dříve, než jsou známy výsledky screeningu. Pro deficit CPS I a NAGS je typický nález nízké plazmatické hladiny citrulinu, u OTC deficitu nalézáme nízkou hladinu citrulinu a zároveň v moči zvýšenou hladinu kyseliny orotové. U ASS deficitu je naopak hladina citrulinu zvýšená, u argininjantarové acidurie (ASL deficitu) nacházíme v plazmě mírný vzestup citrulinu a v moči zvýšené množství argininosukcinátu. Deficit ARG I (argininémie) se projevuje kumulací argininu v krvi, zatímco u ostatních poruch cyklu močoviny je hladina argininu snížená [4]. U všech poruch cyklu močoviny kromě ARG I deficitu je přítomna výrazná hyperamonémie. Diagnózu potvrzuje molekulárně-genetické vyšetření nálezem mutace v genu NAGS, CPS I, OTC, ASS, ASL nebo ARG I.
Léčba:
Základem léčby je nízkobílkovinná dieta (s variabilní restrikcí v rozptylu 0,6–1,1 g/kg/den) doplněná o PZLÚ s esenciálními aminokyselinami, vitaminy, minerálními látkami a stopovými prvky (směs esenciálních aminokyselin poskytuje méně N ve srovnání s intaktním proteinem) [4, 12, 13]. Pacientům je doporučeno suplementovat esenciální mastné kyseliny. Dávky vitaminů a minerálních látek by měly mírně překračovat DRI pro daný věk, monitorovat je třeba především hladiny draslíku, fosforu a hořčíku. U všech poruch cyklu močoviny kromě ARG I deficitu je podáván L-arginin. Pacientům s CPS I, NAGS a OTC deficitem je podáván i L-citrulin [4]. Nutné je také zvýšit exkreci nadbytečného dusíku, například podáváním fenylbutyrátu nebo benzoátu sodného. Rozdělení přirozené bílkoviny do jednotlivých jídel je možné přizpůsobit dávkování N-vazačů, tím se zajistí stabilnější hladina dusíkatých metabolitů [12]. Podávání N-vazačů však snižuje hladinu větvených aminokyselin, proto je nutné je suplementovat, například z PZLÚ s jejich zvýšeným obsahem [12]. V době akutního onemocnění nebo při akutní dekompenzaci s hyperamonémií je nutné nejprve maximálně omezit příjem bílkovin a zajistit dostatečný energetický příjem k zabránění katabolismu [12]. V případě velmi vysoké hladiny amoniaku je nutná hemodialýza či hemodiafiltrace. U nemocných s poruchami cyklu močoviny je často přítomno nechutenství, averze k jídlu, reflux, zvracení nebo neurologické potíže s příjmem stravy a pacienti často potřebují zavedení PEG. Velká část pacientů s obtížemi s perorálním příjmem potřebuje také doplnit energii ve formě maltodextrinu nebo rostlinných olejů [13].
Těhotné ženy s poruchou cyklu močoviny musí být často monitorovány, zejména u OTC deficitních žen je riziko hyperamonemického kómatu po porodu. Transplantace jater je krajním řešením u pacientů s těžkým deficitem některého z enzymů cyklu močoviny a obtížnou kompenzací nebo u pacientů s progresivním poškozením jater [4].
PORUCHY METABOLISMU SACHARIDŮ
1. Klasická galaktosémie
Klasická galaktosémie je AR dědičné onemocnění, způsobené deficitem galaktóza-1-fosfát-uridyltransferázy (GALT), který přeměňuje galaktózu-1-fosfát a UDP--glukózu na glukóza-1-fosfát a UDP-galaktózu. V důsledku deficitu GALT se hromadí galaktóza (Gal), galaktóza-1-fosfát, galaktitol a galaktonát, které působí toxicky na játra, mozek a ledviny, akumulace v oční čočce způsobuje kataraktu. Vysoká koncentrace galaktóza-1-fosfátu zasahuje i do glukoneogeneze a glykogenolýzy a vede k hypoglykémii. Narušena je také glykosylace proteinů a lipidů a metabolismus fosforu [1].
Klasická galaktosémie se projevuje již 3.–10. den po narození potížemi s pitím, hmotnostním úbytkem, ikterem, zvracením, hepatomegalií, někdy i krvácením a sepsí, nejčastěji způsobenou E. coli [2]. Stav progreduje do edému mozku, selhání jater. V laboratorním nálezu dominuje výrazná hyperbilirubinémie, hypoglykémie, elevace jaterních aminotransferáz, koagulopatie, mírná hyperamonémie. K dlouhodobým potížím, a to i přes nutriční intervenci, patří poruchy učení, snížení intelektu, dysartrie, poruchy řeči, osteoporóza a hypergonadotropní hypogonadismus, intenzita potíží není přímo úměrná kompenzaci [1].
Klasická galaktosémie není v ČR součástí novorozeneckého screeningu. Pro klasickou galaktosémii svědčí nález zvýšené koncentrace galaktózy a galaktóza-1-fosfátu v krvi a zvýšená hladina galaktitolu v moči. Na enzymatické úrovni zjišťujeme sníženou aktivitu galaktóza-1-fosfát uridyltransferázy v erytrocytech. Molekulárně-genetické vyšetření nalézá mutaci v genu GALT.
Léčba:
Základem léčby je, zejména před ukončením puberty, přísná bezmléčná a nízkogalaktózová dieta, doplněná o PZLÚ s mikronutrienty. Nutná je suplementace vápníku, vitaminu D, pozornost je nutno věnovat hladinám vitaminu A, přívodu vlákniny a antioxidantů. Jídelníček novorozence a kojence do 4.–6. měsíce je založen na PZLÚ bez laktózy a galaktózy. Nemléčné příkrmy se zařazují podle obsahu galaktózy. Bez omezení je možno zařazovat pouze potraviny s obsahem galaktózy <13 mg/100 g, například maso, ryby, jablka, kukuřici, vaječný bílek, některé tvrdé sýry, některé produkty z nefermentovaných sójových bobů. Omezen je kromě mléčných výrobků příjem i mnoha druhů ovoce a zeleniny, luštěnin, žloutku. Obsah galaktózy ve vybraných potravinách ukazuje tabulka 4. Pozornost je nutno věnovat i obsahu laktózy a galaktózy v léčivech.

Celkový přívod dietární galaktózy má být u kojence 50–200 mg/den, u starších dětí <300 mg/den a u dospělých <400–500 mg/den [1]. Novější zdroje uvádí, že přísná restrikce galaktózy z ovoce, zeleniny nebo tvrdých sýrů není zejména po ukončení puberty nezbytná vzhledem k tomu, že endogenní produkce galaktózy je vyšší než dietární přívod [14]. Část galaktózy, obsažená v rostlinných potravinách, je navíc v nevyužitelné formě a nemá vliv na hladiny toxických metabolitů (obsah volné galaktózy však stoupá skladováním a některými technologickými úpravami). Doporučuje se zcela vyloučit pouze mléko a mléčné výrobky s výjimkou kaseinátů a některých tvrdých sýrů, vnitřnosti, fermentované luštěniny a výrobky z nich [14]. Díky endogenní produkci galaktózy není vždy možné dosáhnout optimální kompenzace [2].
2. Hereditární fruktózová intolerance
Hereditární intolerance fruktózy (HFI) je AR dědičné onemocnění s deficitem enzymu aldolázy B (fruktóza-1,6-bisfosfát aldoláza B). Enzym se exprimuje především v játrech, ledvinách a tenkém střevě. Při jeho deficitu dochází ke kumulaci fruktóza-1-fosfátu, k narušení glykolýzy i glukoneogeneze (nedostatkem substrátu i inhibicí dalších enzymů), ke snížené produkci ATP a k toxickému působení fruktóza-1-fosfátu na jaterní a ledvinné buňky [1].
Protože mateřské mléko a většina kojeneckých formulí fruktózu ani sacharózu neobsahuje, první projevy onemocnění jsou obvykle spojeny až se zařazením nemléčných příkrmů, především ovocných, a s podáním sladkých nápojů. Po konzumaci i poměrně malého množství fruktózy (kolem 5 g), sacharózy nebo sorbitolu dochází u pacientů s HFI k rozvoji nauzey, zvracení, hypoglykémie, při větších dávkách se rozvíjí hepatopatie s hepatomegalií až jaterní selhání, metabolická a laktátová acidóza, renální postižení, někdy až šok nebo Reye-like syndrom. Dlouhodobá zátěž nižšími dávkami fruktózy vede k poruchám růstu a Fanconiho syndromu [1]. Laboratorně je přítomna fruktosurie, zvýšená hladina fruktózy v krvi, elevace jaterních enzymů, hypoglykémie, někdy hypofosfatémie a hypermagnezémie.
Onemocnění není součástí novorozeneckého screeningu, diagnóza se stanovuje na základě klinického podezření a laboratorního nálezu. Molekulárně-genetické vyšetření nachází mutaci v genu ALDOB (gen pro aldolázu B).
Léčba:
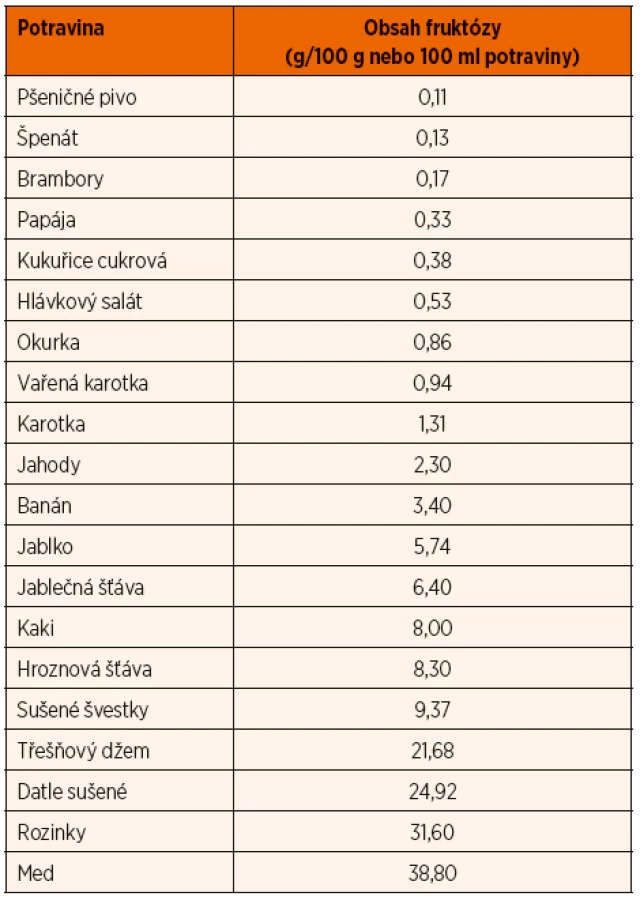
Je nutné převést pacienta na dietu s vyloučením sacharózy, sorbitolu a s velmi výrazným omezením fruktózy. Celkový denní příjem fruktózy by neměl přesáhnout 2–3 g, rozdělené do více denních dávek. Počítat je nutné s obsahem fruktózy nejen v ovoci, cukru, medu a běžných slazených potravinách a nápojích, ale také v zelenině a obilovinách, luštěninách, ovocných a zeleninových šťávách a výrobcích z nich. Vzhledem k výraznému omezení ovoce, ale i zeleniny ve stravě je nutné suplementovat vitamin C. Je třeba zamezit deficitu zinku. Zeleninu podáváme přednostně vařenou (má nižší obsah fruktózy), celkový denní příjem fruktózy ze zeleniny nemá přesáhnout 1,5 g, z obilovin 1 g. Sladit je možné glukopurem a sladidly bez obsahu polyolů, například aspartamem, stévií, sukralózou, sacharinem. Nízký obsah (do 0,5 g fruktózy na 100 g) má většina obilovin, chřest, cukrová kukuřice, papája, avokádo, některá konzervovaná zelenina. Střední obsah (0,51 až 1,5 g fruktózy/100 g) mají mandarinky, cukrový meloun, citrony a většina druhů zeleniny. Vysoký obsah fruktózy (nad 1,5 g/100 g potraviny) má většina ovoce a výrobky z něj, bílé zelí, černý kořen, artyčok, med. Obsah fruktózy v některých potravinách ukazuje tabulka 5.
3. Glykogenózy
Glykogenóza Ia – nemoc von Gierke
Glykogenóza Ia (GSD Ia) je AR dědičné onemocnění, způsobené deficitem enzymu glukóza-6-fosfatáza (G-6-Páza), který štěpí glukóza-6-fosfát na glukózu. G-6--Páza je spolu s transportním proteinem glukóza-6-fosfát translokázou klíčovým enzymem v regulaci glykémie [15]. V důsledku deficitu G-6-Pázy dochází k rozvoji závažné hypoglykémie při delší pauze mezi jídly nebo při zvýšených nárocích organismu, například při infektu nebo tělesné námaze. Deficit vede dále k hromadění laktátu, který přispívá k rozvoji hyperlipidémie vlivem zvýšené transkripce lipidogenních genů a k hyperurikémii vlivem kompetitivní inhibice exkrece kyseliny močové [1]. Při deficitu G-6-Pázy vzniká také jaterní rezistence k STH a pokles IGF-1. Glykogen nemůže být účinně degradován na glukózu a hromadí se v játrech a ledvinách a rovněž ve střevní mukóze [15, 16].
První projevy GSD Ia mohou být v novorozeneckém nebo časném kojeneckém věku v závislosti na prodloužení intervalu mezi krmením. Těžká hypoglykémie je spojena s atakami neklidu, apnoickými pauzami, křečemi, poruchou vědomí [15, 16]. Hromadění glykogenu v játrech, ledvinách a střevní mukóze vede k hepatomegalii, nefromegalii a k neprospívání vlivem malabsorpce. Přítomna je typická „tvář panenky“ (kulatý obličej s plnými tvářemi) a porucha růstu. Motorický vývoj bývá opožděn, intelekt je většinou normální, pokud není narušen opakovanými těžkými hypoglykémiemi [15, 16]. V laboratorním nálezu dominuje nalačno závažná hypoglykémie, přítomna je laktátová acidóza, hyperlipidémie, hyperurikémie, hyperkalciurie. Na rozdíl od ostatních glykogenóz je u GSD Ia hypoglykémie neketotická nebo hypoketotická [1, 15]. S věkem se hepatomegalie stává méně výraznou, u některých pacientů ale vznikají jaterní adenomy a méně často i hepatocelulární karcinomy. U nedostatečně kompenzovaných pacientů se rozvíjí dysfunkce proximálních renálních tubulů, která může vyústit v renální selhání. U dívek může být přítomna hemorhagie a polycystická ovaria [17].
GSD Ia není součástí novorozeneckého screeningu, diagnostikuje se na základě klinického podezření a laboratorních nálezů. Potvrzuje se na molekulárně-genetické úrovni nálezem mutace v genu G6PC [1].
Léčba:
Dietoterapie spočívá především v dodržování zásad antihypoglykemického režimu. Stravu podáváme přes den frekventně podle individuální tolerance, někteří pacienti vyžadují podávání stravy každých 75–90 minut, pauza mezi jídly by neměla přesáhnout 3 hodiny. Dávka sacharidů má být cca 8–10 mg glukózy/kg/min u novorozenců a kojenců a 4–8 mg/kg/min u starších dětí [1, 15]. Samozřejmostí je pravidelná monitorace glykémií. Součástí každého jídla musí být buď škrob z přirozených zdrojů (obiloviny, luštěniny, rýže, brambory), nebo podáváme u kojenců a malých batolat maltodextrin a u větších dětí nevařený kukuřičný škrob. V jídelníčku je potřeba co nejvíce omezit laktózu (novorozenci a kojenci dostávají bezlaktózové umělé mléčné výživy), ale také sacharózu a fruktózu, podáváme přednostně ovoce s nižším obsahem fruktózy, například avokádo, citrusy, papáju, švestky. Kromě ovoce jsou v jídelníčku jednoduché sacharidy omezeny s výjimkou glukózy, která je součástí noční kontinuální výživy. Přes noc jsou děti živeny kontinuálně enterální pumpou cestou PEG (nejprve nazogastrickou sondou), kontinuální výživu je nutné zahájit hodinu po posledním denním jídle a ihned po jejím skončení musí následovat snídaně. U starších dětí a dospělých se přechází na podávání stravy s nevařeným kukuřičným škrobem 2x za noc. Novinkou je speciální modifikovaný škrob s voskovaným povrchem zrn umožňující udržení glykémie až 8 hodin [1]. Protože pacienti s GSD Ia mají tendenci k hyperlipidémii, je potřeba omezovat v jídelníčku také tuk se suplementací vícenenasycených mastných kyselin s dlouhým řetězcem (LC-PUFA). Kromě toho je nutné dodávat vápník a vitaminy D i C. Při výraznějším omezení přívodu ovoce je nutné podávat multivitaminový přípravek s minerálními látkami. Celkový přívod energie má být ze sacharidů 60–70 %, z proteinů 10–15 %, zbytek energie představují tuky. U některých glykogenóz, zejména u typů III, VI a IX, se doporučuje dieta s vysokým obsahem proteinů [17]. Doporučená dávka nevařeného kukuřičného škrobu je 1,6 g/kg ideální hmotnosti u malých dětí a 1,7–2,5 g/kg u starších dětí a dospělých [15]. Další léčba zahrnuje podávání allopurinolu k redukci hladin kyseliny močové, terapie růstovým hormonem není vzhledem k rezistenci k STH indikována, vliv na konečnou dosaženou výšku má spíše míra kompenzace (čím lepší je kompenzace, tím vyšší vzrůst lze očekávat).
4. Poruchy glukózového a energetického metabolismu s indikací ketogenní diety
GLUT1 deficit
Deficit glukózového přenašeče GLUT1 je příkladem metabolického onemocnění s AD přenosem. Je způsoben mutací v genu SLC2A1. Deficit vede k poruše přenosu glukózy z krve přes hematoencefalickou bariéru do mozku.
Klinické projevy jsou především neurologické. Typickým projevem je časně nastupující farmakorezistentní epilepsie, psychomotorická retardace, mikrocefalie, poruchy učení i chování, spasticita, dyskineze, ataxie [1].
Diagnostika vychází z klinického podezření a laboratorních vyšetření s typickým nálezem nízké hladiny glukózy a laktátu v likvoru [1]. GLUT 1 deficit lze potvrdit molekulárně-genetickým vyšetřením.
Léčba:
V léčbě deficitu GLUT1 se uplatňuje především ketogenní dieta, která obsahuje vysoké množství tuků, odpovídající příjem bílkovin a výraznou restrikci sacharidů. Poměr tuků k bílkovinám a sacharidům je 4 : 1. Energetický příjem je redukován na 90 % DRI pro daný věk. Dietu obvykle nasazujeme postupnou změnou jídelníčku se zvyšujícím se poměrem tuků k bílkovinám a sacharidům (1 : 1; 2 : 1; 3 : 1 a nakonec 4 : 1 v intervalu změny á 3 dny). U kojenců je dieta složena hlavně z PZLÚ s vhodným poměrem tuků k bílkovinám a sacharidům a s přídavkem mikronutrientů. Větším dětem je nutno sestavit přesný dietní plán z běžných potravin. Ketogenní dieta zvyšuje nabídku mastných kyselin a ketolátek jako zdroje energie na úkor glykolýzy. Ketolátky mohou pokrýt až 2/3 energetických potřeb mozku [1, 18]. U mírnějších forem onemocnění je možno využít méně přísnou modifikovanou Atkinsovu dietu nebo dietu s omezením monosacharidů a disacharidů.
Příklad režimu: 7letý chlapec s diagnózou GLUT1, na ketogenní dietě s poměrem 4 : 1. Hmotnost 20 kg (8. percentil, proporčně 4. percentil), výška 123 cm (34. percentil).
Přirozená strava: 1160 kcal/den (58 kcal/kg/den), 20 g bílkovin/den (1,0 g bílkovin/kg/den), 9 g sacharidů/den (0,45 g sacharidů/kg/den), 116 g tuků/den (5,8 g tuků/kg/den).
5. Poruchy pyruvátdehydrogenázového komplexu
Pyruvátdehydrogenázový komplex (PDHc) je multienzymový komplex, orientovaný na vnitřní membráně mitochondrií. Dochází zde k přeměně pyruvátu na acetyl-CoA. Poruchy PDHc patří do skupiny mitochondriálních onemocnění [1]. Dědičnost poruchy PDHc je X-vázaná, zatímco u poruch kofaktorů a přenašečů asociovaných s PDHc je přenos AR.
Do klinického obrazu deficitu PDHc patří opoždění vývoje, hypotonie nebo hypertonie, křeče, mikrocefalie, ataxie, ataky hyperventilace při laktátové acidóze, dysmorfie, spasticita, polyneuropatie, atrofie optiku, nystagmus, ptóza, dystonie či strabismus [1]. U těžkých forem je popisován Leighův syndrom.
Diagnózu stanovujeme enzymatickým vyšetřením PDHc v izolovaných lymfocytech, svalové biopsii, kultivovaných fibroblastech. U dívek je třeba stanovit množství a složení PDHc pomocí elektroforézy a imunochemické analýzy [1].
Léčba:
Laktátovou acidózu je třeba kompenzovat alkalinizační léčbou. U části pacientů se nasazuje ketogenní dieta. Asi u 5 % pacientů, u nichž je porucha na úrovni thiaminového přenašeče (kofaktoru PDHc), dochází ke zmírnění projevů při podávání thiaminu v dávce 50 mg//kg/den a biotinu [1].
PORUCHY LIPIDOVÉHO METABOLISMU
1. Familiární hypercholesterolémie
Jako familiární hypercholesterolémii označujeme skupinu poruch vychytávání LDL částic pomocí LDL receptoru. Poruchy vedou k hromadění LDL částic v krvi a k izolované LDL-hypercholesterolémii, přičemž hladiny HDL cholesterolu a triacylglycerolů mohou být normální. Familiární hypercholesterolémie zahrnuje 3 autosomálně dominantně přenosné poruchy: poruchu LDL-receptoru, poruchu apolipoproteinu B-100 (ApoB) a poruchu proprotein konvertázu subtilisin/kexin 9 (PCSK9) [19]. Závažnější průběh má onemocnění u homozygotů. Výskyt familiární hypercholesterolémie (FH) je v evropské a severoamerické populaci až 1 : 200.
K projevům FH patří podkožní a šlachové xantomy a arcus senilis corneae před 45. rokem věku a projevy předčasné aterosklerózy (ischemická choroba srdeční, infarkt myokardu či cévní mozková příhoda před 50. ro-kem). U heterozygotů se první příznaky objevují v dospívání, u homozygotů již v dětství, infarkt myokardu se u nich může vyskytovat i před 20. rokem.
Onemocnění diagnostikujeme na základě klinického podezření a nálezu hypercholesterolémie, která u heterozygotů dosahuje 7–15 mmol/l a u homozygotů dokonce až 16–23 mmol/l celkového cholesterolu. Diagnózu můžeme potvrdit molekulárně-genetickou analýzou genů LDLR, APOB nebo PCSK9 [19].
Léčba:
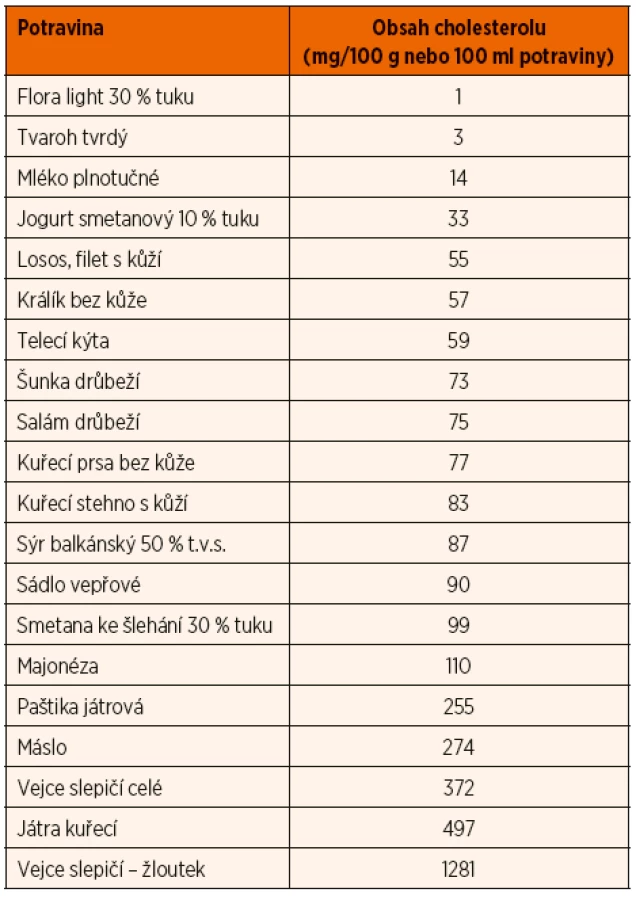
Základem léčby je nízkotuková dieta, režimová opatření se zvýšenou fyzickou zátěží a farmakoterapie založená na podávání statinů. U heterozygotů se může částečně uplatňovat pouze dieta s restrikcí tuku a cholesterolu (pokles LDL o 5–10 %). Příjem cholesterolu by měl být maximálně 200–300 mg/den. V dietě omezujeme nejen volné tuky, především živočišné tuky a kokosový a palmový olej, ale také potraviny s vysokým obsahem nasyceného tuku, jako jsou uzeniny, masné výrobky, tučné mléčné výrobky, žloutky a výrobky z nich, polotovary, lahůdky, sladké a jemné pečivo, sušenky, rychlé občerstvení apod. Přehled obsahu cholesterolu ve vybraných potravinách přináší tabulka 6. Hladinu LDL-cholesterolu pomáhá naopak snížit příjem tuků s polynenasycenými mastnými kyselinami, zejména ω-3. Jejich zdrojem jsou především tučné mořské ryby jako losos, makrela, tuňák či sleď, z rostlinných zdrojů řepkový olej, lněné a konopné semínko, avokádo. Příznivý vliv má ale i příjem ω-6 polynenasycených mastných kyselin (ω-6 PUFA), například ze slunečnicového oleje, semen a ořechů. Aby byl zachován příznivý vliv, měl by být příjem ω-3 polynenasycených mastných kyselin alespoň 0,5–2 % energetického příjmu a ω-6 PUFA 2,5–9 % příjmu energie.
2. Smithův-Lemliho-Opitzův syndrom
Smithův-Lemliho-Opitzův syndrom (SLOS) je nejčastější z poruch endogenní tvorby cholesterolu [1]. Je AR dědičný a je způsoben deficitem enzymu 7-dehydrocholesterolreduktázy.
V novorozeneckém věku se SLOS obvykle projevuje intrauterinní růstovou retardací a kraniofaciální dysmorfií. Změny postihují i morfologii prstů na rukou a nohou, časté jsou vrozené vývojové vady srdce a urogenitálního ústrojí, rozštěp patra či jazyka, malformace CNS a další vrozené vývojové vady. Míra postižení je mírná až těžká, výjimkou není porod mrtvého plodu nebo úmrtí novorozenců či kojenců [1]. Děti se SLOS velmi často neprospívají, mají poruchy růstu, potíže s pitím a častým zvracením. Často je nutné zavedení PEG či gastrostomie s fundoplikací. SLOS se projevuje rovněž výrazně opožděným psychomotorickým vývojem, mentální schopnosti jsou obvykle v pásmu středně těžké až těžké mentální retardace. Přítomna bývá hyperaktivita, hetero - či autoagresivita, úzkost, afektivní porucha. I při mírné mentální retardaci jsou přítomny poruchy učení a pozornosti. Časté jsou poruchy autistického spektra. Laboratorně je u většiny těchto dětí významně snížená hladina cholesterolu, naopak se kumulují prekurzory cholesterolu, zejména 7-dehydrocholesterol v krvi [1].
Diagnostika je založena na klinickém podezření a typickém laboratorním nálezu, potvrzuje se molekulárně-genetickým vyšetřením.
Léčba:
Léčba spočívá především v symptomatické terapii, chirurgické či ortopedické léčbě jednotlivých vývojových vad a v sociální pomoci. Pacientům je třeba suplementovat cholesterol v dávce 20–150 mg/kg/den pomocí PZLÚ nebo podáváním žloutku [1]. Podávání cholesterolu vede nejen ke zvýšení hladiny celkového cholesterolu, ale zpětnou vazbou také ke snížení hladiny toxického 7-dehydrocholesterolu. Tento efekt je posílen i podáváním statinů.
3. Sitosterolémie
Sitosterolémie je AR dědičné onemocnění, způsobené mutací v některém z tandemových genů pro ATP-cassette vazebné proteiny – ABCG5 nebo ABCG8. Tyto transportní proteiny umožňují exkreci především noncholesterolových sterolů z hepatocytů a epitelových buněk žlučníku do žluče nebo z enterocytů do střevního lumen. Defekt v ABCG5/ABCG8 genu vede ke zvýšené absorpci noncholesterolových sterolů ve střevě, ke snížení jejich exkrece žlučí, a tím k 30–100násobnému zvýšení plazmatických hladin fytosterolů. Narušena je i absorpce a exkrece cholesterolu.
Onemocnění se projevuje šlachovými a podkožními xantomy, hemolytickou anémií, makrotrombocytopenií, abnormálním krvácením, splenomegalií, předčasnou aterosklerózou, artralgiemi, artritidou, neprospíváním, hepatopatií [20, 21, 22]. Kromě hyperfytosterolémie je u dětí většinou přítomna i hypercholesterolémie.
Diagnostika je založena na klinickém podezření a laboratorním nálezu. Diagnózu potvrzuje molekulárně-genetické vyšetření nálezem mutace v genu ABCG5 nebo ABCG8.
Léčba:
Dieta s nízkým obsahem cholesterolu a fytosterolů přináší výrazný efekt, přesto je k normalizaci cholesterolémie nutná kombinace s léčbou ezetimibem, případně cholestyraminem [22, 23]. Ve stravě omezujeme rostlinné i živočišné tuky, nepodáváme uzeniny a masné výrobky, žloutky, tučné mléčné výrobky, avokádo, ořechy a semena, obilné klíčky, měkkýše. Podáváme pouze velmi libové maso, ryby s nízkým obsahem tuku, nízkotučné nebo v menším množství polotučné mléčné výrobky, obiloviny, luštěniny, ovoce, zeleninu, vaječný bílek. Hladiny fytosterolů zůstávají i při důsledné dietě a kombinované léčbě alespoň mírně zvýšené.
4. Poruchy β-oxidace mastných kyselin
β-oxidace mastných kyselin je proces, při kterém dochází v mitochondriální matrix ke zkracování řetězce mastných kyselin, tvorbě acetyl-CoA (který vstupuje do Krebsova cyklu a je využíván k tvorbě ketolátek) a redukčních ekvivalentů (NADH + H+ a FADH2), které se dále využívají k oxidativní fosforylaci a tvorbě energie. β-oxidace mastných kyselin je v době hladovění hlavním zdrojem energie. Probíhá pouze za aerobních podmínek a je katalyzována 4 enzymy: acyl-CoA dehydrogenázou, enoyl-CoA hydratázou, 3-hydroxyacyl-CoA dehydrogenázou a β-ketothiolázou [1]. Acyl-CoA dehydrogenázy dělíme podle délky uhlíkatého řetězce na dehydrogenázu velmi dlouhých acyl-CoA (VLCAD), dehydrogenázu dlouhých 3-hydroxyacyl-CoA (LCHAD), dehydrogenázu středně dlouhých acyl-CoA (MCAD) a dehydrogenázu mastných kyselin s krátkým řetězcem (SCAD). Deficity těchto jednotlivých dehydrogenáz mají za následek poruchu β-oxidace mastných kyselin o konkrétní délce řetězce. Mastné kyseliny s dlouhým a velmi dlouhým řetězcem navíc potřebují k transportu přes vnější a vnitřní mitochondriální membránu specifické přenašeče karnitin palmitoyltransferázu I (CPT I), karnitinacylkarnitin translokázu (CACT) a karnitin palmitoyltransferázu II (CPT II), jejichž deficit také způsobuje poruchu zpracování mastných kyselin s dlouhým a velmi dlouhým řetězcem. Mastné kyseliny s krátkým a středně dlouhým řetězcem prostupují mitochondriální membrány bez potřeby specifických transportních proteinů.
Porucha β-oxidace mastných kyselin se středně dlouhým řetězcem (MCAD deficit)
U MCAD deficitu je narušen metabolismus mastných kyselin se sudým počtem uhlíků o délce C6-C12. Jedná se o nejčastější poruchu β-oxidace mastných kyselin.
Projevy hypoglykémie se nejčastěji objevují mezi 3.–24. měsícem při prodloužení pauzy mezi krmením nebo při horečnatém infektu, zvracení nebo průjmu. Hypoglykémie jsou neketotické nebo hypoketotické [1] a projevují se změnou chování, pocením s třesem, progredují do poruchy vědomí s křečemi. U nediagnostikovaných pacientů končí až 20 % prvních atak úmrtím [24]. Při opakovaných hypoglykémiích může dojít k opoždění psychomotorického vývoje nebo k rozvoji sekundární epilepsie. U pozdní formy MCAD deficitu mohou být ataky rhabdomyolýz. Laboratorně je v atace přítomna těžká hypoglykémie, metabolická acidóza, někdy elevace laktátu, mohou být zvýšené aminotransferázy, hladina amoniaku a kyseliny močové. Pacienti mohou mít hepatomegalii s malokapénkovou steatózou [1].
MCAD deficit je od r. 2009 součástí novorozeneckého screeningu. Laboratorně ho lze potvrdit nálezem zvýšené koncentrace oktanoyl-karnitinu a zvýšených poměrů acyl-karnitinů [24]. V moči jsou přítomny zvýšené hladiny kyseliny sebakové, suberové a adipové a glycinových konjugátů. Na molekulárně-genetické úrovni potvrzuje diagnózu MCAD deficitu nález mutace v genu ACADM.
Léčba:
Léčba spočívá v antihypoglykemickém režimu – podávání frekventní stravy s dostatečným obsahem škrobů s nižším glykemickým indexem, včetně podávání škrobů v noci. Do nočních dávek stravy přidáváme u batolat a větších dětí nevařený kukuřičný škrob, přes den u větších dětí a ve dne i v noci u novorozenců a kojenců přidáváme do stravy maltodextrin. U novorozenců a kojenců do 6 měsíců věku by interval mezi jídly neměl být delší než 3 hodiny, u starších kojenců 4 hodiny, u batolat 5 hodin, u předškolních dětí 6 hodin, u školních dětí 7 hodin a u mladistvých a dospělých 8 hodin. Ve snaze omezit prudké výkyvy glykémie omezujeme příjem jednoduchých cukrů, výjimkou je jejich podávání při odvracení hypoglykémie. Omezení jednoduchých sacharidů je vhodné také z důvodu prevence nadváhy, která je u pacientů s poruchami β-oxidace mastných kyselin častá. Energetický příjem a mikronutrienty jsou doplňovány pomocí PZLÚ s obsahem tuku <0,1 g/100 g.
Kontraindikováno je podávání MCT olejů a ve stravě rovněž omezujeme jejich přirozené zdroje, jako je např. olivový olej. Podávání L-karnitinu se u MCAD nedoporučuje, a to ani při jeho mírně snížené hladině [1]. Při zvýšené fyzické aktivitě se podávají 10–25% roztoky maltodextrinu, u některých pacientů preventivní infuze glukózy.
Porucha β-oxidace mastných kyselin s dlouhým řetězcem (LCHAD deficit)
Jedná se o AR dědičnou metabolickou poruchu, při které je narušen metabolismus dlouhých mastných kyselin se sudým počtem uhlíků o délce C14-C22.
První projevy se objevují v novorozeneckém až batolecím věku při delším lačnění nebo při zvýšených nárocích organismu (infekt, fyzická zátěž). LCHAD deficit se projevuje Reye-like syndromem – poruchou vědomí s hypoketotickou nebo neketotickou hypoglykémií, mírnou hyperamonémií, hepatopatií. Sonografické vyšetření nalézá hepatomegalii s mikrovezikulární steatózou v cytoplazmě. Přítomna může být hypertrofická kardiomyopatie, hromadící se toxické metabolity 3-hydroxykyselin, 3-hydroxynenasycených kyselin a jejich karnitinových derivátů poškozují také kosterní svaly za vzniku myopatie a atak rhabdomyolýzy. Komplikací onemocnění je také retinitis pigmentosa a polyneuropatie, zejména dolních končetin [1]. Ataky rhabdomyolýzy jsou poměrně časté i u dobře kompenzovaných pacientů, a to buď při zvýšené fyzické námaze, nebo při akutních infektech. Akutní komplikací rhabdomyolýzy může být renální selhání. U gravidních heterozygotek s plodem s LCHAD deficitem se může ve druhé polovině gravidity rozvinout syndrom AFLP (acute fatty liver of pregnancy), syndrom akutní steatózy jater nebo HELLP syndrom (hemolysis, elevated liver enzymes and low platelets) [1].
Od roku 2009 je LCHAD deficit diagnostikován z novorozeneckého screeningu. V moči jsou přítomny 3-hydroxy-dikarboxylové a dikarboxylové kyseliny (adipová, sebaková, suberová), může být snížena hladina volného karnitinu. Molekulárně-genetické vyšetření nachází mutaci v genu HADHA. V případě, že se mutace nachází také v genu HADHB, mluvíme o deficitu mitochondriálního trifunkčního proteinu (MTP), který se skládá z podjednotek α a β a je lokalizovaný na vnitřní mitochondriální membráně.
Léčba:
Základem léčby je antihypoglykemický režim, stejně jako u deficitu MCAD. Shodné je frekventní podávání výživy s nočními dávkami, podáváním maltodextrinů a později nevařeného kukuřičného škrobu. Během dne by pacient měl jíst každé 3 hodiny a v noci by intervaly mezi jídly neměly být další než 4 hodiny. Podává se rovněž PZLÚ s obsahem tuku <0,1 g/100 g s obsahem mikronutrientů. Navíc je nutná restrikce přirozených tuků, obvykle na cca 1 g/kg/den, příjem tuků a energie se navyšuje podáváním MCT olejů, jejichž metabolismus není u deficitu LCHAD narušen. MCT tuky se podávají obvykle rozděleně s jednotlivými dávkami stravy, jejich podávání je možné přizpůsobit fyzické zátěži. Pokud pacient netoleruje větší dávky MCT oleje, je možné část nahradit podáváním MCT tuků v práškové formě. Nutná je suplementace LC-PUFA a vitaminu E, nedoporučuje se suplementace karnitinu. V případě akutního infektu s netolerancí stravy, se zvracením nebo průjmem a při rhabdomyolýze je nutná hospitalizace a podávání vysokých dávek glukózy (6–12 mg//kg/min, dávka klesá s věkem) a inzulinu až do úpravy laboratorních parametrů a do obnovení plné tolerance stravy. Ani velmi dobrá dietní kompenzace nezabrání občasným metabolickým rozvratům při akutních infektech nebo zvýšené fyzické námaze, nelze se proto zcela vyhnout dlouhodobým komplikacím onemocnění (polyneuropatie, retinitis pigmentosa, myopatie). Nejnovější léčbou, která je však zatím předmětem výzkumu, je podávání oleje s triheptanoátem (mastné kyseliny s lichým počtem uhlíků).
PORUCHY METABOLISMU PURINŮ
Většina poruch metabolismu purinů je autosomálně recesivně přenosná. Patří sem ale i X-vázaná onemocnění (např. Leschův-Nyhanův syndrom). Poruchy metabolismu purinů rozdělujeme na poruchy de novo syntézy, poruchy degradace a poruchy zpětné syntézy [1]. Hyperurikémie může být způsobena i poruchou exkrece kyseliny močové do moči.
Klinický obraz je velmi rozmanitý, projevy mohou být od velmi mírných až po těžké stavy s neurologickým postižením. Mezi časté symptomy poruch purinového metabolismu s hyperurikémií patří dnavá artritida, vznik dnavých tof v podkoží, ledvinné a močové urátové kameny. Neurologické postižení zahrnuje hypotonii nebo hypertonii, psychomotorickou retardaci, dystonii, choreoatetózu. U Leschova-Nyhanova syndromu je typická autoagrese.
Diagnostika vychází z klinického podezření. Laboratorní vyšetření nalézá hyper - nebo hyporurikémii, zvýšenou exkreci kyseliny močové, zvýšené může být vylučování hypoxantinu a xantinu. Diagnózu potvrzuje molekulárně-genetické vyšetření příslušných genů.
Léčba:
Pacienti s hyperurikémií musí dodržovat přísnou nízkopurinovou dietu s obsahem purinů nepřekračujícím 1000 mg/týden. Ze stravy jsou eliminovány především vnitřnosti, kvasnice, alkohol, silná káva, silný čaj, kolové nápoje, sardinky, ančovičky. Snížený musí být příjem masa a ryb, luštěnin, hub, ale i některého sušeného ovoce (rozinky) a některé zeleniny (hrášek, brokolice). Dieta je založena především na čerstvém ovoci a zelenině, mléčných výrobcích, obilovinách, podávat lze vejce a výrobky z nich (např. šmakoun), tofu. Z masa a ryb mají nižší obsah purinů krůtí, kuřecí a kachní maso, zajíc, vepřové a hovězí maso, úhoř, candát, štika. Denní porce masa by neměla přesáhnout 100 g (hmotnost před tepelnou úpravou). Naopak nevhodné jsou pro vysoký obsah purinů telecí a jehněčí maso, koňské maso, šproty, sardinky, tuňák. Z technologických úprav je vhodné vaření a dušení ve větším množství tekutin pro vyplavení purinů.
Přehled obsahu purinů ve vybraných potravinách přináší tabulka 7. Velmi důležitý je i zvýšený pitný režim, a to v průběhu celého dne. Hladina kyseliny močové se ovlivňuje i pomocí purinových nebo nepurinových inhibitorů.

ZÁVĚR
Nutriční terapie má zásadní význam v léčbě řady dědičných poruch metabolismu, především u tzv. nemocí malých molekul. Základem léčby je dieta s restrikcí látky, jejíž metabolismus je narušen. Energetický příjem udržujeme tak, aby nedocházelo ke katabolismu ani k rozvoji nadváhy. Dieta je většinou doplněna podáváním potravin pro zvláštní lékařské účely, modulárními dietetiky, případně suplementací živin, které jsou v dietě nedostatečně zastoupeny.
Cílem nutriční terapie je celoživotní udržení normálních hladin metabolitů a dosažení normálního růstu a vývoje pacienta. Přinesli jsme přehled hlavních zásad nutričního managementu u nejčastěji se vyskytujících nutričně ovlivnitelných dědičných metabolických poruch.
Podpořeno: MZ ČR – RVO VFN 64165.
Seznam použitých zkratek:
AD – autosomálně dominantní
AMK – aminokyseliny
Apo B – apolipoprotein B-100
AR – autosomálně recesivní
ARG – argináza
ASL – argininosukcinát lyáza
ASS – argininosukcinát syntáza
BH4 – tetrahydrobiopterin
CACT – karnitinacylkarnitin translokáza
CBS – cystathionin-β-syntáza
CPS I – karbamoylfosfát syntáza I
CPT I – karnitin palmitoyltransferáza I
CPT II – karnitin palmitoyltransferáza II
DHA – kyselina dokosahexanová
DHPR – dihydropteridinreduktáza
DPM – dědičné poruchy metabolismu
DRI – doporučený referenční příjem
G-6-Páza – glukóza-6-fosfatáza
Gal – galaktóza
GALT – galaktóza-1-fosfát uridyltransferáza
GMP – glykomakropeptid
GSD Ia – glycogen storage disease Ia (glykogenóza Ia)
Hcy – homocystein
HFI – hereditární fruktózová intolerance
HMG-CoA – 3-hydroxy-3-metylglutaryl-Co A
HPA – hyperfenylalaninémie
Ile – isoleucin
LC-PUFA – vícenenasycené mastné kyseliny s dlouhým řetězcem
Leu – leucin
LCHAD – dehydrogenáza dlouhých 3-hydroxyacyl-CoA
LNAA – velké neutrální aminokyseliny
MCAD – dehydrogenáza středně dlouhých acyl-CoA
Met – methionin
MMA – methylmalonová acidurie
MSUD – maple syrup urine disease (leucinóza)
MTP – mitochondriální trifunkční protein
NAGS – N-acetylglutamát syntáza
OTC – ornitintranskarbamyláza
PA – propionová acidurie
PAH – fenylalaninhydroxyláza
PCSK9 – proprotein konvertáza subtilisin/kexin9
PDHc – pyruvátdehydrogenázový komplex
PEG – perkutánní endoskopická gastrostomie
PEJ – perkutánní endoskopická jejunostomie
Phe – fenylalanin
PKU – fenylketonurie
PZLÚ – potravina pro zvláštní lékařské účely
SCAD – dehydrogenáza mastných kyselin s krátkým řetězcem
SLOS – Smithův-Lemliho-Opitzův syndrom
T – tuky
tHcy – celkový homocystein
Tyr – tyrosin
UDP – uridindifosfát
Val – valin
VLCAD – dehydrogenáza velmi dlouhých acyl-CoA
ω-6 PUFA – omega-6 vícenenasycené mastné kyseliny
Korespondující autor:
Doc. MUDr. RNDr. Pavel Ješina, Ph.D.
Metabolické centrum
Klinika dětského a dorostového lékařství
1. LF UK a VFN
Ke Karlovu 2
128 08 Praha 2
e-mail: pavel.jesina@vfn.cz
Zdroje
1. Honzík T, Zeman J, a kol. Dědičné poruchy metabolismu v kazuistikách. 1. vyd. Praha: Mladá fronta a.s., 2016 : 1–278. ISBN: 978-80-204-4178-4.
2. Boyer SW, Barclay LJ, Burrage LC. Inherited metabolic disorders: Aspect of chronic nutritional management. Nutr Clin Pract 2015 August; 30 (4): 502–510.
3. Honzík T, Zeman J. Dědičné poruchy metabolismu v dětském věku. 1. vyd. Praha: IPVZ, 2013 : 1–96. ISBN: 978-80-87023-10-5.
4. Acosta PB (ed), et al. Nutrition Management of Patients with Inherited Metabolic Disorders. 1st ed. Sudbury, Massachusetts: Jones and Bartlett Publishers, 2010 : 1–476. ISBN-13 : 978-0-7637-5777-9.
5. Nützenadel W. Failure to thrive in childhood. Dtsch Arztebl Int 2011; 108 (38): 642–649.
6. MacLeod EL, Ney, DM. Nutritional management of phenylketonuria. Ann Nestle Eng 2010; 68 : 58–69.
7. Singh RH, Rohr F, Frazier DM, et al. Recommendations for the nutrition management of phenylalanine hydroxylase deficiency. Genet Med 2014; 16 (2): 121–131.
8. Singh RH, Cunningham AC, Mofidi S, et al. Updated, web-based nutrition management guideline for PKU: An evidence and consensus based approach. Mol Genet Metab 2016; 118 : 72–83.
9. Morris AAM, Kožich V, Santra S, et al. Guidelines for the diagnosis and management of cystathionine beta-synthase deficiency. J Inherit Metab Dis 2017; 40 : 49–74.
10. Fernandes J, Saudubray JM, van der Berghe G, et al. Diagnostika a léčba dědičných metabolických poruch. 4. vyd. Praha: Triton, 2008 : 1–607. ISBN: 80-738-7096-6.
11. Frazier DM, Allgeier C, Homer C, et al. Nutrition management guideline for maple syrup urine disease: An evidence-and konsensus-based approach. Mol Genet Metab 2014; 112 : 210–217.
12. Singh RH. Nutritional management of patients with urea cycle disorders. J Inherit Metab Dis 2007; 30 : 880–887.
13. Adam S, Almeida MF, Assoun M, et al. Dietary management of urea cycle disorders: European practice. Mol Genet Metab 2013; 110 : 439–445.
14. Van Calcar SC, Bernstein LE, Rohr FJ, et al. A re-evaluation of life-long severe galactose restriction for the nutrition management of classic galactosemia. Mol Genet Metab 2014; 112 : 191–197
15. Kishnani PS, Austin SL, Abdenur JE, et al. Diagnosis and management of glycogen storage disease type I: a practice guideline of the American College of Medical Genetics and Genomics. Genet Med 2014; 16 (11): 1–29.
16. Heller S, Worona L, Consuelo A. Nutritional therapy for glycogen storage diseases. J Pediatr Gastroenterol Nutr 2008; 47 (1): 15–21.
17. Goldberg T, Slonim AE. Nutrition therapy for hepatic glycogen storage diseases. J Am Diet Assoc 1993 Dec; 93 (12): 1423–30.
18. Brožová K, Hadač J. Ketogenní dieta. Neurol praxi 2013; 14 (2): 89–91.
19. Youngblom E, Pariani M, Knowles JW. Familial hypercholesterolemia. GeneReviews Seattle. https://www.ncbi.nlm.nih.gov/books/NBK174884, cit. 14. 3. 2018.
20. Nghiem-Rao TH, Patel SB. Investigating sitosterolemia to understand lipid physiology. Clin Lipidol 2013; 8 (6): 649–658.
21. Yoo E-G. Sitosterolemia: a review and update of pathophysiology, clinical spectrum, diagnosis, and management. Ann Pediatr Endocrinol Metab 2016; 21 : 7–14.
22. Floriánková M, Urbanová Z, Bláhová Š, a kol. Sitosterolémie: klinická, biochemická a molekulárně genetická charakteristika 3letého chlapce s významnou hypercholesterolémií. Čes-slov Pediat 2017; 72 (8): 495–503.
23. Merkens LS; Myrie SB; Steiner RD, et al. Sitosterolemia. GeneReviews – NCBI Bookshelf online. http://www.ncbi.nim.nih.goc/books/NBK131810/, cit. 3. 5. 2017.
24. Purevsuren J, Hasegawa Y, Fukuda S, et al. Clinical and molecular aspects of Japanese children with medium chain acyl-CoA dehydrogenase deficiency. Mol Genet Metab 2012; 107 : 237–240.
Štítky
Neonatologie Pediatrie Praktické lékařství pro děti a dorostČlánek vyšel v časopise
Česko-slovenská pediatrie

2018 Číslo 6
- Efektivita kartáčku Sonicare For Kids u dětí předškolního věku
- Inosin pranobex v léčbě HPV infekcí děložního čípku
- Cytomegalovirové infekce u novorozenců a dětí
Nejčtenější v tomto čísle
- Charakteristické klinické příznaky a laboratorní odchylky dědičných poruch metabolismu
- Komplexný pohľad na deficit vitamínu B12 v detskom veku
- Novorozenecký screening dědičných metabolických poruch v České republice
- Lyzozómové choroby – vývoj diagnostiky a liečby na Slovensku
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy