
Deficit fosfomanomutázy 2: klinická, biochemická a molekulárně-genetická charakteristika 22 pacientů diagnostikovaných v České republice
Phosphomannomutase 2 deficiency: clinical, biochemical and molecular analyses in 22 Czech patients
Introduction:
PMM2-CDG is the most common autosomal recessive N-glycosylation disorder with more than 900 patients described worldwide. It is caused by a deficiency of the phosphomannomutase 2 enzyme (PMM2) which catalyzes the second step of the mannose pathway, namely the conversion of mannose-6-phosphate to mannose-1-phosphate. The clinical presentation is characterised by encephalopathy, neuropathy, typical dysmorphism, cerebellar atrophy and coagulopathy. We present the results of clinical, biochemical and molecular analyses in patients diagnosed in the Czech Republic.
Results:
Since 2002, a total of 22 Czech patients from 18 families with PMM2 deficiency have been diagnosed. The age range of the patients spans from 9 months to 29 years with a median of 14 years, except two patients who died during infancy. Muscle hypotonia, intellectual disability of varying severity, strabismus, ataxia, bone deformities, and coagulopathy were observed in all patients. Cerebellar atrophy was documented in 94% of the investigated patients. The characteristic dysmorphism (inverted nipples and atypical fat pads) were present in 82% of the patients. Nine patients suffered from seizures, and three patients showed transient neurological deterioration after stroke-like episodes. In all the patients, increased amount of hypoglycosylated transferrin was found by isoelectric focusing. The diagnosis of the PMM2-CDG was confirmed at enzymatic and/or at molecular levels. Molecular analyses revealed that all patients are compound heterozygotes for a total of 10 different mutations in PMM2, and that 71% of our patients´ alleles have one of the two most frequent genetic variants (c.422G>A, c.338C>T).
Conclusion:
The estimated incidence of PMM2-CDG is 1 : 20,000, suggesting that this disorder is underdiagnosed in the Czech Republic. PMM2-CDG must be considered in differential diagnosis of patients with cerebellar atrophy even if they do not manifest characteristic dysmorphism. We plan to include our patients in a longitudinal international multicenter observational study and potentially the upcoming clinical trial with LipoM1P (lipomised mannose-1-phosphate).
KEY WORDS:
CDG syndrome, phosphomannomutase 2, PMM2-CDG, isoelectric focusing, cerebellar atrophy, coagulopathy
Autoři:
A. Čechová; N. Ondrušková; M. Tesařová; H. Hansíková; J. Zeman; T. Honzík
Působiště autorů:
Klinika dětského a dorostového lékařství 1. LF UK a VFN, Praha
Vyšlo v časopise:
Čes-slov Pediat 2018; 73 (6): 365-374.
Kategorie:
Sympozium: dědičné poruchy metabolismu
Souhrn
Úvod:
Deficit fosfomanomutázy 2 (PMM2-CDG) je nejčastějším typem poruch N-glykosylace s popsanými >900 pacienty. Onemocnění je autosomálně recesivně dědičné a biochemickou podstatou je porucha přeměny manóza-6-fosfátu na manóza-1-fosfát (M1P). Onemocnění se projevuje encefalopatií, neuropatií, typickou dysmorfií, atrofií mozečku a koagulopatií. Práce prezentuje výsledky klinických, biochemických a molekulárních vyšetření pacientů diagnostikovaných v ČR.
Výsledky:
Od roku 2002 bylo v ČR diagnostikováno 22 pacientů z 18 rodin. Dvě děti zemřely v kojeneckém věku. Věk žijících 20 pacientů je v rozpětí 9 měsíců až 29 let (medián 14 let). U všech pacientů je přítomný hypotonický a mozečkový syndrom, strabismus, deformity skeletu, koagulopatie a mentální retardace, která je v pásmu od lehkého až po hluboké postižení. U 94 % pacientů jsme prokázali atrofii mozečku. Typická dysmorfie (atypické rozložení tuku a vpáčení bradavek) byla zjištěna u 82 % dětí. U 9 pacientů se projevila epilepsie a 3 pacienti prodělali iktu podobné příhody. Isoelektrická fokusace transferinu v séru prokázala u všech pacientů zvýšené zastoupení nízkosialovaných forem. Diagnóza byla potvrzena enzymologicky detekcí snížené aktivity PMM2 v lymfocytech či fibroblastech a/nebo molekulárně-geneticky. V našem souboru pacientů bylo zastoupeno 10 mutací v genu PMM2, všichni pacienti jsou složení heterozygoti a 71 % mutovaných alel neslo jednu ze dvou prevalentních patogenních variant (c.422G>A, c.338C>T).
Závěr:
S ohledem na odhadovanou incidenci PMM2-CDG 1 : 20 000 se jedná o onemocnění v ČR poddiagnostikované. PMM2-CDG patří do diferenciální diagnostiky všech dětí s atrofií mozečku, a to i bez přítomnosti charakteristických dysmorfických rysů. V plánu je zařazení českých pacientů do prospektivní multicentrické mezinárodní studie hodnotící přirozený průběh onemocnění a eventuálně zařazení do klinické studie s novou experimentální léčebnou molekulou LipoM1P (manóza-1-fosfát inkorporovaná do liposomu).
KLÍČOVÁ SLOVA:
CDG syndrom, fosfomanomutáza 2, PMM2-CDG, isoelektrická fokusace, atrofie mozečku, koagulopatie
ÚVOD
Glykosylace je jednou z nejběžnějších a nejkomplexnějších posttranslačních úprav proteinů a lipidů [1], která se týká více než poloviny buněčných bílkovin [2]. Odhaduje se, že na syntéze glykokonjugátů se podílí >2 % našich genů [2]. Její procesy se odehrávají v cytoplazmě, endoplazmatickém retikulu a Golgiho aparátu. Glykosylace ovlivňuje stabilitu, konformaci, lokalizaci a odolnost proteinů vůči proteázám, podílí se na buněčných interakcích, účastní se vazby cytoskeletu s extracelulární matrix [3]. Glykoproteiny se podle typu vazby připojující oligosacharid (glykan) dělí na N-, O- a C-vázané glykoproteiny; z dalších glykosylovaných sloučenin zmiňme proteoglykany, glykosfingolipidy a proteiny s glykosylfosfatidylinositolovou (GPI) kotvou.
Dědičné poruchy glykosylace (Congenital Disorders of Glycosylation, CDG) jsou širokou skupinou geneticky podmíněných onemocnění. Od svého objevení v roce 1980 [4] se podařilo popsat více než 125 typů glykosylačních poruch [5]. Vzhledem k velké rozmanitosti glykosylovaných molekul zahrnují i CDG syndromy široké spektrum klinických projevů od monosymptomatického postižení po těžké multiorgánové dysfunkce a fetální hydrops [6].
Základem diagnostiky pro poruchy N-glykoproteinů je isoelektrická fokusace (IEF) transferinu [7] a pro defekty O-glykoproteinů IEF apolipoproteinu CIII [8], dále je využívaná analýza celkových glykanů v séru/plazmě hmotnostní spektrometrií [9]. Potvrzení diagnózy probíhá na úrovni průkazu snížené aktivity postiženého enzymu a/nebo molekulárně-geneticky průkazem patogenní mutace v příslušeném genu. Od dřívějšího dělení N-glykosylačních poruch na CDG typ I (porucha syntézy oligosacharidu vázaného přes lipid dolichol lokalizovaná v cytoplazmě a endoplazmatickém retikulu) a II (porucha modifikace N-glykoproteinů v Golgiho aparátu) [10] se dnes upouští a jednotlivá onemocnění se nazývají podle postiženého enzymu/genu [11]. Léčba je zatím známá jen u několika typů CDG, a to buď ve formě perorální suplementace (např. galaktóza u PGM1-CDG, manóza u MPI-CDG), nebo transplantací orgánů (např. játra u MPI-CDG, srdce u DOLK-CDG) [12].
Deficit fosfomanomutázy 2 (PMM2-CDG) je nejčastějším typem poruch N-glykosylace s popsanými >900 pacienty na světě. Biochemickou podstatou onemocnění je porucha přeměny manóza-6-fosfátu na manóza-1-fosfát v cytoplazmě. Manóza-1-fosfát je prekurzor GDP-manózy a dolichol-fosfát-manózy, které slouží jako donor manózy v endoplazmatickém retikulu pro syntézu oligosacharidu vázaného přes lipid (LLO). Onemocnění se projevuje obvykle v kojeneckém věku encefalopatií, neuropatií, typickou dysmorfií, atrofií mozečku a koagulopatií. Prognóza tohoto onemocnění může být závažná (až 15–20 % dětí umírá do 2 let věku), účinná léčba dosud není známá, experimentální terapie manóza-1-fosfátem inkorporovaným do liposomu (LipoM1P) je v předfázi I/II klinické studie.
V této práci prezentujeme výsledky klinických, biochemických a molekulárních vyšetření pacientů s PMM2-CDG diagnostikovaných na Klinice dětského a dorostového lékařství 1. LF UK a VFN.
METODIKA
Pacienti
Na našem pracovišti bylo od roku 2002 s PMM2-CDG diagnostikováno 22 pacientů z 18 rodin, 13 z nich jsou dívky, 9 chlapci. Věk žijících 20 pacientů je v rozpětí 9 měsíců až 29 let (medián 14 let), dvě děti zemřely v kojeneckém věku. Všichni pacienti jsou kavkazského etnika, příbuzenské sňatky se v žádné rodině nevyskytly.
Mentální retardace (intelektuální disabilita) byla hodnocena na základě DSM-4 (Diagnostic and Statistical Manual of Mental Disorders) klasifikace Americké psychiatrické společnosti.
Trombotické/tromboembolické a krvácivé komplikace vyžadující antikoagulační léčbu definují těžkou koagulopatii.
Metody
K potvrzení defektu na úrovni biosyntézy N-glykoproteinů jsme použili isoelektrickou fokusaci sérového transferinu s následnou imunofixací dle dříve publikované metodiky [8]. Aktivita PMM2 byla měřena spektrofotometricky při 340 nm jako přeměna NADP+ na NADPH v izolovaných lymfocytech z periferní krve a/nebo v kultivovaných fibroblastech také podle dříve publikované metodiky (Hansíková H, Ondrušková N, Honzík T, Veselá K, Horová E, Švecová Š, Tesařová M, Zeman J. Aktivita fosfomanomutázy 2 u pacientů s podezřením na dědičnou poruchu glykosylace. Klin Biochem Metab 2016; 24 (sv. 45, 2): 67‒74).
Molekulárně-geneticky jsme kauzální mutace prokazovali přímou sekvencí osmi exonů a přilehlých intronových oblastí PMM2 genu. Mutace byly u všech pacientů vždy potvrzeny analýzou DNA obou rodičů.
Etika
Všechny odběry a vyšetření byly provedeny po vyjádření informovaného souhlasu zákonnými zástupci pacienta. Studie byla provedena v souladu s Helsinskou deklarací Světové lékařské asociace.
VÝSLEDKY
První projevy onemocnění
U všech našich pacientů se první projevy onemocnění objevily při narození (n = 4) či v časném kojeneckém věku (n = 18), průměrný věk při nástupu onemocnění byl 2,3 měsíce (rozptyl 0 dní – 6 měsíců).
Dva novorozenci měli při nástupu onemocnění dominantní příznaky panhypopituitarismu s iontovým rozvratem a opakovanými hypoglykémiemi. Třetí chlapec manifestoval 14. den života jaterní selhání se smíšenou koagulační poruchou a následným multi-orgánovým selháním. Odmítání kojení, opakované zvracení a alterace stavu byly u čtvrtého novorozence projevem rozvoje post-hemorhagického hydrocefalu.
Typickými projevy při manifestaci v kojeneckém věku bylo opoždění psychomotorického vývoje (n = 17), hypotonie (n = 16), strabismus (n = 11), typická dysmorfie (atypická distribuce tuku, vpáčení prsních bradavek, n = 7), neprospívání (n = 7) a mikrocefalie (n = 5). U jedné dívky byl psychomotorický vývoj při prvním vyšetření hodnocen v mezích širší normy a vyšetřovaná byla pro epizody stáčení bulbů s hypotonií a náhodným nálezem hepatopatie a hepatomegalie. Průměrná prodleva diagnostikování nemoci u našich pacientů je 4,5 roku (rozptyl 2 měsíce – 15 let), ale významně se liší podle data narození: u pacientů narozených před rokem 2000 byla prodleva 10,6 roku, u pacientů narozených po roce 2000 jen 1 rok.
Klinické projevy
Klinické projevy a četnost jednotlivých příznaků u našich pacientů jsou shrnuty v obrázku 1. Typický fenotyp našich pacientů zahrnuje psychomotorickou retardaci, hypotonii, strabismus a typickou dysmorfii (obr. 2). Mentální retardace byla hodnocena u devatenácti dosud žijících pacientů starších 2 let: tři pacienti měli hlubokou, pět těžkou, šest středně závažnou a pět mírnou mentální retardaci. Inteligenční kvocient byl stanoven u dvanácti pacientů s maximální hodnotou 66 bodů. Mikrocefalii jsme zaznamenali v 73 % případů. Z neurologických projevů se u našich pacientů dále vyskytuje periferní neuropatie a mozečkový syndrom. Epilepsie se vyskytla u devíti pacientů, jedná se o atonické i tonicko-klonické záchvaty, první záchvat se projevil od 6 měsíců do 8 let, u jednoho pacienta jsou záchvaty vázané na horečnaté stavy. Záchvaty jsou u pěti pacientů dobře kompenzované na monoterapii, u tří pacientů kompenzované na dvojkombinaci antiepileptik, u jednoho pacienta se ani dvojkombinací léků nepodařilo docílit ideální kompenzace a 1x měsíčně se stále objevují epiparoxysmy. Iktu podobné příhody se vyskytly u tří pacientů ve věku 2–29 let (průměrný věk 11,8 roku). U dvou dívek se vyskytly opakovaně jako hemiparéza trvající několik dní až měsíc, z toho jedna z dívek se při atace chabosti 1x topila, u jednoho chlapce se vyskytla jen jedna epizoda s projevy 4 dny trvající alterace vědomí se spasticitou končetin při horečce.

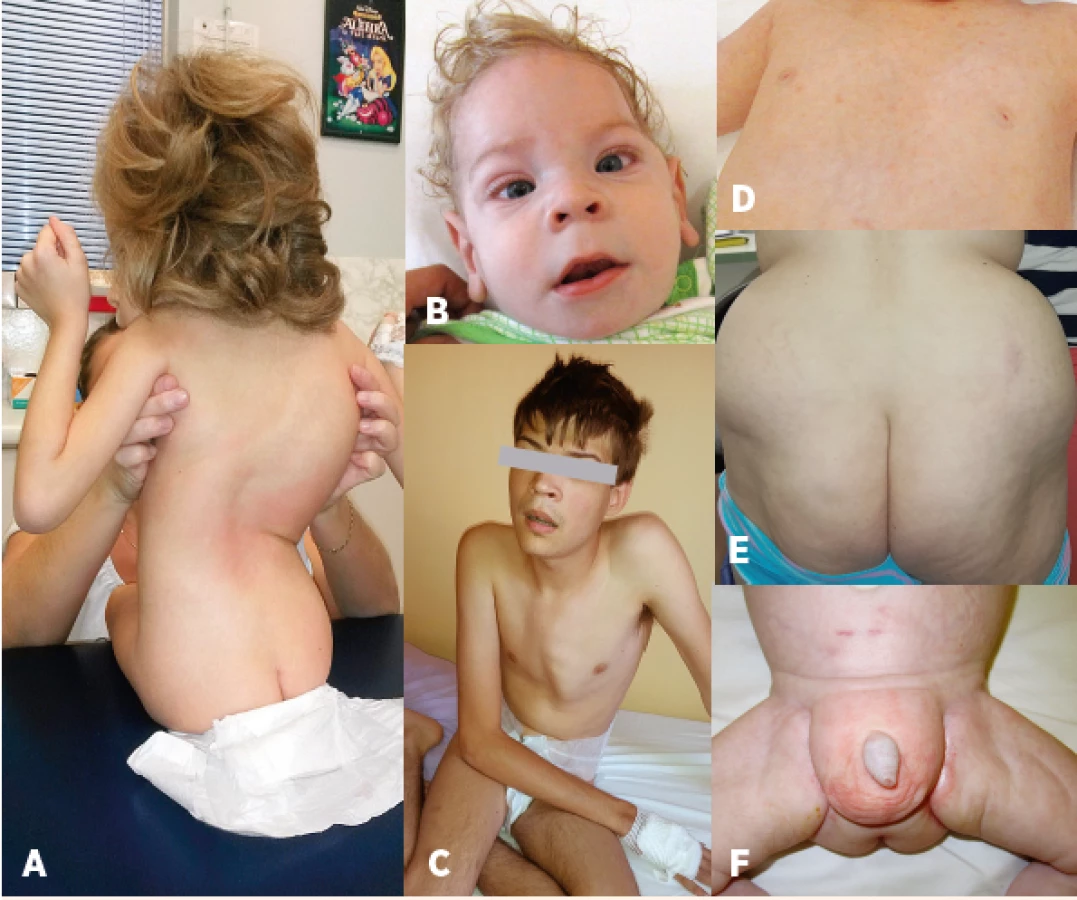
Fig. 2. Characteristic phenotype: A – bone deformities – scoliosis, B – craniofacial
dysmorphism, strabismus, C – pectus carinatum, D – nipples inversion,
E – atypical supragluteal fat distribution, F – atypical perigenital fat
distribution.
Z extraneuronálních projevů jsou nejběžnější endokrinologické, gastroenterologické, oční, nefrologické a kardiologické abnormity. Z endokrinologických nálezů jsme u našich pacientů nejčastěji pozorovali hypothyreózu (u 50 %), která je velmi často subklinická. Hypogonadismus jsme zachytili u všech sledovaných dívek (6/6) v pubertálním věku, ale nevyskytl se u žádného z chlapců (0/3) v daném věku. Sekundární pohlavní znaky se u těchto chlapců vyvinuly normálně. Z dalších endokrinologických patologií se u dvou dětí od novorozeneckého věku rozvinul panhypopituitarismus.
Dalšími častými příznaky byla růstová retardace a neprospívání (u 59, resp. 41 % pacientů). Tyto příznaky se vyvíjejí postnatálně, naprostá většina dětí má při narození normální antropometrická data. Často se vyskytují v kombinaci: 2/3 neprospívajících pacientů mají zároveň i růstovou retardaci.
Kardiologické obtíže jsme pozorovali u 38 % pacientů a zahrnovaly především vrozené vývojové vady srdce (19 %, z toho polovina významných), kardiomyopatii (10 %, z toho polovina vymizela) a nekonstantní perikardiální výpotek (10 %).
Oční patologie se vyskytla až u 85 % pacientů: kromě výše zmíněného strabismu jsme poměrně často dokumentovali retinitis pigmentosa (37 %), refrakční vady různé tíže (30 %), těžké kombinované vady (15 %, atrofie terčů zrakových nervů, katarakta) a u jednoho pacienta byla přítomná bilaterální amauróza.
Abnormální nefrologický nález byl přítomný u 45 % pacientů: nejčastěji se vyskytovala nespecifická nefromegalie se setřelou kresbou bez poruchy funkce, u tří pacientů byla zachycena signifikantní proteinurie (u jednoho pacienta se rozvinul nefrotický syndrom), u tří pacientů se vyskytla cystická přestavba ledvin a u dvou pacientů bylo postižení ledvin spojeno s obtížně kontrolovanou hypertenzí.
Výsledky zobrazovacího vyšetření mozku byly dostupné u 18 pacientů (17x MR, 1x CT), z toho u 17 dětí byl nález atrofie mozečku. U jedné dívky byl na MR mozku ve věku 1 měsíce normální nález bez mozečkové atrofie. U jednoho chlapce byla provedena magnetická rezonance dvakrát, a to ve věku 10 měsíců a téměř 6,5 let: při srovnání snímků je patrná progrese atrofizace mozečku (obr. 3).
Fig. 3. Brain MRI (T2W) showing progressive cerebellar atrophy
in a boy with PMM2-CDG: the same patient at the
age of A – 10 months, B – 6.5 years.

Klinické známky neuropatie (areflexie/hyporeflexie) byly přítomné u 12 pacientů. EMG vyšetření bylo provedeno u deseti těchto pacientů a u šesti s potvrzením axonálně-demyelinizační periferní neuropatie. Medián věku pacientů s EMG patologií byl 20 let, zatímco při fyziologickém nálezu 7 let.
Symptomy koagulopatie se vyskytly u 36 % pacientů: velmi často se jednalo o benigní nález snadnější tvorby modřin, jen u čtyř pacientů byly příznaky závažnější a byla u nich nutná dlouhodobá antikoagulační terapie. Dvě pacientky prodělaly ischemickou cévní mozkovou příhodu – u první se projevila ve věku 11 let těžkou levostrannou hemiparézou a deviací bulbů, druhá dívka měla ve věku 2,5 let pravostrannou parézu horní končetiny. U první bylo zvoleno 4měsíční antikoagulační zajištění, u druhé bylo vzhledem k současné heterozygotní mutaci pro faktor V Leiden zvoleno dlouhodobé antitrombotické zajištění. Opakované periferní žilní trombózy (více než 10 trombóz na horních i dolních končetinách) byly u třetí pacientky, která je zároveň heterozygotkou mutace pro faktor V Leiden, důvodem pro zavedení kombinované antitrombotické a antikoagulační terapie. Poslední pacient neprodělal jasnou trombotickou příhodu, ale přesto byl vzhledem k závažnému celkovému stavu a koagulační patologii přechodně zajištěn antikoagulancii.
Prognóza – mortalita
V současné době je osm našich pacientů dospělých, tři jsou v adolescentním věku, šest je školního věku a tři jsou v batolecím věku. Klinický obraz našich pacientů v dospělém věku je charakteristický především stabilizací stavu včetně úrovně intelektuální disability, neurologického nálezu a laboratorních nálezů (hladina jaterních testů a koagulačních parametrů).
Dva chlapci zemřeli v kojeneckém věku (9 a 11 měsíců), jednalo se o děti manifestující se v novorozeneckém věku, příčinou smrti byla dekompenzace jaterního postižení při probíhajícím akutním respiračním infektu a náhlé úmrtí při horečnatém infektu v domácím prostředí u chlapce s prakticky zastaveným psychomotorickým vývojem, těžkou smíšenou koagulační poruchou, hepatopatií a mikrocystózou ledvin s významnou renoparenchymatózní hypertenzí.
Laboratorní výsledky
Nejčastější laboratorní odchylky jsou shrnuty v tabulce 1; jedná se o příznaky koagulopatie, hepatopatie, myopatie, thyreopatie a hypergonadotropního hypogonadismu u adolescentních dívek.

2ALT: rozptyl 0,4–10,3 μkat/l, průměr 1,2 μkat/l; AST: rozptyl
0,4–3,9 μkat/l, průměr 1,1 μkat/l
Koagulopatie se vyskytla u všech našich pacientů: u osmnácti pacientů byla mírné a u čtyř závažnější tíže s klinickým dopadem (viz výše). Ve všech případech se jednalo o smíšenou koagulopatii, nejčastěji se alterace týkala koagulačního faktoru XI, proteinu C a S a anti-trombinu. U devíti pacientů se vyskytlo prodloužení aPTT, z toho u čtyř také prodloužené INR.
Hepatopatie se vyskytla u šestnácti pacientů, u šesti pacientů byla zachycena závažná elevace sérových trans-amináz nad desetinásobek normy. U čtyř pacientů byla náhodně zjištěná hepatopatie jedním z prvních příznaků, pro které bylo dítě vyšetřované.
Myopatie byla laboratorně zachycena u čtrnácti pacientů, nicméně elevace kreatinkinázy a myoglobinu byla u všech mírná a žádný z pacientů nikdy neprodělal ataku rhabdomyolýzy.
Thyreopatii jsme zaznamenali u jedenácti pacientů, u většiny (n = 8) se jednalo o subklinickou formu periferní hypothyreózy bez alterace fT4. Častým nálezem byla také nižší hladina TBG (u 7 z 10 vyšetřených pacientů). U dvou pacientů se jednalo o centrální hypothyreózu v rámci panhypopituitarismu. Všichni pacienti jsou na terapii levothyroxinem a v jednom případě jodidem.
Hypergonadotropní hypogonadismus byl prokázán u všech šesti dívek v pubertálním věku s klinickými známkami pubertas tarda. Tři postpubertální chlapci měli normální hladiny pohlavních hormonů a známky rozvoje puberty.
Denzitometrie prokázala u všech pacientů (n = 9), u kterých byly výsledky dostupné, sníženou kostní denzitu. U čtyř pacientů byly dostupné výsledky celotělové denzitometrie s průměrným Z-skóre -2,8 (rozptyl -2 až -3), u ostatních pacientů byl výsledek celotělové denzitometrie hodnocen jako nevalidní. U osmi pacientů byla dostupná denzitometrie bederní oblasti s průměrným Z-skóre -3,4 (rozptyl -1,1 až -6,8), nejvýraznější byl nález u 10leté dívky s těžkou formou nemoci s výraznými kostními deformitami a trvale upoutanou na lůžko.
Biochemické a molekulárně-genetické vyšetření
U všech pacientů v souboru byl pomocí IEF nalezen patologický profil sialovaných forem sérového transferinu (TRF), konkrétně snížený tetrasialo-TRF a zvýšený obsah disialo - a asialo-TRF, typický profil CDG typu I. Ukázka profilu typu I je uvedena na obrázku 4.
K potvrzení diagnózy při pozitivní IEF TRF byla provedena enzymologická a/nebo molekulárně-genetická analýza. Snížená aktivita PMM2 pod 30 % průměru kontrol byla prokázána v izolovaných lymfocytech u 15 ze 17 a v kultivovaných fibroblastech u 6 z 8 testovaných vzorků (tab. 2). Molekulárně-genetické vyšetření u všech dětí potvrdilo diagnózu průkazem mutace v PMM2 genu. 71 % mutovaných alel nese jednu ze dvou nejčastějších patogenních variant (tab. 2). Nebyla nalezena žádná korelace mezi klinickým fenotypem, reziduální enzymatickou aktivitou a genotypem.

DISKUSE
Klinický průběh
Celkový průběh onemocnění u 22 pacientů diagnostikovaných na naší klinice se významně neliší od průběhu popsaného v literatuře [13]. Většina našich pacientů postupně rozvíjí příznaky typické pro čtyři jednotlivá stadia onemocnění [13]: multisystémové onemocnění v kojeneckém a batolecím věku, stadium ataxie a mentální retardace v dětství, stadium neuropatie a svalové atrofie v adolescentním věku a hypogonadální období v dospělosti [14]. Skupina pacientů s neonatálním nástupem onemocnění a závažnými multisystémovými příznaky je spojena s vysokou mortalitou až 20 % [15], v naší skupině pacientů došlo k úmrtí 2/4 těchto pacientů.
Prenatální projevy a typická dysmorfie
Prenatální projevy (hydrops plodu, hydrops placenty, polyhydramnion), které jsou popisované v literatuře jen vzácně [16], se u našich pacientů nevyskytly. Kraniofaciální dysmorfii jsme u našich pacientů pozorovali často (71 %), ale byla povětšinou velmi mírná. Z vrozených malformací se vyskytuje kryptorchismus (u 50 % našich pacientů) a vrozené srdeční vady (19 % našich pacientů).
Atypická distribuce tuku a s tím spojené vpáčené bradavky (podmíněné depozitem tuku při minimu glandulární tkáně v oblasti mamily) je přítomna u 25–100 % pacientů s PMM2-CDG dle jednotlivých studií, u našich pacientů nebyla přítomná pouze u čtyř dětí. Etiopatogeneze atypického ukládání tuku není zcela jasná, nicméně předpokládá se souvislost se zkráceným biologickým poločasem IGF-1 a hypoglykosylací inzulinových a leptinových receptorů s následným ovlivněním adipogeneze [17]. Vpáčené bradavky jsou popisovány i jednostranně [16], toto jsme zaznamenali u 2 pacientů. S věkem má tento nález tendenci regredovat [18]. V našem souboru vpáčené bradavky vymizely u 5 pacientů (tj. 26 %), atypická depozita tuku u 6 pacientů (tj. 40 %).
Neurologické příznaky
PMM2-CDG se může projevovat izolovanými neurologickými projevy či jako multiviscerální forma. Neurologická manifestace nejčastěji zahrnuje psychomotorickou retardaci, mentální retardaci (intelektuální disabilitu), hypotonii, mozečkový syndrom a periferní neuropatii. Psychomotorická retardace se vyskytuje u naprosté většiny pacientů, včetně pacientů naší kohorty. V literatuře bylo dosud popsáno jen 20 pacientů bez psychomotorické retardace a s normálním IQ [15, 19–24]. Intelektuální disabilita byla v naší skupině posunuta směrem k těžším formám – zastoupení hraničního intelektu (podprůměr) či normální inteligence nebylo v naší skupině pozorováno, naopak hluboká mentální retardace byla u 23 % našich pacientů. Periferní neuropatie se vyskytla u 63 % pacientů, i když jen u 2/3 vyšetřených pacientů byl nález potvrzen na EMG. Rozvíjí se obvykle v druhé dekádě života: popisujeme ho u 85 % pacientů starších 10 let, ale jen u 17 % mladších 10 let. Málo časté jsou poruchy hybnosti: u několika málo pacientů byly popsané dystonické dyskinetické pohyby rukou či cervikální dystonie [25, 26]. Žádné podobné obtíže se u našich pacientů nevyskytly.
Z akutních neurologických projevů se nejčastěji vyskytují epilepsie a iktu podobné příhody, oboje s poměrně nízkou incidencí (dokumentována pouze u 68 resp. 36 pacientů v literatuře). Křeče se mohou objevit i v neonatálním období [27], což se mezi našimi pacienty neobjevilo. Mohou mít charakter tonicko-klonický i atonický a u naprosté většiny pacientů jsou dobře kompenzované na antiepileptické terapii. Za nejčastější spouštěč ataky iktu podobných příhod se považuje febrilní infekt a trauma hlavy [28]. Předpokládá se, že patofyziologickým podkladem by mohla být kanálopatie podobně jako u CACNA1A – asociované familiární hemiplegické migrény s hypoglykosylací týchž napěťově řízených CaV2.1 kanálů [29]. V naší skupině se iktu podobné příhody vyskytly u tří pacientů: u dvou pacientů k atakám došlo v dětském věku, ale jedna pacientka prodělala opakované ataky v adolescenci a dokonce v dospělosti, což je v kontrastu s literaturou [30].
Atrofie mozečku je také velmi častým nálezem (94 % našich pacientů) a je možno ji detekovat i prenatálně [31] či v novorozeneckém věku [24]. Přesto se nejedná o hypoplazii, neboť zmenšení objemu mozečku má progresivní charakter v časném dětském věku [32]. To se v naší skupině podařilo prokázat zobrazením mozku téhož pacienta ve věku 10 měsíců a 6,5 roku (obr. 3). U jednoho pacienta byl na MR mozku ve věku 1 měsíce normální nález na mozečku, což můžeme vysvětlit časným věkem, ve kterém bylo vyšetření provedeno (srovnávací snímek v pozdějším věku bohužel není dostupný), jak bylo popsáno v některých pracích [20, 21].
Strabismus a pigmentová degenerace sítnice, dokumentovaná u 85 % resp. 22 % z 535 PMM2 pacientů z literatury s popisem očních projevů, byly nalezeny v našem souboru u 81 % resp. 41 % jedinců. Neuropatie optiku je vzácnějším projevem PMM2-CDG s popsaným pouze jedním pacientem s poruchou zrakové ostrosti v tíži praktické slepoty.
Viscerální postižení
Ledvinné postižení je součástí většinou těžkých multiviscerálních fenotypů PMM2-CDG a bylo popsáno u 85 % z 56 pacientů z literatury s důkladnějším popisem renálních funkcí. Charakteristickým nálezem je cystická dysplazie ledvin, tubulární proteinurie, méně často hydronefróza. Nefrotická proteinurie, přítomná u jednoho našeho chlapce, byla dokumentována ve světě pouze u šesti pacientů [24, 33–36].
Jaterní selhání bylo příčinou úmrtí u našeho 11měsíčního chlapce s PMM2-CDG. Hepatopatie se dále vyskytla u šestnácti pacientů, u šesti z nich byla zachycena závažná elevace sérových transamináz s přechodnou poruchou jaterních funkcí. Těžké jaterní onemocnění v kojeneckém věku je většinou asociováno s multiviscerálním fenotypem. Autopsie v publikovaných případech ukázala cholestázu s dilatací dobrých žlučovodů, periportální fibrózu, steatózu až po portoportální přemosťující fibrózu anebo cirhózu [13, 31, 37–42]. I u mírnějších fenotypů onemocnění je hepatomegalie a hepatopatie přítomná u velkého počtu pacientů (12,5–100 %) [18, 30, 43, 44], nejedná se však o progresivní jaterní onemocnění. Jaterní testy výrazněji stoupají v době horečnatých stavů, nicméně laboratorní i klinická patologie má tendenci se mírnit či dokonce ustupovat po druhé dekádě života. Jaterní biopsie je zřídkakdy u těchto pacientů prováděna a ukazuje steatózu [23, 45, 46] či lysosomální inkluze v hepatocytech [47].
Endokrinopatie
Mezi charakteristické endokrinologické abnormity u pacientů s PMM2-CDG patří postnatální růstová porucha, thyreopatie, opožděná puberta, hypoglykémie a osteo-poróza. Více než 50 % našich pacientů má významnou růstovou poruchu, což je v souladu s literaturou, kde 49 % pacientů z 53 hodnocených subjektů v různých studiích se prezentovalo postnatálním růstovým selháním. Pacienti mají normální či dokonce vyšší koncentraci růstového hormonu v krvi, ale snížená je koncentrace IGF-1, IGFBP3 a acid-labile subunit (ALS) [45, 48]. IGF-1 není sice glykosylován, ale více než 90 % IGF-1 molekul je vázaných na transportní glykoproteiny IGFBP3 a ALS. Jejich abnormní glykosylace vede ke zkrácenému biologickému poločasu IGF-1. Jen jedna práce dokládá úspěšnou terapii rekombinantním lidským IGF-1 [49]. Abnormální thyreoidální funkce byla nalezena u 255 jedinců s PMM2-CDG v literatuře. Podobně jako u našich pacientů, většinou se jednalo o subklinickou formu periferní hypothyreózy na základě měření hladiny TSH. Částečná deficience hypoglykosylovaného TBG je přítomná u 75 % pacientů při zkrácení jeho biologického poločasu na 15 %, nicméně jeho deficit nejspíše nevede k poruše thyroidálních funkcí [50]. S ohledem na časté nadbytečné léčení PMM2-CDG pacientů substitucí levothyroxinem jen na základě vyšší hladiny TSH, se doporučuje sledovat výhradně parametry FT4 – hormonu, který není glykosylován. U dvou našich pacientů se rozvinula centrální hypothyreóza v rámci panhypopituitarismu, což zatím nebylo v literatuře popsáno.
Hypoglykémie v novorozeneckém a kojeneckém věku jsme zjistili u čtyř pacientů s PMM2-CDG. U dvou pacientů se jednalo o hypoglykémie v rámci deficitu ACTH a kortizolu, u jednoho chlapce v rámci jaterního selhání a u čtvrtého pacienta při multiviscerálním postižení s komplexní symp-tomatologií. Hladina inzulinu nebyla u těchto pacientů stanovena. U popsaných 24 novorozenců a kojenců s PMM2--CDG s opakovanými hypoglykémiemi v literatuře byl u 40 % z nich popsán hyperinzulinismus s dobrou odpovědí na diazoxid. Všechny adolescentní a dospělé dívky z našeho souboru mají dokumentovanou opožděnou pubertu v rámci hypergonadotropního hypogonadismu. Jen 3 dívky z 85 žen s PMM2-CDG s dokumentovanými gonadálními funkcemi měly normální nástup puberty a menarché [24, 45, 51]. Patofyziologie ovariálního selhání není přesně známa. Abnormální glykosylace gonadotropinů, jejich receptorů a pohlavních hormonů může vyústit v hypergonadotropní hypogonadismus, ale FSH in vitro měřená aktivita nebyla významně snížena [48]. U třech vyšetřovaných chlapců v adolescentním věku jsme zaznamenali normální pohlavní vývoj, i když v literatuře jsou popsané ojedinělé případy testikulární atrofie a hypogonadismu u mužů [40, 48, 52]. Snížená kostní denzita, v rozsahu osteopenie až po osteoporózu se začátkem již v dětství, je nalezena až u 60 % PMM2-CDG pacientů [30] a vede ke zvýšenému riziku fraktur (až ve 26 % případů) [18]. Denzitometrie prokázala u všech našich devíti vyšetřených pacientů sníženou kostní denzitu, dosahující u jedné pacientky v oblasti bederní páteře až hodnoty Z-skóre -6,8.
Koagulopatie
Charakteristickou laboratorní patologií u pacientů s PMM2-CDG je smíšená koagulopatie. Je přítomná u všech našich pacientů, s klinickým dopadem u přibližně jedné třetiny z nich. Postižené jsou jak prokoagulační, tak antikoagulační faktory. Nejčastější a nejvýznamnější patologie je popisována pro koagulační faktor IX, XI, antitrombin, protein C a S. V rámci poddiagnostikovanosti PMM2-CDG pacientů je nutné zdůraznit, že někteří pacienti s mírným fenotypem mohou být sledováni právě pro izolovaný deficit antitrombinu [53]. Trombotické příhody arteriální i venózní jsou popsány u 12,5 % pacientů, u čtyř pacientů došlo k trombóze mozkových žil, mezi závažné rizikové faktory trombóz patří operační zákrok a katetrizace [54]. Zajímavým nálezem je fluktuace tíže koagulační poruchy s věkem [55] a horečkou, pravděpodobně kvůli termolabilitě PMM2 [24, 56].
Diagnostika
Jako screeningovou metodu v diagnostice jsme používali IEF sérového transferinu, která je rychlá a senzitivní, ale nespecifická metoda k průkazu nemoci. Je založena na separaci transferinových isoforem na základě odlišného náboje vyplývajícího z různého počtu přítomných negativně nabitých terminálních zbytků sialové kyseliny. U PMM2-CDG obecně, jako i u všech našich pacientů v souboru, typicky nacházíme profil typu I se sníženým množstvím tetrasialovaných forem transferinu a zvýšením di - a asialovaných forem transferinu (obr. 4). V našem souboru jsme potvrdili poruchu PMM2 (10–30 % reziduální aktivity) u 84 % testovaných vzorků, pouze u dvou vzorků lymfocytů (P13 a P20) byla reziduální aktivita hraničně patologická na 52–63 % a u dvou vzorků fibroblastů 32–41 % (P7, P9) průměru kontrol. Až u 18 % publikovaných pacientů s PMM2-CDG byla v izolovaných lymfocytech a/nebo kultivovaných fibroblastech nalezena normální aktivita PMM2 [43, 57], proto nelze na enzymologické vyšetření spoléhat z důvodu rizika falešně negativního výsledku a vždy je nutno provést i molekulárně-genetické vyšetření.

Molekulárně-genetické vyšetření genu PMM2 je k dispozici na našem pracovišti. Gen PMM2 je lokalizován na chromosomu 16p13.2, má osm exonů a jeho proteinový produkt je o velikosti 246 aminokyselin. V genu pro PMM2 bylo nalezeno již 129 mutací, zahrnujících >95 bodových mutací, několik nonsense mutací, 8 mutací měnící čtecí rámec, >10 sestřihových mutací a jedna delece celého exonu 8 o velikosti 28 kb [58]. V našem souboru pacientů bylo zastoupeno 10 mutací v PMM2 (tab. 2), všichni pacienti jsou složení heterozygoti. Mutaci c.422G>A (p.Arg141His), která je nejpočetnější a je v literatuře popisována u 60 % pacientů, jsme nalezli i v našem souboru u 18 pacientů. Mutace c.422G>A nebyla dosud nalezena v homozygotní formě, a proto se předpokládá, že je v homozygotní formě neslučitelná se životem [59–61]. Mutace c.338C>T (p.Phe119Leu) byla nalezená u 13 našich subjektů, ve složené heterozygotní formě s mutací c.422G>A u 10 z nich. Obě mutace jsou tedy v České republice prevalentní.
ZÁVĚR
Domníváme se, že PMM2-CDG je v České republice, i 38 let od svého objevení, diagnózou málo známou a často opomíjenou. Důkazem pro to může být rozpor mezi odhadovanou prevalencí 1 : 20 000 [62] a tím, že na našem území bylo za 16 let od zavedení diagnostiky potvrzeno jen 22 pa-cientů s touto nemocí. Dále také mezi našimi pacienty nenacházíme pacienty s normálním či hraničním intelektem a mírnou neurologickou symptomatologií, jak je popsáno v literatuře [19–22]. Příčinou může být fakt, že zatímco dosud tuto diagnózu zvažujeme především v diferenciální diagnostice výrazné psychomotorické retardace a mozečkové atrofie, jedná se zřejmě o onemocnění s mnohem širším spektrem příznaků a heterogenní tíží fenotypu.
Věříme, že tato práce přispěje k lepšímu popisu přirozeného průběhu PMM2-CDG. Tato znalost je pro nás důležitá kvůli urychlení diagnostiky a možnostem co nejpřesnější informovanosti pacientů a jejich rodin. Dosud nebylo publikováno mnoho prací charakterizujících dlouhodobý vývoj pacientů s PMM2-CDG a jeho projevy v dospělosti. Proto je v plánu zařazení našich pacientů do prospektivní multicentrické mezinárodní studie hodnotící přirozený průběh onemocnění a eventuálně zařazení do klinické studie s novou léčebnou molekulou LipoM1P, která by měla zajistit dostupnost produktu narušené biochemické reakce přímo v buňkách.
Zkratky:
ALS (acid-labile subunit) – kyselá labilní podjednotka
aPTT – aktivovaný parciální tromboplastinový čas
CDG (congenital disorder of glycosylation) – dědičné poruchy glykosylace
CMP – cévní mozková příhoda
DOLK-CDG (dolichol kinase deficiency) – deficit dolicholkinázy
EMG – elektromyografie
GPI – glykosylfosfatidylinositol
IGF-1 (insulin-like growth factor 1) – inzulinu podobný růstový faktor
IGFBP3 (IGF binding protein 3) – IGF vázající protein 3
INR (international normalized ratio) – mezinárodní normalizovaný poměr
LipoM1P – manóza-1-fosfát inkorporovaná do liposomu
LLO (lipid-linked oligosaccharide) – oligosacharid vázaný na lipid
M1P – manóza-1-fosfát
MPI-CDG (mannosophosphate isomerase deficiency) – deficit fosfomanoisomerázy
NADP+, NADPH – nikotinamidadenindinukleotidfosfát a jeho redukovaná forma
PGM1-CDG (phosphoglucomutase deficiency type 1) – deficit fosfoglukomutázy 1
PMM2 – fosfomanomutáza 2
PMR – psychomotorická retardace
SLE (stroke-like episodes) – iktu podobné příhody
TBG (thyroxine-binding globulin) – tyroxin vázající globulin
IEF TRF (isoelectric focusing of transferrin) – isoelektrická fokusace transferinu
Podpořeno: MZ ČR – RVO VFN 64165, MZ ČR – AZV 16-31932A.
Korespondující autor:
Doc. MUDr. Tomáš Honzík, Ph.D.
Klinika dětského a dorostového lékařství
1. LF UK a VFN
Ke Karlovu 2
128 08 Praha 2
e-mail: tomas.honzik@vfn.cz
Zdroje
1. Corfield A, Berry M. Current aspects of eukaryotic glycosylation. Trends Biochem Sci 2015; 40 : 351–359.
2. Apweiler R, Hermjakob H, Sharon N. On the frequency of protein glycosylation, as deduced from analysis of the SWISS-PROT database. Biochim Biophys Acta (BBA) – General Subjects 1999; 1473 (1): 4–8.
3. Moremen KW, Tiemeyer M, Nairn AV. Vertebrate protein glycosylation: diversity, synthesis and function. Nat Rev Mol Cell Biol 2012; 13 (7): 448–462.
4. Jaeken J, Vanderschueren-Lodeweyckx M, Casaer P, et al. Familial psychomotor retardation with markedly fluctuating serum prolactin, FSH and GH levels, partial TBG-deficiency, increased serum arylsulphatase A and increased CSF protein: a new syndrome? Pediatr Res 1980; 14 : 179.
5. Ng BG, Freeze HH. Perspectives on glycosylation and its congenital disorders. Trends Genet 2018.
6. Hennet T, Cabalzar J. Congenital disorders of glycosylation: a concise chart of glycocalyx dysfunction. Trends Biochem Sci 2015; 40 (7): 377–384.
7. Jaeken J, Van Eijk H, Van der Heul C, et al. Sialic acid-deficient serum and cerebrospinal fluid transferrin in a newly recognized genetic syndrome. Clin Chim Acta 1984; 144 (2–3): 245–247.
8. Wopereis S, Grünewald S, Huijben KM, et al. Transferrin and apolipoprotein C-III isofocusing are complementary in the diagnosis of N - and O-glycan biosynthesis defects. Clin Chem 2007; 53 (2):1 80–187.
9. Lacey JM, Bergen HR, Magera MJ, et al. Rapid determination of transferrin isoforms by immunoaffinity liquid chromatography and electrospray mass spectrometry. Clin Chem 2001; 47 (3): 513–518.
10. Aebi M, Helenius A, Schenk B, et al. Carbohydrate-deficient glycoprotein syndromes become congenital disorders of glycosylation: An updated nomenclature for CDG. Glycoconj J 1999; 16 (11): 669–671.
11. Jaeken J, Hennet T, Matthijs G, et al. CDG nomenclature: time for a change! Biochim Biophys Acta – Mol Basis of Dis 2009; 1792 (9): 825–826.
12. Thiel C, Körner C. Therapies and therapeutic approaches in congenital disorders of glycosylation. Glycoconj J 2013; 30 (1): 77–84.
13. Grünewald S. The clinical spectrum of phosphomannomutase 2 deficiency (CDG-Ia). Biochim Biophys Acta – Mol Basis of Dis 2009; 1792 (9): 827–834.
14. Hagberg BA, Blennow G, Kristiansson B, et al. Carbohydrate-deficient glycoprotein syndromes: peculiar group of new disorders. Pediatr Neurol 1993; 9 (4): 255–262.
15. de Lonlay P, Seta N, Barrot S, et al. A broad spectrum of clinical presentations in congenital disorders of glycosylation I: a series of 26 cases. J Med Genet 2001; 38 (1): 14–19.
16. van de Kamp JM, Lefeber DJ, Ruijter GJ, et al. Congenital disorder of glycosylation type Ia presenting with hydrops fetalis. J Med Genet 2007; 44 (4): 277–280.
17. Wolthuis D, van Asbeck E, Kozicz T, et al. Abnormal fat distribution in PMM2-CDG. Mol Genet Metab 2013; 110 (3): 411–413.
18. Kjaergaard S, Schwartz M, Skovby F. Congenital disorder of glycosylation type Ia (CDG-Ia): phenotypic spectrum of the R141H/F119L genotype. Arch Dis Child 2001; 85 (3): 236–239.
19. Vuilleumier-Barrot S, Isidor B, Dupré T, et al. Expanding the spectrum of PMM2-CDG phenotype. In: JIMD Reports – Case and Research Reports. Vol. 5, edn.: Springer, 2011 : 123–125.
20. Giurgea I, Michel A, Le Merrer M, et al. Underdiagnosis of mild congenital disorders of glycosylation type Ia. Pediatr Neurol 2005; 32 (2): 121–123.
21. Pancho C, Garcia-Cazorla A, Varea V, et al. Congenital disorder of glycosylation type Ia revealed by hypertransaminasemia and failure to thrive in a young boy with normal neurodevelopment. J Pediatr Gastroenterol Nutr 2005; 40 (2): 230–232.
22. Shanti B, Silink M, Bhattacharya K, et al. Congenital disorder of glycosylation type Ia: Heterogeneity in the clinical presentation from multivisceral failure to hyperinsulinaemic hypoglycaemia as leading symptoms in three infants with phosphomannomutase deficiency. J Inherit Metab Dis 2009; 32 (1): 241–251.
23. Damen G, de Klerk H, Huijmans J, et al. Gastrointestinal and other clinical manifestations in 17 children with congenital disorders of glycosylation type Ia, Ib, and Ic. J Pediatr Gastroenterol Nutr 2004; 38 (3): 282–287.
24. Schiff M, Roda C, Monin M-L, et al. Clinical, laboratory and molecular findings and long-term follow-up data in 96 French patients with PMM2-CDG (phosphomannomutase 2-congenital disorder of glycosylation) and review of the literature. J Med Genet 2017; 54 (12): 843–851.
25. Antoun H, Villeneuve N, Gelot A, et al. Cerebellar atrophy: an important feature of carbohydrate deficient glycoprotein syndrome type 1. Pediatr Radiol 1999; 29 (3): 194–198.
26. Neumann LM, von Moers A, Kunze J, et al. Congenital disorder of glycosylation type 1a in a macrosomic 16-month-old boy with an atypical phenotype and homozygosity of the N216I mutation. Eur J Pediatr 2003; 162 (10): 710–713.
27. Funke S, Gardeitchik T, Kouwenberg D, et al. Perinatal and early infantile symptoms in congenital disorders of glycosylation. Am J Med Genet Part A 2013; 161 (3): 578–584.
28. Barone R, Carrozzi M, Parini R, et al. A nationwide survey of PMM2-CDG in Italy: high frequency of a mild neurological variant associated with the L32R mutation. J Neurol 2015; 262 (1):154–164.
29. Izquierdo-Serra M, Martinez-Monseny AF, Lopez L, et al. Stroke-like episodes and cerebellar syndrome in phosphomannomutase deficiency (PMM2-CDG): evidence for hypoglycosylation-driven channelopathy. Int J Mol Sci 2018; 19 (2): 619.
30. Monin M-L, Mignot C, De Lonlay P, et al. 29 French adult patients with PMM2-congenital disorder of glycosylation: outcome of the classical pediatric phenotype and depiction of a late-onset phenotype. Orphanet J Rare Dis 2014; 9 (1): 207.
31. Edwards M, McKenzie F, O‘callaghan S, et al. Prenatal diagnosis of congenital disorder of glycosylation type Ia (CDG–Ia) by cordocentesis and transferrin isoelectric focussing of serum of a 27–week fetus with non–immune hydrops. Prenat Diagn 2006; 26 (10): 985–988.
32. de Diego V, Martínez-Monseny AF, Muchart J, et al. Longitudinal volumetric and 2D assessment of cerebellar atrophy in a large cohort of children with phosphomannomutase deficiency (PMM2-CDG). J Inherit Metab Dis 2017; 40(5):709–713.
33. Hutchesson A, Gray R, Spencer D, et al. Carbohydrate deficient glycoprotein syndrome; multiple abnormalities and diagnostic delay. Arch Dis Child 1995; 72 (5): 445–446.
34. van der Knaap MS, Wevers RA, Monnens L, et al. Congenital nephrotic syndrome: a novel phenotype of type I carbohydrate-deficient glycoprotein syndrome. J Inherit Metab Dis 1996; 19 (6): 787–791.
35. Coman D, McGill J, MacDonald R, et al. Congenital disorder of glycosylation type 1a: three siblings with a mild neurological phenotype. J Clin Neurosci 2007; 14 (7): 668–672.
36. Jamroz E, Adamek D, Paprocka J, et al. CDG type Ia and congenital cytomegalovirus infection: two coexisting conditions. J Child Neurol 2009; 24 (1): 13–18.
37. Silva MG, De Castro J, Stibler H, et al. Prenatal hypertrophic cardiomyopathy and pericardial effusion in carbohydrate-deficient glycoprotein syndrome. J Inherit Metab Dis 1996; 19 (2): 257–259.
38. Marquardt T, Hülskamp G, Gehrmann J, et al. Severe transient myocardial ischaemia caused by hypertrophic cardiomyopathy in a patient with congenital disorder of glycosylation type Ia. Eur J Pediatr 2002; 161 (10): 524–527.
39. Aronica E, van Kempen A, Van der Heide M, et al. Congenital disorder of glycosylation type Ia: a clinicopathological report of a newborn infant with cerebellar pathology. Acta Neuropathol 2005; 109 (4): 433–442.
40. Schoffer KL, O‘sullivan JD, McGill J. Congenital disorder of glycosylation type Ia presenting as early–onset cerebellar ataxia in an adult. Mov Disord 2006; 21 (6): 869–872.
41. Wurm D, Hänsgen A, Kim Y-J, et al. Early fatal course in siblings with CDG-Ia (caused by two novel mutations in the PMM2 gene): clinical, molecular and autopsy findings. Eur J Pediatr 2007; 166 (4): 377–378.
42. Ong BB, Gole GA, Robertson T, et al. Retinal hemorrhages associated with meningitis in a child with a congenital disorder of glycosylation. Forensic Sci Med Pathol 2009; 5 (4): 307–312.
43. Grünewald S, Schollen E, Van Schaftingen E, et al. High residual activity of PMM2 in patients’ fibroblasts: possible pitfall in the diagnosis of CDG-Ia (phosphomannomutase deficiency). Am J Hum Genet 2001; 68 (2): 347–354.
44. Arnoux J, Boddaert N, Valayannopoulos V, et al. Risk assessment of acute vascular events in congenital disorder of glycosylation type Ia. Mol Genet Metab 2008; 93 (4): 444–449.
45. Perez-Duenas B, García-Cazorla A, Pineda M, et al. Long-term evolution of eight Spanish patients with CDG type Ia: typical and atypical manifestations. Eur J Paediatr Neurol 2009; 13 (5): 444-451.
46. Enns GM, Steiner RD, Buist N, et al. Clinical and molecular features of congenital disorder of glycosylation in patients with type 1 sialotransferrin pattern and diverse ethnic origins. J Pediatr 2002; 141 (5): 695–700.
47. Grünewald S, De Vos R, Jaeken J. Abnormal lysosomal inclusions in liver hepatocytes but not in fibroblasts in congenital disorders of glycosylation (CDG). J Inherit Metab Dis 2003; 26 (1): 49–54.
48. de Zegher F, Jaeken J. Endocrinology of the carbohydrate-deficient glycoprotein syndrome type 1 from birth through adolescence. Pediatr Res 1995; 37 (4 Pt 1): 395–401.
49. Miller BS, Duffy MM, Addo OY, et al. rhIGF-1 therapy for growth failure and IGF-1 deficiency in congenital disorder of glycosylation Ia (PMM2 deficiency). J Investig Med High Impact Case Rep 2013 Sep 5; 1 (3): 2324709613503316.
50. Jaeken J, Hagberg B, Strømme P. Clinical presentation and natural course of the carbohydrate–deficient glycoprotein syndrome. Acta Pædiatrica 1991; 80 : 6–13.
51. Pineda M, Pavia C, Vilaseca M, et al. Normal pubertal development in a female with carbohydrate deficient glycoprotein syndrome. Arch Dis Child 1996; 74 (3): 242.
52. Krasnewich D, O‘Brien K, Sparks S. Clinical features in adults with congenital disorders of glycosylation type Ia (CDG–Ia). Am J Med Genet Part C Semin Med Genet 2007; 145c (3): 302–306.
53. de la Morena-Barrio ME, Hernández-Caselles T, Corral J, et al. GPI-anchor and GPI-anchored protein expression in PMM2-CDG patients. Orphanet J Rare Dis 2013; 8 (1): 170.
54. Linssen M, Mohamed M, Wevers R, et al. Thrombotic complications in patients with PMM2-CDG. Mol Genet Metab 2013; 109 (1): 107–111.
55. Ono H, Sakura N, Yamashita K, et al. Novel nonsense mutation (R194X) in the PMM2 gene in a Japanese patient with congenital disorder of glycosylation type Ia. Brain Dev 2003; 25 (7): 525–528.
56. Le Bizec C, Vuillaumier–Barrot S, Barnier A, et al. A new insight into PMM2 mutations in the French population. Hum Mutat 2005; 25 (5): 504–505.
57. Westphal V, Peterson S, Patterson M, et al. Functional significance of PMM2 mutations in mildly affected patients with congenital disorders of glycosylation Ia. Genet in Medicine 2001; 3 (6): 393.
58. Schollen E, Keldermans L, Foulquier F, et al. Characterization of two unusual truncating PMM2 mutations in two CDG-Ia patients. Molec Genet Metab 2007; 90 (4): 408–413.
59. Kjaergaard S, Skovby F, Schwartz M. Absence of homozygosity for predominant mutations in PMM2 in Danish patients with carbohydrate-deficient glycoprotein syndrome type 1. Eur J Hum Genet 1998; 6 (4): 331.
60. Matthijs G, Schollen E, Van Schaftingen E, et al. Lack of homozygotes for the most frequent disease allele in carbohydrate-deficient glycoprotein syndrome type 1A. Am J Hum Genet 1998; 62 (3): 542–550.
61. Pirard M, Matthijs G, Heykants L, et al. Effect of mutations found in carbohydrate-deficient glycoprotein syndrome type IA on the activity of phosphomannomutase 2. FEBS Letters 1999; 452 (3): 319–322.
62. Schollen E, Kjaergaard S, Legius E, et al. Lack of Hardy-Weinberg equilibrium for the most prevalent PMM2 mutation in CDG-Ia (congenital disorders of glycosylation type Ia). Eur J Hum Genet 2000; 8 (5): 367.
Štítky
Neonatologie Pediatrie Praktické lékařství pro děti a dorostČlánek vyšel v časopise
Česko-slovenská pediatrie

2018 Číslo 6
- Efektivita kartáčku Sonicare For Kids u dětí předškolního věku
- Inosin pranobex v léčbě HPV infekcí děložního čípku
- Cytomegalovirové infekce u novorozenců a dětí
Nejčtenější v tomto čísle
- Charakteristické klinické příznaky a laboratorní odchylky dědičných poruch metabolismu
- Komplexný pohľad na deficit vitamínu B12 v detskom veku
- Novorozenecký screening dědičných metabolických poruch v České republice
- Lyzozómové choroby – vývoj diagnostiky a liečby na Slovensku
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy