
Charakteristické klinické příznaky a laboratorní odchylky dědičných poruch metabolismu
Characteristic clinical features and laboratory findings of inborn errors of metabolism
Introduction:
Inborn Errors of Metabolism (IEM) represent a group of >1000 rare genetic disorders that are caused by a defect of single/multiple enzymes or changes in structural, assembling and transporting proteins that participate in metabolic pathways. Since any system can be affected, variable clinical symptoms may occur, leading to a diagnostic delay.
Aim:
To provide clear overview of the main key clinical and laboratory findings of IEM encountered by both primary health care or hospital physicians – pediatricians.
Results:
Main clinical features of IEM are represented by developmental regression, pharmacoresistant epilepsy, cardiomyopathy, myopathic syndrome or muscle pain in rhabdomyolysis and hepato-/splenomegaly. As much as 70% of a childhood urolithiasis are caused by one of the IEM. Detailed ophthalmic examination may reveal corneal clouding or cataract, ophthalmoplegia, pigmentary retinopathy and optic neuropathy. Some of the symptoms may be helpful in making the right diagnosis faster at a glance, such as craniofacial dysmorphism, hypertrichosis, atypical structure of adnexa and some bone changes. The main laboratory abnormalities include hypoglycemia, hyperammonemia, dyslipidemia and (cholestatic) hepatopathy. Standardized cerebrospinal fluid examination is crucial for the diagnosis of some of the IEM that can be potentially treatable.
Conclusion:
Although individual IEM are considered rare, their estimated total prevalence is higher than 1 : 200. It is therefore very likely, that most physicians will experience at least one of the IEM in their lifetime. Setting up the correct diagnosis is of utmost importance for initiating therapy as for genetic and prenatal counselling.
Key words:
inborn errors of metabolism, developmental regression, cardiomyopathy, myopathy, rhabdomyolysis, hepatopathy, hepato-/splenomegaly, urolithiasis, optic neuropathy, dysostosis multiplex, hypoglycaemia, hyper - ammonemia, dyslipidemia
Autoři:
H. Kolářová; T. Honzík
Působiště autorů:
Klinika dětského a dorostového lékařství 1. LF UK a VFN, Praha
Vyšlo v časopise:
Čes-slov Pediat 2018; 73 (6): 348-364.
Souhrn
Úvod:
Dědičné poruchy metabolismu (DPM) představují skupinu >1000 vzácných geneticky podmíněných onemocnění, jejichž společným rysem je porucha funkce jednoho či více enzymů nebo změny strukturálních, asemblačních a transportních proteinů, které se účastní metabolických pochodů. DPM mohou postihovat jakýkoliv systém, což vysvětluje velkou variabilitu klinické manifestace a často opožděné stanovení správné diagnózy.
Cíl:
Podat přehledný souhrn klíčových klinických příznaků a laboratorních odchylek DPM, se kterými se mohou lékaři – pediatři setkat jak v rámci primární péče, tak i v nemocnicích.
Výsledky:
Mezi hlavní klinické projevy DPM patří regres psychomotorického vývoje, farmakorezistentní epilepsie, kardiomyopatie, myopatický syndrom či myalgie při atakách rhabdomyolýz a hepato-/splenomegalie. Až 70 % urolitiázy v dětském věku je způsobeno některou z DPM. Podrobné oční vyšetření může odhalit zákal rohovky a čočky, obrnu okohybných svalů, pigmentovou retinopatii a neuropatii optiku. V rámci rychlé diagnostiky mohou být nápomocné i na první pohled patrné příznaky, jako např. kraniofaciální dysmorfie, hypertrichóza, atypická struktura adnex a některé kostní změny. Mezi časté laboratorní odchylky DPM patří hypoglykémie, hyperamonémie, dyslipidémie a (cholestatická) hepatopatie. V diagnostice některých potenciálně léčitelných DPM je zásadní standardizované vyšetření mozkomíšního moku.
Závěr:
Přestože se jednotlivé DPM vyskytují jen velmi vzácně, jejich odhadovaná souhrnná prevalence je vyšší než 1 : 200. Je tedy pravděpodobné, že se během své praxe potká s některou z DPM každý lékař. Včasná diagnóza je důležitá jak pro rychlé zahájení léčby postižených dětí, tak pro potřebu genetického poradenství a prenatální diagnostiky v rodině.
Klíčová slova:
dědičné poruchy metabolismu, regres vývoje, kardiomyopatie, hepatopatie, hepato-/splenomegalie, myopatie, rhabdomyolýza, urolitiáza, neuropatie optiku, dysostosis multiplex, hypoglykémie, hyperamonémie, dyslipidémie
ÚVOD
Dědičné poruchy metabolismu (DPM) tvoří skupinu >1000 geneticky podmíněných onemocnění, jejichž společným rysem je porucha funkce jednoho či více enzymů nebo změny strukturálních, asemblačních a transportních proteinů, které se účastní metabolických pochodů. Přestože se jednotlivá onemocnění vyskytují jen velmi vzácně, jejich odhadovaná souhrnná prevalence je vyšší než 1 : 200 [1]. Je tedy pravděpodobné, že se během své praxe potká s některou z DPM každý lékař.
DPM mohou postihovat jakýkoliv systém, což má za následek velkou variabilitu klinických projevů. Některé příznaky však bývají tak typické, že by jejich zjištění mělo vést přinejmenším k pomyšlení na to, že by se mohlo jednat o některé z těchto onemocnění. První klinické příznaky DPM se mohou projevit v kterémkoliv věku; velká část z nich se však manifestuje již v novorozeneckém období a během raného dětství. Včasná diagnóza je důležitá jak pro rychlé zahájení léčby postižených dětí, tak pro potřebu genetického poradenství a prenatální diagnostiky v rodině. K rychlejší diagnóze DPM přispívá rozšíření celopopulačního novorozeneckého screeningu.
Smyslem této práce není podat podrobný výčet patobiochemických mechanismů konkrétních onemocnění, které jsou podrobně zpracovány jinde [2, 3]. Hlavní snahou je podat přehledný souhrn klíčových příznaků a laboratorních abnormalit DPM, se kterými se mohou lékaři ‒ pediatři setkat jak v rámci primární péče, tak i v nemocnicích. Vzhledem k tomu, že se uvedené symptomy mohou v rámci jednoho onemocnění kombinovat, uvádíme zde příznak hlavní a dominující, a další, přidružené symptomy, jsou zmíněny v textu či v tabulce. Hlavní zástupci onemocnění jsou u každého příznaku rozebráni podrobněji.
Práce je rozdělena do dvou podkapitol: první část představuje klinické příznaky rozdělené dle jednotlivých postižených systémů a v části druhé jsou uvedeny nejčastější laboratorní nálezy a jejich diferenciální diagnostika. Pro přehlednost a rychlou orientaci je v rámci textu i několik obrázků a diferenciálně diagnostických tabulek nejčastějších klinických a laboratorních nálezů.
KLINICKÉ PŘÍZNAKY
Postižení centrálního nervového systému
Neurologické projevy jsou součástí klinického spektra u 2/3 všech DPM; jejich podrobný výčet je mimo rámec tohoto sdělení. Na DPM budí podezření především regres psychomotorického vývoje a projevy zejména farmakorezistentní epilepsie. Velmi podezřelá je i kombinace neurologického (mozečkový syndrom, extrapyramidové postižení, poruchy zraku a sluchu) a systémového postižení. DPM obvykle vedou ke generalizovanému postižení všech vývojových oblastí. Alespoň základní metabolické vyšetření by však mělo být prováděno u všech pacientů s psychomotorickou retardací i s obrazem charakteru dětské mozkové obrny bez jednoznačné anamnézy peripartální hypoxie. Metabolické poruchy s regresem původně normálního psychomotorického vývoje či intelektu jsou shrnuty v tabulce 1.

X-vázaná adrenoleukodystrofie je peroxisomální onemocnění, které způsobuje progresivní poškození myelinu na podkladě ukládání toxických mastných kyselin s velmi dlouhým řetězcem. Jedním z nejčastějších typů je dětská cerebrální forma, která postihuje chlapce mezi 4. a 10. rokem. Obtíže většinou začínají zhoršením prospěchu ve škole, změnami chování a poruchou chůze. Postupně dochází k rozvoji spasticity a těžké kognitivní deteriorace. Pacienti obvykle umírají ve vegetativním stavu v průměru 2‒6 let od začátku prvních příznaků. Onemocnění je v přibližně polovině případů doprovázeno primární adrenokortikální insuficiencí, která se může projevit Addisonskou krizí.
Z onemocnění manifestujících se epilepsií zmiňme alespoň dvě velmi dobře léčitelná onemocnění: pyridoxin-dependentní křeče a deficit glukózového transportéru typu 1 (GLUT-1), a závažnou glycinovou encefalopatii, která má i přes určité terapeutické pokusy nepříznivou prognózu. Likvorologická diagnostika těchto onemocnění je v části Laboratorní nálezy. Pyridoxal-5-fosfát, derivát vitaminu B6, je důležitý pro syntézu některých neurotransmiterů (např. dopaminu a GABA). Jeho deficit se projeví jako farmakorezistentní epilepsie, která se může objevit již během intrauterinního období. U neléčených pacientů dochází k postupnému rozvoji těžké encefalopatie. Terapie spočívá v podávání vysokých dávek vitaminu B6. GLUT-1 umožňuje transport glukózy přes hematoencefalickou bariéru. Onemocnění se manifestuje v 90 % případů již během kojeneckého věku farmakorezistentní epilepsií, ataxií a spasticitou. Pacienti mají těžkou mikrocefalii s mentální retardací. Onemocnění představuje jednu z indikací zahájení ketogenní diety. Glycinová encefalopatie nebo také neketotická hyperglycinémie se v 85 % případů projevuje již v novorozeneckém věku hypotonií, apnoickými pauzami a rozvojem tonicko-klonických křečí se závažným korelátem na EEG. Psychomotorický vývoj je od počátku prakticky zastaven a nemocní umírají během kojeneckého nebo časného batolecího věku.
Postižení srdce
DPM mohou postihnout celý kardiovaskulární systém. Nejtypičtějším projevem, který by měl vést k podezření na některou z DPM, je kardiomyopatie (KMP). KMP jsou u dětí vzácné ‒ jejich odhadovaná incidence u dětí mladších 18 let je pouze 1,1 až 1,5 : 100 000 [4, 5]. KMP mají závažnou prognózu s obrazem srdečního selhání i náhlé smrti s často neúspěšnou kardiopulmonální resuscitací. Udává se, že až 26 % hypertrofických a 16 % dilatačních kardiomyopatií je způsobeno některou DPM [6]. KMP doprovázená zvracením, letargií, hypoglykémií, rhabdomyolýzou a dalšími známkami metabolické dekompenzace by vždy měla vést k podezření na DPM a pacient by měl být neprodleně odeslán do specializovaného centra. Obdobně i projevy multisystémového postižení, opoždění psychomotorického vývoje/vývojový regres, neprospívání a obtíže s pitím, kraniofaciální dysmorfie a bolesti svalů vyžadují podrobné metabolické vyšetření [7].
Nástup KMP a dalších tkáňově specifických příznaků závisí na základní diagnóze DPM i na věku dítěte. Hypertrofická KMP je typická pro většinu mitochondriálních onemocnění (např. deficit proteinu TMEM70 a SCO2, které představují asemblační faktory ATP syntázy, resp. cytochrom c oxidázy [8, 9], Danonovu nemoc a některé glykogenózy (GSD) ‒ typu III a II (Pompeho nemoc). Posledně zmíněná se řadí mezi lysosomální onemocnění (obr. 1) a KMP bývá spolu s těžkou svalovou hypotonií prvním příznakem onemocnění. Naopak KMP dilatační je častěji způsobena GSD typu IV a kombinovanými poruchami systému oxidativní fosforylace včetně Barthova syndromu, který je způsoben poruchou syntézy kardiolipinu tvořícího významnou součást mitochondriální vnitřní membrány [10]. Ve více než 10 % může být prvním projevem mukopolysacharidózy (MPS) typu I [11]. Poruchy ß-oxidace mastných kyselin, dědičné poruchy glykosylace a organické acidurie mohou vést jak k hypertrofickému, tak k dilatačnímu typu KMP nebo k obrazu smíšené formy.
Fig. 1. Chest X-ray of four-month old child with Pompe
disease with evidence of cardiomegaly due
to hypertrophic cardiomyopathy decompensation.

Srdeční selhání se však může rozvinout i jakožto následek převodní poruchy rytmu, která může být izolovaným symptomem nebo doprovází výše uvedené KMP. Typickým zástupcem primární poruchy rytmu je atrioventrikulární blok se synkopou u pacientů s Kearnsovým-Sayreovým syndromem. Poruchy ß-oxidace mastných kyselin s dlouhým řetězcem se mohou projevit celou řadou poruch srdečního rytmu (nejčastěji atrioventrikulární blok, komorová tachykardie), které vznikají ukládáním toxických acylkarnitinů s dlouhým řetězcem do kardiomyocytů.
Postižení srdečních chlopní bývá projevem alkaptonurie a některých střádavých onemocnění, typicky např. MPS. Porucha endotelu s rozvojem tromboembolických příhod v mladém věku může být známkou homocystinurie.
Postižení svalů, myopatie
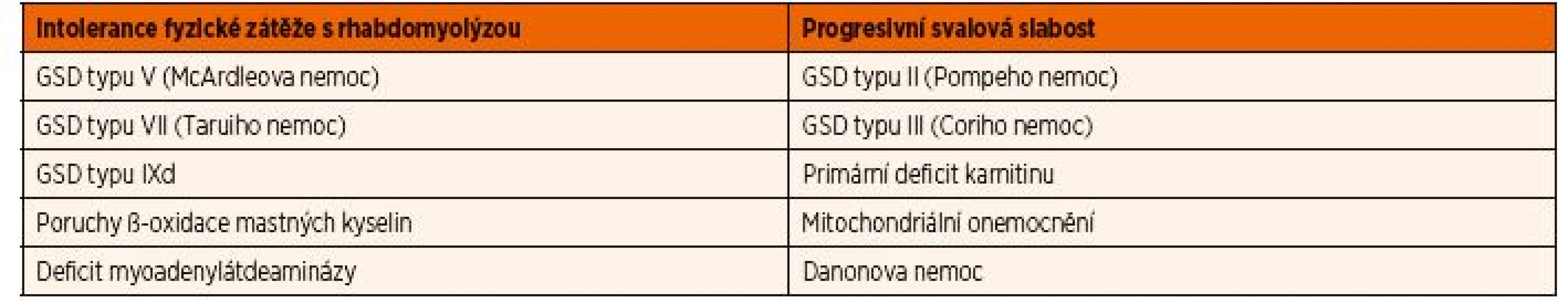
Hlavními skupinami onemocnění, které se manifestují postižením kosterních svalů, jsou svalové GSD, poruchy ß-oxidace mastných kyselin a mitochondriální onemocnění. Na základě klinických projevů lze rozlišit 2 hlavní skupiny (tab. 2): i) metabolické myopatie manifestující se akutní nebo subakutní netolerancí fyzické zátěže (rozvoj myalgií, svalové únavy, slabosti, křečí a rhabdomyolýzy v návaznosti na zvýšenou fyzickou aktivitu) a ii) progresivní svalové slabosti a hypotonie, které jsou stálé a obvykle bývají asociovány se systémovým postižením. U těchto onemocnění bývá obvykle hladina kreatinkinázy (CK) v normě, ale může se zvýšit i po menší fyzické námaze (max. desítky µkat/l). Onemocnění spojená s rhabdomyolýzou jsou podrobně popsána v části Laboratorní nálezy.
Infantilní forma Pompeho nemoci se typicky projevuje těžkou svalovou hypotonií a hypertrofickou KMP. Nenápadně se může projevovat především u starších dětí a dospělých, kde onemocnění začíná pomalu progredující svalovou slabostí, která může odpovídat klinickému obrazu pletencové formy svalové dystrofie.
Myopatie patří spolu s encefalopatií mezi nejčastější projevy mitochondriálních poruch. Tato onemocnění mají za následek nedostatečné zpracování substrátů (monosacharidů, aminokyselin, mastných kyselin a glycerolu) aerobním systémem oxidativní fosforylace, který je největším zdrojem energie v buňce. Obvykle se jedná o závažná multisystémová onemocnění, která nejvíce postihují energeticky náročné tkáně. Projevy postižení kosterního svalu jsou typické pro syndrom Mitochondriální encefalopatie, laktátové acidózy a iktu podobných příhod (MELAS), syndrom Myoklonické epilepsie s ragged-red fibres (MERRF), chronické progresivní zevní oftalmoplegie (CPEO) a Kearnsův-Sayreův syndrom. U mitochondriálních poruch bývá aktivita CK jen mírně zvýšená (do 10 µkat/l).
Hepatomegalie
Fyziologicky je zevní jaterní okraj hmatný pod pravým žeberním obloukem do 1‒2 cm, a to pouze do 1. roku života. Pokud během fyzikálního vyšetření zjistíme významnější hepatomegalii do 1 roku života, anebo játra hmatáme i po prvním roce života, mělo by být indikováno sonografické vyšetření břicha. Je nutné zmínit, že v případě velké hepatomegalie můžeme při fyzikálním vyšetření palpovat levý jaterní lalok pod levým žeberním obloukem (obr. 2), což může vést k nesprávné diagnóze splenomegalie.
Fig. 2. Extreme hepatomegaly in 8-year-old child with
Glycogenosis Type III. Both right and left liver lobes
were enlarged with the latter reaching left hypochondriac
region.

Hepatomegalie může být: i) asociovaná s nekrózou jaterních buněk způsobující hepatopatii, která může vyústit až v cirhózu, ii) asociovaná s cholestatickou žloutenkou a iii) izolovaným příznakem. První dvě skupiny jsou podrobně probrány v části Laboratorní nálezy. Důležitým vodítkem k určení diagnózy je zhodnocení konzistence a povrchu jater. Až kamenně tvrdá játra s nerovným povrchem (na podkladě hepatocelulárního karcinomu nebo jaterní cirhózy) bývají u tyrosinémie typu I, GSD typu IV, neonatální hemochromatózy, deficitu α1-antitrypsinu, Wilsonovy nemoci a dědičné poruchy glykosylace (např. deficit fosfomanoisomerázy, MPI-CDG). Pokud je jaterní konzistence normální, povrch jater je hladký a onemocnění je asociováno se splenomegalií, mělo by být zvažováno lysosomální onemocnění. V těchto případech obvykle zůstávají jaterní funkce v normě. V případě izolované, resp. dominantní hepatomegalie pomůže v diferenciální diagnostice především hodnota glykémie a další přidružené projevy (tab. 3). U části onemocnění může být do jisté míry vyjádřena i porucha jaterních funkcí, která však většinou nebývá významná a nekončí jaterním selháním.

Splenomegalie
Fyziologicky není slezina v dětském věku hmatná. Pokud slezina při hlubokém nádechu naráží na prsty vyšetřujícího, je zvětšená minimálně o více než jednu třetinu, a když ji hmatáme pod žeberním obloukem, může být až dvojnásobně velká. Po vyloučení hematoonkologické a infekční příčiny splenomegalie je vždy třeba pomyslet na lysosomální (střádavá) onemocnění. Lékaři prvního kontaktu bývají méně obeznámeni s těmito vzácnějšími (a potenciálně léčitelnými) jednotkami, které mohou splenomegalii způsobit, a diagnóza se tak někdy může opozdit i o několik let nebo vést, ze strachu z ruptury, k nesprávné indikaci invazivní splenektomie. Splenomegalie u střádavých onemocnění obvykle doprovází hepatomegalii. Izolovaná splenomegalie může být příznakem Niemannovy-Pickovy, Gaucherovy a Tangierovy nemoci.
Niemannova-Pickova nemoc typu C (NPC) je způsobena poruchou transportu cholesterolu a glykolipidů uvnitř buňky v lysosomech, kde dochází k jejich akumulaci. Část pacientů umírá již v dětském věku v důsledku těžkého jaterního selhání. Ostatní pacienti rozvíjí neurologické postižení s epilepsií, cereberelární ataxií, dystonií, dysfagií a obrnou vertikálního pohledu (viz dále). Nejčastější juvenilní forma onemocnění má bez terapie prognózu dožití menší než 25 let. Pravidelnou součástí průběhu onemocnění je psychiatrické postižení s progresivní demencí a různým stupněm psychózy nebo psychotické deprese s halucinacemi. Splenomegalie (event. i hepatomegalie) bývá vyjádřena již během časného dětství (obr. 3A) a, na rozdíl od Niemannovy-Pickovy nemoci typu A/B, není progresivní ani (kromě již výše zmíněného jaterního selhání, které část pacientů rozvíjí; obr. 3B) symptomatická. NPC je třeba zvážit u všech pacientů se splenomegalií a neuropsychiatrickými symptomy.
Fig 3. A. Extreme hepatosplenomegaly in 2-month-old child
with Niemann-Pick disease Type C.
B. Dystrophic 2-year-old boy with progressive hepatosplenomegaly
and liver failure based on Niemann-
-Pick disease Type A/B.

Nejčastější non-neuronopatická forma Gaucherovy nemoci typu I se manifestuje v průběhu dětského věku výraznou splenomegalií a mírnější hepatomegalií (obr. 4). V krevním obraze bývá typický nález trombocytopénie a anémie. Pacienti mívají bolesti dolních končetin na podkladě osteonekrózy kostí. Prognóza je u většiny pacientů díky dostupné enzymové substituční terapii uspokojivá. Mnohem závažnější je Gaucherova nemoc typu II, kde je již od prvních měsíců života patrné opoždění až kompletní zástava psychomotorického vývoje se spastickou kvadruplegií a těžkým neprospíváním s hepatosplenomegalií.
Fig. 4. Hepatosplenomegaly in
3.5-year-old child with
Gaucher disease Type I.

Urolitiáza, nefrolitiáza
Urolitiáza a nefrolitiáza, která se projeví již během dětského věku, je vzácná, ale až v 70 % je způsobena některou z DPM. Část pacientů má izolované rekurentní ataky urolitiázy, které jsou doprovázeny renálními kolikami s nauzeou a zvracením (např. cystinurie). Někteří pacienti však mohou mít kromě ledvinného postižení i další systémové či neurologické postižení (např. primární oxalurie typu I, Leschův-Nyhanův syndrom). Uro - a nefrolitiáza může být pozdním příznakem některých dalších DPM, u kterých hypersaturace moči kamenotvornou látkou vzniká na podkladě sekundárního defektu, jak je tomu např. u GSD typu I způsobující hyperurikémii. Nefrolitiáza se také může vyvinout jako následek DPM, které vedou k rozvoji Fanconiho syndromu (např. cystinóza, tyrosinémie typu I, Wilsonova nemoc). Litiáza u DPM bývá prakticky vždy bilaterální; diferenciálně diagnosticky může pomoci přítomnost kontrastu litiázy na RTG snímcích (tab. 4).

Postižení oka
Oko je velmi různorodým orgánem z hlediska složení a energetické náročnosti jeho jednotlivých struktur. Různé DPM proto vedou k rozvoji celé řady klinických projevů podle typu postižené oblasti. Podrobné oční vyšetření by proto mělo být součástí každého vyšetřovacího schématu u pacienta s podezřením na DPM. Kromě zhodnocení funkčních vyšetření je vhodné i provedení fundoskopie k zobrazení sítnice a zhodnocení předního segmentu oka pomocí štěrbinové lampy. Toto vyšetření dokáže velmi dobře zobrazit strukturu rohovky a čočky, které bývají u DPM často postiženy, neboť obě tyto tkáně jsou velmi citlivé na ukládání cizorodého materiálu. Zákal rohovky bývá pravidelnou součástí lysosomálních onemocnění (např. MPS) a některých poruch metabolismu lipidů. Katarakta se může rozvinout u pacientů s klasickou galaktosémií, Smithovým-Lemliho-Opitzovým syndromem, Wilsonovou a Menkesovou nemocí a některými mitochondriálními a peroxisomálními onemocněními. K patologické kumulaci krystalů v rohovce dochází u pacientů s cystinózou (obr. 5).
Fig. 5. Cystine crystals in cornea
of 2.5-year-old boy with
infantile cystinosis and
photophobia.

Dislokace čočky s těžkou krátkozrakostí je jedním z hlavních příznaků pacientů s homocystinurií. Tato porucha metabolismu aminokyselin se může fenotypově podobat Marfanovu syndromu – pacienti bývají vysocí, štíhlí a mají skoliózu. Dislokace čočky je však v případě homocystinurie kaudálně a v případě Marfanova syndromu kraniálně. Hlavní příčinou úmrtí pacientů s homocystinurií jsou tromboembolické komplikace v časném věku.
Ptóza a obrna zevního pohledu jsou typické pro některá mitochondriální onemocnění (Kearnsův-Sayreův syndrom, CPEO, MELAS, syndrom Mitochondriální neurogastrointestinální encefalopatie). U izolované ptózy je vždy třeba vyloučit kongenitální myastenické syndromy. Poměrně typickým příznakem Niemannovy-Pickovy nemoci typu C je vertikální supranukleární oftalmoplegie (obrna vertikálního pohledu). Oproti tomu u Gaucherovy nemoci jsou jako první vyjádřeny omezené pomalé horizontální sakadické pohyby.
Onemocnění, která způsobují abnormality na očním pozadí, jsou shrnuta v tabulce 5. Tzv. třešňová nebo také třešňově červená skvrna na očním pozadí vzniká typicky u střádavých onemocnění, která způsobují akumulaci nejčastěji lipidů a jejich derivátů v buňkách různých vrstev sítnice. Makula je naopak relativně bezbuněčná a vzhledem ke její průhlednosti proto v případě těchto onemocnění způsobí viditelnost červeně prosvítající cévnatky. Pigmentová retinopatie je způsobená poruchou fotoreceptorů, což se klinicky projevuje jako šeroslepost. Je typickým projevem celé řady DPM, nejčastěji mitochondriálních onemocnění a poruch ß-oxidace mastných kyselin na úrovni 3-hydroxy-acyl-koenzym A-dehydrogenázy pro mastné kyseliny s dlouhým řetězcem a při deficitu mitochondriálního trifunkčního proteinu.

Neuropatie optiku někdy také nesprávně označovaná jako atrofie optiku podle konečného stadia nemoci, je způsobena postižením kdekoliv na dráze od těl a axonů retinálních buněk až po corpus geniculatum laterale thalamu. Klinicky se onemocnění projevuje rozvojem centrálních skotomů, které se postupně rozšiřují do periferie; při fundoskopickém vyšetření jsou patrné bledé zrakové terče. Primární hereditární neuropatie optiku jsou nejčastěji způsobené mitochondriálními onemocněními, a to z důvodu velké energetické náročnosti vedení akčního potenciálu optickým nervem. Leberova hereditární neuropatie optiku je nejčastějším mitochondriálním onemocněním vůbec. Vzniká na podkladě mutací v mitochondriální DNA, a proto je jeho dědičnost maternální. Typickým pacientem je muž mezi 20. a 30. rokem věku, který náhle rozvine rychlou progresivní ztrátu zrakové ostrosti, která nejpozději do 12 měsíců vyústí v obraz praktické slepoty. Nemocní s dominantní optickou atrofií způsobenou mutacemi v nukleárním genu OPA1 se naopak manifestují v dětství, ale onemocnění je obvykle jen pomalu progresivní s dlouho zachovalými zrakovými funkcemi. U více než jedné čtvrtiny pacientů dochází k rozvoji extraokulárních příznaků v podobě percepční poruchy sluchu, myopatie a periferní neuropatie. Sekundární neuropatie optiku je součástí klinického spektra u celé řady dalších metabolických onemocnění (např. peroxisomální poruchy, Krabbeho nemoc, neuronální ceroidní lipofuscinózy). Je zajímavé, že se i u velké části z nich předpokládá podíl sekundární mitochondriální dysfunkce.
Kraniofaciální a jiné dysmorfické rysy
Mírné izolované dysmorfické rysy (atypický tvar a odstup ušních boltců, větší vzdálenost očí a jiný sklon očních štěrbin, nápadný tvar rtů či heterochromie duhovek) jsou přítomny u asi 10 % zdravých dětí a obvykle nepředstavují indikaci k vyšetření DPM nebo genetického syndromu. Jejich kombinace (2 a více příznaků) je však fyziologicky vyjádřena jen u asi 1 % dětí [12]. Nespecifická kraniofaciální dysmorfie může být vyjádřená u celé řady DPM (např. panenkovitý obličej u GSD typu I a III). Pouze některá onemocnění však mají tak typické rysy, že je možné již na jejich základě pomyslet na konkrétní klinickou jednotku.
Kraniofaciální dysmorfie je typická pro pacienty s MPS. U typu I, II a VII je kromě typických hrubých rysů (obraz chrliče, tzv. gargoylismus) patrné těžké systémové postižení. Tito pacienti jsou při narození, na rozdíl od mukolipidózy typu II, normálního vzezření a k rozvoji typických rysů u nich dochází až od konce kojeneckého věku. V kontrastu s tím je těžké postižení centrálního nervového systému s mentální retardací bez dramatického somatického postižení (a obvykle jen mírně vyjádřenými hrubými rysy) u MPS typu III (Sanfilippova nemoc). U MPS typu III bývá přítomna hyperaktivita, porucha spánku, stereotypní chování a auto - i heteroagresivita, která je v případě MPS ještě umocněna prakticky normální fyzickou silou. Naopak MPS typu IV, VI a některé přechodné typy MPS typu I (např. Scheieho a Hurlerové-Scheieho nemoc) obvykle nepostihují centrální nervový systém (CNS). U pacientů s MPS typu I, II a VII dominuje velká hlava, někdy s hyperostózou sagitálních švů a vyklenuté čelo s hypertelorismem. Oční víčka působí edematózně a stejně tak i rty jsou velké a ztluštělé. Jazyk může vyčnívat ven z úst; dásně jsou hypertrofické a zuby dysplastické a daleko od sebe. Kořen nosu je sedlovitý s antevertovanými nostrilami (obr. 6). Pacienti s MPS mívají hirsutismus s nízkou vlasovou hranicí a široké, husté obočí a povšechnou hypertrichózu.
Fig. 6. Craniofacial dysmorphism and skeletal changes in 6-year-old child with
Mucopolysaccharidosis Type II. Facial features are coarse, the eyes are prominent with
bushy eyebrows, and the nasal bridge is depressed. There is frontal bossing, widened
anteverted alae nasi and thick lips. The boy is short and has a chest deformity with
marked thoracolumbar gibbus. He has phalangeal contractures and there is a limitation
of motion in the position of elbows and knees.
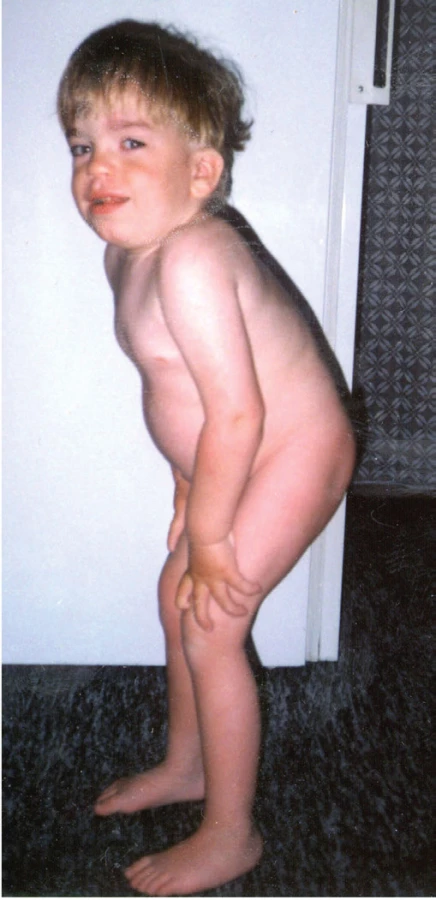
Přestože je psychomotorický vývoj pacientů v prvních měsících života bez pozoruhodností, některé typické projevy nás mohou upozornit na MPS již v časném kojeneckém věku. V anamnéze můžeme dohledat operaci ingvinálních, nebo později i umbilikálních hernií a chronickou rýmu. Po prvním roce života se rozvíjí recidivující respirační infekty a otitidy, které si často vyžádají aplikaci gromet. V tomto období dochází i k progresivnímu zhoršování mentálního vývoje – vrchol intelektových schopností se pohybuje mezi 2.–4. rokem a následně postupně klesá. Období lineárního růstu se u pacientů zastavuje kolem 2.–3. roku života. Krk a trup jsou krátké; na páteři je obvykle patrná hrudní kyfóza s gibbem a bederní lordóza (obr. 6), která přispívá spolu s extrémní hepatosplenomegalií k vyklenutí bříška. Nálezy na rentgenových snímcích odpovídají tzv. dysostosis multiplex (rozšíření diafýz, krátké a silné kosti ruky se sešikmením a angulací distálních konců radia a ulny vzhledu tzv. klepetové ruky, srdčitá pánev, platyspondylie a ovoidní, rybí obratle; obr. 7A-C). Kostní změny v oblasti krční, ale i jiných částí páteře mohou vest k závažné komplikaci útlaku míchy a rozvoji parézy.
Fig. 7. Typical X-ray findings in dysostosis multiplex. A. Radiograph of the
left hand, illustrating broadened metacarpals at their distal ends
and tapered at the proximal ends with a hook-like deformity; digits
are „bullet-shaped“. B. Pelvis radiograph in 13-month-old girl with
Mucopolysaccharidosis Type I showing flared iliac wing and flat acetabular
roof (Mickey mouse ear pelvis). C. Roentgenographic appearance of
the spine. The anterior-posterior distance was diminished in the vertebral
bodies and there was marked posterior scalloping (platyspondyly
and „fish vertebra“) in a 5-year-old child with Mucopolysaccharidosis
Type IVA. The pedicles of the spine were elongated with inferior beaking.

U peroxisomálního onemocnění je kraniofaciální dysmorfie vyjádřena především u poruch Zellwegerovského spektra (Zellwegerův syndrom, neonatální adrenoleukodystrofie a infantilní Refsumova nemoc). Tato onemocnění vznikají na podkladě poruch biogeneze, což vede k nedostačné oxidaci velmi dlouhých mastných kyselin a porušené syntéze etherů lipidů (tzv. plasmalogenů), které jsou důležitou součástí myelinu. Pacienti se obvykle manifestují již v novorozeneckém věku křečemi, těžkou svalovou hypotonií a praktickou zástavou psychomotorického vývoje. Nápadná je velká, široce otevřená fontanela s rozestouplými švy. Čelo je vysoké a vyklenuté; nadočnicové oblouky jsou naopak hypoplastické. Může být přítomen i atypický tvar a odstup ušních boltců a šikmé postavení očí s epikanty. Některé formy peroxisomálních onemocnění mají rhizomelické zkrácení končetin s patrnými kožními řasami. Na rentgenových snímcích bývají v novorozeneckém a kojeneckém věku na epifýzách vidět tečkovité kalcifikace (tzv. calcific stippling), nejčastěji na bočním snímku patelly a/nebo paty.
Kůže a adnexa
Nápadně hrubá a tvrdá kůže, drsné vlasy a husté obočí jsou spolu s charakteristickou kraniofaciální dysmorfií typické pro pacienty s MPS. Rozvoj angiokeratomů (červených až namodralých cévních útvarů, které vznikají na podkladě ektatického rozšíření cév s hyperkeratózou (obr. 8)), je typický pro sfingolipidózy a bývá často prvním příznakem Fabryho nemoci. Samotná hyperkeratóza dlaní a plosek nohou se typicky vyskytuje u tyrosinémie typu II a některých typů porfyrií. Fotosenzitivita u posledně zmíněných dala za vznik legendám o upírech. Je způsobena excitací porfyrinů nahromaděných v kůži, vytvářejících velké množství kyslíkových radikálů. Po vystavení kůže slunečnímu záření se objevují tekutinou naplněné rozsáhlé puchýře, které se hojí za vzniku jizev a hyperpigmentací. Kůže je při opakovaném poškození atrofická a často se objevuje i hypertrichóza obličeje.
Fig. 8. Angiokeratomas in 10-year-old boy with Fabry
disease.

Změny vlasů by měly upozornit na možný průběh poruchy cyklu močoviny či poruchy metabolismu kovů. Hypertrichóza se typicky objevuje u některých mitochondriálních onemocnění (obr. 9A, B); naopak alopecie včetně absence řas a obočí může doprovázet acrodermatitis enteropathica a nepoznaný deficit biotinidázy. Pacienti s Menkesovou nemocí (porucha transportéru mědi přes střevní sliznici) mají zvláštní, obvykle kudrnaté, jemné a lámavé vlasy šedavé barvy (obr. 10A), které při pozorování pod mikroskopem vytvářejí charakteristický obraz zákrutů v různě dlouhém intervalu (tzv. Pilli torti; obr. 10B). Přítomen může být i obraz Cutis laxa. Onemocnění je charakterizováno těžce opožděným mentálním vývojem a malým vzrůstem. Neléčení pacienti mají velmi špatnou prognózu a obvykle umírají nejpozději do 3. roku věku. Vzácněji tito pacienti mohou rozvinout i obraz tzv. Trichorrhexis nodosa (nodózní rozšíření části vlasu, který se pak snadněji láme). Tato klinická jednotka je však častější u některých poruch cyklu močoviny (např. citrulinémie a deficitu argininosukcinátlyázy či arginázy).
Fig. 9. Hypertrichosis on the back (A) and lower limbs (B) in 3.5-year-old girl with
Mitochondrial encephalopathy, Lactic acidosis and Stroke-like episodes (MELAS)
syndrome.

Fig. 10. A. Detail of hair in 3-months-old boy with Menkes disease. The scalp hair was sparse with dry head skin. Hair are short,
hypopigmented and kinky with brittle tips. B. Microscopic hair examination shows hair twisting (Pilli torti).

Dědičné poruchy glykosylace tvoří heterogenní soubor více než 125 onemocnění [13], které jsou způsobeny poruchou vazby cukerných zbytků na proteiny či lipidy. Nejčastější typ Ia (PMM2-CDG) se typicky projevuje v novorozeneckém věku hypotonií a strabismem. Později se může rozvinout atypická distribuce tuku v oblasti hýždí, genitálu a invertace mamil (obr. 11A, B). Pacienti mají různý stupeň mentální retardace a v laboratorním nálezu obraz smíšené koagulopatie.
Fig. 11. A. Strabism, failure to thrive and atypical supragluteal fat distribution in 12-month-old girl with congenital disorder of glycosylation
(CDG) type phosphomannomutase deficiency (PMM2). B. Persisting nipple inversions in 17-year-old girl with PMM2-CDG.
LABORATORNÍ NÁLEZY
Hypoglykémie
Hypoglykémie je příznakem celé řady metabolických i endokrinologických onemocnění. Za hypoglykémii považujeme u donošených novorozenců v prvních 24 hodinách života hladinu glykémie <2,5 mmol/l a u větších dětí <3 mmol/l. Na rozdíl od kosterního svalu a srdce využívá CNS glukózu jako prakticky výhradní zdroj energie. Klinické příznaky hypoglykémie u batolat a větších dětí se obecně rozdělují do dvou skupin:i) akutní příznaky z aktivace adrenergního systému a ii) příznaky způsobené sníženou utilizací glukózy v CNS. Klinický obraz hypoglykémie v novorozeneckém a časném kojeneckém věku je však velmi nespecifický a často bývá podhodnocený (tab. 6).

Závažné či opakované hypoglykémie mohou narušit vývoj CNS a vést k rozvoji mentální retardace nebo k sekundární epilepsii. Laboratorně nezachycené či asymptomatické hypoglykémie v novorozeneckém a kojeneckém věku mohou zpomalit růst obvodu hlavičky a způsobit mikrocefalii s atrofií CNS.
Klinický přístup k hypoglykémii je založen na 4 hlavních kritériích – ta lze zjistit vyšetřením, které má k dispozici každý lékař: velikost jater, časový nástup hypoglykémie a přítomnost laktátové acidózy a ketolátek. Především asociace hypoglykémie a hepatomegalie budí velké podezření, že by se mohlo jednat o DPM. Pečlivě odebraná anamnéza je klíčová v diferenciálně diagnostickém rozhodování o příčině hypoglykémie. Hypoglykémie po krátkém lačnění (2–3 hodiny) mohou svědčit pro jaterní GSD. Nízká hladina glukózy objevivší se až po delší době lačnění při nočním hladovění (>6 hodin) je podezřelá z průběhu poruchy ß-oxidace mastných kyselin. Nepředvídatelné postprandiální hypoglykémie se objevují u dětí s hyperinzulinismem. Návaznost hypoglykémií na konzumaci mateřského mléka či ovocných příkrmů jsou typické pro galaktosémii, resp. hereditární intoleranci fruktózy. Diagnostický přístup k hypoglykémii je přehledně uveden na obrázku 12.
Akutní jaterní selhání
Akutní jaterní selhání se u dětí projeví ikterem, koagulopatií s prodloužením protrombinového času (nízké hladiny vitamin K-dependentních faktorů II, VII, IX, X, proteinu C a S), hyperamonémií a nízkou syntetickou funkcí jater (nízká hladina albuminu, prealbuminu a nízká aktivita cholinesterázy). Hepatocelulární ne - króza způsobí uvolnění jaterních transamináz. Relativně pozdním příznakem jaterního selhání u dětí s DPM je hypoglykémie. Vzhledem k tomu, že při hepatocelulárním poškození může docházet i k poškození žlučového pólu jaterní buňky, má část pacientů více či méně vyjádřenou i složku cholestatickou. Důležitým vodítkem při rychlém rozhodování o možné etiologii jaterního selhání je věk dítěte. Hlavní DPM a další onemocnění vedoucí k jaternímu selhání jsou uvedeny v tabulce 7.

£ Vzhledem k absenci fruktózy v současných mléčných formulích se onemocnění manifestuje až po zavedení ovocných příkrmů.
Vysvětlivky: AFP – α-fetoprotein; CESD – nemoc se střádáním esterů cholesterolu; CK – kreatinkináza; mtDNA – mitochondriální DNA
Klasická forma galaktosémie se projevuje ve většině případů u novorozence mezi 3.–7. dnem života, kdy již dítě vypilo minimálně celkem 300 ml mléka. Stav je dramatický – u dítěte se rozvíjí rychle progredující jaterní selhání se zvracením, hepatomegalií, ikterem a edémem mozku s poruchou vědomí. Onemocnění bývá zprvu mylně vyhodnoceno jako novorozenecká sepse. Vzhledem k velké endogenní produkci galaktózy nemusí být i při správně vedené bezlaktózové a nízkogalaktózové dietě prognóza onemocnění příznivá a postupně může docházet k rozvoji mentální retardace a hypergonadotropního hypogonadismu.
Neonatální hemochromatóza je dnes považována za jednu z nejčastějších příčin neonatálního jaterního selhání (60–90 %; [14]). Onemocnění je charakterizováno hromaděním železa v játrech i v extrahepatálních tkáních. V krvi je vysoká hladina železa, ferritinu (>2000 mg/l) a α-fetoproteinu. I když patogenetický mechanismus není do dnešního dne přesně objasněn, předpokládá se, že onemocnění je způsobeno aloimunitním poškozením jater mateřskými protilátkami proti specifickému hepatocytárnímu antigenu. Je možné, že nadbytek železa je způsoben nižší hladinou játry produkovaného hepcidinu [15], který tlumí transport železa placentou. Název hemochromatóza není správný, neboť onemocnění není způsobeno primárním defektem v metabolismu železa jako stejně nazvaná klasická forma. Nově používaný termín je gestational alloimune liver disease/hepatitis (tj. gestační aloimunitní hepatitida).
Tyrosinémie typu I je způsobená poruchou odbourávání aminokyseliny tyrosinu, který se spolu s dalšími toxickými metabolity hromadí v těle nemocných a způsobují dominantní poškození jater a ledvin. Akutním jaterním selháním se manifestují především kojenci; v pozdějším věku se přidává těžké neprospívání a postižení ledvin. Chronická forma onemocnění je charakterizována pomalu progresivním poškozením ledvin a jater do obrazu cirhózy nebo hepatocelulárního karcinomu.
Klinické projevy hereditární intolerance fruktózy závisí na době zavedení výživy s obsahem fruktózy či sacharózy. Pacienti rozvíjejí akutní jaterní selhání s hypoglykémií, zvracením, průjmy a neprospívání. U dětí s mírnějším průběhem onemocnění může docházet k podvědomému vyhýbání se stravě s vyšším obsahem výše uvedených sacharidů.
Cholestatická hepatopatie
Cholestatická hepatopatie se projeví konjugovanou hyperbilirubinémií s ikterem (>20 % celkového bilirubinu), zvýšenými aktivitami gamma-glutamyltransferázy, alkalické fosfatázy a zvýšenou koncentrací žlučových kyselin v krvi. Obvykle je současně vyjádřen i různý stupeň hepatomegalie s hepatopatií. U novorozenců je vždy třeba nejprve vyloučit kongenitální cytomegalovirovou infekci, biliární atrézii, cystu choledochu a některé další vývojové vady žlučového systému. Přehled DPM, které se mohou manifestovat cholestatickou hepatopatií, a to již během novorozeneckého období, je uveden v tabulce 8.

Cholestatická žloutenka a těžké neprospívání jsou hlavními příznaky deficitu α1-antitrypsinu. Jedná se o proteázový inhibitor, který inaktivuje řadu enzymů včetně elastázy neutrofilů v plicní tkáni. Jeho nedostatek je způsoben neschopností jeho sekrece z hepatocytu, který svým lokálním působením poškozuje. V plicích vede nedostatečná aktivita α1-antitrypsinu k proteolytickému poškození alveolárních stěn a rozvoji emfyzému. Část pacientů se manifestuje v dětském věku hepatomegalií a cholestatickou hepatopatií, která může progredovat až do obrazu jaterního selhání s rozvojem cirhózy. Deficit α1-antitrypsinu je v současnosti jednou z nejčastějších indikací transplantace jater u dětí. Dospělí pacienti jsou naopak ohroženi rozvojem plicního emfyzému.
Hyperamonémie
Hyperamonémie je hodnocena jako hladina amoniaku >60 µmol/l (u novorozenců >80 µmol/l). Jedná se o velmi závažný laboratorní nález celé řady DPM, který má při nedostatečně rychle zahájené léčbě za následek ireverzibilní poškození CNS pacientů. Určitý problém představuje především obtížná preanalytická fáze s nutností speciálního odběru a rychlého transportu do laboratoře. Nejčastější příčinou hyperamonémie tak stále představuje laboratorní chyba. Vzhledem k závažnosti dopadů hyperamonémie je však nutností standardně zajistit pacienta na specializovaném pracovišti (parenterální přívod glukózy, zastavení perorálního příjmu bílkovin) před objasněním její etiologie. Přehled DPM, které se manifestují hyperamonémií, je znázorněn v tabulce 9. Podrobněji bude zmíněna skupina poruch cyklu močoviny.

Poruchy cyklu močoviny tvoří skupinu enzymatických poruch, které mohou nastat v každém kroku syntézy močoviny, neboli urey, kterou ureotelní organismy vylučují přebytečný dusík. Klinické příznaky jsou velmi obdobné všem onemocněním z tohoto okruhu. Nejčastějším zástupcem je X-vázaný deficit ornitintranskarbamylázy (OTC). Neonatální forma onemocnění je typická přítomností krátkého bezpříznakového období po porodu, kdy se dítě jeví jako naprosto zdravé (několik hodin – 2 dny). Po něm následuje spavost, odmítání stravy a zvracení. Vysoká hladina amoniaku přímo stimuluje dýchací centrum, což vede k rozvoji tachypnoe a k primární respirační alkalóze. Mohou být přítomny i křeče s abnormálním nálezem na EEG. Pokud dítě není včas léčeno, rozvíjí se mozkový edém s centrální apnoe a těžkým kómatem a multiorgánovým selháním, většinou s hladinou amoniaku 250 µmol/l a více. V laboratorním nálezu bývá v akutní fázi mimo hyperamonémii přítomna i hepatopatie jakožto projev jaterního poškození, které může vést až do jaterního selhání. Mimo tyto těžké případy s manifestací v neonatálním období se u některých chlapců se zachovanou parciální aktivitou OTC onemocnění manifestuje až během dětství, nebo dokonce až v dospělosti.
Dyslipidémie
Dyslipidémie představují nejčastější dědičné poruchy metabolismu se souhrnnou incidencí až 1 : 200 živě narozených dětí. Jejich detailní rozbor přesahuje rozsah této publikace. V tabulce 10 uvádíme diferenciální diagnostiku dyslipidémie v závislosti na biochemických nálezech a dalších přidružených příznacích.

Elevace kreatinkinázy (CK) a myoglobinu
Aktivity CK jsou závislé na věku a pohlaví (tab. 11) a jejich interpretace u dětí může být svízelná. I u zdravých dětí bývá hladina CK zvýšená (obvykle do 10 µkat/l) po větší námaze v rámci sportovních aktivit i několik dní. Navíc u netrénovaných lidí může po velké zátěži a nadměrných ztrátách tekutin docházet ke klinicky nepoznané ponámahové rhabdomyolýze, kdy hladiny CK mohou vystoupat až do stovkových hodnot [16]. Hladiny CK obvykle stoupají do 12 hodin po akutním svalovém poškození, dosahují vrcholu mezi 24.–36. hodinou a následně klesají o 35–40 % za den [17]. Trvale zvýšená hodnota CK poukazuje na pokračující svalové poškození. V tomto případě je nutno pomyslet na onemocnění ze skupiny svalových dystrofií.

Pochopení jednoduchého principu svalového metabolismu pomůže již na základě pečlivě odebrané anamnézy pojmout silné podezření na konkrétní příčinu myopatie. Svaly při krátkém intenzivním cvičení využívají ve svalových vláknech typu II jako zdroj energie adenosintrifosfát (ATP) z kreatinfosfátu (1–5 minut) a glukózu (>5 minut) ze svalového glykogenu. V průběhu delšího cvičení je svalovými vlákny typu I využíván především acetyl-koenzym A z ß-oxidace mastných kyselin (>40 minut). Porucha syntézy a transportu kreatinu byla do dnešního dne popsána pouze v souvislosti s těžkým postižením CNS. Některé poruchy metabolismu sacharidů a tuků však mohou mít při vyšší fyzické námaze za následek rozvoj metabolické dekompenzace s akutním rozpadem svalů (rhabdomyolýzou), které vede k uvolnění svalových proteinů – CK a myoglobinu – do krve a následně i do moči, kde je můžeme stanovit. Hodnoty CK dosahují během ataky rhabdomyolýzy sto - až tisícinásobku µkat/l. K rozvoji myopatie však nepřispívá jen nedostatek ATP, ale i strukturální změny svalů na podkladě např. akumulace abnormálního glykogenu nebo produkce reaktivních kyslíkových radikálů.
Rozvoj svalových křečí a myalgií již během krátkého namáhavého cvičení je velmi podezřelý z průběhu GSD typu V (McArdleova nemoc). Toto onemocnění je způsobeno poruchou svalové myofosforylázy, která uvolňuje glukózu z glykogenu pro akutní potřeby svalu. Klinické příznaky se obvykle rozvíjejí v období dětství či adolescence. Vysoká fyzická námaha vede k rychlému rozvoji myalgií, únavy a svalových křečí, které rychle ustupují při odpočinku či po požití jednoduchých cukrů. GSD typu V je typická náhlým zlepšením tolerance a ústupem bolesti po asi 10 minutách po začátku cvičení (tzv. fenomén druhého dechu). Odhadovaná prevalence je asi 1 : 100 000 živě narozených, nicméně přepokládá se, že se jedná o významně poddiagnostikované onemocnění, neboť odhadovaná prevalence šesti nejčastějších mutací v homozygotním stavu je až 1 : 7650 [16]. Mírněji postižení pacienti totiž mimo ataky rhabdomyolýzy nemusí působit nikterak nemocně, a bývají vzhledem k sporé aktivitě omylem považováni za neaktivní. U pacientů s GSD typu V bývá vždy zvýšená hladina CK i v období mezi cvičením a dále se zvyšuje během fyzické aktivity.
Naopak ponámahová rhabdomyolýza rozvíjející se až v návaznosti na prolongované cvičení a zhoršující se při hladovění, resp. horečce (typicky během akutní gastroenteritidy nebo respiračního infektu) je charakteristická pro poruchy ß-oxidace mastných kyselin. Tato skupina onemocnění vede prostřednictvím snížené produkce ATP k energetické deprivaci buněk. Nejzávažnějším projevem těchto onemocnění je hypoketotická hypoglykémie u onemocnění MCAD (porucha ß-oxidace mastných kyselin na úrovni 3-hydroxy-acyl-koenzym A-dehydrogenázy pro mastné kyseliny se středním řetězcem). Ostatní poruchy (porucha ß-oxidace mastných kyselin na úrovni 3-hydroxy-acyl-koenzym A-dehydrogenázy pro mastné kyseliny s dlouhým řetězcem a na úrovni acyl-CoA-dehydrogenázy mastných kyselin s velmi dlouhým řetězcem, deficit mitochondriálního trifunkčního proteinu a poruchy transportu karnitinu) se častěji projevují myopatií s atakami rhabdomyolýzy a později se komplikují rozvojem hypertrofické kardiomyopatie a těžké periferní neuropatie. Diagnostika poruch ß-oxidace mastných kyselin se v ČR provádí od října 2009 v rámci novorozeneckého screeningu pomocí hmotnostní spektrometrie ze suché krevní kapky.
Důležité likvorologické nálezy
Odběr mozkomíšního moku je nezbytným vyšetřením hned u několika DPM. Provedení lumbální punkce je třeba zvážit zejména u jinak nevysvětlitelných křečových projevů u dětí s hypotonií a poruchami vědomí, neboť část z těchto onemocnění má při správně zahájené léčbě velmi dobrou prognózu. Vždy s odběrem mozkomíšního moku musí být současně odebrána také krev a event. i moč.
Materiál je nutné ihned odeslat ke speciálnímu vyšetření aminokyselin a dalších metabolitů (např. neuro-transmiterů, 5-metyltetrahydrofolátu) po domluvě s konzultujícím lékařem s uložením ihned do -80 °C. I z tohoto důvodu bývá někdy vhodnější pacienta ve stabilizovaném stavu přeložit na specializované pracoviště, abychom se vyhnuli opakování tohoto invazivního vyšetření. Přehled nejdůležitějších likvorologických nálezů u DPM a jejich diferenciální diagnostika jsou uvedeny v tabulce 12.
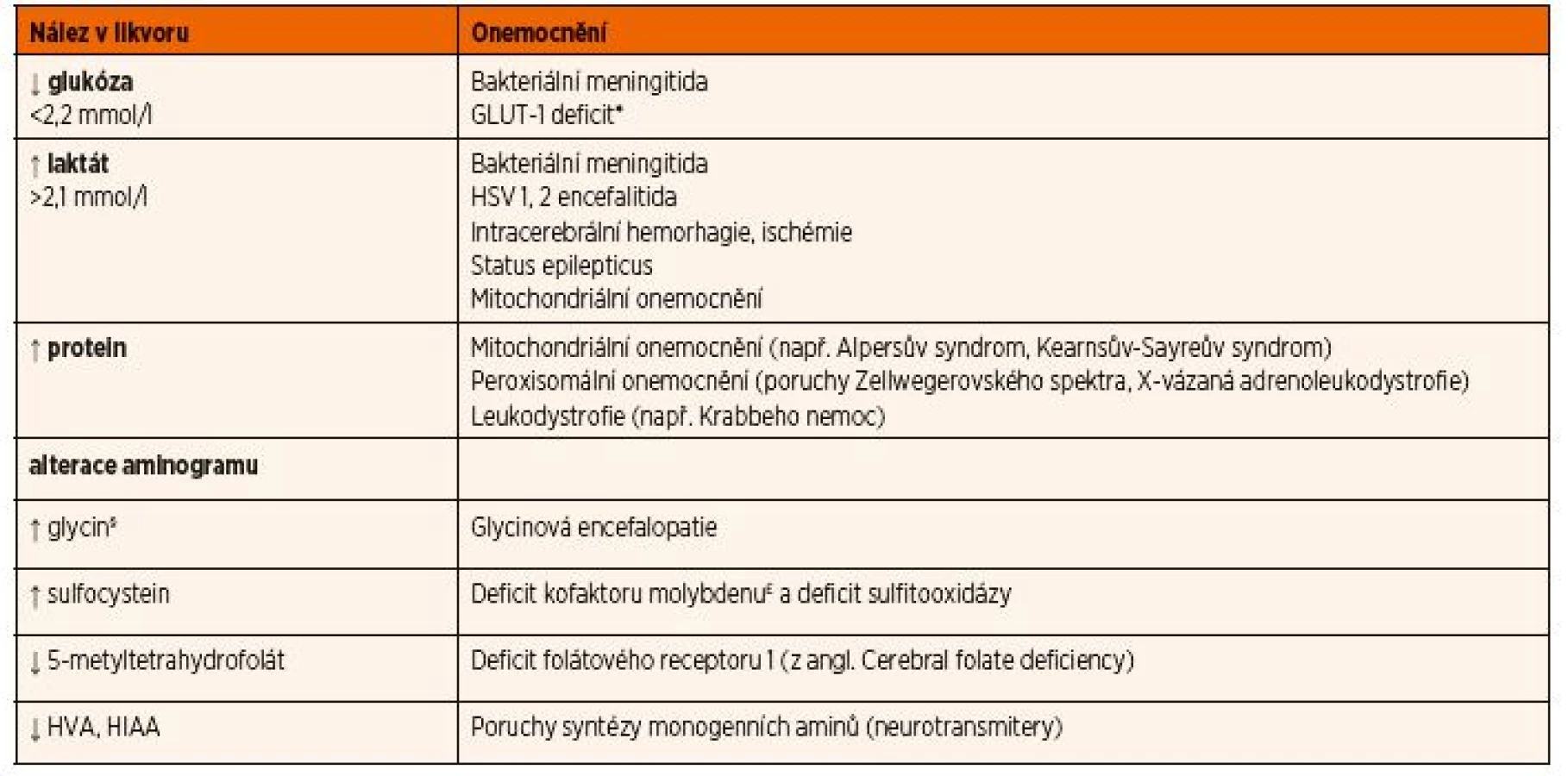
§Dalším diagnostickým znakem glycinové encefalopatie je zvýšený poměr koncentrace glycinu v mozkomíšním moku/koncentrace glycinu
v krvi >0,08.
£Dalším laboratorním nálezem bude nízká hladina kyseliny močové v séru a v moči.
Vysvětlivky: GLUT-1 – glukózový transportér typu I; HSV – herpes simplex virus; HIAA – kyselina hydroxyindoloctová; HVA – kyselina homovanilmandlová

Zkratky:
AFP – α-fetoprotein; ATP – adenosintrifosfát; CESD – nemoc se střádáním esterů cholesterolu; CK – kreatinkináza; CDG – dědičné poruchy glykosylace; CNS – centrální nervový system; CPEO – chronická progresivní zevní oftalmoplegie; DOA – dominantní optická atrofie; DPM – dědičné poruchy metabolismu; GLUT-1 – glukózový transportér typu 1; GSD – glykogenóza; HIAA – kyselina hydroxyindoloctová; HSV – herpes simplex virus; HVA – kyselina homovanilmandlová; KMP – kardiomyopatie; LCHAD – porucha ß-oxidace mastných kyselin na úrovni 3-hydroxy-acyl-koenzym A-dehydrogenázy pro mastné kyseliny s dlouhým řetězcem; LHON – Leberova hereditární neuropatie optiku; MCAD – porucha ß-oxidace mastných kyselin na úrovni 3-hydroxy-acyl-koenzym A-dehydrogenázy pro mastné kyseliny se středním řetězcem; MELAS – syndrom Mitochondriální encefalopatie, laktátové acidozy a iktu podobných příhod; MERRF – syndrom Myoklonické epilepsie s ragged-red fibres; MPI-CDG – dědičné poruchy glykosylace na úrovni manózafosfátizomerázy; mtDNA – mitochondriální DNA; MTP – porucha mitochondriálního trifunkčního proteinu; MPS – mukopolysacharidóza; NCL – neuronální ceroidní lipofuscinóza; OTC – deficit ornitintranskarbamylázy; VLCAD – porucha ß-oxidace mastných kyselin na úrovni acyl-koenzym A-dehydrogenázy mastných kyselin s velmi dlouhým řetězcem.
Podpořeno: MZ ČR – RVO VFN 64165, MZ ČR – AZV 16-32341A.
Korespondující autor:
Doc. MUDr. Tomáš Honzík, Ph.D.
Klinika dětského a dorostového lékařství
1. LF UK a VFN
Ke Karlovu 2
128 08 Praha 2
e-mail: tomas.honzik@vfn.cz
Zdroje
1. Nordestgaard BG, Chapman MJ, Humphries SE, et al.; European Atherosclerosis Society Consensus Panel. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur Heart J 2013; 34 (45): 3478–3490a.
2. Honzik T, Zeman J. Dědičné metabolické poruchy v dětském věku. 1. vyd. Praha: Institut postgraduálního vzdělávání ve zdravotnictví, 2013 : 13–93.
3. Honzik T, Zeman J. Dědičné poruchy metabolismu v kazuistikách. In: Honzik T, Zeman J et al (Eds). Dědičné poruchy metabolismu v kazuistikách. 1. vyd. Praha: Mladá fronta, 2016 : 3–27.
3. Lipshultz SE, Sleeper LA, Towbin JA, et al. The incidence of pediatric cardiomyopathy in two regions of the United States. N Engl J Med 2003; 348 : 1647–1655.
4. Nugent AW, Daubeney PE, Chondros P, et al. National Australian Childhood Cardiomyopathy Study. The epidemiology of childhood cardiomyopathy in Australia. N Engl J Med 2003; 348 : 1639–1646.
5. Kindel SJ, Miller EM, Gupta R. Pediatric cardiomyopathy: importance of genetic and metabolic evaluation. J Card Fail 2012 May; 18 (5): 396–403.
6. Byers SL, Ficicioglu C. Infant with cardiomyopathy: When to suspect inborn errors of metabolism? World J Cardiol 2014 November 26; 6 (11): 1149–1155.
7. Honzik T, Tesarova M, Mayr J, et al. Mitochondrial encephalocardiomyopathy with early neonatal onset due to TMEM70 mutation. Arch Dis Child 2010 Apr; 95 (4): 296–301.
8. Honzik T, Tesarova M, Magner M, et al. Neonatal onset of mitochondrial disorders in 129 patients: clinical and laboratory characteristics and a new approach to diagnosis. J Inherit Metab Dis 2012; 35 (5): 749–759.
9. Mazurová S, Tesařová M, Magner M, et al. Novel mutations in the TAZ gene in patients with Barth syndrome. Liver Transpl 2016; 22 (5): 677–685.
10. Wiseman DH, Mercer J, Tylee K, et al. Management of mucopolysaccharidosis type IH (Hurler‘s syndrome) presenting in infancy with severe dilated cardiomyopathy: a single institution‘s experience. J Inherit Metab Dis 2013 Mar; 36 (2): 263–270.
11. Seemanová E. Dysmorfické příznaky – klíč v diagnostice genetických poruch. Pediatr pro Praxi 2008; 9 (5): 305–308.
12. Ng BG, Freeze HH. Perspectives on glycosylation and its congenital disorders. Trends Genet 2018 Jun; 34 (6): 466–476. doi: 10.1016/j.tig.2018.03.002. Epub 2018 Mar 29. Review.
13. Taylor SA, Whitington PF. Neonatal acute liver failure. Liver Transpl 2016; 22 (5): 677–685.
14. Bonilla S, Prozialeck JD, Malladi P, et al. Neonatal iron overload and tissue siderosis due to gestational alloimmune liver disease. J Hepatol 2012; 56 (6): 1351–1355.
15. De Castro M, Johnston J, Biesecker L. Determining the prevalence of McArdle disease from gene frequency by analysis of next-generation sequencing data. Genet Med 2015; 17 (12): 1002–1006.
16. Clarkson PM, Eichner ER. Exertional rhabdomyolysis: does elevated blood creatine kinase foretell renal failure? Curr Sports Med Rep 2006 Apr; 5 (2): 57–60.
17. Lappalainen H, Tiula E, Uotila L, et al. Elimination kinetics of myoglobin and creatine kinase in rhabdomyolysis: implications for follow-up. Crit Care Med 2002; 30 (10): 2212–2215
Štítky
Neonatologie Pediatrie Praktické lékařství pro děti a dorostČlánek vyšel v časopise
Česko-slovenská pediatrie

2018 Číslo 6
- Efektivita kartáčku Sonicare For Kids u dětí předškolního věku
- Inosin pranobex v léčbě HPV infekcí děložního čípku
- Cytomegalovirové infekce u novorozenců a dětí
Nejčtenější v tomto čísle
- Charakteristické klinické příznaky a laboratorní odchylky dědičných poruch metabolismu
- Komplexný pohľad na deficit vitamínu B12 v detskom veku
- Novorozenecký screening dědičných metabolických poruch v České republice
- Lyzozómové choroby – vývoj diagnostiky a liečby na Slovensku
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy