
Poruchy funkce nadledvin
Autoři:
J. Lebl; S. Koloušková; M. Šnajderová; Z. Šumník
Působiště autorů:
Pediatrická klinika UK 2. LF a FN Motol, Praha
Vyšlo v časopise:
Čes-slov Pediat 2012; 67 (4): 275-282.
Kategorie:
Vybrané kapitoly z nové učebnice Klinická pediatrie
1. FYZIOLOGIE FUNKCE NADLEDVIN
Nadledviny se skládají z kůry a dřeně – dvou histologicky i funkčně odlišných částí, které se v embryonálním vývoji derivují z různých zárodečných tkání.
Dřeň nadledvin pochází z ektodermu, vývojově se odštěpuje z nervové tkáně. Produkuje katecholaminy, zejména adrenalin a noradrenalin. Funkčně je tedy blízká sympatickým nervovým gangliím. Hlavní klinicky významnou poruchu funkce dřeně nadledvin představují hyperfunkční nádory, především feochromocytom.
Kůra nadledvin vzniká současně s gonádami ze společného mezodermálního základu. Proto jsou si obě tyto žlázy funkčně blízké – jsou jedinými tkáněmi organismu schopnými vyrábět steroidní hormony. Kůra nadledvin produkuje tři skupiny steroidních hormonů – mineralokortikoidy (aldosteron), glukokortikoidy (kortisol) a pohlavní hormony (androgeny, v menší míře estrogeny). Jednotlivé skupiny hormonů se tvoří v histologicky poněkud odlišných vrstvách tkáně – zona glomerulosa, zona fasciculata a zona reticularis. Tyto tři tzv. definitivní zóny nadledvin se diferencují až po narození; u fétu převažuje podíl tzv. fetální zóny, která v prvních dvou týdnech života regreduje.
Společným prekurzorem steroidních hormonů je cholesterol, který je v řadě enzymaticky katalyzovaných kroků přeměňován na jednotlivé aktivní hormony. Syntéza a sekrece glukokortikoidů a nadledvinových androgenů je řízena adrenokortikotropním hormonem (ACTH) na principu jednoduché zpětné vazby mezi kortisolem a ACTH. Produkce aldosteronu je řízena především systémem renin-angiotenzin, který přináší informaci o prokrvení ledvin a tedy o objemu cirkulující krve.
2. PATOFYZIOLOGIE PORUCH FUNKCE NADLEDVIN
V dětském věku členíme poruchy funkce nadledvin do tří skupin:
- (1) kongenitální adrenální hyperplazie – skupina vrozených enzymatických poruch steroidogeneze, které vedou k deficitu části spektra steroidních hormonů, ale vlivem zpětné vazby přes ACTH k nadprodukci jiných steroidů;
- (2) adrenální insuficience – snížená schopnost syntézy a sekrece steroidních hormonů; může mít příčinu vrozenou, zpravidla geneticky podmíněnou, nebo získanou – k destrukci tkáně dochází vlivem krvácení, infekce nebo autoimunitního procesu (Addisonova nemoc);
- (3) nadprodukce nadledvinových hormonů v důsledku tumoru kůry nebo dřeně nadledvin, v případě Cushingovy nemoci je nadměrná sekrece kortisolu způsobena nadprodukcí ACTH.
3. KONGENITÁLNÍ ADRENÁLNÍ HYPERPLAZIE
Jako kongenitální adrenální hyperplazii (dřívější název „adrenogenitální syndrom“) označujeme skupinu poruch, které jsou důsledkem geneticky podmíněného nedostatku některého z enzymů steroidogeneze. Všechna tato onemocnění se dědí autozomálně recesivně. Je pro ně příznačný enzymatický blok, který vede k deficitu části spektra steroidních hormonů a k nadbytku jiné skupiny hormonů, která je vyvolána nadprodukcí ACTH při uvolnění zpětné vazby (obr. 1).
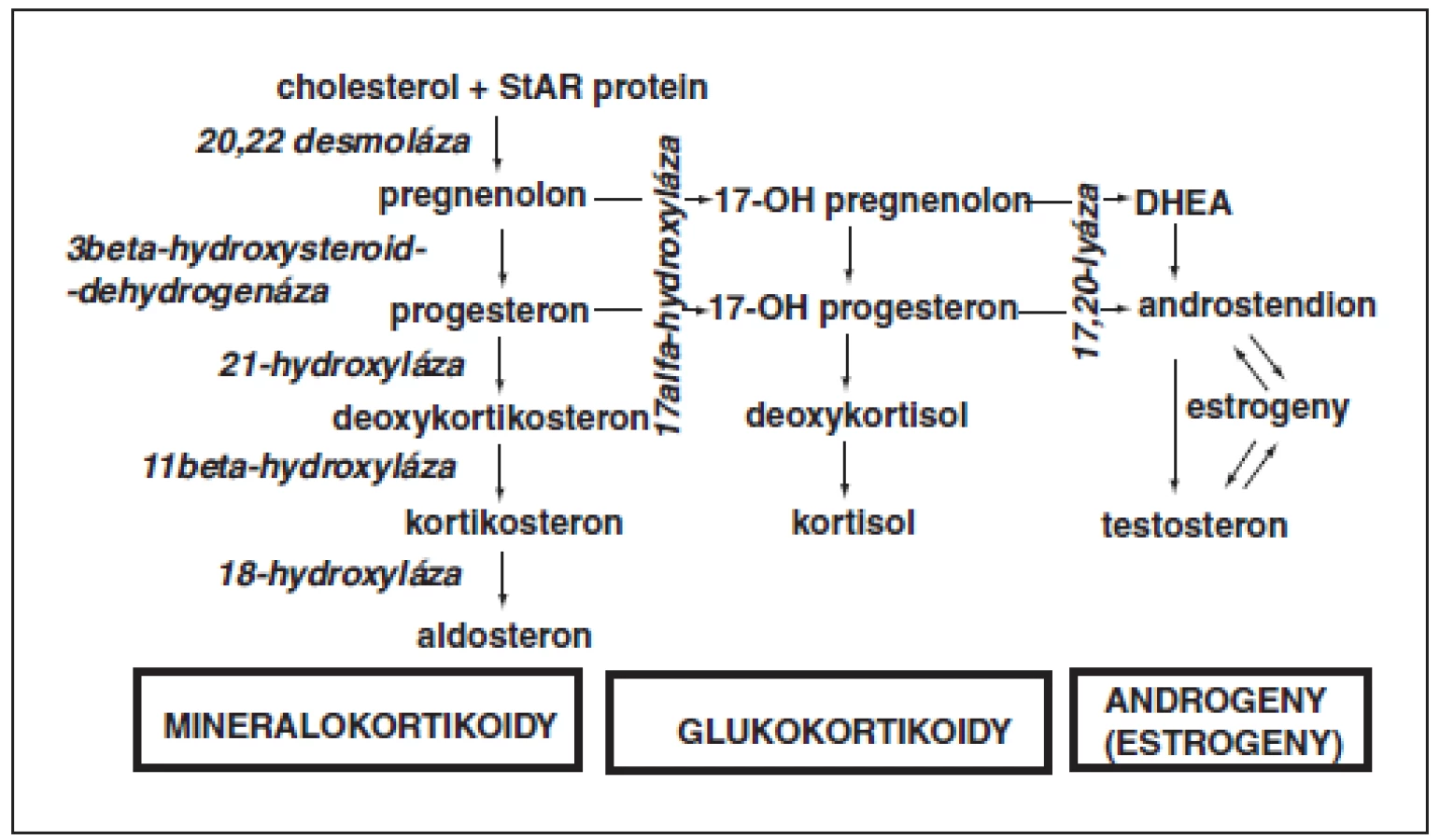
Jedná se tedy o poruchy hypofunkčně-hyperfunkční. Klinický obraz je pro každý typ enzymatického defektu specifický a zahrnuje příznaky z hormonálního deficitu spolu s projevy hormonálního nadbytku (tab. 1).

3.1. Deficit 21-hydroxylázy
Přibližně 95 % nemocných s kongenitální adrenální hyperplazií představují pacienti s deficitem 21-hydroxylázy. Porucha se u nás vyskytuje s četností přibližně 1 : 10 000. Vázne sekrece kortisolu i aldosteronu, proto stoupá výdej ACTH, který stimuluje produkci adrenálních metabolitů s androgenní aktivitou.
Klinické projevy deficitu 21-hydroxylázy závisejí na typu genové mutace, která určuje zbytkovou aktivitu enzymu (tab. 2). Podle tíže poruchy se rozlišuje forma neklasická a klasická, která se dále člení na formu se solnou poruchou (salt-wasting) s klinicky významným deficitem kortisolu i aldosteronu a formu prostou virilizující (simple virilising), u které převažují příznaky z nadprodukce androgenů.

Děti se solnou poruchou mají zachovanou enzymatickou aktivitu minimální. Nedostatek glukokortikoidů a mineralokortikoidů ovlivňuje dítě již během nitroděložního života. Sonograficky lze prokázat u novorozenců obou pohlaví hypertrofické nadledviny. Chlapci se rodí bez klinické stigmatizace a bezprostředně po narození proto nelze diagnózu klinicky rozpoznat. Ve 2.–4. týdnu života se rozvine těžká nadledvinová nedostatečnost a metabolický rozvrat („solná krize“), který může vést k náhlému úmrtí. U dívek je již při narození patrný různý stupeň malformace zevního genitálu. Vnitřní genitál dívek (vaječníky, vejcovody, děloha a horní dvě třetiny pochvy) je utvářen normálně, dolní třetina pochvy a zevní genitál jsou však in utero vlivem vysokých hladin androgenů virilizovány. Proto je diagnóza u dívek zpravidla stanovena již na novorozeneckém oddělení, ještě před rozvojem metabolického rozvratu.
U klasické formy bez solné poruchy je u většiny dívek přítomna virilizace zevního genitálu, což vede ke stanovení diagnózy a zahájení léčby. U chlapců se ale tato porucha projeví až později jako pseudopubertas praecox, většinou ve věku 2–5 let. Zvětšuje se genitál, objeví se pubické ochlupení, ale testes zůstávají dětská (velikosti do 3 ml). U obou pohlaví nadbytek androgenů akceleruje růst a urychluje kostní zrání, což v konečném důsledku vede k malé postavě.
Neklasickou formu deficitu 21-hydroxylázy (tzv. late-onset) diagnostikujeme v adolescenci nebo mladé dospělosti u dívek a žen s hyperandrogenním stavem. Pacientky mají primární amenoreu, akné, hirsutismus, poruchy menstruačního cyklu. U chlapců se tato forma zpravidla klinicky neprojeví.
Laboratorní diagnostika je založena na průkazu zvýšené hladiny 17-hydroxyprogesteronu (17OHP). Hladiny kortisolu jsou nízké a hladiny ACTH vysoké. Pro potvrzení diagnózy je optimální provést před zahájením léčby ACTH („Synacthenový“) test, pokud to klinický stav dítěte dovoluje. Stanovení dalších nadledvinových steroidů pomáhá odlišit jiné formy kongenitální adrenální hyperplazie.
V akutním stavu podáváme i.v. Hydrocortison v dávce 50–100 mg a pokračujeme v jeho parenterálním podávání v intervalu 6 hodin. Pacienta parenterálně rehydratujeme s cílem korigovat iontogram a předejít hypoglykemii. Po několika dnech přecházíme na p.o. léčbu. Dlouhodobou léčbou klasické formy deficitu 21-hydroxylázy je podávání Hydrocortisonu a syntetického mineralokortikoidu. Cílem léčby je substituovat tyto hormony a potlačit nadprodukci ACTH a tím i sekreci androgenů. Dlouhodobým cílem léčby je zajistit fyziologický růst a vývoj, optimální dospělou tělesnou výšku a fertilitu. I v současné době je však dosažení těchto cílů někdy obtížné. U dívek s virilizací zevního genitálu je potřebné provést chirurgickou korekci.
Vzhledem ke klinické závažnosti deficitu 21-hydroxylázy a k riziku náhlého úmrtí před stanovením diagnózy byl zaveden celoplošný novorozenecký screening této poruchy. Je založen na stanovení hladiny 17-hydroxyprogesteronu ze suché krevní kapky odebrané každému novorozenci mezi 48. a 72. hodinou života na novorozeneckém oddělení. Tento screening pravděpodobně zachytí téměř všechny pacienty s nejtěžší formou poruchy. Část pacientů s prostou virilizující formou a většina pacientů s neklasickou formou ale rozpoznání tímto screeningem uniká. Klinická diagnostika proto neztrácí na významu.
V rodinách, kde se již narodilo dítě s deficitem 21-hydroxylázy, je v dalším těhotenství možné nabídnout mamince prenatální terapii plodu. Je založena na podávání Dexamethazonu matce nejpozději od 8. týdne těhotenství. Následuje prenatální diagnostika prvního trimestru a léčba poté pokračuje až do porodu u postižených fétů ženského pohlaví. Cílem terapie je suprimovat fetální nadledviny a předejít virilizaci zevního genitálu u dívek. Tato léčba má být prováděna výhradně v centrech s dostatečnou zkušeností.
3.2. Deficit 17-alfa-hydroxylázy
Při tomto enzymatickém bloku dokáže nadledvina vyrábět jen mineralokortikoidně aktivní prekurzory. Chybí kortisol, androgeny i estrogeny. U chlapců nedochází in utero k virilizaci zevního genitálu, při úplném bloku mají jedinci s karyotypem 46,XY dívčí zevní genitál a zevní třetinu slepě zakončené pochvy. Testes jsou uložena intraabdominálně. U obou pohlaví chybí pubertální vývoj.
Mineralokortikoidně aktivní substance vedou k hypokalemii, hraniční hypernatremii a k hypertenzi. Šokový stav z těžkého deficitu kortisolu se rozvíjí jen v zátěžových situacích. Porucha se u dětí zpravidla diagnostikuje buď při náhodném změření krevního tlaku, nebo při vyšetření pro nepřicházející dospívání.
3.3. Lipoidní adrenální hyperplazie
Příčinou lipoidní adrenální hyperplazie je porucha tvorby steroidogenního akutního regulačního proteinu (StAR), který je aktivním transportérem cholesterolu přes vnitřní mitochondriální membránu. Tvorba StAR v nadledvinách je řízena ACTH a zajišťuje rychlou syntézu steroidních hormonů. Mutace genu pro StAR způsobuje kompletní chybění adrenální a gonadální steroidogeneze. Nevyužitý cholesterol se hromadí v nadledvinách, které jsou hyperplastické a vakuolizované s depozity esteru cholesterolu. Postižení chlapci mají ženský zevní genitál v důsledku poruchy syntézy fetálních androgenů. Po narození se po krátkém asymptomatickém intervalu projeví závažná adrenální insuficience.
4. ADRENÁLNÍ INSUFICIENCE
4.1. Klinické příznaky adrenální insuficience
Nejdůležitější příznaky adrenální insuficience shrnuje tabulka 3.

4.2. Vrozená adrenální insuficience
Přehled významných příčin vrozené adrenální insuficience (s výjimkou enzymatických defektů steroidogeneze ze skupiny kongenitální adrenální hyperplazie) shrnuje tabulka 4.

Uvedené stavy jsou závažné – fatální nebo potenciálně fatální. I když jsou způsobeny vrozenými genovými defekty, věk při prvních příznacích se u jednotlivých poruch liší. U všech poruch jsou známé i méně závažné varianty s pozdějšími nebo neúplně vyjádřenými klinickými projevy.
Kongenitální adrenální hypoplazie je důsledkem poruchy vývoje nadledvin při chybění DAX-1 transkripčního regulátoru. Jde o X-vázanou dědičnost. Projeví se zpravidla brzy po narození neprospíváním a „solnou krizí“ s hyponatremií, hyperkalemií a zvracením, hypoglykemií a metabolickou acidózou. Postižení chlapci mohou náhle zemřít v prvních týdnech života vlivem hyperkalemie. Hladiny všech steroidních hormonů jsou nízké, proto onemocnění uniká novorozeneckému screeningu kongenitální adrenální hyperplazie, založenému na detekci vysokých hladin 17-hydroxyprogesteronu (17OHP). Onemocnění se léčí substituční léčbou Hydrocortisonem a mineralokortikoidy. DAX-1 reguluje také gonadotropní buňky hypofýzy a spermatogenezi, proto pacienti zpravidla spontánně nedospívají a jsou neplodní.
Vzácně může být delece DAX1 genu spojena s delecí delšího úseku chromozomu X, který obsahuje několik genů. V tomto případě se současně může vyskytovat deficit glycerol-kinázy a těžká forma Duchenneovy muskulární dystrofie, která je zpravidla fatální.
Familiární glukokortikoidní deficience je skupina vrozených poruch funkce receptoru pro ACTH nebo postreceptorové signalizace. Hladiny ACTH jsou vysoké, ale hladiny kortisolu nízké. Mineralokortikoidy se v nadledvinách vyrábějí správně, proto nedochází k „solné krizi“. Děti jsou brzy po narození ohroženy fatální hypoglykemií. Po správném stanovení diagnózy je léčba snadná – substituční podávání Hydrocortisonu.
Tripple A syndrom je komplexní porucha způsobená defektem aladinu – proteinu s dosud nejasnou funkcí. Postižené děti trpí adrenální insuficiencí, alakrimií (nevytvářejí slzy), achalázií a progresivním neurologickým postižením, které je zpravidla fatální i přes optimální substituční léčbu adrenální insuficience.
Adrenoleukodystrofie je dalším X-vázaným onemocněním. Projeví se v předškolním nebo mladším školním věku neurologickými příznaky (nejistou chůzí, svalovou slabostí, emoční labilitou, poruchami chování, snížením intelektu, poruchami zraku a sluchu, ale také např. poruchou porozumění řeči). O málo později se rozvíjí adrenální insuficience s vysokými hladinami ACTH a hyperpigmentací. Onemocnění je fatální – postižení chlapci umírají vlivem progresivního neurologického postižení v důsledku demyelinizace zpravidla ve druhé dekádě života pod obrazem kvadruparézy a demence, a to i při správné substituční léčbě adrenální insuficience. Příčinou je akumulace mastných kyselin s velmi dlouhým řetězcem v bílé hmotě CNS, nadledvinách a testes. Léčebné pokusy s vyloučením mastných kyselin s velmi dlouhým řetězcem se bohužel ukázaly jako neúčinné – dochází k jejich novotvorbě v organismu. Určitou nadějí je transplantace kostní dřeně ještě před rozvojem významné neurologické poruchy.
4.3. Získaná adrenální insuficience
Přehled příčin získané adrenální insuficience u dětí shrnuje tabulka 5.

Krvácení do nadledvin může nastat u novorozence vlivem větší zranitelnosti nadledvin v tomto věku – žlázy jsou velké a jejich podstatnou část tvoří silně vaskularizovaná fetální zóna. Krvácení výjimečně postihuje obě nadledviny a vede k akutním příznakům adrenální insuficience.
Nejzávažnější formou je krvácení do nadledvin v průběhu těžkých septických stavů (meningokokové nebo pseudomonádové infekce), při úrazech nebo po operačních výkonech. Klinické příznaky adrenální insuficience (hypotenze, minerální rozvrat, hypoglykemie) vznikají náhle, ale mohou být maskovány jinými závažnými symptomy základního onemocnění.
Poškození nadledvin autoimunitním procesem (Addisonova nemoc) je nejčastější příčinou adrenální insuficience u předškolních a školních dětí, adolescentů a dospělých. Vývoj klinických příznaků je plíživý a projeví se často až v zátěžové situaci (interkurentní onemocnění, operační výkon). Nerozpoznaná Addisonova nemoc může zejména u menších dětí v zátěžové situaci bezprostředně ohrozit život, zejména hypoglykemií.
Diagnózu Addisonovy nemoci potvrdí typické laboratorní nálezy (vysoká hladina ACTH, nízká hladina kortisolu, hypoglykemie, hyponatremie a hyperkalemie). V séru lze prokázat protilátky proti nadledvinovým antigenům, např. proti 21-hydroxyláze, rutinně se ale nevyšetřují. Kůra nadledvin je infiltrována lymfocyty a postupně atrofuje.
Addisonova nemoc se vzácně vyskytuje izolovaně, u dětí a adolescentů je častěji součástí jednoho z autoimunitních polyglandulárních syndromů – APS-1 (zvaný také APECED), který se typicky rozvíjí u menších dětí, nebo APS-2 (Addisonova nemoc a autoimunitní tyreoiditis a/nebo diabetes mellitus 1. typu), který je charakteristický pro adolescenty a mladé dospělé. Pokud má již pacient jinou složku APS, je třeba na riziko Addisonovy nemoci pomýšlet, a to zejména u dětí s APS-1.
Iatrogenně vyvolaná adrenální insuficience je zpravidla důsledkem dlouhodobé léčby kortikoidy ve farmakologických dávkách podávaných perorálně. Potlačí se sekrece ACTH a utlumí vlastní funkce kůry nadledvin, které atrofují. Přerušení léčby může vést k projevům adrenální insuficience, proto je nezbytné dávky kortikoidů redukovat postupně a při jejich úplném vysazení vlastní funkci nadledvin dobře sledovat. Zcela výjimečně byly příznaky adrenální insuficience pozorovány i po přerušení dlouhodobé léčby inhalačními kortikoidy, i když tato cesta podávání kortikoidů je mnohem bezpečnější.
5. NADPRODUKCE NADLEDVINOVÝCH HORMONŮ
5.1. Tumory kůry nadledvin a Cushingova nemoc
Adenom a adenokarcinom naledvin patří u dětí mezi vzácné tumory. V 90 % případů jsou hormonálně aktivní. Nejčastěji produkují androgeny, případně v kombinaci s glukokortikoidy. Projevují se obvykle předčasnou pseudopubertou – isosexuální u chlapců, heterosexuální u dívek, v adolescenci u dívek virilizací (obr. 2). Pokud převažuje produkce kortisolu, dochází ke vzniku Cushingova syndromu (obr. 3). Jen vzácně tumor produkuje mineralokortikoidy (Connův syndrom) a zcela raritně estrogeny.


Zatímco prognóza adenomů nadledvin je po chirurgické resekci velmi dobrá, adenokarcinom nadledviny je i dnes velmi obtížně léčitelný.
Cushingova nemoc je důsledkem nadměrné produkce ACTH při hyperfunkčním adenomu hypofýzy. Jedná se zpravidla o mikroadenom (s průměrem do 10 mm). Důsledkem je oboustranná hyperplazie nadledvin s nadprodukcí výlučně kortisolu. Postižené děti se téměř zastavují v růstu, opožďuje se jim kostní zrání, rychle přibývá hmotnost a objevuje se cushingoidní habitus. Současně se rozvíjí osteoporóza, která může vést ke kompresivním patologickým frakturám obratlů. Laboratorně nacházíme natremii kolem horní hranice normy a kalemii při dolní hranici normy, může být vyšší glykemie a porucha glukózové tolerance. Denní odpad kortisolu v moči a hladiny ACTH v krvi jsou výrazně zvýšené, ztrácí se přirozený cirkadiální rytmus sekrece kortisolu.
K průkazu a lokalizaci mikroadenomu je někdy třeba použít speciální diagnostické postupy, např. vyšetření hladiny ACTH v krvi získané katetrizací sinus petrosus inferior. Po průkazu tumoru následuje zpravidla neurochirurgické řešení.
5.2. Tumory dřeně nadledvin
Nejčastějším nádorem nadledvin u dětí je neuroblastom či jeho různě diferencované formy (ganglioneurom, ganglioneuroblastom). Vyskytuje se již v novorozeneckém věku. Klinické příznaky bývají kromě hmatné rezistence nespecifické (neprospívání, teploty, anemie, poruchy střevní pasáže, metastatická postižení jiných orgánů). Jen výjimečně se tento typ nádorů projeví hormonální symptomatologií z nadměrné sekrece katecholaminů. O jeho hormonální aktivitě však svědčí vyšetření volných katecholaminů a jejich metabolitů v krvi, resp. v moči, které se používá v diagnostice neuroblastomu a jeho neinvazivním odlišení od jiných typů nádorů.
Feochromocytom vychází z chromafinních buněk dřeně nadledvin. Secernuje extrémní množství katecholaminů a projevuje se proto paroxyzmální či stálou hypertenzí, prchavými vyrážkami kůže, pocením a tachykardií. Hypertenze může dosahovat extrémních hodnot (až 250/180 mmHg), ale i takto vysoké hodnoty mohou být u některých dětí nebo dospívajících asymptomatické a mohou být náhodným nálezem při preventivní prohlídce.
Pro feochromocytom platí „pravidlo deseti“ – 10 % je umístěno mimo nadledviny a vychází ze sympatických nervových ganglií, 10 % je vícečetných a 10 % má maligní charakter (feochromoblastom). Feochromocytom může být součástí syndromů mnohočetné endokrinní neoplazie (MEN 2a a MEN 2b) a někdy je jejich prvním projevem. K diagnostice se vedle zobrazovacích vyšetření také používá stanovení volných katecholaminů a jejich metabolitů v krvi, resp. v moči. Při obtížné lokalizaci tumoru při jeho extraadrenálním uložení lze využít scintigrafii pomocí MIBG či oktreotidu nebo pozitronovou emisní tomografii.
Terapií feochromocytomu je chirurgická resekce, která musí být prováděna velmi šetrně, a to při dokonalém oběhovém zajištění pacienta. Před výkonem předchází 10–14denní farmakologická příprava blokátory adrenergních alfa-receptorů a volum-expandery. Během výkonu hrozí kritická hypertenze, pokud dojde k nárazovému vyplavení katecholaminů, a následně hypotenzi po definitivním odstranění zdroje katecholaminů. Proto je v prvních dnech po výkonu zpravidla nutná oběhová podpora s podáváním katecholaminů v klesajících dávkách.
Štítky
Neonatologie Pediatrie Praktické lékařství pro děti a dorostČlánek vyšel v časopise
Česko-slovenská pediatrie

2012 Číslo 4
- Efektivita kartáčku Sonicare For Kids u dětí předškolního věku
- OZP pečuje o zdraví svých pojištěnců již 30 let. V čem je lepší a jaké výhody nabízí?
- Cytomegalovirové infekce u novorozenců a dětí
- Cytomegaloviróza a spalničky u dětí
Nejčtenější v tomto čísle
- Poruchy funkce nadledvin
- Znamená postnatální záchyt dilatace ledvinové pánvičky větší riziko infekce močových cest?
- Poruchy kalciofosfátového metabolismu
- Totální anomální návrat plicních žil – méně obvyklá příčina neprospívání u kojence
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy