
Vybrané kapitoly z nové učebnice Klinická pediatrie
Vyšlo v časopise:
Čes-slov Pediat 2011; 66 (2): 103-114.
Kategorie:
Vybrané kapitoly z nové učebnice Klinická pediatrie
Nakladatelství Galén připravuje k vydání novou učebnici „Klinická pediatrie“, která bude určena jak pro pregraduální studium na lékařských fakultách, tak i v rámci postgraduálního vzdělávání pro přípravu na atestaci z dětského lékařství a z praktického dětského lékařství. Autoři věří, že učebnici ocení i dětští lékaři v praxi.
Ve spolupráci nakladatelství Galén a redakce Česko-slovenské pediatrie na stránkách našeho časopisu v letošním roce postupně uveřejňujeme jednotlivé stati z nové učebnice – a jako bonus také některé kapitoly, které se do nové učebnice už „nevešly“, protože učebnice má přesně stanovený rozsah.
Čes-slov Pediat 2011; 66(2): 103–118.
Poruchy růstu
Lebl J., Koloušková S., Šnajderová M., Šumník Z.
Pediatrická klinika UK 2. LF a FN Motol, Praha
1. Fyziologie dětského růstu
V okamžiku vzniku nového života má budoucí lidský jedinec rozměr jedné oplodněné zárodečné buňky. Po devíti měsících intrauterinního vývoje měří donošený novorozenec kolem 50 cm. Za dalších 18 let dosáhne u nás mladý muž tělesné výšky v průměru 180,2 cm, mladá žena 167,3 cm.
Tělesný růst je zákonitý a přísně regulovaný proces. Pro posuzování růstu dětí používáme percentilové grafy tělesné výšky, které jsou sestrojeny na základě celostátních antropologických výzkumů. Ty se konají každých deset let (naposledy 2001) a poskytují tak stále aktuální přehled o tělesném vývoji české dětské a adolescentní populace. Podle Karlbergova modelu lidského růstu rozlišujeme tři růstová období, která na sebe navazují a částečně se překrývají:
- infantilní (od narození do cca 2 let věku)
- dětské (od cca 2 let věku do počátku pubertálního vývoje)
- pubertální (od počátku pubertálního vývoje do dosažení dospělé výšky).
1.1. Infantilní růstové období
Infantilní růstové období začíná in utero a pokračuje první dva roky po narození. Rychlost růstu je nejvyšší z celého lidského života. Plod roste nejrychleji ve 2. trimestru. Od 3. trimestru růstové tempo mírně klesá. Rychlý růst pokračuje i postnatálně, zdravé dítě vyroste v prvním roce života o 23 cm (přibližně o 50 % porodní délky), ve druhém roce o dalších 12 cm (přibližně o 50 % přírůstku prvního roku). Při druhých narozeninách dítě dosáhne asi poloviny své budoucí dospělé výšky.
Do konce druhého roku měříme děti vleže a stanovujeme tedy tělesnou délku. Ke správnému měření používáme bodymetr („korýtko“).
Úloha růstového hormonu v prenatálním růstu je okrajová. V první fázi gestace (embryonální období) je zřejmě významným regulátorem růstu inzulinu podobný růstový faktor-II (insulin-like growth factor-II, IGF-II), ve druhé fázi gestace (fetální období) inzulinu podobný růstový faktor-I (IGF-I). Fetální IGF-I je regulován také prostřednictvím transportu glukózy placentou, který řídí i sekreci fetálního inzulinu. Osa glukóza-inzulin-IGF-I se tak primárně uplatňuje v prenatálním růstu.
IGF-I si zachovává klíčovou roli v regulaci růstu i po narození. V řízení jeho sekrece se během prvního roku života začíná uplatňovat růstový hormon.
1.2. Dětské růstové období
Nejdelší vývojovou fází je dětské růstové období. Trvá přibližně od druhých narozenin do začátku puberty. Růstová rychlost mírně klesá ze 7,5 cm/rok (ve 3 letech) až na 5 cm/rok (v posledním roce před nástupem pubertálního růstového výšvihu). Dětské růstové období přispívá asi 30 % k budoucí dospělé výšce.
Od dvou let měříme děti vstoje, stanovujeme tělesnou výšku. Ke správnému měření používáme stadiometr, zařízení s posuvnou hlavicí upevněné na stěně.
V dětském růstovém období je růst řízen osou růstový hormon–IGF-I. Hladinu IGF-I spoluovlivňují další faktory, zejména výživa, metabolické parametry, celkový zdravotní stav a některé další hormony (obr. 1).

1.3. Pubertální růstové období
Pubertální růstové období trvá 4–5 let a začíná o dva roky dříve u dívek (ve věku 10,5 ± 2 roky) než u chlapců (ve věku 12,5 ± 2 roky). Růstová rychlost postupně stoupá a vrcholí v roce před dosažením plné pohlavní zralosti (menarché u dívek; první ejakulace u chlapců). Na vrcholu puberty vyroste dívka v průměru 9 cm/rok, chlapec 10,3 cm/rok. Poté růstová rychlost klesá. Růst končí u českých dívek v průměru v 15 letech, u chlapců v 17–18 letech. Během pubertálního růstového období vyroste mladý člověk o posledních 20 % své konečné dospělé výšky.
Základem regulace růstu v dospívání zůstává osa růstový hormon–IGF-I. Na urychlení růstu se podílejí pohlavní hormony, které stimulují sekreci růstového hormonu i IGF-I. Produkce růstového hormonu a IGF-I je v dospívání nejvyšší z celého lidského života. Pohlavní hormony navíc přímo ovlivňují růstové zóny dlouhých kostí („růstové chrupavky“). Nejdříve urychlují proliferaci nezralých kostních elementů (chondroblastů a osteoblastů) a tím stimulují růst kosti do délky. V závěru puberty dochází k apoptóze buněk v růstových zónách; růst se zpomaluje a končí (obr. 2).
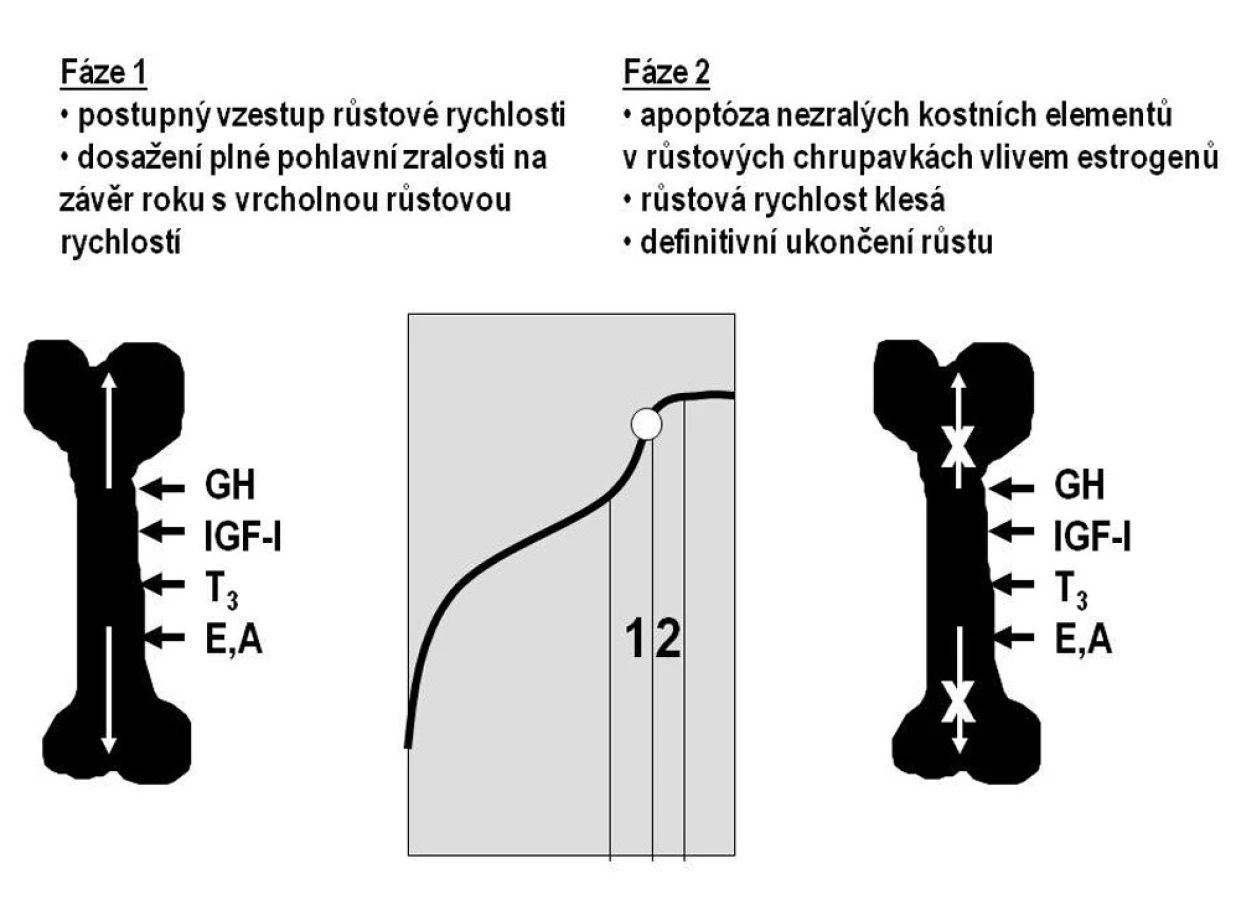
1.4. Sekulární akcelerace
V průběhu 20. století se růstová dynamika dětí ve vyspělých zemích postupně změnila. Díky zásadnímu zlepšování socioekonomického a zdravotního stavu celých populací se růst urychlil, klesl věk při začátku dospívání a zvýšila se dospělá tělesná výška. Tento jev označujeme jako sekulární akceleraci tělesného růstu (saeculum = století) (obr. 3). Proto k posouzení růstu konkrétního dítěte používáme recentní normativní data (percentilové grafy).

V současné době je socioekonomický a zdravotní stav dětských populací ve vyspělých zemích stabilizovaný a téměř optimální. Proto už neočekáváme další významné urychlení růstu a dospívání ani změny dospělé výšky.
2. Růstová retardace
Růstovou retardací (poruchou růstu ve směru minus) rozumíme tělesnou výšku dítěte pod 3. percentilem pro daný věk a/nebo růstovou rychlost dítěte pod 25. percentilem pro daný věk, spočítanou ze dvou přesných měření v odstupu alespoň 6 měsíců. K výpočtu růstové rychlosti lze použít tabulky či percentilové grafy růstové rychlosti. V praxi se snížená růstová rychlost projeví poklesem v grafu tělesné výšky o více než jedno pásmo.
2.1. Příčiny růstové retardace
Z etiopatogenetického hlediska lze děti s růstovou retardací etiologicky rozdělit do pěti skupin:
A. Idiopatický menší vzrůst (idiopathic short stature, ISS) je nejčastější. Tělesná výška je nízká, ale růstová rychlost normální. Tyto děti byly v minulosti označovány jako „short-normal“ (malé, ale zdravé). Tento pojem se již nepoužívá, protože alespoň část z nich je nositelem určité patologické vlohy, kterou dosud nedokážeme rozpoznat.
Mezi dětmi s ISS tvoří dvě významné podskupiny děti s familiárně menším vzrůstem (familiar short stature, FSS) a s konstitučním opožděním růstu a puberty (constitutional delay of growth and adolescence, CDGA). Pro FSS je příznačná malá výška v souladu s predikcí podle výšky rodičů, pro CDGA opoždění biologického věku (kostního věku a/nebo pubertálního vývoje). Zatímco u FSS bude dospělá výška přibližně odpovídat současné výškové pozici dítěte mezi ostatnímiu vrstevníky, u CDGA bude významně lepší – tyto děti porostou déle. Obě odchylky, FSS i CDGA, považujeme za varianty normálu. U některých dětí se mohou FSS a CDGA kombinovat. Jejich společným znakem je (1) nepřítomnost zdravotní poruchy a (2) perspektiva dospělé výšky v souladu s rodičovskou předpovědí.
Předpověď dospělé výšky vypočítáme jako průměr mezi výškou rodiče stejného pohlaví a výškou rodiče druhého pohlaví upravenou o 13 cm (zvýšenou pro chlapce, sníženou pro dívky). Principem tohoto výpočtu je sexuální dimorfismus dospělé výšky – ženy jsou v průměru o 13 cm menší než muži. Matematicky je tento výpočet vyjádřen vzorcem

B. Děti s endokrinní poruchou. Endokrinopatie jsou příčinou růstové retardace jen u malé části dětí s malým vzrůstem (1–2 %). Je pro ně typická nízká růstová rychlost. Rozpoznání těchto poruch je významné, protože jsou zpravidla dobře léčitelné a při včasném zahájení terapie lze tělesnou výšku normalizovat. Přehled endokrinních onemocnění spojených s růstovou retardací shrnuje tabulka 1.

C. Děti s chronickým onemocněním. K růstové retardaci vede většina vážných chronických nemocí (viz též obr. 1). Jejich přehled uvádí tabulka 2. Růstová rychlost je obvykle snížená. Některé chronické nemoci mají chudou klinickou symptomatologii a právě porucha růstu může být prvním příznakem (obr. 4a, b, c, d). Úprava růstové rychlosti u těchto dětí závisí na úspěšném léčení základního onemocnění.


D. Děti s postnatálním růstovým selháním navazujícím na intrauterinní růstovou retardaci. Jako SGA/IUGR (small for gestational age/intrauterine growth retardation) označujeme děti, které se narodily malé na svůj gestační věk – s porodní délkou a/nebo hmotností menší než -2 směrodatné odchylky (SD) pro danou délku těhotenství (tab. 3). Příčinou mohou být faktory vnitřní (ze strany plodu) a faktory zevní (ze strany prostředí, matky či placenty, tab. 4).


U 85–90 % dětí narozených SGA/IUGR se již během prvního roku života růst urychluje („catch-up“). Malou výškou po celé dětství i v dospělosti jsou ohroženy ty děti, jejichž výška do 3 let věku nepřesáhla hodnotu -2,5 SD pro daný věk.
E. Děti s primární poruchou růstu skeletu (kostní dysplazií). Tyto děti mají normální hladiny hormonů včetně IGF-I. Jejich vzrůst je disproporcionální – je zkrácen dolní segment těla proti hornímu segmentu. To znamená, že dlouhé kosti rostou méně. Disproporcionalitu můžeme prokázat různými technikami měření. Nejsnazší je posouzení výšky vsedě ve srovnání s výškou vstoje – výška vsedě je v tomto případě normální, zatímco vyška vstoje snížená.
Klasickým příkladem kostní dysplazie je achondroplazie a její mírnější varianta – hypochondroplazie, které jsou způsobeny aktivační mutací genu pro FGFR-3 (3. typu receptoru pro fibroblastový růstový faktor). Aktivace FGFR-3 pak potlačuje proliferaci chondrocytů růstové chrupavky. Růst skeletu je narušen také u většiny chromozomální aberací, např. u Downova syndromu a u Turnerova syndromu. U Turnerova syndromu (viz kap. 2.3) je příčinou poruchy růstu chybění jedné kopie SHOX genu v pseudoautozomální oblasti X chromozomu. Lériho-Weillův syndrom je mesomelická kostní dysplazie s disproporcionálně malou postavou, způsobená mutací či delecí SHOX genu. Růst a skeletální příznaky (např. Madelungova deformita předloktí) jsou proto podobné jako u Turnerova syndromu. Dědí se ale dominantně a postihuje obě pohlaví. Růst skeletu je narušen u dětí s řadou dalších genetických syndromů.
2.2. Hypopituitarismus
Hypofýza má klíčovou roli v růstu, reprodukci i v kontrole vnitřního prostředí. Selhání hypofýzy má zásadní dopad zejména na rostoucí a vyvíjející se organismus.
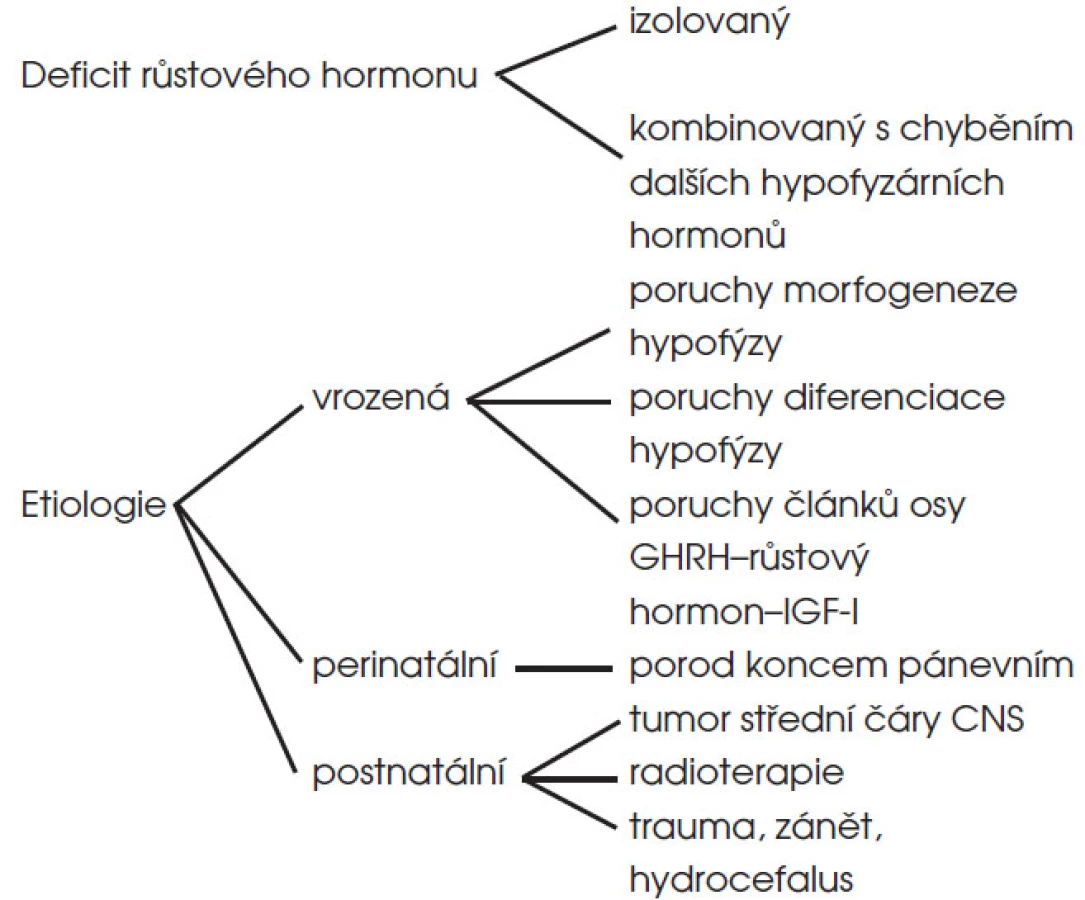
Hypopituitarismus je poruchou funkce adenohypofýzy s deficitem jednoho nebo více pituitárních hormonů. Vzhledem k vývojovému charakteru dětského období je zpravidla klinicky nejvýznamnější deficit růstového hormonu, který je buď izolovaný, nebo sdružený s deficitem dalších hormonů (kombinovaný deficit hypofyzárních hormonů). Izolované deficity ostatních hypofyzárních hormonů jsou u dětí vzácné.
Z hlediska etiologie může být hypopituitarismus (1) vrozený, (2) důsledkem perinatálního traumatu nebo (3) získaný postnatálně (obr. 5).
U cca 20–25 % pacientů s vrozeným hypopituitarismem lze při současné úrovni diagnostiky prokázat genový defekt, buď na úrovni transkripčních faktorů (porucha morfogeneze, resp. diferenciace hypofýzy) nebo na úrovni funkčních proteinů příslušné hormonální osy (defekt receptoru pro GHRH, defekt genu pro růstový hormon). Při MRI zobrazení je ve většině případů hypofýza hypoplastická; při defektu transkripčního faktoru PROP1 ale může být přechodně zvětšená až do obrazu adenomu (obr. 6). V tomto případě může molekulárně genetické vyšetření pacienta uchránit před zbytečným neurochirurgickým výkonem, neboť tato hypofyzární masa je vždy benigní a zpravidla spontánně regreduje.

U některých pacientů s vrozeným hypopituitarismem mohou být přítomny další vývojové anomálie středočárových mozkových či obličejových struktur, z nichž relativně nejčastější je septo-optická dysplazie. Pro tuto diagnózu svědčí alespoň dva ze tří základních projevů: (1) hypopituitarismus, (2) částečná nebo úplná atrofie optických nervů s poruchou zraku, (3) morfologické abnormality CNS – ageneze septum pellucidum, abnormality corpus calosum a jiné (obr. 7a, b).
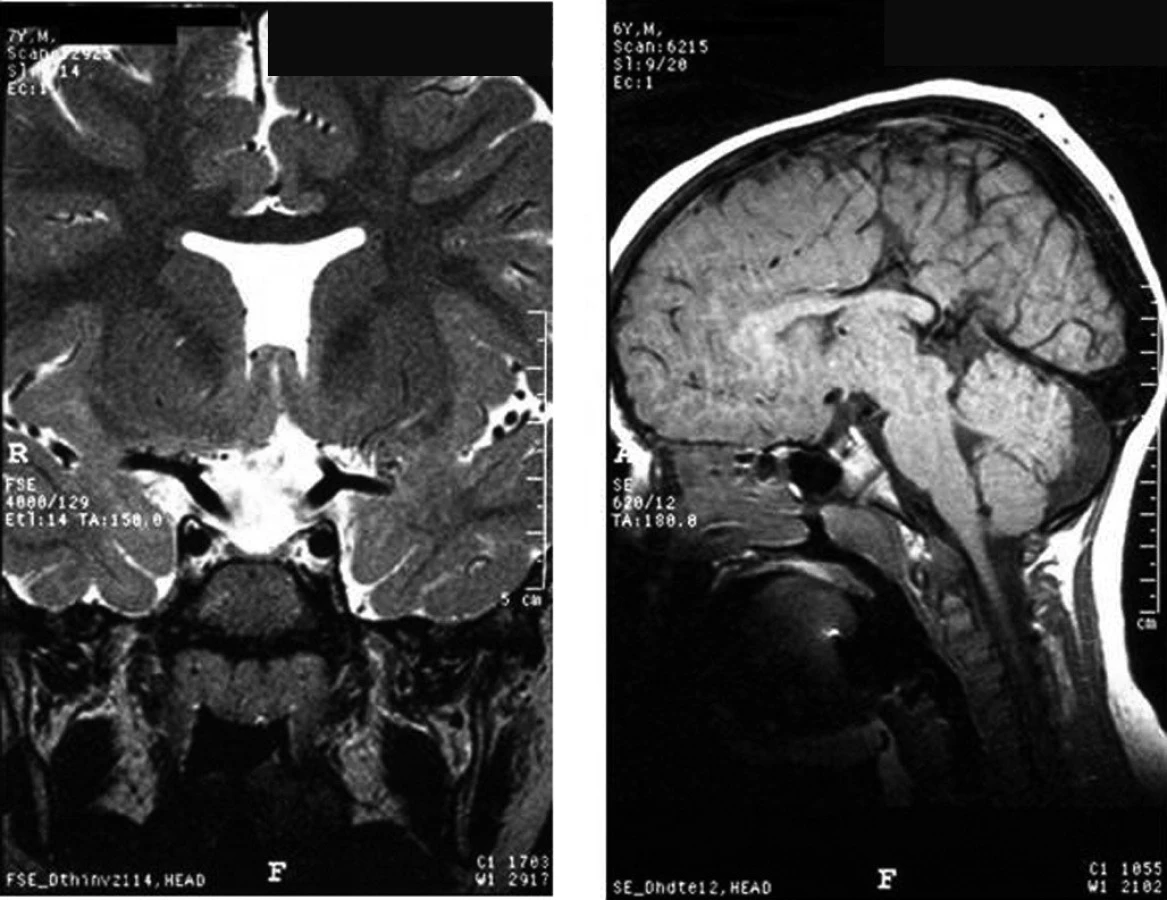
Perinatální trauma vedoucí následně k hypopituitarismu je nejčastěji důsledkem porodu koncem pánevním. V tomto případě je riziko hypopituitarismu cca 7krát zvýšené. Může dojít ke zhmoždění stopky hypofýzy, drobnému krvácení a následnému přerušení spojení mezi hypotalamem a hypofýzou. Typický je obraz tzv. ektopické neurohypofýzy – v MRI obrazu je hypersignální okrsek neurohypofýzy umístěn kraniálně, v místě novotvořeného zakončení neuronů dříve vedoucích k neurohypofýze (obr. 8); adenohypofýza bývá hypoplastická, případně obraz připomíná „empty sella“.

Z postnatálních příčin hypopituitarismu u dětí jsou nejzávažnější tumory oblasti střední čáry, zejména kraniofaryngeom (obr. 9), germinom a gliom optiku či chiasmatu. U dětí s kraniofaryngeomem se růst zpravidla opožďuje řadu let před diagnózou tumoru. V tomto případě může pravidelné sledování a vyhodnocování růstu dítěte doslova zachránit život, protože dříve odhalený kraniofaryngeom lze snáze a s menšími následky radikálně odstranit. Rozsáhlé kraniofaryngeomy ohrožují zrakové dráhy a v důsledku svých komplikací mohou být fatální.

Hypopituitarismus může být pozdním důsledkem infiltrace hypotalamu a stopky hypofýzy při histiocytóze z Langerhansových buněk a je pravidelným důsledkem radioterapie pro tumory vzdálené od střední čáry, extrakraniální solidní tumory a lymfomy, pokud byla hypotalamo-hypofyzární oblast zasažena ložiskovou dávkou vyšší než 30 Gy. I nižší dávky záření u části pacientů poškozují funkci hypofýzy.
Vzácně je u dětí hypopituitarismus důsledkem traumatu CNS, hydrocefalu či neuroinfekce.
Klinický obraz hypopituitarismu se liší v jednotlivých věkových obdobích. U novorozence se může projevit hypoglykémiemi (důsledek deficitu růstového hormonu, případně ACTH), protrahovaným novorozeneckým ikterem (důsledek deficitu TSH), u chlapců někdy bývá mikropenis a testikulární retence (důsledek deficitu gonadotropinů).
Děti s těžkou formou deficitu růstového hormonu mají typickou facies (obr. 10). Hlavním projevem hypopituitarismu u dětí je progresivní růstová retardace, nejčastěji s počátkem mezi 6–12 měsíci života, provázená opožděným prořezáváním mléčné dentice a následně opožděnou výměnou dentice i retardací kostního zrání. Před érou léčby růstovým hormonem dosahovali pacienti s těžkou formu hypopituitarismu dospělé výšky 106–134 cm.

Růstový hormon ovlivňuje také tělesné složení. Neléčené děti s deficitem růstového hormonu vypadají baculaté, aniž by měly nadváhu – mají nadbytek tukové tkáně a nedostatečný vývoj svalové hmoty. Může být patrná akromikrie. Hypoglykémie se mohou vyskytovat i v průběhu dětství, zejména ráno nalačno během interkurentních onemocnění. Nemusejí být správně rozpoznány. V případě deficitu ACTH mohou mít běžné infekční nemoci těžší a protrahovaný průběh. Deficit gonadotropinů je příčinou chybějícího pubertálního vývoje. Projevy deficitu TSH bývají mírné, a to i při těžším stupni centrální hypotyreózy.
Základem diagnostiky hypopituitarismu je stanovení hladin TSH, fT4, kortizolu, IGF-I, prolaktinu, u dětí ve věku puberty také FSH, LH a estradiolu, resp. testosteronu. Teprve při nízkých hladinách IGF-I (hladiny vždy interpretujeme ve vztahu k věku, nejlépe ve formě směrodatné odchylky od normy) zahajujeme vyšetřování růstového hormonu. Vzhledem k pulzatilitě jeho sekrece a krátkému poločasu lze sekreci růstového hormonu zhodnotit výlučně na základě stimulačních testů, kterých bylo vyvinuto několik (např. test s inzulinovou hypoglykémií, clonidinový test, argininový test, glukagonový test a jiné). Pro průkaz deficitu růstového hormonu je podmínkou nedostatečný vzestup (méně než 20 mIU/l nebo 10 ng/ml) alespoň ve dvou stimulačních testech.
2.3. Turnerův syndrom
Turnerův syndrom je častou chromozomální aberací s četností 1 : 2000–2500 živě narozených dívek. Přibližně v polovině případů je zjištěn karyotyp 45,X, u ostatních je příčinou chromozomální mozaika a/nebo strukturální anomálie X chromozomu. U menší části děvčat (cca u 10 %) je součástí karyotypu Y chromozom nebo jeho fragment. V těchto případech se provádí profylaktická gonadektomie pro vysoké riziko gonadoblastomu.
Turnerův syndrom má komplexní klinickou symptomatologii. U novorozených děvčátek mohou na diagnózu upozornit kongenitální lymfedémy nártů nohou a hřbetů rukou, pterygium colli a další dysmorfické známky; výjimečně se již v prvních dnech života může projevit kritická srdeční vada, zejména těsná koarktace aorty. V kojeneckém věku může být nápadné neprospívání. Rodiče si ale mohou všimnout i drobných abnormalit, např. miskovitých nehtů, které mohou být příčinou častých panaritií (obr. 11). Růstová retardace je způsobena chyběním jedné kopie SHOX genu. Rozvíjí se již od nitroděložního období, ale začíná být nápadnější od batolecího věku (obr. 12). V předškolním věku bývají časté otitidy, které často vedou k převodní sluchové poruše a někdy vyžadují zavedení tympanostomií. Jejich příčinou je zřejmě odchylné zakřivení Eustachových trubic. I když intelekt je u dívek s Turnerovým syndromem normální, mají určitý handicap v prostorové představivosti a pohybové koordinaci, což se ve školním věku může projevit v matematice a tělesné výchově. V důsledku ovariální dysgeneze (hypergonadotropního hypogonadismu) u 80 % dívek s Turnerovým syndromem nenastane spontánní dospívání (obr. 13) v případě, že funkce vaječníků je částečně zachovaná, obvykle ještě během dospívání nebo v časné dospělosti dochází k jejich předčasnému vyhasnutí. Přehled a procentuální výskyt základních i vedlejších klinických příznaků Turnerova syndromu shrnuje tabulka 5.




U dívek s Turnerovým syndromem se častěji vyskytují některá orgánově specifická autoimunitní onemocnění, zejména celiakie, lymfocytární tyreoiditida s hypotyreózou a mírná forma autoimunitní hepatitidy. Nejzávažnější komplikací Turnerova syndromu je u některých pacientek disekující aneuryzma aorty, které zpravidla vzniká až v dospělém věku.
Již před 20 lety bylo prokázáno, že léčba růstovým hormonem urychluje růst děvčat s Turnerovým syndromem a zlepšuje jejich tělesnou výšku v dospělosti, i když se nejedná o léčbu substituční, ale farmakologickou. Zatímco neléčené dívky s Turnerovým syndromem vyrostou do výšky v průměru 146 cm (rozmezí 134–158 cm), průměrná dospělá výška po léčbě růstovým hormonem činí u nás cca 158 cm. V kontextu léčby malého vzrůstu probíhá také indukce puberty fyziologickou náhradou estrogenů s cílem napodobit přirozené dospívání a současně optimalizovat dospělou výšku. Také reálná šance na prožití vlastního těhotenství díky in vitro fertilizaci darovaným vajíčkem významně zlepšuje kvalitu života současných mladých žen s Turnerovým syndromem (tab. 6).

2.4. Syndrom Noonanové
Syndrom Noonanové má některé klinické podobnosti s Turnerovým syndromem. Postihuje ale obě pohlaví, je poněkud častější (1 : 1000–1 : 2500 chlapců i dívek) a je monogenně podmíněný. Nejčastější příčinou jsou defekty v genu PTPN11, který kóduje tyrozin-fosfatázu. Ta se podílí na diferenciaci semilunárních srdečních chlopní a ovlivňuje buněčnou proliferaci, diferenciaci a migraci v řadě tkání. U jiných dětí jsou příčinou genové odchylky v dalších genech ze stejné regulační kaskády.
Hlavními příznaky syndromu Noonanové je malý vzrůst (dospělá výška u mužů v průměru 163 cm, u žen 153 cm), pulmonální stenóza nebo hypertrofická kardiomyopatie, poruchy učení až mírná mentální retardace, koagulační poruchy a některé kraniofaciální dysmorfické známky – široké vysoké čelo, hypertelorismus, ptóza, krátký široký krk a nízko nasedající antevertované ušní boltce. U chlapců je častá testikulární retence.
2.5. Syndrom Pradera-Williho
Syndrom Pradera-Williho je způsoben abnormalitou 15. chromozomu v oblasti 15q11-13. U jednotlivých pacientů lze nalézt mikrodeleci, maternální disomii nebo translokaci. V některých případech se jedná o klasický příklad genetického imprintingu.
Pro děti se syndromem Pradera-Williho je v kojeneckém věku typická centrální hypotonie, obtížné krmení a neprospívání. V dalších letech se však rozvine hyperfagie až jídelní obsese, která vede u většiny pacientů k rozvoji velmi významné obezity.
Z drobnějších projevů je nápadná akromikrie, kraniofaciální dysmorfie (převislý horní ret, oči tvaru mandlí), hypogonadismus (prakticky všichni chlapci mají určitý stupeň testikulární retence) a mírná až střední mentální retardace. Postupně se rozvíjí růstové opožďování, které vede u neléčených pacientů ke ztrátě 18–24 cm dospělé výšky. Je provázeno porušeným tělesným složením s nadbytkem tukové tkáně a nedostatkem svaloviny. Tyto změny vedou k celoživotně zvýšenému metabolickému riziku.
Příčinou růstové retardace i hyperfagie u syndromu Pradera-Williho je hypotalamická dysfunkce, která jednak narušuje sekreci růstového hormonu, podílí se na vzniku hypogonadismu a zároveň mění jídelní chování.
Děti se syndromem Pradera-Williho velmi dobře reagují na nízké (substituční) dávky růstového hormonu urychlením růstového tempa a úpravou tělesného složení. Pokud se rodičům daří bránit jejich nadměrnému energetickému příjmu, přispěje růstový hormon ke snížení metabolického rizika a posílení fyzické výkonnosti. To pomáhá i k lepší sociální integraci (obr. 14a, b).

2.6. Léčba růstové retardace
Základem terapie růstové retardace je léčení vyvolávající příčiny. V některých případech je léčebný postup jednoznačný (např. substituce L-tyroxinem u hypotyreózy, bezlepková dieta u celiakie). U těchto dětí dojde v prvních měsících po zahájení léčby k růstovému výšvihu („catch-up“ růst) a během 1–2 let se vracejí výškou do svého původního růstového pásma. U jiných onemocnění (např. Crohnova nemoc, cystická fibróza, chronická renální insuficience) je léčení složitější a řadu projevů nemoci lze zvládat obtížně. Snažíme se o optimální kontrolu nemoci také proto, aby dětský pacient co nejlépe využil svůj růstový potenciál.
Léčba růstovým hormonem je v současné době na základě studií účinnosti a bezpečnosti indikována u šesti skupin dětských pacientů a vedle toho i u dospělých osob s těžkou formou deficitu růstového hormonu. Přehled indikací léčby růstovým hormonem shrnuje tabulka 7. Růstový hormon se podává injekčně s.c. denně doma večer před spaním. Pro jeho s.c. podávání jsou k dispozici injekční pera nebo transdermální tryskový aplikátor. Dávkování při substituční léčbě (u deficitu růstového hormonu nebo u syndromu Pradera-Williho) je zpravidla nižší než při farmakologické léčbě, jejímž cílem je překonat určitou rezistenci cílových tkání (Turnerův syndrom, jiné formy deficitu SHOX, chronická renální insuficience).

K optimálním výsledkům podávání růstového hormonu při hypopituitarismu je nutné korigovat další hormonální deficity. Centrální hypotyreóza se koriguje L-tyroxinem, centrální hypokortikalismus hydrokortisonem, puberta se při hypogonadotropním hypogonadismu indukuje estradiolem, resp. testosteronem. K dlouhodobé substituci menstruačního cyklu u postmenarchálních dívek a žen se používá estradiol v cyklu s gestagenem.
Pro děti s necitlivostí vůči růstovému hormonu (defekt receptoru pro růstový hormon – Laronův syndrom nebo jiné podobné poruchy) je k dispozici injekční léčba rekombinantním IGF-I (přípravek Increlex). U nás se tyto poruchy téměř nevyskytují; jsou častější v oblastech s hojnými příbuzenskými sňatky, např. na Blízkém východě a v severní Africe.
Některé formy růstové retardace však bohužel zatím léčit neumíme. Patří mezi ně např. achondroplazie a většina dalších skeletálních dysplazií.
3. Nadměrný vzrůst
Nadměrný růst (porucha růstu ve směru plus) je definován jako tělesná výška nad 97. percentilem pro daný věk a/nebo růstová rychlost nad 75. percentilem pro daný věk, spočítaná ze dvou přesných měření v odstupu alespoň 6 měsíců. V praxi snadno rozpoznáme nadměrné růstové tempo podle toho, že výška dítěte stoupla o více než jedno percentilové pásmo mezi druhými narozeninami a věkem obvyklým pro začátek puberty.
Nadměrný růst je důvodem k vyšetření mnohem vzácněji než vzrůst malý, neboť fyziologická varianta vyšší postavy („tall-normal“) bývá s výjimkou extrémních případů vnímána jako společensky výhodná. Zdravotní poruchy spojené s vysokou postavou jsou méně časté než poruchy spojené s postavou malou. Pokud je urychlený růst důsledkem předčasné puberty nebo pseudopuberty, upoutá zpravidla dříve pozornost předčasné pubertální zrání než nadměrný růst.
K diagnostickým účelům vždy posoudíme růstovou rychlost a proporcionalitu.
3.1. Nadměrný vzrůst s vysokou růstovou rychlostí
Nadměrný vzrůst s vysokou růstovou rychlostí je často způsoben hormonální nadprodukcí. Příčinou může být nadprodukce sexuálních steroidů (pubertas praecox nebo pseudopubertas praecox), nadprodukce růstového hormonu (velmi vzácný adenom hypofýzy, který vede ke gigantismu nebo akromegalogigantismu), případně nadprodukce hormonů štítné žlázy (Gravesova-Basedowova tyreotoxikóza, autonomní hyperfunkční uzel štítné žlázy nebo ve vzácných případech ektopická nadprodukce tyreoidálních hormonů – tzv. struma ovarii).
Proto k základnímu vyšetření při nadměrném vzrůstu patří stanovení hladin TSH, fT4, IGF-I a nativní hladiny růstového hormonu a posouzení pubertálního zrání podle Tannerovy klasifikace. Teprve v případě známek předčasného dospívání následuje diagnostika zaměřená na odhalení příčiny.
3.2. Nadměrný vzrůst s narušenou proporcionalitou
Nadměrný vzrůst s narušenou proporcionalitou může být důsledkem primární poruchy metabolismu pojiva nebo důsledkem hypogonadismu.
Mezi poruchy spojené s dysproporcionálně nadměrným vzrůstem (tj. s dlouhými končetinami) patří Marfanův syndrom, homocystinurie, Klinefelterův syndrom či jiné formy hypogonadismu.
Pro diagnostiku Marfanova syndromu (obr. 15) i homocystinurie je důležité oftalmologické vyšetření s posouzením případné subluxace čočky, echokardiografické vyšetření (riziko aortálního aneuryzmatu u Marfanova syndromu), pro Klinefelterův syndrom jsou příznačné vysoké hladiny FSH a LH a tuto diagnózu potvrdí karyotyp.

3.3. Nadměrný vzrůst s normální proporcionalitou
Nadměrný vzrůst s normální proporcionalitou může být důsledkem některé ze vzácných poruch – cerebrálního gigantismu (de Sotova syndromu), Beckwithova-Wiedemannova syndromu, izolovaného deficitu glukokortikoidů, deficitu estrogenů nebo rezistence k estrogenům.
Beckwithův-Wiedemannův syndrom má příznačný fenotyp s umbilikální kýlou, makroglosií a gigantismem, který se projevuje již in utero. Patří mezi syndromy s chromozomálním imprintingem – dochází k nadměrné expresi inzulinového genu a genu pro IGF-II. Zásadním problémem dětí s Beckwithovým-Wiedemannovým syndromem jsou hyperinzulinemické hypoglykémie, vrozené srdeční vady a vysoká prevalence nádorů v útlém věku (např. nefroblastomu, neuroblastomu, adrenálního karcinomu). I po narození pokračuje rychlý, avšak nikoliv extrémní růst.
3.4. Familiárně vysoký vzrůst
Většina dětí, které přicházejí k vyšetření pro vysoký vzrůst, je ve skutečnosti zdravá a má familiárně vysoký vzrůst. Pravidlem je vysoká výška rodičů, normální nebo jen hraničně urychlený pubertální vývoj, normální nebo jen mírně nadprůměrná růstová rychlost, normální nebo hraniční tělesná proporcionalita a nepřítomnost patologických stavů výše uvedených. Jedná se tedy o diagnózu per exclusionem, která je podpořena souladem mezi aktuální výškou dítěte a predikcí výšky podle jeho rodičů.
Štítky
Neonatologie Pediatrie Praktické lékařství pro děti a dorostČlánek vyšel v časopise
Česko-slovenská pediatrie

2011 Číslo 2
- Efektivita kartáčku Sonicare For Kids u dětí předškolního věku
- Cytomegalovirové infekce u novorozenců a dětí
- Cytomegaloviróza a spalničky u dětí
- Kongenitální a perinatální cytomegalová infekce
Nejčtenější v tomto čísle
- Vybrané kapitoly z nové učebnice Klinická pediatrie
- Extraezofágový reflux – otorinolaryngologické komplikácie gastroezofágového refluxu
- Léčíme pacienty s kryptorchismem v doporučeném věku?
- Krikofaryngeálna achalázia ako príčina chronického dráždivého kašľa u dojčaťa
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy