
Porucha růstu a vývoje u chlapce s X-vázanou ichtyózou, protrahovaným porodem a nízkou hladinou estriolu u matky v průběhu těhotenství
Disorder of Growth and Development in a Boy with X-bound Ichthyosis, Protracted Delivery and Low Level of Estriol in the Mother during Pregnancy
X-linked ichthyosis was diagnosed in a 2-year old boy with low maternal estriol serum levels during gestation. The prolonged delivery was terminated by Caesarian section due to fetal hypoxia and turbid amniotic fluid. Apgar score was uneventful, but early postnatal adaptation was complicated by failure to thrive and hypotonia followed on by hypernatremic dehydration and aspiration pneumonia in the second week of life. At this time, cutaneous manifestations of ichthyosis was also observed and severe psychomotor retardation developed since early infancy. Enzymatic investigations in the proband, his mother and her relatives including grandmother, sister and her son revealed steroid sulfatase (STS) deficiency and the cytogenetic analyses using FISH method revealed the microdeletion of STS gene.
The central nervous system impairment is usually not present in patients with X-linked ichthyosis. Although in our patient the role of hypernatremic dehydration and/or eventual hypoxia during aspiration pneumonia cannot be excluded as a cause of the postnatal CNS impairment, we suppose that also the perinatal hypoxia might be important in a boy with prolonged delivery resulting from low maternal estrogens and placental STS deficiency. Because the STS deficiency affects approximately 1 in 2–6000 males, the low estriol level in pregnant woman should be an alerting marker for physicians to give a though to possibility of X-linked ichthyosis.
Key words:
X-linked ichthyosis, steroid sulfatase, estriol, prolonged delivery, psychomotor retardation, growth impairment
Autoři:
E. Flachsová 1; J. Kytnarová 1; J. Ledvinová 2; H. Poupětová 2; E. Kočárek 3; V. Malinová 1; J. Zeman 1
Působiště autorů:
Klinika dětského a dorostového lékařství VFN a UK 1. LF, Praha
přednosta prof. MUDr. J. Zeman, DrSc.
1; Ústav dědičných metabolických poruch VFN a UK 1. LF, Praha
přednosta doc. MUDr. V. Kožich, DrSc.
2; Ústav biologie a lékařské genetiky FN Motol, Praha
přednosta prof. MUDr. M. Macek Jr., DrSc.
3
Vyšlo v časopise:
Čes-slov Pediat 2009; 64 (3): 120-126.
Kategorie:
Kazuistika
Souhrn
U dvouletého chlapce s nízkou hladinou estriolu v séru matky během těhotenství, anamnézou nepostupujícího porodu ukončeného císařským řezem pro hypoxii plodu se zkalenou plodovou vodou, kožními projevy ichtyózy, které se objevily ve druhém týdnu života, a těžkou poruchou psychomotorického vývoje byla diagnostikována X-vázaná ichtyóza. Enzymatická vyšetření u chlapce, jeho matky, babičky, tety a matčina bratrance ukázala poruchu aktivity steroidsulfatázy (STS). Cytogenetické vyšetření metodou FISH u probanda i jeho příbuzných ukázalo mikrodeleci genu pro STS.
Protože postižení CNS obvykle nepatří do klinického obrazu X-vázané ichtyózy, autoři se domnívají, že na vzniku psychomotorické retardace u chlapce se nejspíše podílela perinatální hypoxie při protrahovaném porodu v důsledku snížené hladiny estrogenů při deficitu placentární STS, která se podílí na syntéze estrogenů. Nemohou však vyloučit ani postnatální postižení CNS po prodělané hypernatremické dehydrataci a/nebo hypoxii při aspirační pneumonii v novorozeneckém věku.
V literatuře již byl u chlapců s X-vázanou ichtyózou protrahovaný porod opakovaně popsán. Předpokládá se, že protrahovaný porod u chlapců s X-vázanou ichtyózou je způsoben nízkou hladinou estrogenů při nedostatečné aktivitě placentární STS. Ačkoliv se deficit aktivity STS vyskytuje pouze u 1 chlapce ze 2–6000, měl by být nález nízké hladiny estriolu u těhotné ženy varovným signálem pro ošetřující lékaře, aby v rámci diferenciálně diagnostické rozvahy pomýšleli i na možnost X-vázané ichtyózy.
Klíčová slova:
X-vázaná ichtyóza, steroidsulfatáza, estriol, protrahovaný porod, psychomotorická retardace, růstová porucha
Úvod
X-vázaná ichtyóza je dědičně podmíněné metabolické onemocnění charakterizované poruchou keratinizace. X-vázaná ichtyóza je způsobena poruchou aktivity steroidsulfatázy (STS), která katalyzuje hydrolýzu sulfátové skupiny v 3-β pozici sterolů a steroidů. Mezi nejvýznamnější substráty STS u člověka patří dehydroepiandrosteronsulfát (DHEAS) a cholesterolsulfát [1]. Za ichtyotické změny je pravděpodobně odpovědný zvýšený obsah cholesterolsulfátu ve stratum corneum [2].
První klinické příznaky X-vázané ichtyózy (XLI) se obvykle projevují na kůži již v novorozeneckém nebo časném kojeneckém období ložisky deskvamace (olupování). Charakter kožního postižení se během několika dalších týdnů mění a na kůži se objevují charakteristické polygonální prominující hnědavé šupiny s predilekční lokalizací symetricky na trupu a extenzorových částech zejména dolních končetin. Dlaně, plosky a obličej s výjimkou preaurikulární oblasti většinou nebývají postiženy a také nehty a vlasy jsou intaktní [3]. Přibližně u čtvrtiny pacientů se objeví i mírné opacity na rohovce, které však nevedou k postižení zraku [4].
Prvním klinickým příznakem onemocnění u chlapců s X-vázanou ichtyózou může být i protrahovaný porod, který je způsoben nedostatečnou syntézou estrogenů z dehydroepiandrosteronsulfátu při primární poruše placentární STS a nízkou aktivitou STS v tkáních plodu včetně jater, mozku, kůry nadledvin, testes, kůže [5, 6]. Na poruchu STS je nutno pomýšlet i u těhotných žen, u kterých byla při těhotenském screeningu nalezena snížená hladina estriolu, i když ne každé snížení hladiny estriolu v séru těhotných je způsobeno X-vázanou ichtyózou [7, 8].
V našem sdělení předkládáme výsledky metabolických a molekulárních analýz u chlapce s klinickými příznaky X-vázané ichtyózy doprovázené poruchou růstu a těžkou poruchou psychomotorického vývoje.
Metody
Analýza moči
Na izolaci lipidů z moči byly použity extrakční a chromatografické metody [9]. Současně byly extrahovány sulfáty steroidů a cholesterolsulfát. Detekce byla provedena činidlem obsahujícím octan měďnatý nebo anisaldehyd.
Enzymatické analýzy
Leukocyty byly izolovány z periferní krve metodou dextranové sedimentace [10] a homogenizovány pomocí ponorného ultrazvukového homogenizátoru za chlazení v ledové drti. Aktivita arylsulfatázy C (ASC) byla stanovena použitím fluorescenčního substrátu 4-metylumbelliferylsulfátu bez přidání a po přidání dehydroepiandrosteronsulfátu (DHEAS). Aktivita steroidsulfatázy (STS; EC 3.1.6.2) se vypočítala jako podíl aktivity ASC, inhibovatelné DHEAS [11]. Koncentrace proteinu byla stanovena metodou podle Hartree [12].
Molekulárně cytogenetická analýza
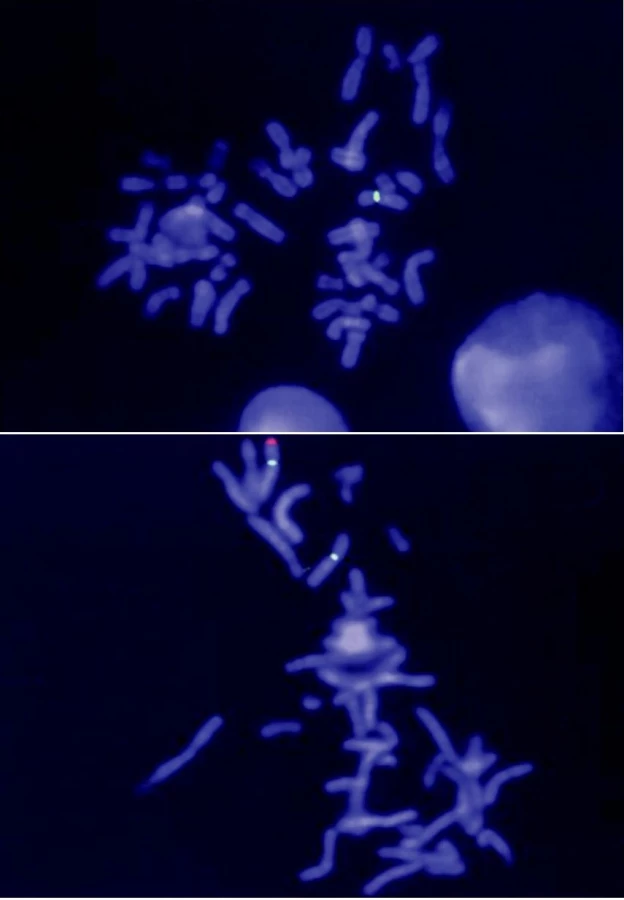
Leukocyty z periferní krve byly kultivovány, fixovány (methanol – kyselina octová; 3 : 1) a naneseny na podložní sklíčko. Mikrodelece chromozomu Xp22.3 v lokusech STS a KAL byla vyšetřena metodou FISH (fluorescenční in situ hybridizací) s využitím lokus-specifických sond značených fluorescenčním barvivem Spectrum Orange (Abbott – Vysis, USA). Chromozom X byl navíc značen kontrolní sondou DXZ1 hybridizující v centromerické oblasti mimo kritické regiony. Pro zpracování vzorků byl použit upravený protokol FISH (podle materiálů fy Abbott – Vysis, USA). Hybridizační signál na chromozomech v mitóze byl dokumentován pomocí epifluorescenčního mikroskopu Olympus BX-60 vybaveného CCD-kamerou a následně zpracován s využitím počítačového systému ISIS (Metasystems, Altlussheim, Germany).
Etika
Studie byla provedena v souladu s Helsinskou deklarací WHO a od rodičů byl získán informovaný souhlas.
Popis případu
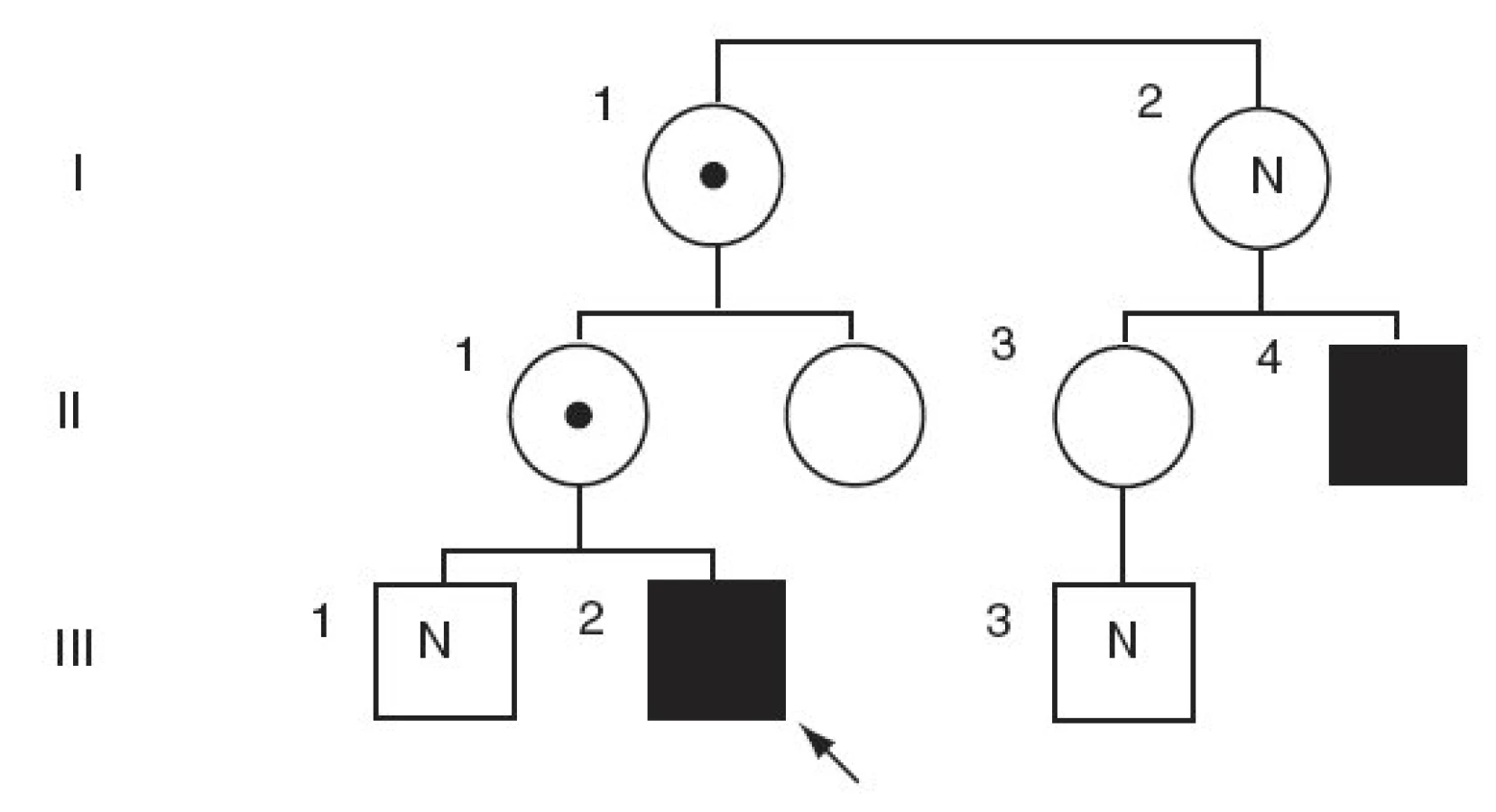
Matka probanda, u které se ve 22 letech manifestovala epilepsie a která prodělala tři záchvaty generalizovaných křečí s poruchou vědomí, byla v průběhu celého těhotenství léčena lamotriginem v dávce 150 mg/den. Během těhotenství byla bez záchvatů, ale protože v rámci biochemického screeningu ve druhém trimenonu byla zjištěna nízká hladina estriolu, byla indikována amniocentéza. Cytogenetické vyšetření plodu ukázalo normální mužský karyotyp 46,XY, také sonografická vyšetření plodu byla v pořádku. Otec, prarodiče a starší bratr jsou zdrávi, pouze 24letý bratranec matky má na bércích nohou světlehnědá symetrická ložiska hyperkeratózy s deskvamací.
Chlapec se narodil v termínu s hmotností 3000 g a délkou 50 cm. Pro nepostupující porod a hrozící hypoxii plodu byl porod ukončen císařským řezem. Plodová voda byla zkalená, skóre podle Apgarové bylo podle zdravotní dokumentace hodnoceno 9-10-10 body, ale podle matky byl chlapec údajně kříšen(?).
V prvních dnech života byl chlapec hypotonický a špatně pil. Pro podezření, že hypotonie je způsobena transplacentárním přestupem antiepileptické léčby na plod a eventuálním přestupem antiepileptika do mateřského mléka, byla u matky zastavena laktace a chlapec byl převeden na umělou výživu. Na hypotonii se však mohla podílet i hyperbilirubinémie, pro kterou byl chlapec 48 hodin na fototerapii. Osmý den života se u chlapce rozvinula hypertonická dehydratace s hypernatrémií 162 mmol/l a osmolalitou séra 338 mOsm/l, která byla korigována infuzní léčbou. Chlapcova rekonvalescence byla komplikována aspirační pneumonií s nutností šestidenní oxygenoterapie. I po úpravě poslechového nálezu na plicích však přetrvával centrální hypotonický syndrom, sonografické vyšetření CNS ukázalo drobné hyperechogenní ložisko periventrikulárně vlevo.
Ve věku 2 měsíců byl chlapec přijat na JIP pro akutní kardiopulomonální selhávání při těžké pneumokokové pneumonii, byla nutná umělá plicní ventilace a farmakologická podpora oběhu. Krátce po přijetí se objevila první ataka tonicko-klonických křečí. Byla zahájena léčba fenobarbitalem, ale objevily se bleskové křeče s korespondujícím patologickým EEG záznamem. Vyšetření MR CNS svědčilo pro redukci gyrifikace v oblasti rhinencefala. V terapii byl přidán Clonazepam, po kterém křeče ustoupily, ale psychomotorický vývoj chlapce zůstal neuspokojivý, trvá centrální hypotonický syndrom a horizontální nystagmus, chlapec fixuje jen sporně.
Aktuální úroveň psychomotorického vývoje ve věku 3 let odpovídá úrovni poloviny II. trimenonu. První kožní projevy onemocnění se objevily na začátku druhého týdne života. Nejdříve to byla mnohočetná ložiska olupující se kůže o velikosti do 20 mm lokalizovaná na trupu i končetinách, ale nikoliv na ploskách či dlaních. Pod oloupanou pokožkou byla kůže makroskopicky čistá, bez mokvání či krvácivých projevů. Ve věku dvou měsíců se objevila symetricky na bércích světlehnědá ložiska deskvamace se šupinami o velikosti 5–10 mm, která přetrvávají dosud.
Růst byl u chlapce zpočátku uspokojivý, v 15 měsících měl délku na 50. percentilu, ale pak se růst zpomalil, ve věku 3 let a 5 měsíců měří 88 cm (-2,91 SDS), hmotnost k délce je -0,52 SDS, obvod hlavy je 49 cm (-1,03 SDS). Byly doplněny dva dynamické testy sekrece růstového hormonu, které nevyloučily možnost parciálního deficitu STH (maximální vzestup STH na 18,8 mIU/l).
Vyšetření moči na přítomnost cholesterolsulfátu a dehydroepiandrosteronsulfátu bylo pozitivní. Enzymatická vyšetření v izolovaných leukocytech ukázala výrazně sníženou aktivitu steroidsulfatázy (STS), sníženou aktivitu STS jsme nalezli i u chlapcovy matky, babičky, tety a matčina bratrance (tab. 1). Cytogenetické vyšetření metodou FISH u chlapce ukázalo mikrodeleci genu pro STS (obr. 2). Stejná delece byla nalezena také u matky a dalších příbuzných z matčiny strany (obr. 1). Naopak cytogenetické vyšetření zaměřené na gen pro Kallmanův syndrom (KAL), který sousedí s genem pro STS, neprokázalo deleci KAL genu u chlapce.

Diskuse
Porucha psychomotorického vývoje u chlapce spolu s kožními projevy ichtyózy, nepostupující porod a nízká hladina estriolu v séru matky během těhotenství vedly k podezření na poruchu metabolismu sulfatidů, především na „multiple sulphatase deficiency“ (mnohočetný sulfatázový deficit) [13]. „Multiple sulfatase deficiency“ je onemocnění způsobené poruchou posttranslační modifikace všech u člověka známých sulfatáz [13]. Klinické příznaky u pacientů s „multiple sulfatase deficiency“ kombinují projevy izolovaných poruch sulfatáz, mezi které patří pět typů mukopolysacharidózy (MPS typ II, IIIA, IIID, IVA a VI), metachromatická leukodystrofie, X-vázaná ichtyóza a X-vázaná chondrodysplasia punctata. Obvykle je přítomna těžká porucha psychomotorického vývoje, kraniofaciální dysmorfie, visceromegalie, ichtyóza a postižení skeletu [14]. U postižených jedinců je současně zvýšené vylučování glykosaminoglykanů i sulfatidů v moči a v mozkomíšním moku je zvýšená proteinorachie.
U našeho probanda jsme diagnózu „multiple sulfatase deficiency“ vyloučili na metabolické i enzymatické úrovni. Enzymatická vyšetření ukázala pouze izolovanou poruchu steroidsulfatázy, která sice vysvětluje etiologii kožních projevů onemocnění, ale nevysvětluje u chlapce poruchu psychomotorického vývoje.
Gen kódující steroidsulfatázu je lokalizován v oblasti Xp22.3 (obr. 3) [15]. U 85 % pacientů s X-vázanou ichtyózou je genetická porucha způsobena delecí celého genu pro STS, parciální delece jsou méně časté [16]. Asi 10 % pacientů má v genu pro STS bodové mutace [17] a cca 5 % nemocných s X-vázanou ichtyózou mívá rozsáhlejší delece, které postihují i sousední geny – tzv. „contiguous gene syndrome“ [18].

V naší rodině jsme deleci genu pro STS nalezli u pacienta, jeho matky, babičky a bratrance. Protože do klasického fenotypu delece STS nepatří neurologická symptomatologie, která je dominujícím postižením našeho probanda, pomýšleli jsme v rámci diferenciální diagnostické rozvahy i na přítomnost rozsáhlejší delece kolem genu pro STS v oblasti chromozomu Xp22.3. Rozsáhlejší deleční syndromy se projevují komplexnějšími klinickými příznaky, které odpovídají delecím jednotlivých genů a poruše funkce jejich proteinových produktů [19]. Centrifugálně od genu pro STS je lokalizován gen pro Kallmannův syndom, gen pro X-vázanou mentální retardaci a gen pro X-vázaný oční albinismus [18]. Centripetálně od genu pro STS se nachází SHOX gen (Short stature homebox, MIM 312865) a gen pro X-vázanou chondrodysplasia punctata.
Kallmannův syndrom (MIM 308700) je onemocnění charakterizované anosmií (nebo hypoosmií), která je způsobená agenezí čichových laloků, a sekundárním hypogonadotropním hypogonadismem s deficitem gonadotropiny stimulujícího hormonu (GnRH) [23]. Pro přítomnost tohoto syndromu u chlapce by mohl svědčit nález redukce gyrifikace v oblasti rhinencephala při vyšetření MR, ale doplněné molekulárně cytogenetické vyšetření KAL genu toto onemocnění nepotvrdilo.
Do oblasti Xp22.3 byl pomocí vazebné analýzy v rodinách s výskytem mentální retardace lokalizován i kandidátní gen pro X-vázanou mentální retardaci (MIM 309530) [22], čemuž odpovídá i častá přítomnost opoždění psychomotorického vývoje u pacientů s většími terminálními delecemi na chromozomu X [18]. Ačkoliv u našeho probanda nemůžeme možnost delece kandidátního genu pro X-vázanou mentální retardaci vyloučit, nepředpokládáme ji, protože jeho porucha CNS je mnohem závažnější a jeho vývojový kvocient (DQ) nedosahuje ani 20 %.
X-vázaný oční albinismus (oční albinismus typ 1, OA1, MIM 300500) je onemocnění charakterizované poruchou zraku, nystagmem, strabismem a fotofobií. Při vyšetření očního pozadí je typický nález depigmentovaného fundu u hemizygotních pacientů, u heterozygotních přenašeček je nález pigmentové mozaiky zejména na periferii fundu [24]. Náš pacient má těžkou poruchu zraku a nystagmus, ale oční vyšetření chlapce i jeho matky ukázalo zcela normální nález na očním pozadí. Také výsledky cytogenetického vyšetření na Kallmanův syndrom, jehož gen se nachází mezi geny pro STS a OA1, toto onemocnění vylučují.
Mutace v lidském SHOX genu se vyskytuje u pacientů s Léri-Weillovým syndromem, který se projevuje těžkou formou skeletální dysplazie s poruchou růstu, ale i u dětí s Langerovým syndromem nebo u dětí s poruchou růstu nejasné etiologie. U dívek s Turnerovým syndromem se předpokládá, že haploinsuficience SHOX genu je příčinou růstové poruchy i skeletálních odchylek [20]. Pro poruchu růstu jsme u našeho pacienta na toto onemocnění pomýšleli, ale delece SHOX genu nebyla nalezena.
X-vázaná chondrodysplasia punctata (MIM 302950) je závažné metabolické onemocnění způsobené deficitem arylsulfatázy E [21], které se sice projevuje hypotonií a těžkou psychomotorickou retardací, především však kostní dysplazií a poruchou růstu s typickými rentgenovými nálezy [22], které nejsou přítomny u našeho pacienta.
Prognóza chlapců s X-vázanou ichtyózou může být příznivá, ale u postižených mužů bývá popisován vyšší výskyt kryptorchismu nebo sekundárního hypogonadismu bez kryptorchismu, který může být způsoben deficitem STS v testikulární tkáni, i když úloha STS v testikulární diferenciaci zůstává nejasná. Genealogické vyšetření v rodině našeho probanda ukázalo, že chlapcova matka má epilepsii a že bratranec matky má podobné kožní projevy na bércích dolních končetin jako náš pacient. Nikdo z chlapcových příbuzných však nemá poruchu funkce CNS.
Enzymatická a cytogenetická vyšetření u našeho probanda sice vysvětlila příčinu kožních projevů onemocnění, ale etiologie těžké psychomotorické retardace zůstává nejasná. Sérologická vyšetření adnátních infekcí byla negativní, cytogenetické vyšetření ukázalo normální karyotyp a podrobná metabolická vyšetření neodhalila jinou metabolickou poruchu. Protože postižení CNS nepatří do klinického obrazu X-vázané ichtyózy, domníváme se, že na vzniku psychomotorické retardace u chlapce se nejspíše podílela perinatální hypoxie při protrahovaném porodu, ale nemůžeme vyloučit ani postnatální postižení CNS při hypernatremické dehydratci a/nebo hypoxii při aspirační pneumonii v novorozeneckém věku. Podíl matce podávaného lamotriginu se ve zvažované etiologii postižení CNS jeví jako velmi nepravděpodobný [25].
V literatuře již byl u chlapců s X-vázanou ichtyózou protrahovaný porod opakovaně popsán. Předpokládá se, že protrahovaný porod u chlapců s X-vázanou ichtyózou je způsoben nízkou hladinou estrogenů při nedostatečné aktivitě placentární STS [7, 8].
Ačkoliv porucha steroidsulfatázy se vyskytuje pouze u 1 chlapce ze 2–6000, měl by nález nízké hladiny estriolu u těhotné ženy vést ošetřující lékaře k tomu, aby v rámci diferenciálně diagnostické rozvahy pomýšleli i na možnost výskytu X-vázané ichtyózy se všemi možnými dalšími riziky komplikací v průběhu porodu a v období časné poporodní adaptace.
Poděkování
Za molekulárně biologické vyšetření SHOX genu děkujeme MUDr. Anně Křepelové z Ústavu biologie a lékařské genetiky FN v Motole.
Práce vznikla s podporou projektu IGA NR 9374-3, IGA MZ ČR NR5497-3 a VZ MSM 0021620806.
Došlo: 9. 12. 2008
Přijato: 26. 1. 2009
Prof. MUDr. Jiří Zeman, DrSc.
Klinika dětského a dorostového lékařství
VFN a UK 1. LF
Ke Karlovu 2
120 00 Praha 2
e-mail: jzem@lf1.cuni.cz
Zdroje
1. Webster D, France JT, Shapiro LJ, Weiss R. X-linked ichthyosis due to steroid-sulphatase deficiency. Lancet 1978 Jan 14;1(8055): 70–72.
2. Elias PM, Crumrine D, Rassner U, Hachem JP, Menon GK, Man W, Choy MH, Leypoldt L, Feingold KR, Williams ML. Basis for abnormal desquamation and permeability barrier dysfunction in RXLI. J. Invest. Dermatol. 2004 Feb;122(2): 314–319.
3. Hoyer H, Lykkesfeldt G, Ibsen HH, Brandrup F. Ichthyosis of steroid sulphatase deficiency. Clinical study of 76 cases. Dermatologica 1986;172(4): 184–190.
4. Costagliola C, Fabbrocini G, Illiano GM, Scibelli G, Delfino M. Ocular findings in X-linked ichthyosis: a survey on 38 cases. Ophthalmologica 1991;202(3): 152–155.
5. Felig P, Baxter JD, Frohman LA. Endocrinology and Metabolism. 3rd ed. Mc Graw Hill, 1995.
6. Sugawara T, Fujimoto S. The potential function of steroid sulphatase activity in steroid production and steroidogenic acute regulatory protein expression. Biochem. J. 2004 May 15;380(Pt1): 153–160.
7. Marshall I, Ugrasbul F, Manginello F, Wajnrajch MP, Shackleton CH, New MI, Vogiatzi MV. Congenital hypopituitarism as a cause of undetectable estriol levels in the maternal triple-marker screen. J. Clin. Endocrinol. Metab. 2003 Sep;88(9): 4144–4148.
8. Schleifer RA, Bradley LA, Richards DS, Ponting NR. Pregnancy outcome for women with very low levels of maternal serum unconjugated estriol on second-trimester screening. Am. J. Obstet. Gynecol. 1995 Oct;173(4): 1152–1156.
9. Berná L, Asfaw B, Conzelmann E, Černý B, Ledvinová J. Determination of urinary sulfatides and other lipids by combination of reversed-phase and thin-layer chromatographies. Anal. Biochem. 1999;269(2): 304–311.
10. Skoog WA, Beck WS. Studies on the fibrinogen, dextran and phytohemagglutinin methods of isolating leukocytes. Blood 1956;11 : 436–454.
11. Diggelen OP, Konstantinidou AE, Bousema MT, Boer M, Bakx Th, Jöbsis AC. A fluorimetric assay of steroid sulphatase in leukocytes: Evidence for two genetically different enzymes with arylsulphatase C activity. J. Inher. Metab. Dis. 1989;12 : 273–280.
12. Hartree EF. Determination of protein: A modification of the lowry method that gives a linear photometric response. Analytical Biochemistry 1972;48 : 422–427.
13. Baenziger JU. A major step on the road to understanding a unique posttranslational modification and its role in a genetic disease. Cell 2003 May 16;113(4): 421–422.
14. Cosma MP, Pepe S, Parenti G, Settembre C, Annunziata I, Wade-Martins R, Di Domenico C, Di Natale P, Mankad A, Cox B, Uziel G, Mancini GM, Zammarchi E, Donati MA, Kleijer WJ, Filocamo M, Carrozzo R, Carella M, Ballabio A. Molecular and functional analysis of SUMF1 mutations in multiple sulfatase deficiency. Hum. Mutat. 2004 Jun;23(6): 576–581.
15. Yen PH, Allen E, Marsh B, Mohandas T, Wang N, Taggart RT, Shapiro LJ. Cloning and expression of steroid sulfatase cDNA and the frequent occurrence of deletions in STS deficiency: implications for X-Y interchange. Cell 1987 May 22;49(4): 443–454.
16. Shapiro LJ, Yen P, Pomerantz D, Martin E, Rolewic L, Mohandas T. Molecular studies of deletions at the human steroid sulfatase locus. Proc. Natl. Acad. Sci. USA. 1989 Nov;86(21): 8477–8481.
17. Alperin ES, Shapiro LJ. Characterization of point mutations in patients with X-linked ichthyosis. Effects on the structure and function of the steroid sulfatase protein. J. Biol. Chem. 1997 Aug 15;272(33): 20756–20763.
18. Ballabio A, Bardoni B, Carrozzo R, Andria G, Bick D, Campbell L, Hamel B, Ferguson-Smith MA, Gimelli G, Fraccaro M. Contiguous gene syndromes due to deletions in the distal short arm of the human X chromosome. Proc. Natl. Acad. Sci. USA. 1989 Dec;86(24): 10001–10005.
19. Schmickel RD. Contiguous gene syndromes: a component of recognizable syndromes. J. Pediatr. 1986 Aug;109(2): 231–241.
20. Rappold GA, Fukami M, Niesler B, Schiller S, Zumkeller W, Bettendorf M, Heinrich U, Vlachopapadoupoulou E, Reinehr T, Onigata K, Ogata T. Deletions of the homeobox gene SHOX (short stature homeobox) are an important cause of growth failure in children with short stature. J. Clin. Endocrinol. Metab. 2002 Mar;87(3): 1402–1406.
21. Brunetti-Pierri N, Andreucci MV, Tuzzi R, Vega GR, Gray G, McKeown C, Ballabio A, Andria G, Meroni G, Parenti G. X-linked recessive chondrodysplasia punctata: spectrum of arylsulfatase E gene mutations and expanded clinical variability. Am. J. Med. Genet. 2003 Mar 1;117A(2): 164–168.
22. Schutz CK, Ives EJ, Chalifoux M, MacLaren L, Farrell S, Robinson PD, White BN, Holden JJ. Regional localization of an X-linked mental retardation gene to Xp21.1–Xp22.13 (MRX38). Am. J. Med. Genet. 1996 Jul 12;64(1): 89–96.
23. Hardelin JP. Kallmann syndrome: towards molecular pathogenesis. Mol. Cell Endocrinol. 2001 Jun 20;179(1–2): 75–81.
24. Shen B, Samaraweera P, Rosenberg B, Orlow SJ. Ocular albinism type 1: more than meets the eye. Pigment Cell Res. 2001 Aug;14(4): 243–248.
25. Cunnington M, Tennis P, and the International Lamotrigine Pregnancy Registry Scientific Advisory Committee. Neurology 2005;64 : 955–960.
Štítky
Neonatologie Pediatrie Praktické lékařství pro děti a dorostČlánek vyšel v časopise
Česko-slovenská pediatrie

2009 Číslo 3
- Efektivita kartáčku Sonicare For Kids u dětí předškolního věku
- Inosin pranobex v léčbě HPV infekcí děložního čípku
- Cytomegalovirové infekce u novorozenců a dětí
Nejčtenější v tomto čísle
- Inzulinóm – príčina recidivujúcich hypoglykémií u 16-ročného pacienta
- Wiskottův-Aldrichův syndrom – onemocnění vyžadující včasnou transplantaci kmenových buněk krvetvorby
- Porucha růstu a vývoje u chlapce s X-vázanou ichtyózou, protrahovaným porodem a nízkou hladinou estriolu u matky v průběhu těhotenství
- Anafylaxe u dětí vyvolaná potravinami
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy