
Praktický přístup k pitvě srdce s vrozenou srdeční vadou
A practical approach to the examination of the congenitally malformed heart at autopsy
Congenital heart defects (CHD) represent the most frequent type of the heart disease in childhood, with incidence up to 1 % of all live-born children. Despite the improving echocardiographic diagnostics, part of CHD remains undiagnosed and can manifest in the later age or may be the cause of the early abortion. On the other hand, some foetuses with prenatally diagnosed severe CHD may be recommended to interruption. Therefore, each pathologist can encounter a malformed heart at the autopsy. Despite the current quality of the echocardiography, the macroscopic assessment of the heart by the pathologist is still considered the best method for evaluation of the structural heart disease. Knowledge of the basic pathologic anatomy thus remains an important prerequisite for adequately performed paediatric autopsy.
Keywords:
pathology – congenital heart disease – sequential segmental analysis
Autoři:
Ondřej Fabián 1; Roman Gebauer 2
Působiště autorů:
Ústav patologie a molekulární medicíny 2. LF UK a FN Motol, Praha
1; Dětské kardiocentrum 2. LF UK a FN Motol, Praha
2
Vyšlo v časopise:
Čes.-slov. Patol., 55, 2019, No. 4, p. 202-208
Kategorie:
Přehledový článek
Souhrn
Vrozené srdeční vady (CHD) představují nejčastější srdeční onemocnění dětského věku s incidencí necelé 1 % živě narozených dětí. Ačkoliv se úroveň echokardiografické diagnostiky stále zlepšuje, část CHD zůstává nediagnostikovaná a mohou být příčinou časného abortu nebo se manifestovat v pozdějším věku. Některá těhotenství s prokázanou závažnou CHD jsou naopak doporučeny k umělému přerušení. S malformovaným srdcem se tak v rámci pitevního provozu může setkat každý patolog. I přes kvalitu současné echokardiografie je makroskopické posouzení srdce patologem stále považováno za nejpřesnější metodou hodnocení strukturální srdeční vady. Znalost základní patologické anatomie srdce tak i dnes zůstává důležitým předpokladem adekvátně provedené pediatrické pitvy.
Klíčová slova:
vrozená srdeční vada – patologie – sekvenční segmentální analýz
Suplementární instruktážní videa jsou dostupná ke stažení na www.cspatologie.cz.
Vrozené srdeční malformace (CHD) s incidencí necelé 1 % živě narozených dětí představují nejčastější srdeční onemocnění dětského věku (1). Se vzrůstající kvalitou echokardiografické diagnostiky je značná část vad diagnostikována již in utero a chirurgicky korigována v prvním roce života. Řadu CHD, které dříve bývaly běžnou součástí pitevního provozu (jmenovitě například Fallotovu tetralogii), tak dnes již téměř nevídáme. Nicméně i když je současná echokardiografie velmi přesná v popisu případných srdečních malformací, makroskopické zhodnocení srdce patologem (v rámci pitvy nebo jako explantovaný orgán) je stále považováno za nejpřesnější metodu definitivního zhodnocení strukturální srdeční vady (2,3). Vzhledem k faktu, že mírnější formy CHD mohou být klinicky němé a naopak závažné komplexní malformace mohou vést k časnému abortu bez adekvátní prenatální diagnózy, měla by být znalost základní srdeční anatomie a morfologie nejčastějších CHD v repertoáru nejenom specializovaných center, ale i periferních patologií, které nemusí mít nutně vazbu na nemocnici s kardiovaskulární operativou či jinými formami intervenční kardiologie (4).
Cílem této práce je poskytnout doporučený postup pitvy malformovaného srdce a shrnout nejčastější srdeční vady zejména s ohledem na jejich morfologii.
SEKVENČNÍ SEGMENTÁLNÍ ANALÝZA
Nomenklatura CHD prošla v průběhu posledních několika desetiletí řadou změn a navržené klasifikace byly pokaždé předmětem větších či menších kontroverzí. Tato přetrvávající nespokojenost byla zapříčiněna množstvím CHD a především pak jejich častým výskytem v rámci komplexních malformací. V srdci, ve kterém „nic není normálně“, se totiž dostáváme do situace, kdy jsme nuceni jednu nekonstantní morfologickou strukturu vztáhnout k jiné jednotce, která sama o sobě podléhá značné variabilitě. Složité CHD jako například funkčně jednokomorová cirkulace tak dlouho zůstávaly nepřesně definované. Zásadní zlom přineslo zavedení tzv. sekvenční segmentální analýzy (5-7), jejíž podstata spočívá v rozčlenění srdce na tří základní oddíly (síňový, komorový a arteriální), přičemž každý z oddílů definuje nikoliv jeho topografické uložení, ale morfologicky nejkonstantnější struktura. Jednotlivé CHD jsou pak definovány právě na základě jejich vztahu k těmto embryonálně zakonzervovaným strukturám. Tento postup je využíván dodnes a ukázal se použitelný i pro značně komplexní malformace, ve kterých by za jiných okolností bylo velmi snadné ztratit orientaci (například korigovaná transpozice velkých tepen se zrcadlovým obrazem celého srdce).
Sekvenční segmentální analýza definuje síně nikoliv dle jejich umístění na levé či pravé straně srdce, ale podle morfologie oušek. Pravé ouško má trojúhelníkovitý tvar s širokou bazí, zatímco levé je štíhlé a tubulární. Nicméně, dlouhotrvající abnormální hemodynamické poměry mohou tvar oušek pozměnit, proto je spolehlivější vycházet spíše z charakteru musculi pectinati, které v levé síni zůstávají omezeny na ouško a v pravé síni přesahují i na její volnou stěnu. Tento zdánlivě drobný detail představuje jednu z vývojově vůbec nejkonstantnějších struktur v lidském těle a lze z ní vycházet i v popisu lateralizačních vad. Standardní uložení oušek označujeme pojmem situs solitus. V případě zrcadlového obrazu hovoříme o situs inversus. Pakliže jsou přítomny dvě morfologicky levá či pravá ouška, jedná se o tzv. levostranný či pravostranný atriální izomerismus, který prakticky vždy doprovází abnormální uložení ostatních orgánů v dutině hrudní a břišní v rámci některého ze syndromů heterotaxe. Jelikož syndromy polysplenie/asplenie ne vždy bývají vyjádřeny plně, je vhodné v jejich definici vycházet právě z atriálního uspořádání. Srdeční komory pak definuje charakter apikální trabekularizace. Trabekuly levé komory jsou jemné a delikátní, zatímco v pravé komoře masivnější, s hlubšími recesy a přítomností trabecula septomarginalis. V nejasných případech (které jsou zcela raritní) mluvíme o indeterminované komoře. Arteriální oddíl (aorta a truncus pulmonalis) určuje jejich větvení pro plicní a systémový oběh a odstupy koronárních arterií. Vlastní napojení jednotlivých oddílů pak popisujeme na atrioventrikulární a ventrikuloarterální úrovni. V případě standardního uspořádání mluvíme o konkordanci. Abnormální napojení (např. u transpozice velkých arterií) používáme termín diskordance. V případě izomerismu síní nebo dvojvtokové či dvojvýtokové komoře je pak spojení smíšené (8,9).
OHLEDÁNÍ SRDCE IN SITU
Pečlivé zevní ohledání srdce ještě před jeho vynětím z hrudníku tvoří důležitou součást pitvy (10). Některé CHD je možné diagnostikovat již in situ nebo na ně lze přinejmenším vyslovit suspekci (tab. 1). Naopak neopatrná manipulace se srdcem může poškodit delikátní anatomické struktury a diagnostiku některých CHD zcela znemožnit. Po rozstřižení perikardu a šetrném odstranění brzlíku, nejlépe tupou preparací tak, aby nedošlo k poškození dorzálně probíhající levostranné v. brachiocephalica, zhodnotíme uložení srdce, jeho tvar a směr apexu. Dle tvaru oušek (s jistými limitacemi, viz výše) určíme atriální uspořádání. Komory zevně definovat nelze, můžeme však alespoň zaznamenat jejich tvar a velikost. Průběh tepenného ramus interventricularis anterior určuje umístění mezikomorového septa a tím i vzájemný poměr velikostí komor. Zhodnotíme morfologii, vzájemný vztah a větvení aorty a truncus pulmonalis. Absence pravostranné a. subclavia v jejím obvyklém místě je suspektní z přítomnosti a. lusoria, která probíhá za jícnem a mohla by být přerušena jeho následným rozstřižením. Všímáme si koronárních arterií, které mohou vykazovat různé odchylky v průbězích i v odstupech z jednotlivých Valsavových sinů. Klinicky nejzávažnější variací je odstup koronární arterie (většinou levé) z truncus pulmonalis (11). V neposlední řadě zhodnotíme návrat plicních žil. Prostým nadzdvihnutím srdečního apexu (tzv. manévr Taussigové) lze totiž se značnou mírou pravděpodobnosti vyslovit podezření na anomální návrat plicních žil. Za normálního stavu lze apex nadzdvihnout jen minimálně, aniž bychom pohnuli plícemi. Pokud jde apex nadzvednout zcela volně, je anomální návrat velmi pravděpodobný. Dělíme jej na parciální, kdy atypicky ústí pouze některé plicní žíly, a totální, kdy jsou takto postiženy žíly všechny (12). Ty se většinou za srdcem nejprve spojují v jednu společnou vénu, která pak ústí anomálně do pravé síně (kardiální návrat), do větví horní duté žíly (suprakardiální návrat) nebo do dolní duté žíly (infrakardiální návrat).

Srdce následně vyjmeme en block spolu s krčními orgány, plícemi, a pro zachování kontinuity dolní duté žíly i s horním okrajem jater. V případě mnohočetných vývojových vad (např. u syndromů heterotaxe) je možná kompletní eviscerace v jednom multiorgánovém bloku.
METODY FIXACE
Pro potřeby kardiopatologické pitvy by měla být srdce fixována alespoň 24 hodin a neměla by být pitvána v nativním stavu. Preventivní fixace je vhodná i v případě pouhého podezření na CHD nebo u stavů se zvýšeným rizikem jejího výskytu (např. geneticky potvrzený Downův syndrom). Naše pracoviště dlouhá léta používala Joresův roztok (13), díky kterému byl fixovaný materiál poddajnější a elastičtější než po standardní 10% formolové fixaci. V posledních několika letech jsme jej po vzoru zahraničních pracovišť nahradili 20% pufrovaným formolem. I přes jeho zdánlivě vysokou koncentraci je výsledná konzistence orgánu překvapivě poddajná, umožňující detailní morfologické zhodnocení, a zároveň dostatečně pevná, aby nedošlo k narušení jemných struktur.
VLASTNÍ PITVA SRDCE A POPIS NEJČASTĚJŠÍCHSRDEČNÍCH VAD
Pitva samotného srdce začíná ve většině případů standardní incizí ve tvaru „W“, reprezentující tok krve. Střih vedeme od ústí dolní duté žíly až do apexu pravého ouška. Otevírání horní duté žíly na začátku pitvy není vhodné, jelikož může snadno dojít k přerušení sinoatriálního uzlu, který je uložený nalevo od ostia duté žíly v místě její junkce s okrajem ouška. Pokračujeme rozstřižením vtokové části pravé komory po margo acutus a následně i výtokového traktu až do větvení plicních arterií. Střih je možné prodloužit přes ductus arteriosus a pokračovat jím až do aorty. Levou síň otevřeme od plicních žil do apexu levého ouška. Incize pokračuje po margo obtusus k apexu a odtud podél interventrikulárního septa do aorty (9,10).
Velmi malá srdce lze pitvat s využitím stereomikroskopu nebo celé srdce zalít do parafínového bloku a hodnotit jednotlivé histotopogramy v sériových řezech. Takto lze zhodnotit anatomii srdce již v 8-10. týdnu gestace (10). U plodů se známkami pokročilé macerace je vhodné srdce pitvat v mělké nádobě pod vodou (9). Některá pracoviště v případech těžké macerace využívají možnost post mortem ultrasonografie či magnetické rezonance (14,15).
Defekt síňového septa (ASD)
Atriální septální defekt vzniká nadměrnou apoptózou v embryonálním septum primum (čili v místě budoucí valvula foraminis ovalis) nebo naopak nedostatečným vývojem septum secundum. Při pitvě je patrný jako otvor v místě fossy ovalis. Tím se odlišuje od foramen ovale patens (v pitevním žargonu často s dovětkem per sondam pervium), které je způsobeno nesplynutím foramen ovale s membránou, která jej zakrývá. Membrána je v tomto případě plně vytvořená a defekt tak za normálních okolností zůstává krytý (díky vyššímu tlaku v levé komoře) (16-18).
Vzácnějším typem ASD je defekt sinus venosus. Nachází se ve stěně duté žíly, v blízkosti jejího vústění do pravé síně. V naprosté většině případů postihuje horní dutou žílu. Ve skutečnosti se jedná o poměrně komplexní malformaci, nikoliv jen prostý otvor. Defekt je způsoben levostranným posunem horní duté žíly mezi pravou a levou síň, která tak „nasedá“ nad síňové septum, v kombinaci s anomálním návratem pravostranné plicní žíly, která ústí do stěny duté žíly nebo do místa kavoatriální junkce. Toto spojení duté žíly s v. pulmonalis tak vytváří kanál, který přemosťuje síňové septum (19).
Nejvzácnějším typem ASD je defekt sinus coronarius, který charakterizuje komunikace mezi levou síní a stěnou sinus coronarius (20). Abnormálně dilatovaný sinus coronarius je mimo jiné suspektní i z perzistující levostranné horní duté žíly, po které je v těchto případech zapotřebí aktivně pátrat (21).
Defekt komorového septa (VSD)
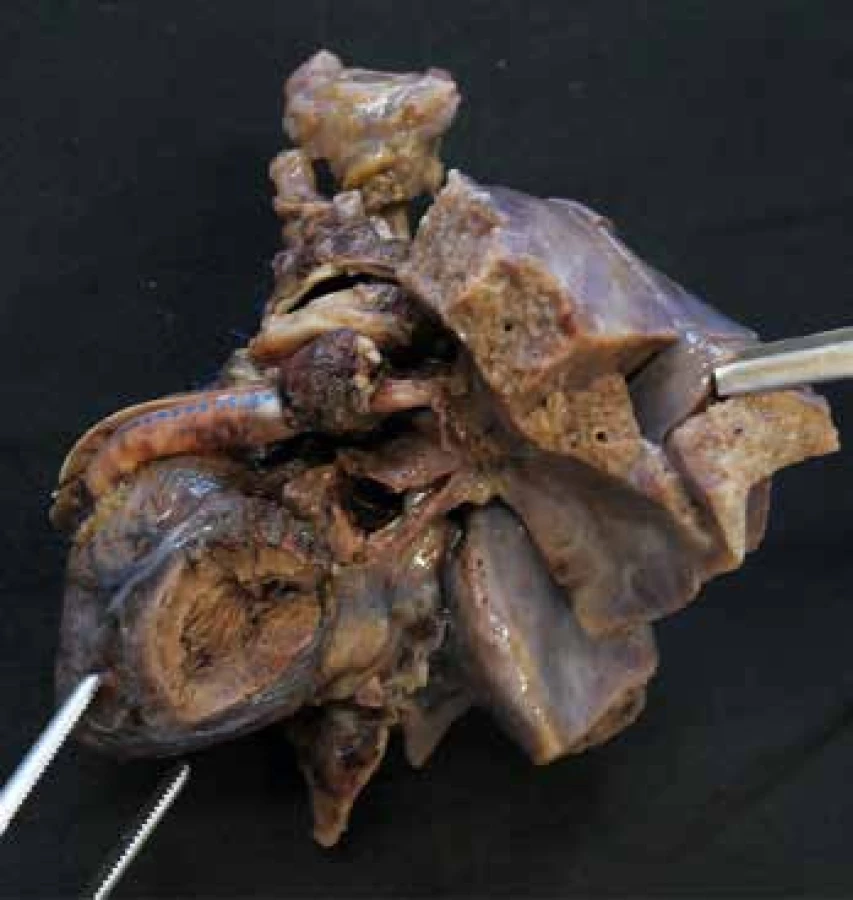
VSD představuje komunikaci na komorové úrovni. Může být solitární i mnohotný, s průměrem od milimetru až po několik centimetrů. Často bývá součástí komplexnějších srdečních malformací. Nejčastější je perimembranózní typ (obr. 1), který postihuje pars membranacea mezikomorového septa a definuje jej fibrózní dolní okraj (zbytek membranózního septa). Většinou se nachází ukrytý pod septálním cípem trikuspidální chlopně, při pitvě je proto vhodné toto místo aktivně sondovat. Méně častý je muskulární typ, který postihuje svalovou část septa a má tak okraje po celém obvodu svalové. Nejvzácnější je juxtaarteriální typ, umístěný v bezprostřední blízkosti aortální a plicní chlopně a definovaný fibrózním stropem, který je kontinuální s odstupem poloměsíčitých chlopní. Velmi rozsáhlé defekty pak mohou být kombinované (například kombinovaný perimembranózní a juxtaarteriální defekt) (22-24).

Atrioventrikulární septální defekt (AVSD)
Tento typ defektu může být obtížnější na představu zejména s ohledem na definici, kterou není komunikace na síňové a komorové úrovni, nýbrž jedna společná atrioventrikulární junkce. Právě společný anulus fibrosis s atypickou pěticípou chlopní namísto oddělené mitrální a trikuspidální chlopně jsou tím, co AVSD charakterizuje, zatímco případný defekt sept není podmínkou. Ve většině případů je přítomná komunikace na síňové i komorové úrovni (obr. 2), ale může být pouze na komorové, pouze na síňové (dříve nesprávně označovaný jako defekt síňového septa typu ostium primum) nebo mohou být obě septa zcela intaktní. Společná atypická chlopeň má dva cípy uložené vpravo, jeden vlevo a dva tzv. přemosťující, které přepažují případný septální defekt a upínají se do papilárních svalů obou komor. V případě podtypu AVSD s intaktním síňovým i komorovým septem může pohled do levé komory imponovat jako rozštěp mitrální chlopně, ten však bývá umístěný v předním cípu směrem k výtokové části levé komory, nikoliv směrem ke komorovému septu jako v případě AVSD (25-28).

Společná atrioventrikulární junkce způsobuje posun aorty z jejího obvyklého umístění v centru srdce směrem ventrálním, čímž dochází k protažení a zúžení výtokového traktu levé komory (dávající obraz tzv. „husího krku“ na zobrazovacích metodách) (29).
Fallotova Tetralogie (ToF)
Stenóza plicnice, hypertrofie pravé komory, defekt komorového septa a nasedající aorta jsou charakteristickými komponentami ToF, které však všechny představují až sekundární změny vyvolané jedinou kongenitální malformací, kterou je abnormální tvar a pozice výtokového septa pravé komory. Toto septum je za normálních okolností drobným výběžkem crista supraventricularis, muskulárního valu přepažujícího vtokový a výtokový trakt pravé komory. V případě ToF je toto septum hypertrofické a posunuté ventrokraniálním směrem. Výsledkem je subvalvární obstrukce truncus pulmonalis, která je posléze umocněna sekundární hypertrofií volné stěny komory. Samotný truncus pulmonalis bývá také obvykle hypoplastický, může však mít normální průměr nebo být i dilatovaný. Doprovodný VSD bývá většinou perimembranózní. Plicní chlopeň bývá v různé míře stenotická nebo i zcela atretická. V případě pulmonální atrézie může chybět i ductus arteriosus a krev se pak dostává do plicní cirkulace výrazně dilatovanými systémovými kolaterálami z descendentní aorty (obdobně jako v případě truncus solitarius, viz níže) (30-33).
Od ToF je potřeba odlišovat pulmonální atrézii s intaktním septem. Neprůchodnost truncus pulmonalis spolu s intaktním komorovým septem může představovat účinnou překážku v toku krve z pravostranného srdečního oddílu a část chorob, v závislosti na velikosti pravé komory, tak lze zařadit do skupiny hypoplastického pravého srdce (34).
Dvojvýtoková pravá komora (DORV)
DORV rozumíme stav smíšeného ventrikuloarteriálního uspořádání, kdy obě velké tepny (přesněji více než 50 % jejich průměrů) odstupují z pravé komory. Tato komora bývá dominantní, hypertrofická a pravidlem je VSD, který může být lokalizovaný pod odstupem aorty, pod plicnicí, mezi nimi nebo v muskulární části komorového septa. DORV s defektem pod odstupem truncus pulmonalis nese název Taussig-Bingova anomálie. DORV se subaortálně lokalizovaným defektem je pak velmi podobný Fallotově tetralogii, a to jak klinickou prezentací, tak i morfologií. Nasedající aorta u TOF totiž může věrně imponovat jako dvovýtoková komora a odlišení těchto dvou chorob spočívá v pečlivém zhodnocení, zda „nasedání“ aorty nepřesahuje 50 % jejího průměru (pak by se jednalo o DORV) nebo jestli není přítomno hypertrofické výtokové septum (které definuje ToF). V případě, že nalezneme obě tyto změny společně, jedná se o kombinovanou vadu (DORV + ToF)(35-37).
Společný arteriální trunkus (PTA)
Tato vada byla diskutována již v sekci o zevním ohledání srdce. Namísto aorty a truncus pulmonalis z baze srdce odstupuje společný arteriální kmen, který dává odstup koronárním arteriím, plicním arteriím a velkým tepnám aortálního oblouku (obr. 3). Chlopeň trunku bývá prakticky vždy abnormální, s variabilním počtem cípů a dysplastická. Téměř vždy vadu doprovází VSD (38,39).

Standardně byl PTA dělen na 4 subtypy podle odstupu plicních arterií. Typ I charakterizují plicní arterie odstupující z jednoho společného kmene, v případě typu II mají dvě blízko uložená, ale separátní ostia na dorzální straně trunku, u typu III pak odstupují plicní arterie nezávisle na sobě. Typ IV, kdy „plicní arterie“ odstupovaly z descendentní aorty, je dnes vyčleněn. Jedná se totiž o formu pulmonální atrézie a „společný trunkus“ je tak ve skutečnosti aortou. Distálně odstupující větve jsou pouze systémové kolaterály (40,41).
Transpozice velkých arterií (TGA)
TGA představuje formu ventrikuloarteriální diskordance, kdy aorta odstupuje z morfologicky pravé a truncus pulmonalis z morfologicky levé komory, nezávisle na jejich uložení v hrudníku. Téměř vždy vadu doprovází nějaká z forem defektu síňového nebo komorového septa (42,43). Velmi časté jsou i přidružené anomálie, zejména obstrukční malformace výtokové části komor v podobě fibrózních a muskulárních valů či prstenců (44,45). Z klinického pohledu jsou pak důležité anomální průběhy koronárních arterií, které u TGA také bývají poměrně frekventované (46).
Vrozeně korigovaná transpozice velkých arterií (CCTGA)
U CCTGA je přítomná diskordance na ventrikuloarteriální i atrioventrikulární úrovni. Čistě z hemodynamického pohledu by se srdce mohlo jevit plně funkční, z pohledu morfologie se však téměř nikdy nejedná o prostou inverzi komor. Prakticky ve všech případech lze nalézt doprovodné malformace, které tuto vadu nějakým způsobem „dekorigují“. Pro CCTGA je obzvlášť typická triáda VSD, pulmonální stenóza a malformace trojcípé chlopně s kaudálním posunem septálního a dorzálního cípu do vtokové části pravé komory, velmi podobná Ebsteinově anomálii (tzv. ebsteinoidní posun trikuspidální chlopně) (47).
Koarktace aorty (CoA)
Koarktací rozumíme zúžení aorty, nacházející se téměř vždy v těsné blízkosti ductus arteriosus. CoA s absencí duktu bývá izolovaná vada, způsobená excesivní retrakcí kolabujícího duktu. Mívá charakter zářezu v blízkosti ligamentum arteriosum. CoA s přítomným duktem, ať už v preduktální nebo postduktální lokalizaci, má komplexnější etiopatogenezu a často přichází v rámci složitých srdečních malformací jako např. syndrom hypoplastického levého srdce. Zúžení může nabývat podoby až extenzivní tubulární hypoplázie aortálního oblouku. V krajním případě může jít až o úplné přerušení (interrupci) aorty, které téměř vždy postihuje isthmickou část oblouku (úsek mezi levostrannou a. carotis communis a levostrannou a. subclavia) (obr. 4). Méně často je zevní tvar aorty normální a koarktaci způsobuje intraluminální membrána. Často je přítomna dvojcípá aortální chlopeň (48-50).

Funkčně jednokomorové srdce
Funkčně jednokomorová cirkulace představuje skupinu závažných kongenitálních malformací, jejichž společným rysem je absence dvou plně vyvinutých a funkčních komor. Termín „jednokomorová“ je tak myšlen ve smyslu funkce, nikoliv morfologie, neboť v drtivé většině případů jsou komory přítomny dvě. Jedna z komor je však různými mechanismy vyřazená z funkce. Skutečně jednokomorové srdce je zcela raritní (51).
Funkčně jednokomorové srdce lze rozdělit do dvou skupin: 1) s jednokomorovým atrioventrikulárním spojením, kam by patřila dvojvtoková komora a atrioventrikulární valvární atrézie, 2) s dvojkomorovým atrioventrikulárním spojením, kam se řadí plicní a aortální atrézie s intaktním komorovým septem. Atretické semilunární či atrioventrikulární chlopně přicházejí často jako součást hypoplastického levého nebo pravého srdce.
Dvojvtoková komora
Dvojvtokovou komoru charakterizuje smíšené atrioventrikulární uspořádání, kdy obě síně ústí do společné komory, která má ve většině případů morfologii komory levé. Druhá komora je zcela rudimentální a s dominantní komorou může komunikovat přes VSD. Síně mohou do komory ústit přes dvě oddělené chlopně. Druhou možností je společné atrioventrikulární spojení v podobě AVSD. Odstupy velkých tepen jsou pak variabilní, mohou být v konkordanci i v diskordanci. V případě dominantní levé komory může být pravá komora umístěna vlevo i vpravo, vždy se však od levé komory nachází kraniálně a ventrálně. Při pitvě srdce, které se na první pohled jeví jako jednokomorové, je po přítomnosti rudimentální komory zapotřebí aktivně pátrat v těchto místech. Pakliže je dominantní komora morfologicky pravá, levá komora může být opět vlevo i vpravo, ale vždy je dorzokaudálně (52-54).
Atrioventrikulární valvární atrézie
Atretická nebo imperforovaná atrioventrikulární chlopeň i v izolované podobě též účinně vyřazuje komoru z funkce a proto spadá do skupiny jednokomorové cirkulace. V případě imperforace je chlopeň přítomna, je však neprůchozí. V případě atrézie pak atrioventrikulární junkce zcela chybí a síň tak lze volně nadzdvihnout od komory. Mnohem častější je atrézie trikuspidální chlopně. Odstupy velkých tepen jsou obdobně jako v případě dvojvtokové komory variabilní. Vadu ve většině případů doprovází alespoň mírná hypoplázie příslušné komory a představuje tak plynulý přechod do syndromu hypoplastického levého či pravého srdce (55,56).
Syndrom hypoplastického levého srdce
Jedná se o skupinu malformací, pro které je společná různá míra hypoplázie levostranného srdečního oddílu. Obvykle je zde zachována atrioventrikulární i ventrikuloarteriálníkonkordance. Jinými slovy, srdce je napojeno normálně, ale levostranný oddíl je variabilně hypoplastický. Změny mohou postihnout jakoukoliv část levého srdce (např. v podobě izolované mitrální atrézie, viz výše), většinou však bývá v různé míře postižený celý levostranný oddíl. Levá síň bývá menší, komora těžce hypoplastická, s endokardiální fibroelastózou a mitrální atrézií (nebo přinejmenším těžkou mitrální stenózou). Aorta bývá užší, se stenotickou či atretickou aortální chlopní a někdy bývá přítomna koarktace. Komorové septum bývá většinou intaktní, VSD bývá vzácný. Častěji je přítomný perzistující ductus arteriosus nebo ASD (57-59).
Pulmonální atrézie s intaktním komorovým septem a syndrom hypoplastického pravého srdce
Jak již bylo zmíněno v kapitole o ToF, pulmonální atrézie s intaktním komorovým septem taktéž vyřazuje pravou komoru z funkce a část případů lze zařadit do skupiny hypoplastického pravého srdce v závislosti na velikosti pravé komory a případnému postižení dalších pravostranných oddílů (např. trikuspidální atrézie). Cca v 10 % případů se trikuspidální atrézie kombinuje s Ebsteinovou anomálií (tzv. imperforovaný Ebstein). Většinou vadu doprovází ASD (60,61).
ZÁVĚR
Moderní diagnostika CHD je dnes převážně klinická, zejména díky stále se zkvalitňující echokardiografii, nicméně úloha patologa i tak zůstává významná. Současné zobrazovací metody totiž zejména prenatálně v nižších gestačních týdnech nedokáží zhodnotit strukturální srdeční vadu s takovou přesností jako adekvátně provedená kardiopatologick á pitva. Část CHD navíc zůstává nediagnostikovaná nebo naopak vedou k doporučení k umělému přerušení těhotenství pro infaustní prognózu. Tyto případy se tak stávají součástí pitevního provozu i na periferních pracovištích. S nutností makroskopického posouzení základní patologické anatomie srdce se tak může setkat každý praktikující patolog.
PROHLÁŠENÍ
Autor práce prohlašuje, že v souvislosti s tématem, vznikem a publikací tohoto článku není ve střetu zájmů a vznik ani publikace článku nebyly podpořeny žádnou farmaceutickou firmou.
Práce byla podpořena MZ ČR – RVO, FN v Motole 00064203.
PODĚKOVÁNÍ
Autoři práce by rádi poděkovali MUDr. Miroslavu Koblížkovi za neocenitelnou technickou pomoc při pořizování audiovizuální dokumentace.
MUDr. Ondřej Fabián
Ústav patologie a molekulární medicíny 2. LF UK a FN Motol
V Úvalu 84, 150 06, Praha 5
tel.: +420 224 435 645
e-mail: Ondrej.Fabian2@fnmotol.cz
Zdroje
1. Krasuski RA, Bashore TM. Congenital Heart Disease Epidemiology in the United States. Circulation 2016; 134(2): 110-113.
2. Ernst LM. A pathologists perspective on the perinatal autopsy. Semin Perinatol 2015; 39(1): 55–63.
3. Taylor GP, Faye-Petersen OM, Ernst L et al. Small patients, complex challenging cases: a reappraisal of the professional efforts in perinatal autopsies. Arch Pathol Lab Med 2014; 138(7): 865-868.
4. Thiene G, Veinot JP, Angelini A et al. AECVP and SCVP 2009 recommendations for training in cardiovascular pathology. Cardiovasc Pathol 2010; 19(3): 129-135.
5. Anderson RH, Shirali G. Sequential segmental analysis. Ann Pediatr Cardiol 2009; 2(1): 24–35.
6. Craatz S, Künzel E, Spanel-Borowski K. Classification of a collection of malformed human hearts: practical experience in the use of sequential segmental analysis. Pediatr Cardiol 2002; 23(5): 483–490.
7. Devine WA, Debich DE, Anderson RH. Dissection of congenitally malformed hearts, with comments on the value of sequential segmental analysis. Pediatr Pathol 1991;11(2): 235–259.
8. Anderson RH, Ho SY. Continuing Medical Education. Sequential segmental analysis - description and catergorization for the millenium. Cardiol Young 1997; 7 : 98-116.
9. Khong TY, Malcombson RDG. Keelings Fetal and Neonatal Pathology (5th edn). Switzerland: Springer; 2015 : 486-488.
10. Erickson LK. An Approach to the examination of the fetal congenitally malformed heart at autopsy. J Fetal Med 2015; 2(3): 135-141.
11. Arey JB. Cardiovascular pathology in infants and children. Philadelphia: WB Saunders Company; 1984 : 77-111, 204–217.
12. DeLisle G, Ando M, Calder AL et al. Total anomalous pulmonary venous connection: Report of 93 autopsied cases with emphasis on diagnostic and surgical considerations. Am Heart J 1976; 91(1): 99–122.
13. Jores L. Die Konservirung anatomischer Präparate in Blutfarbe mittelst Formalin. Centralbl F Allgemeine Path Und Path Anat 1896; 7 : 134.
14. Cain MA, Guidi CB, Steffensen T, Whiteman VE, Gilbert-Barness E, Johnson DR. Postmortem ultrasonography of the macerated fetus complements autopsy following in utero fetal demise. Pediatr Dev Pathol 2014; 17(3): 217–220.
15. Arthurs OJ, Thayyil S, Olsen OE et al. Diagnostic accuracy of post-mortem MRI for thoracic abnormalities in fetuses and children. Eur Radiol 2014; 24(11): 2876–2884.
16. Hagen PT, Scholz DG, Edwards WD. Incidence and size of patent oval foramen during the first decades of life; an autopsy study of 965 normal hearts. Mayo Clin Proc 1984; 59(1): 1489–1494.
17. Fisher DC, Fisher EA, Budd JH et al. The incidence of patent oval foramen in 1,000 consecutive patients. A contrast transesophageal echocardiography study. Chest 1995; 107(6): 1504–1509.
18. Ferreira Martins JD, Anderson RH. The anatomy of interatrial communications - what does the interventionist need to know? Cardiol Young 2000; 10(5): 464-473.
19. Al Zaghal AM, Li J, Anderson RH et al. Anatomic criteria for the diagnosis of sinus venosus defects. Heart 1997; 78(3): 298–304.
20. Lee ME, Sade RM. Coronary sinus septal defect. Surgical considerations. J Thorac Cardiovasc Surg 1979; 78(4): 563–569.
21. Goyal SK, Punnam SR, Verma G, Ruberg FL. Persistent left superior vena cava: a case report and review of literature. Cardiovasc Ultrasound 2008; 6 : 50.
22. Anderson RH, Lennox CC, Zuberbuhler JR. The morphology of ventricular septal defects. Perspect Pediatr Pathol 1984; 8(3): 235–268.
23. Soto B, Becker AE, Moulaert AJ, Lie JT, Anderson RH. Classification of ventricular septal defects. Br Heart J 1980; 43(3): 332-343.
24. Anderson RH, Wilcox BR. The surgical anatomy of ventricular septal defect. J Card Surg 1992; 7(1): 17-35.
25. Becker AE, Anderson RH. Atrioventricular septal defects. What’s in a name? J Thorac Cardiovasc Surg 1982; 83(3): 461–469.
26. Anderson RH, Ho SY, Falcao S, Daliento L, Rigby ML. The diagnostic features of atrioventricular septal defect with common atrioventricular junction. Cardiol Young 1998; 8(1): 33-49.
27. Ebels T, Meijboom EJ, Anderson RH et al. Anatomic and functional “obstruction” of the outflow tract in atrioventricular septal defects with separate valve orifices (“ostium primum atrial septal defect”): a echocardiographic study. Am J Cardiol 1984; 54(7): 843-847.
28. Sigfùsson G, Ettedgui JA, Silverman NH, Anderson RH. Is a cleft in the anterior leaflet of an otherwise normal mitral valve an atrioventricular canal malformation? J Am Coll Cardiol 1995; 26(2): 508-515.
29. Espinola-Zavaleta N, Muñoz-Castellanos L, Kuri-Nivón M, Keirns C. Understanding atrioventricular septal defect: anatomoechocardiographic correlation. Cardiovasc Ultrasound 2008 : 6: 33.
30. Anderson RH, Allwork SP, Ho SY et al. Surgical anatomy of tetralogy of Fallot. J Thorac Cardiovasc Surg 1981; 81(6): 887–896.
31. Emmanouilides GC, Thanopoulos B, Siassi B et al. „Agenesis“ of ductus arteriosus associated with the syndrome of tetralogy of Fallot and absent pulmonary valve. Am J Cardiol 1976; 37(3): 403–409.
32. Liao PK, Edwards WD, Julsrud PR et al. Pulmonary blood supply in patients with pulmonary atresia and ventricular septal defect. J Am Coll Cardiol 1985; 6(6): 1343–1350.
33. Anderson RH, Devine WA, del Nido P. The surgical anatomy of tetralogy of Fallot with pulmonary atresia rather than pulmonary stenosis. J Card Surg 1991; 6(1): 41-59.
34. Daubeney PE, Delaney DJ, Anderson RH et al. Pulmonary atresia with intact ventricular septum: range of morphology in a population based study. J Am Coll Cardiol 2002; 39(10): 1670–1679.
35. Brandt PWT, Calder AL, Barratt-Boyes BG, Neutze JM. Double outlet left ventricle. Morphology, Cineangiography, diagnosis and surgical treatment. Am J Cardiol 1976; 38(7): 897-909.
36. Stellin G, Ho SY, Anderson RH, Zuberbuhler JR, Siewers RD. The surgical anatomy of double-outlet right ventricle with concordant atrioventricular connection and non-committed ventricular septal defect. J Thorac Cardiovasc Surg 1991; 102(6): 849-855.
37. Stellin G, Zuberbuhler JR, Anderson RH, Siewers RD. The surgical anatomy of the Taussig-Bing malformation. J Thorac Cardiovasc Surg 1987; 93(4): 560-569.
38. Crupi G, Macartney FJ, Anderson RH. Persistent truncus arteriosus. A study of 66 autopsy cases with special reference to definition and morphogenesis. Am J Cardiol 1977; 40(4): 569-578.
39. Suzuki A, Ho SY, Anderson RH, Deanfield JE. Coronary arterial and sinusal anatomy in hearts with a common arterial trunk. Ann Thorac Surg 1989; 48(6): 792-797.
40. Collett RW, Edwards JE. Persistent truncus arteriosus: a classification according to anatomic types. Surg Clin N Am 1949; 29(4): 1245–1270.
41. Rossi M, Rossi Filho R, Ho SY. Solitary arterial trunk with pulmonary atresia and arteries with supply to the left lung from both an arterial duct and systemic-pulmonary collateral arteries. Int J Cardiol 1988; 20(1): 145-148.
42. Becker AE, Anderson RH. How should we describe hearts in which the aorta is connected to the right ventricle and the pulmonary trunk to the left ventricle? A matter for reason and logic. Am J Cardiol 1983; 51(5): 911-912.
43. Anderson RH, Henry GW, Becker AE. Morphological aspects of complete transposition. Cardiol Young 1991; 1(1): 41-53.
44. Allwork SP, Bentall HH, Becker AE et al. Congenitally corrected transposition of the great arteries: morphologic study of 32 cases. Am J Cardiol 1976; 38(7): 910–923.
45. Anderson RH, Becker AE, Gerlis LM. The pulmonary outflow tract in classically corrected transposition. J Thorac Cardiovasc Surg 1975; 69(5): 747-757.
46. Massoudy P, Baltalarli A, de Leval MR et al. Anatomic variability in coronary arterial distribution with regard to the arterial switch procedure. Circulation 2002; 106(15): 1980-1984.
47. Anderson RK, Lie JT. Pathologic anatomy of Ebstein’s anomaly of the heart revisited. Am J Cardiol 1978; 41(4): 739–745.
48. Ho SY, Anderson RH. Coarctation, tubular hypoplasia and the ductus arteriosus. Histological study of 35 specimens. Br Heart J 1979; 41(3): 268-274.
49. Pellegrino A, Deverall PB, Anderson RH et al. Aortic coarctation in the first three months of life. An anatomopathological study with respect to treatment. J Thorac Cardiovasc Surg 1985; 89(1): 121-127.
50. Ho SY, Wilcox BR, Anderson RH, Lincoln JCR. Interrupted aortic arch--anatomical features of surgical significance. Thorac Cardiovasc Surgeon 1983; 31(4): 199-205.
51. Jacobs JP, Maruszewski B. Functionally univentricular heart and the fontan operation: lessons learned about patterns of practice and outcomes from the congenital heart surgery databases of the European association for cardio-thoracic surgery and the society of thoracic surgeons. World J Pediatr Congenit Heart Surg 2013; 4(4): 349-355.
52. Van Praagh R, Ongley PA, Swan HJC. Anatomic type of single or common ventricle in man. Morphologic and geometric aspects of 60 necropsied cases. Am J Cardiol 1964; 13(3): 367-386.
53. Anderson RH, Becker AE, Tynan M et al. The univentricular atrioventricular connection: getting to the root of a thorny problem. Am J Cardiol 1984; 54(7): 822–828.
54. Keeton BR, Macartney FJ, Hunter S et al. Univentricular heart of right ventricular type with double or common inlet. Circulation 1979; 59(2): 403-411.
55. Anderson RH, Becker AE, Macartney FJ, Shinebourne EA, Wilkinson JL, Tynan MJ. Is “tricuspid atresia” a univentricular heart? Pediatr Cardiol 1979; 1(2): 51-56.
56. Rigby ML, Carvalho JS, Anderson RH et al. The investigation and diagnosis of tricuspid atresia. Int J Cardiol 1990; 27(1): 1–17.
57. Van Son JAM, Edwards WD, Danielson GK. Pathology of coronary arteries, myocardium and great arteries in supravalvular aortic stenosis. Report of five cases with implications for surgical treatment. J Thorac Cardiovasc Surg 1994; 108(1): 21–28.
58. Elzenga NJ, Gittenberger de Groot AC. Coarctation and related aortic arch anomalies in hypoplastic left heart syndrome. Int J Cardiol 1985; 8(4): 379–393.
59. Aiello VD, Ho SY, Anderson RH, Thiene G. Morphologic features of the hypoplastic left heart syndrome-A reappraisal. Pediatr Pathol 1990; 10(6): 931-943.
60. Bull C, de Leval M, Mercanti C, Macartney FJ, Anderson RH. Pulmonary atresia with intact ventricular septum: a revised classification. Circulation 1982; 66(2): 266-271.
61. Stamm C, Anderson RH, Ho YS. Clinical anatomy of the normal pulmonary root compared with that in isolated pulmonary valvular stenosis. J Am Coll Cardiol 1998; 31(6): 1420–1425.
Štítky
Patologie Soudní lékařství ToxikologieČlánek vyšel v časopise
Česko-slovenská patologie

2019 Číslo 4
Nejčtenější v tomto čísle
- Myokarditida a kardiomyopatie z pohledu kardiologa
- Fumarát hydratáza deficientní karcinom z renálních buněk a jemu podobný karcinom z renálních buněk: Komparativní studie 23 geneticky testovaných případů
- Inflamatorní myofibroblastický tumor dělohy – kazuistika
- Praktický přístup k pitvě srdce s vrozenou srdeční vadou
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy