
Vaskulitidy z pohledu dermatologa
Vasculitis from the Perspective of a Dermatologist
Vasculitides are a heterogeneous group of diseases of various etiologies. The inflammatory process primarily affects the vessel wall, which leads to the occlusion of vascular lumen. As a result of ischemia of the tissue supplied by the affected vessel, clinical conditions with frequent skin lesions arise. Vasculitis may be systemic or affect only one organ. It may be primarily cutaneous, or the skin condition develops secondarily, e.g. due to vasculitis associated with systemic connective tissue disease. The basic classification represents the anatomical pathological differentiaton according to the size of the affected vessel, including arteries, veins, capillaries or postcapillary venules. We distinguish vasculitides of large vessels, representing the aorta and its stem branches, vasculitides of middle vessels represented by vessels in muscles, subcutaneous tissue and deeper dermis. Small vessels vasculitis affecting the skin is generally referred to as leukocytoclastic vasculitis and affects the vessels of the middle and papillary parts of the dermis. One of the most common cutaneous manifestations of vasculitis is palpable purpura, which may be manifestation of small and medium vessel vasculitis. Due to the diverse clinical picture interdisciplinary approach is necessary. The author presents the current state of classification of vasculitides and a brief description of individual vasculitic diseases.
Keywords:
vasculitis – leukocytoclastic vasculitis – palpable purpura – IgA vasculitis – ANCA antibodies
Autoři:
O. Kodet 1,2,3
Působiště autorů:
Dermatovenerologická klinika 1. LF UK a VFN, přednosta prof. MUDr. Jiří Štork, CSc.
1; Anatomický ústav 1. LF UK, přednosta prof. MUDr. Karel Smetana, DrSc.
2; BIOCEV – Biotechnologické a biomedicínské centrum Akademie věd a Univerzity Karlovy ve Vestci u Prahy, vedoucí laboratoře/senior researcher prof. MUDr. Karel Smetana, DrSc.
3
Vyšlo v časopise:
Čes-slov Derm, 96, 2021, No. 3, p. 99-122
Kategorie:
Souborné referáty (doškolování lékařů)
Souhrn
Vaskulitidy představují heterogenní skupinu onemocnění různorodé etiologie primárně postihující zánětlivým procesem cévní stěnu, který vede k uzávěru cévního lumen. V důsledku ischemie tkáně zásobené postiženou cévou vznikají klinické stavy s častými kožními projevy. Vaskulitidy mohou být systémové, či postihující pouze jeden orgán, mohou být primárně kožní, nebo kožní postižení se rozvíjí sekundárně, např. u vaskulitid doprovázející systémová onemocnění pojiva. Základní klasifikace představuje anatomicko-patologické dělení dle kalibru postižené cévy zahrnující arterie, vény, kapiláry či postkapilární venuly. Rozlišujeme vaskulitidy velkých cév, které představují aortu a její kmenové větve, vaskulitidy středních cév představují cévy svalové, podkožní a hlubší dermis. Vaskulitidy malých cév postihující kůži se obecně označují jako leukocytoklastické vaskulitidy a postihují cévy střední a papilární části dermis. Jedním z nejčastějších kožních projevů vaskulitid je palpovatelná purpura, která může být přítomna u vaskulitid malých i středních cév. Vzhledem k rozmanitému klinickému obrazu je nutné klást důraz na mezioborovou spolupráci. Autor uvádí současný stav klasifikace vaskulitid a stručný popis jednotlivých klinických jednotek.
Klíčová slova:
vaskulitidy – leukocytoklastické vaskulitidy – palpovatelná purpura – IgA vaskulitida – ANCA protilátky
ÚVOD
Samotný termín vaskulitida označuje zánětlivý proces poškozující cévní stěnu, doprovázený následnou agregací trombocytů, koagulací a částečným nebo úplným uzávěrem cévního lumen. V důsledku uzávěru cévního řečiště vznikají klinické stavy či syndromy vycházející z ischemie okrsku tkáně zásobeného danou cévou a v důsledku případných zánětlivých změn doprovázejících tato onemocnění. Vedle termínu vaskulitida je nutné odlišit ještě termín vaskulopatie, a to zejména vaskulopatie obliterující, kde dochází primárně k uzávěru cévního lumen trombem (např. patologickou bílkovinou, paraproteinem) a cévní stěna sama o sobě postižená není a zánětlivé změny jsou druhotné. Klinické stavy u obliterujících vaskulopatií mohou být podobné jako u vaskulitidy, jelikož vycházejí primárně z ischemie tkáně zásobené danou cévou. Samotné vaskulitidy se liší svou patogenezí a kalibrem postižené cévy, tyto rozdíly se odráží v klasifikaci těchto onemocnění. Vaskulitidy mohou postihovat malé cévy, středně velké cévy a cévy velkého kalibru arteriálního i žilního systému (obr. 1). Malé cévy představují arterioly, kapiláry a postkapilární venuly, které nacházíme v povrchové a střední části dermis. Středně velké cévy nacházíme v hluboké části dermis a v podkoží. Pod termínem velké cévy rozumíme aortu a její větve zásobující danou topografickou oblast či orgány. Pro kožní vaskulitidy je typické postižení malých nebo středně velkých cév, ale kožní projevy v podobě např. ulcerací lze diagnostikovat i u postižení velkých cév. Vaskulitidy mohou vznikat bez známé etiologie de novo, jako tzv. primární vaskulitidy (např. primární kožní vaskulitidy) nebo se objevují sekundárně v rámci infekčních onemocnění, po podání léčiv (lékové reakce), nebo mohou být projevem systémových onemocnění. Jednotlivé klinické projevy těchto vaskulitid se liší svým původem a etiologií, rozsahem samotného postižení (kožní vaskulitidy, nebo systémové vaskulitidy) a kalibrem postižených cév. Kožní projevy vaskulitid patří mezi nejčastější klinické příznaky i u systémových vaskulitid, proto je nutné problematiku těchto onemocnění akcentovat v rámci dermatologické péče s důrazem na jednotlivé klinické manifestace a diferenciální diagnostiku. Tento přehledový článek shrnuje komplexní problematiku vaskulitid se zahrnutím doplněné klasifikace kožních vaskulitid z roku 2018 [143].
Převzato podle Sunderkötter et al., 2018 [143].
![Schéma postižení cév, rozlišení podle postiženého kalibru cévy<br>
Převzato podle Sunderkötter et al., 2018 [143].](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image_pdf/4a3cfd0c1bafd9220702142a77b58ea2.png)
KLASIFIKACE VASKULITID
V současné době jsou akceptována dvě klasifikační schémata, the American College of Rheumatology z roku 1990 a revize těchto kritérií International Chapel Hill Consensus Conference Nomenclature system z roku 2012 (CHCC2012), které odrážejí rozdíly podle kalibru postižené cévy, a pomáhají tak predikovat a hodnotit klinické projevy jednotlivých vaskulitid [59, 72]. V nedávné době byla publikována dermatologická revize kožních vaskulitid vycházející z CHCC2012, která zohledňuje zejména kožní formy primárních vaskulitid, standardizuje názvosloví, jednotlivé typy, ale již ne diagnostická kritéria [143]. Další kritéria, která patří ke klasifikaci kožních vaskulitid, představují přítomnost celkových příznaků, pozitivita či negativita v přímé imunoflorescenci (PIF) a přítomnost či absence pozitivity protilátek proti cytoplazmě neutrofilních leukocytů (ANCA). Samotnou klasifikaci CHCC2012 s akcentací kožního postižení shrnuje tabulka 1. V této práci bude následně diskutována problematika jednotlivých kožních vaskulitid a systémových vaskulitid s možnou kožní manifestací.
![Vaskulitidy – modifikováno podle Sunderköttera et al. [143]](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image_pdf/dee48a2ad55751e14ba2d7bbac7f476d.png)
Vaskulitidy představují poměrně heterogenní skupinu onemocnění. Podle současné klasifikace se třídí na:
a) vaskulitidy velkých cév,
b) vaskulitidy středních cév,
c) vaskulitidy malých cév,
d) vaskulitidy postihující variabilní cévy,
e) vaskulitidy asociované se systémovými onemocněními,
f) vaskulitidy spojené s pravděpodobnou etiologií a
g) vaskulitidy postihující jeden orgán – kůži (chybí v klasifikaci CHCC2012), viz tabulka 1 [143, 72].
Kožní manifestace mohou doprovázet systémové vaskulitidy (např. IgA vaskulitidu), dále na kůži limitované vaskulitidy či kožně dominantní systémové vaskulitidy (např. kožní forma IgA vaskulitidy) a vaskulitidy postihující jeden orgán s postižením kůže (např. nodulární vaskulitida). Tato poslední jmenovaná skupina vaskulitid není zařazena jako samostatná skupina ve CHCC2012 klasifikaci, ale dokládá vhodný důraz na rozlišení a klasifikaci kožních vaskulitid [143]. Jako systémovou vaskulitidu označujeme vaskulitidu postihující více orgánů či systémů (např. kůže a ledviny u IgA vaskulitidy). U kožně dominantních forem systémových vaskulitid může dojít v průběhu onemocnění a úvodní čistě kožní manifestace k rozvoji plně vyjádřené systémové vaskulitidy s postižením jiných or-gánů, což dokazuje nutnost pečlivého vyšetření pacie-nta a následné sledování. Doprovodné nespecifické celkové příznaky systémových zánětlivých onemocnění, jako je febrilní stav, třesavky, zimnice, artralgie a myalgie, únava, nechutenství či elevace C-reaktivního proteinu (CRP) a trombocytóza, nejsou jednoznačným dokladem systémové vaskulitidy a mohou doprovázet i jen na kůži limitované vaskulitidy.
Epidemiologie
Incidence biopticky verifikovaných vaskulitid se udává zhruba 4,5 postižených na 100 tis. obyvatel/rok a zahrnuje systémové i kožní vaskulitidy [6]. Kožní vaskulitidy postihují všechny věkové skupiny u dospělých průměrně věku 47 let a u dětí v průměru 7 let. Mírně více postihují ženy než muže a jsou daleko častější u dospělých pacientů než u dětí. Nejčastější vaskulitidou v dětském věku je IgA vaskulitida (Henoch-Schönleinova purpura) [20, 131].
Vaskulitidy velkých cév
Velké tepny jsou definovány jako aorta a její hlavní větve, ale vaskulitidy velkých cév mohou postihnout i střední cévy, a dokonce i malé cévy. V dermis a podkožní tkáni se velké cévy nevyskytují, a proto primárně tento typ vaskulitid není řazen mezi typické kožní vaskulitidy, i když při těchto onemocněních mohou být diagnostikovány kožní změny (ulcerace) vyplývající z ischemie daného okrsku tkáně. Dalším příznakem mohou být nespecifické změny v podobě projevů charakteru erythema nodosum. Mezi vaskulitidy velkých cév řadíme Takayasuovu arteriitidu (TAK) a obrovskobuněčnou arteriitidu (GCA). Kožní projevy vaskulitid velkých cév shrnuje tabulka 2.

Takayasuova arteritida (TAK)
Jedná se o vzácnou formu vaskulitidy také známou jako syndrom aortálního oblouku, nebo nespecifická aortoarteritida s postižením aorty a jejích kmenových větví, včetně např. plicních tepen. Etiologie onemocnění je neznámá. Typický je nález granulomatózního zánětu, resp. vaskulitidy se segmentálním postižením velkých cév s masivní intimální fibrózou a zúžením lumen postižené cévy. Onemocnění postihuje nejčastěji asijské ženy mladého až středního věku. Ženy jsou postiženy asi 8–9krát častěji než muži.
V klinickém obraze u některých pacientů dominují nespecifické příznaky, jako je zimnice, třesavka, horečky, bolesti kloubů, úbytek na hmotnosti či noční pocení. Mohou se objevit i synkopy, nebo tzv. „fáze bez pulsu“, která vzniká na podkladě vaskulární nedostatečnosti způsobené zúžením tunica intima postižené cévy. Tento stav se projevuje klaudikací paží nebo dolních končetin, stenózou renální arterie způsobující hypertenzi a neurologickými projevy v důsledku sníženého průtoku krve mozkem. Nejčastějším kožním příznakem je tvorba podkožních nodulů či projevů charakteru erythema nodosum. V těchto případech jsou ale postiženy středně velké arterioly dermis a subcutis, které mohou být doprovázeny navíc i septální či lobulární panikulitidou. U některých pacientů byly popsány projevy charakteru pyoderma gangrenosum [61].
Diagnostika se opírá o zobrazovací metody, zejména pak vyšetření pomocí počítačové tomografie (CT) a magnetické rezonance (MRI). Na CT a MRI je patrné difuzní zesílení arteriální stěny, tzv. „příznak makaronů“. K diagnostice aktivního onemocnění může přispět i vyšetření PET CT s použitím fluor-18-deoxyglukózy (FDG PET CT) [124]. TAK není spojena s pozitivitou protilátek ANCA, antinukleárních (ANA), revmatoidního faktoru (RF), nebo anti-kardiolipinu (ACLA) [16].
U většiny pacientů jsou lékem první volby celkové steroidy (prednison 1 mg/kg/den), z dalších možností byl popsán efekt metotrexátu, azathioprinu, cyklofosfamidu, leflunomidu, mykofenolát mofetilu, inhibitorů tumor necrosis factor (TNFi) nebo tocilizumabu (humanizovaná monoklonální protilátka proti receptoru pro interleukin 6) [8, 134].
Obrovskobuněčná arteriitida, GCA (temporální, Hortonova arteriitida)
Jedná se o vaskulitidu neznámé etiologie postihující velké cévy. Jednou z typicky/nejčastěji postižených arterií je arteria temporalis superficialis, z čehož plyne i historický název temporální arteriitida [72]. Tento typ vaskulitidy postihuje spíše ženy než muže v poměru 2 : 1, starší 50 let.
Příznaky lze rozdělit na kraniální a extrakraniální. Mezi nejčastější příznaky patří bolest hlavy, bolesti spánků, dvojité vidění/poruchy vidění a čelistní klaudikace. Závažnou komplikací GCA může být okluze arteria oftalmica s následnou slepotou, u kmenových tepen pak disekce aorty či její aneuryzma nebo klaudikační bolesti při postižení tepen končetin [153]. GCA se často vyskytuje s revmatickou polymyalgií [152]. Kožní postižení představují nekrózy a ulcerace z ischemie tkáně zásobené poměrně velkou tepnou [61].
Diagnostika se opírá o biopsii postižené tepny (např. a. temporalis superficialis) nebo o vyšetření zobrazovacími metodami. K dostatečné diagnostice je doporučována excize cca 1,5–3cm části postižené tepny k histologickému vyšetření a detekci obrovskobuněčného granulomu ve stěně cévy. Histologické vyšetření může být až ve 30 % neprůkazné [14, 108]. K pomocným vyšetřením opět patří vyšetření CT nebo MRI vyšetření aorty, FDG PET CT či ultrazvukové vyšetření temporálních a krčních tepen. Z laboratorních vyšetření může být zvýšená sedimentace erytrocytů (FW), elevace CRP, destiček (PLT) nebo jaterních enzymů [153].
V terapii se nejvíce uplatňují systémové kortikoidy perorálně v dávce 40–60 mg na den nebo v pulzní intravenózní léčbě. Z dalších léku je asi nejvíce dokumentován efekt metotrexátu v monoterapii či v kombinaci s celkovými kortikoidy a s prokázaným kortikoidy šetřícím efektem. Z biologických léčiv byl popsán efekt při použití tocilizumabu [140].
Vaskulitidy středně velkých cév
Vaskulitidy středně velkých cév postihují primárně středně velké cévy, ale mohou postihovat i cévy jakékoliv velikosti s dominantním kožním postižením. Do této skupiny patří polyarteriitis nodosa (PAN) s možným postižením kůže a Kawasakiho nemoc (Kawasaki disease, KD) s rozvojem nevaskulitických kožních projevů označovaných jako mukokutánní uzlinový syndrom. Kožní postižení vaskulitid středně velkých cév shrnuje tabulka 3.

Polyarteriitis nodosa (PAN)
Polyarteriitis nodosa je systémová vaskulitida charakterizovaná segmentální nekrotizující vaskulitidou postihující středně velké cévy. Kožní forma PAN je na kůži limitované postižení s benigním, ale chronickým průběhem. Některé práce ale uvádějí, že se jedná spíše o onemocnění s možným postižením nervové a svalové tkáně než jen čistě kožní variantu PAN [4].
Incidence klasické (systémové) formy PAN se uvádí 0,4–1,6 na 100 tis. obyvatel/rok, častěji postihuje muže než ženy v poměru 4 : 1 mezi 40–60 roky věku života. Kožní forma představuje asi 10 % všech PAN a je častější u dětí než dospělých pacientů.
V patogenezi onemocnění se uplatňují prodělané infekce, zánětlivá onemocnění, léky nebo maligní, zejména hematoonkologická onemocnění (leukemie z vlasatých buněk) [147]. Infekce virem hepatitidy B (HBV) je popsána asi u 7 % PAN a vzhledem k častějšímu následnému postižení gastrointestinálního traktu, nervového systému či ledvin představuje zvláštní klinickou formu [141]. Pacienti s HBV a PAN mají významně snížené celkové přežití než pacienti s PAN bez HBV [76, 129]. Z dalších infekcí je popsána asociace s infekcemi hepatitidou C (HCV) či lidským virem imunodeficience (HIV) nebo parvovirem B19 [7, 126, 141, 146]. U kožních forem, zejména u dětí, je PAN spojována se streptokokovou infekcí. Z léků byla PAN popsána po podání např. minocyklinu [1].
V klinickém obraze, i u systémové varianty, může dominovat postižení kůže, což dokládá studie na 348 pacientech, kde bylo kožní postižení detekováno u 50 % pacientů [110]. Nejčastější kožní projevy představuje palpovatelná purpura, dále livedo racemosa, zánětlivá forma retiformní purpury, ostře vyseknuté vředy, dále mohou být patrné podkožní zánětlivé noduly nebo nekrózy prstů doprovázející častěji systémovou variantu [61, 110] (obr. 2a). Palpovatelná purpura je nejčastější kožní příznak u PAN a představuje až 19 % kožních projevů [82]. Z celkových příznaků dominuje horečka, bolesti kloubů a svalů, parestezie, úbytek na váze či bolesti břicha, nebo varlat, které v menší míře mohou doprovázet i kožně limitovanou variantu onemocnění. U systémové formy (klasické) může být multiorgánová manifestace s typickým postižením ledvin v podobě uzávěru interlobární arterie. Toto postižení vede k renovaskulární hypertenzi a renálnímu selhání. U PAN není přítomna glomerulonefritida [95]. Postižení gastrointestinálního traktu v podobě ischemie mezenterické tepny je spojené se špatnou prognózou a přežitím jednoho roku méně jak u 50 % pacientů s touto manifestací [129, 132]. U pacientů s infekcí HBV byla popsána asociace s postižením varlete [15].

Kožní forma představuje 10 % všech PAN a je charakterizovaná projevy livedo racemosa, kožními nekrózami a podkožními noduly postihujícími zpravidla bérce. Postižení je častější u dětí a je asociované se streptokokovou infekcí. Kožní forma má navíc výraznější tendenci k chronickému průběhu [36]. Doposud není zřejmé, zda tato limitovaná forma může přecházet v systémové postižení, vzhledem k doposud jasně dokumentovanému jedinému případu [4].
U klasické PAN nacházíme v laboratoři nespecifické změny charakteru leukocytózy, trombocytózy a elevace CRP. ANCA protilátky jsou u klasické formy negativní, ale u kožní formy je lze detekovat až ve 20 % případů – může se jednat v 10–20 % jak o ANCA protilátky s perinukleární fluorescencí (pANCA) většinou proti myeloperoxidáze (MPO), tak i v 10–20 % o ANCA protilátky s cytoplazmatickou fluorescencí (cANCA) většinou proti proteináze-3 (PR3), mohou být přítomny i antifosfolipidové protilátky (aPL), nebo protilátky proti kardiolipinu (ACLA) ve třídě IgG [79].
Histologicky lze prokázat nekrotizující vaskulitidu středně velké arterie, v případě kožního postižení lokalizovanou v hlubší dermis či podkožní tukové tkáni (obr. 3). Přímá imunoflorescence může vykazovat depozita komplementu C3, imunoglobulinu typu M (IgM) a fibrinu v nebo v okolí cévní stěny. Do diferenciální diagnostiky kožních projevů charakteru livedo racemosa patří cholesterolové embolizace, kalcifylaxe, antifosfolipidový syndrom a další (obliterující) vaskulopatie [141].

V léčbě u systémové formy se uplatňují s dobrým efektem systémové kortikoidy v dávce 1 mg/kg. U pacientů bez terapeutické odpovědi lze vyzkoušet v kombinaci s celkovými kortikoidy i terapii cyklofosfamidem [102, 110]. Ve třetí linii léčby lze využít terapii azathioprinem, mykofenolát mofetilem, metotrexátem nebo ojediněle interferonem alfa. Z biologik byl popsán efekt u mepolizumabu, rituximabu (anti-CD20), infliximabu (anti-TNF), tocilizumabu či tofacitinibu (inhibitor Janus kináz, JAKi) [33, 44, 85, 121]. U kožně limitované PAN s mírným až středně závažným průběhem nejsou celkové kortikoidy doporučovány a preferují se nesteroidní antirevmatika či kolchicin [100, 102].
Kawasakiho nemoc (KD)
Jedná se o vaskulitidu neznámé etiologie postihující především kojence a malé děti. U 80 % pacientů je věk nižší než 5 let [138]. I když onemocnění postihuje pacien-ty všech ras, existují mezi jednotlivými populacemi výrazné rozdíly. V Japonsku, kde bylo onemocnění popsáno, je incidence uváděna až 265 případů na 100 tis. obyvatel ročně [94]. Ve Spojených státech a Evropě pak okolo 19 případů na 100 tis. obyvatel ročně [144].
V etiopatogenezi jsou diskutovány vlivy infekční odpovědné za katar horních cest dýchacích, nebo antigeny z prostředí. Především o této teorii se hojně diskutuje, jsou zvažovány velké troposférické větry vanoucí ze severovýchodní provincie v Číně, která slouží jako zdrojový region pro sezonní shlukování alergenů a každoroční epidemii případů KD v Japonsku, na Havaji a v jižní Kalifornii [122, 125]. V současné době je nutné zmínit obdobné postižení zahrnující mukokutánní příznaky v rámci multisystémového zánětlivého syndromu (multisystem inflammatory syndrome in children, MIS-C), postihující zejména děti při infekci SARS-CoV-2 [120].
KD postihuje středně velké cévy, typicky koronární arterie, z čehož pramení závažné postižení myokardu. V klinickém obraze onemocnění začíná jako několik dní trvající (5 dní) horečnatý stav, který omezeně reaguje na běžná antipyretika. Mezi další běžné příznaky patří zvětšené lymfatické uzliny na krku, exantém v genitální oblasti s konjunktivitidou, začervenání rtů, dlaní nebo plosek. Na kůži a sliznicích dominují příznaky označované jako mukokutánní uzlinový syndrom. Nejběžnějším projevem na kůži je difuzní makulopapulózní exantém trupu s progresí na končetiny, obličej a anogenitální oblast [47]. Byl popsán i urtikariální exantém, multiformní projevy nebo pustulózní až vezikulózní exantém [87].
Vedle klinického nálezu se diagnostika opírá o laboratorní vyšetření s nálezem normocytární anemie, trombocytózy, elevace CRP, jaterních enzymů a nízké hladiny albuminu [86]. Elektrokardiogram (EKG) může vykazovat známky komorové dysfunkce nebo příležitostně arytmie způsobené myokarditidou. Echokardiografické vyšetření může vykazovat jemné změny koronárních tepen nebo později skutečné aneuryzma, pro jejichž diagnostiku se provádí rutině angiografie [47, 105].
V léčbě se nejvíce uplatňují intravenózně aplikované imunoglobuliny, kyselina acetylsalicylová a celkově podávané kortikoidy, případně cyklosporin [55]. Zásadní je včasné rozpoznání příznaků a zahájení léčby, což snižuje možnost rozvoje koronárního aneurysmatu [105].
Vaskulitidy malých cév
Vaskulitidy malých cév postihují převážně malé cévy, definované jako malé intraparenchymatózní tepny, arterioly, kapiláry a postkapilární venuly. Mohou být postiženy i střední tepny a vény. V základním schématu rozlišujeme dvě imunopatologické jednotky, a to ANCA asociované vaskulitidy a imunokomplexové vaskulitidy. Kožní vaskulitidy malých cév jsou označované jako leukocytoklastické vaskulitidy. Leukocytoklasie označuje histologicky odlišný typ rozpadu leukocytů charakterizovaný karyorrhexis. Světelné a elektronové mikroskopické rysy leukocytoklasie připomínají apoptotickou buněčnou smrt s kondenzací chromatinu, fragmentací jáder, smrštěním degranulované cytoplazmy a neporušenými membránami a organelami. I když tento termín podle některých autorů zahrnuje pouze imunokomplexové vaskulitidy, je plně akceptován v současné klasifikaci vaskulitid [72, 143]. Kožní projevy těchto vaskulitid shrnuje tabulka 4.

Dále se můžeme setkat i s termínem lymfocytární vaskulitida, resp. vaskulitida zprostředkovaná lymfocyty, která zůstává do značné míry nedefinována. To může být způsobeno udržováním stereotypního obrazu lymfocytární vaskulitidy jako obrazu spojeného s vaskulární nekrózou, depozity fibrinu, hemoragií a pouze nahrazením neutrofilů lymfocyty. Vymezení lymfocytární vaskulitidy může být rozšířeno určením klinického prostředí, ve kterém se takové reakce mohou vyskytnout, jako jsou autoimunitní kožní onemocnění, nebo např. kožní projevy reakce štěpu proti hostiteli (GvHD). Lymfocytární endovaskulitida, lymfocytární lichenoidní vaskulitida a granulomatózní vaskulitida jsou potenciálními příklady lymfocytární vaskulitidy, které se liší od histologického vzorce pozorovaného v souvislosti s imunokomplexovou leukocytoklastickou vaskulitidou především výrazným lymfocytárním infitrátem [84].
ANCA asociované vaskulitidy
Pro tento typ vaskulitid je typické postižení jak malých cév, tak i možné postižení středně velkých cév, které spojuje pozitivita ANCA protilátek a překrývající se spektrum orgánového postižení. Mezi ANCA asociované vaskulitidy patří mikroskopická polyangiitida (MPA), granulomatóza s polyangiitidou (GPA, dříve označované jako Wegenerova granulomatóza) a eozinofilní granulomatóza s polyangiitidou (EGPA, dříve označovaná jako Churg-Straussové syndrom, nebo alergická granulomatóza).
Mikroskopická polyangiitida (MPA)
Jedná se o vaskulitidu postihující malé a středně velké cévy s typickým postižením kůže, plic a ledvin. U některých případů s postižením pouze středně velkých cév bez případné glomerulonefritidy nelze postižení zcela odlišit od PAN [150].
Incidence se pohybuje mezi 0,3–2,4 postiženými na 100 tis. obyvatel za rok, s výraznějším postižením mužů než žen, průměrného věku 57 let. U starších pacientů je častější postižení ledvin [70]. Etiologie není známá, je popsána souvislost s infekční endokarditidou, nebo nádorovými onemocněními (lymfomy, karcinomatóza) a léky (levamizol, kokain, amfetaminy) [70, 139]. Zásadní roli hraje pravděpodobně tvorba ANCA protilátek. První krok zahrnuje vystavení neutrofilů zánětlivým cytokinům, což vede k povrchové expozici kryptogenních antigenů, jako je myeloperoxidáza (MPO). Dále predisponující genetické, environmentální a další faktory vedou k produkci specifických protilátek proti MPO. Ve druhém kroku tyto protilátky proti MPO způsobují poškození cévní stěny vazbou neutrofilů na endoteliální epitel [148].
V klinickém obraze dominují u většiny pacientů celkové příznaky, jako je horečka, úbytek hmotnosti, artralgie a myalgie, které samotnému onemocnění předchází měsíce až roky. Kožní postižení se objevuje asi u poloviny pacientů (20–70 %) v podobě palpovatelné purpury nejčastěji na dolních končetinách. Některé práce uvádějí i rozvoj erytematózních makul (cca u 50 % pacientů), livedo racemosa (17 %), třískovitých hemoragií nehtů, akrálních erytematózních makul, tvorbu ulcerací nebo urtikariálního exantému (cca u 6 % pacientů) – obr. 4, 5, 6. Kožní projevy se u většiny pacientů rozvíjí až po postižení ledvin nebo plic [78, 107]. Rozvoj kožních projevů je spojován se zvýšeným výskytem artralgií, očním postižením a rozvojem mononeuritis multiplex [82]. Postižení ledvin je diagnostikováno prakticky u všech pacientů s MPA s rozvojem srpkovité a nekrotizující glomerulonefritidy. Plicní postižení (asi do 50 % pacientů) je doprovázeno dušností a tvorbou plicních infiltrátů na podkladě kapilaritidy, která může být až zdrojem difuzního alveolárního krvácení. Progresivní klinický průběh MPA obvykle vede k selhání ledvin, a/nebo plicnímu krvácení. Plicní postižení MPA je patrné asi u poloviny pacientů. Časté je také neurologické postižení, které se obvykle projevuje jako periferní neuropatie nebo mononeuritida u třetiny pacientů [66, 77].



Typicky detekujeme pozitivitu ANCA protilátek (vzácně mohou být pacienti negativní). Častěji jsou pacienti pozitivní na protilátky proti MPO (50–80 %, s perinu-kleární fluorescencí ANCA protilátek, pANCA), méně často pak na protilátky proti proteináze-3 (PR3, 10–20 % s cytoplazmatickou fluorescencí ANCA protilátek, cANCA). Mezi pomocná vyšetření patří CT vyšetření hrudníku a břicha, případně sonografické vyšetření ledvin. Hodnoty IgE jsou v normě, což napomáhá v diferenciální diagnostice jiných ANCA asociovaných vaskulitid [49]. V močovém sedimentu nacházíme hematurii. Při bioptickém vyšetření je patrna segmentální nekrotizující vaskulitida (bez granulomatózního zánětu) malých cév (kapilár, venul, arteriol), vzácněji poté i středně velkých tepen [49, 77].
Léčba MPA je rozdělena do dvou fází: indukce remise a udržovací terapie. K navození remise se zpočátku používají glukokortikoidy (např. 1 mg/kg/den prednisonu) v kombinaci s cyklofosfamidem u pacientů s významným postižením ledvin nebo plic. Cyklofosfamid lze podávat po dobu 6 měsíců buď perorálně (1,5–2 mg/kg/den),nebo jako intravenózní pulzní terapie (0,5–1 g/m2, každé 4 týdny). Další alternativou je kombinace celkových glukokortikoidů s rituximabem, který má obdobný efekt jako cyklofosfamid [142]. V udržovací terapii se uplatňuje metotrexát, azathioprin, mykofenolát mofetil, nebo intravenózní imunoglobuliny (IVIg), které navíc prokázaly kortikoid šetřící efekt [49, 77, 111].
Granulomatóza s polyangiitidou (GPA)
Granulomatóza s polyangiitidou, dříve označována jako Wegenerova granulomatóza, je popisována jako triáda postižení charakterizovaná granulomatózním zánětem horních a dolních cest dýchacích, systémovou nekrotizující vaskulitidou malých cév a rychle progredující glomerulonefritidou (pauciimunitní, při nichž je imunofluorescenční histochemický bioptický nález negativní, ačkoliv jde o autoimunitní onemocnění). Od míry postižení orgánů se odvíjí i prognóza onemocnění a jeho průběh. Při systémovém postižení je GPA spojena i s vysokou úmrtností. Limitovaná forma je spojena s postižením plic (dýchacích cest) bez projevů vaskulitidy [51].
Incidence se pohybuje od 0,5 do 1,2 případů na 100 tis. obyvatel/rok, s mírně častějším postižením žen ve 4. až 6. decenniu [93]. V dětském věku je GPA považována za mírně častější systémovou vaskulitidu než u dospělých, dosahující incidence až 3,2 na 100 tis. obyvatel/rok [18].
Patogeneze GPA se odráží ve dvou klinicko-patologických charakteristikách onemocnění, a to při tvorbě granulomů a při vzniku vaskulitidy postihující malé a střední cévy. Přítomnost ANCA protilátek, vlivy zevního prostředí a přítomnost predisponujících genetických polymorfismů přispívá k imunitní dysregulaci. Dalším zjištěným faktorem je snížená aktivita inhibitoru proteinázy alfa-1-antitrypsinu [101]. Přestože patogenní role anti-PR3 protilátek zůstává nejasná, povrchová exprese PR3 na apoptotických a aktivovaných neutrofilech může stimulovat zrání dendritických buněk s následným uvolněním prozánětlivých cytokinů antigen prezentujícími buňkami. Posléze může docházet k neutrofilní aktivaci s podporou tvorby granulomů pomocí Th1-lymfocytů. Exprese PR3 na povrchu neutrofilů omezuje fagocytózu, což může potencovat tvorbu autoprotilátek. V další imunopatologické reakci dochází k vazbě ANCA protilátek na PR3 na neutrofilech a tím dochází k jejich degranulaci a poškození cévní stěny [71, 103].
Diagnostická kritéria pro GPA požadují přítomnost alespoň 3 kritérií z 6 a zahrnují: zánět horních cest dýchacích, typické radiologické změny na RTG plic či CT hrudníku, abnormální biochemické vyšetření močového sedimentu, biopticky verifikovaný granulomatózní proces; stenózu dýchacích cest (zejména u dětí) a sérologické nálezy (např. anti-PR3 protilátky) [18, 93].
V klinickém obraze mohou být u 10–40 % pacientů přítomny mukokutánní projevy. V kožním postižení dominuje palpovatelná purpura. V dutině ústní nacházíme aftózní projevy s překrvením gingivální sliznice (jahodové dásně) [29]. U některých pacientů byly popsány bolestivé podkožní vředy a noduly, které se podobají pyoderma gangrenosum, nebo papulonekrotické léze vyskytující se na končetinách (zejména loktech), které však mohou postihnout i obličej [29, 42]. U dětí s lokalizovanou GPA může být přítomen edém a infiltrace horních víček, či akneiformní projevy [155].
Postižení horních nebo dolních cest dýchacích se vyskytuje až u 90 % pacientů. Při postižení horních cest dýchacích mohou být přítomné recidivující epistaxe, ulcerace sliznice nosní, vedoucí až v perforaci nosní přepážky či deformaci nosního sedla. Pacienti s plicním postižením vykazují dušnost, kašel, hemoptýzu nebo až pleuritidu. Při RTG hrudníku jsou patrné nepravidelné zánětlivé infiltráty a hilová lymfadenopatie. Postižení ledvin se rozvíjí asi u 20 % pacientů, ale u většiny z nich je následně přítomna glomerulonefritida [38, 57]. Mezi další orgánové systémy postižené při GPA patří manifestace muskuloskeletální (70 %), oční (30–60 %), neurologické (20–50 %), gastrointestinální (5–10 %) a kardiální (5–40 %) [38, 57]. V laboratorním vyšetření je patrna elevace CRP, anemie, leukocytóza. Naopak eozinofily a sérová hladina IgE jsou v normě. Častěji jsou přítomny anti-PR3 protilátky (cANCA, 80 až 90 %) než anti-MPO (pANCA, 10–20 %) protilátky. Pacienti s postižením ledvin mají přítomnou proteinurii, hematurii s glomerulární erytrocyturií v močovém sedimentu [133]. Ačkoli většina bioptických vzorků může vykazovat nespecifické histopatologické změny (např. perivaskulární lymfocytární infiltráty), až u 50 % je přítomna leukocytoklastická vaskulitida nebo granulomatózní forma zánětu [96].
Základem léčby je systémová léčba glukokortikoidy (1 mg/kg/den) v kombinaci s cyklofosfamidem, podávaným v perorální formě nebo pulzní terapii, která vykazuje nižší toxicitu. Tato kombinovaná léčba navozuje remisi onemocnění až u 75 % pacientů s mírou přežití až 90 % [12, 18, 57]. Podobného efektu je dosaženo i při kombinaci celkových kortikoidů a rituximabu, který však nemá tak výrazný efekt na granulomatózní zánětlivé projevy [58, 142]. Rychlé navození remise a kortikoid šetřící efekt byl pozorován v jedné prospektivní studii u terapie s infliximabem [10], avšak pro doporučení infliximabu v léčebném algoritmu GPA je zatím málo důkazů. Azathioprin se používá spíše v udržovací terapii remise onemocnění a metotrexát je uváděn jako lék volby u limitované formy GPA [98].
Eozinofilní granulomatóza s polyangiitidou, EGPA
Eozinofilní granulomatóza s polyangiitidou, označovaná dříve jako Churg-Straussové syndrom, nebo Churg-Straussové vaskulitida, je charakterizována tvorbou vaskulárních a extravaskulárních granulomů, dále tvorbou plicních infiltrátů s eozinofily a nekrotizující vaskulitidou, která postihuje malé a středně velké cévy. Jedná se o systémové onemocnění, které bývá spojeno s astma bronchiale a eozinofilií [106].
Incidence onemocnění se pohybuje do 0,3 případů na 100 tis. obyvatel/rok, bez predilekce pohlaví, průměrného věku 48 let. Výskyt EGPA u pacientů s astma bronchiale se pohybuje od 3,5 do 6,5 astmatiků na 100 tis. obyvatel ročně [41].
Vznik onemocnění je spojován s různorodými spouštěcími faktory, jako je stav po očkování, desenzibilizační terapie, terapie inhibitory leukotrienů či rychlé vysazení celkových glukokortikoidů. V samotné patogenezi se uplatňuje role T-lymfocytů, eozinofilů a tvorba ANCA protilátek. Pravděpodobně klíčovou roli sehrávají eozinofily, které po degranulaci poškozují tkáň a následně Th2-lymfocyty přispívají k tvorbě granulomů. Aktivace neutrofilů vede prostřednictvím ANCA protilátek, podobně jako v předchozích případech, k poškození cévní stěny a vzniku vaskulitidy [48, 106].
Průběh onemocnění a klinický obraz lze rozdělit do tří po sobě následujících fází:
1. fáze – dominují příznaky alergické rýmy, nosní polypy a zhoršení nebo rozvoj astmatických příznaků. Tyto projevy mohou přetrvávat i roky;
2. fáze – je patrna periferní eozinofilie, recidivující infekce dýchacích cest a gastrointestinální příznaky, jako je nechutenství, nevolnosti a průjmovité sto-lice;
3. fáze – již se rozvíjí systémová nekrotizující vaskulitida s typickým granulomatózním zánětem. Třetí fáze onemocnění může nastat až několik let (i desetiletí) po úvodních nespecifických příznacích onemocnění [34, 41].
Kožní projevy se vyskytují u 40–75 % pacientů, zpravidla až ve třetí fázi onemocnění. Opět je nejčastějším příznakem palpovatelná purpura s postižením dolních končetin, s možným rozvojem až nekrotických projevů a ulcerací [11]. Na končetinách se mohou utvářet podkožní noduly, livedo racemosa, retiformní purpura či papulonekrotické léze. Méně často lze pozorovat urtikariální projevy [27]. Kromě postižení dýchacích cest mají pacienti s EGPA často neurologické obtíže, které se projevují jako mononeuritida. Kardiální postižení se projevuje jako kardiomyopatie nebo perikarditida s výraznou dušností a atypickými bolestmi na hrudi. Kardiální postižení, pro které je dominantní granulomatózní forma zánětu, je přítomno až u poloviny pacientů ve třetím stadiu onemocnění a je hlavní příčinou úmrtí [17, 52]. Nekrotizující glomerulonefritida a plicní kapilaritida s hemoptýzou v důsledku difuzního alveolárního krvácení se vyskytují méně často než u jiných ANCA asociovaných vaskulitid (asi u 30–35 %). Vzácnější je i gastrointestinální a oční postižení [41, 48].
Laboratorní nálezy jsou obdobné jako u předchozích ANCA asociovaných vaskulitid. Dominantním nálezem je však výrazná eozinofilie (> 109/l) a zvýšená sérová hladina IgE, které toto onemocnění odlišuje od MPA a GPA. Anti-MPO (pANCA, v 60 %) protilátky jsou pozorovány častěji než anti-PR3 protilátky (cANCA, v 10 %) na rozdíl od GPA. Navíc pozitivita ANCA protilátek byla pozorována více u pacientů se srdečním postižením, rozvojem pleuritidy a tvorbou livedo racemosa. U ANCA negativních pacientů (30 %) byla častěji pozorována palpovatelná purpura, postižení neurologické a ledvin. Histopatologický průkaz nekrotizující vaskulitidy je častěji pozorován u pacientů ANCA pozitivních (cca 70 %) oproti pacientům ANCA negativních (30 %), kde byla pozorována jen asi u 40 % pacientů. Granulomatózní forma zánětu je přítomna bez asociace s ANCA pozitivitou či negativitou. V histologii dominují infiltráty eozinofilů, tvorba extravaskulárních granulomů a nekrotizující vaskulitida malých až středních cév s možným postižením tepen i žil. Bioptické vzorky získané z papulonekrotických lézí vykazují infiltraci eozinofilů, s tvorbou granulomů a eozinofilní nekrobiózou [27, 41, 45, 106]. V diferenciální diagnostice je nutné odlišit hypereozinofílii a hypereozinofilní syndrom.
Většina pacientů reaguje na léčbu celkovými glukokortikoidy, i když relapsy se mohou objevit až u čtvrtiny pacientů dosahujících remise. Pacienti s těžkým systémovým postižením jsou léčeni cyklofosfamidem v pulzním podání [22]. Jako alternativní (udržovací a kortikoidy šetřící) terapii lze použít azathioprin (případně metotrexát, leflunomid, mykofenolát mofetil), mepolizumab (anti-IL-5) [41], IVIg nebo interferon-α. Rituximab v terapii tohoto onemocnění neprokázal takový efekt jako u předchozích ANCA asociovaných vaskulitid [23]. V jedné studii byl prokázán i efekt omalizumabu (anti-IgE), navzdory možnému časnému relapsu onemocnění po této terapii [21].
IgA vaskulitida
IgA vaskulitida, dříve označovaná jako Henoch-Schönleinova purpura (HSP), je charakterizovaná depozity IgA imunokomplexů ve stěně malých cév. Pro tuto vaskulitidu je typická tetráda, skládající se z palpovatelné purpury, artritidy, bolestí břicha a hematurie. IgA vaskulitida postihuje typicky děti po infektu respiračních cest, ale může se vyskytovat v jakémkoliv věku. Incidence IgA vaskulitidy v dětském věku je odhadována na 3 až 27 případů na 100 tis. dětí ročně, zatímco u dospělých pacientů je tato vaskulitida pozorována s incidencí jen 0,8 až 1,8 na 100 tis. obyvatel ročně [88, 113, 128 ]. Výskyt u dětí dominuje v 90 % ve věku do 10 let, s mírnou prevalencí mužského pohlaví jak u dětí, tak dospělých pacientů [88, 128].
Zejména v dětském věku hraje roli v patogenezi onemocnění parainfekční etiologie. Projevy vaskulitidy se často objevují 1 až 2 týdny po infekci horních cest dýchacích. Nejvíce je zvažována infekce beta-hemolytickým streptokokem skupiny A, především pro přítomné vyšší hladiny antistreptolysinu O [16, 128]. Zásadní roli IgA (především IgA1) dokazují práce poukazující na zvýšené hladiny IgA v séru pacientů s touto vaskulitidou, i když zásadnější je vazba ve tkáních než cirkulující hladiny IgA [19]. Nedostatek glykosylace vazebné oblasti IgA může podporovat tvorbu makromolekulárních komplexů, které se ukládají v mezangiu cév a aktivují alternativní cestu komplementu [128]. Určitou roli hrají pravděpodobně i genetické faktory v podobě některých polymorfismů, jako je pozitivita HLA-B35, která inklinuje k postižení ledvin, či polymorfismus ICAM-1 469K/E, u kterého je pozorováno mírnější gastrointestinální postižení [16, 74, 128].
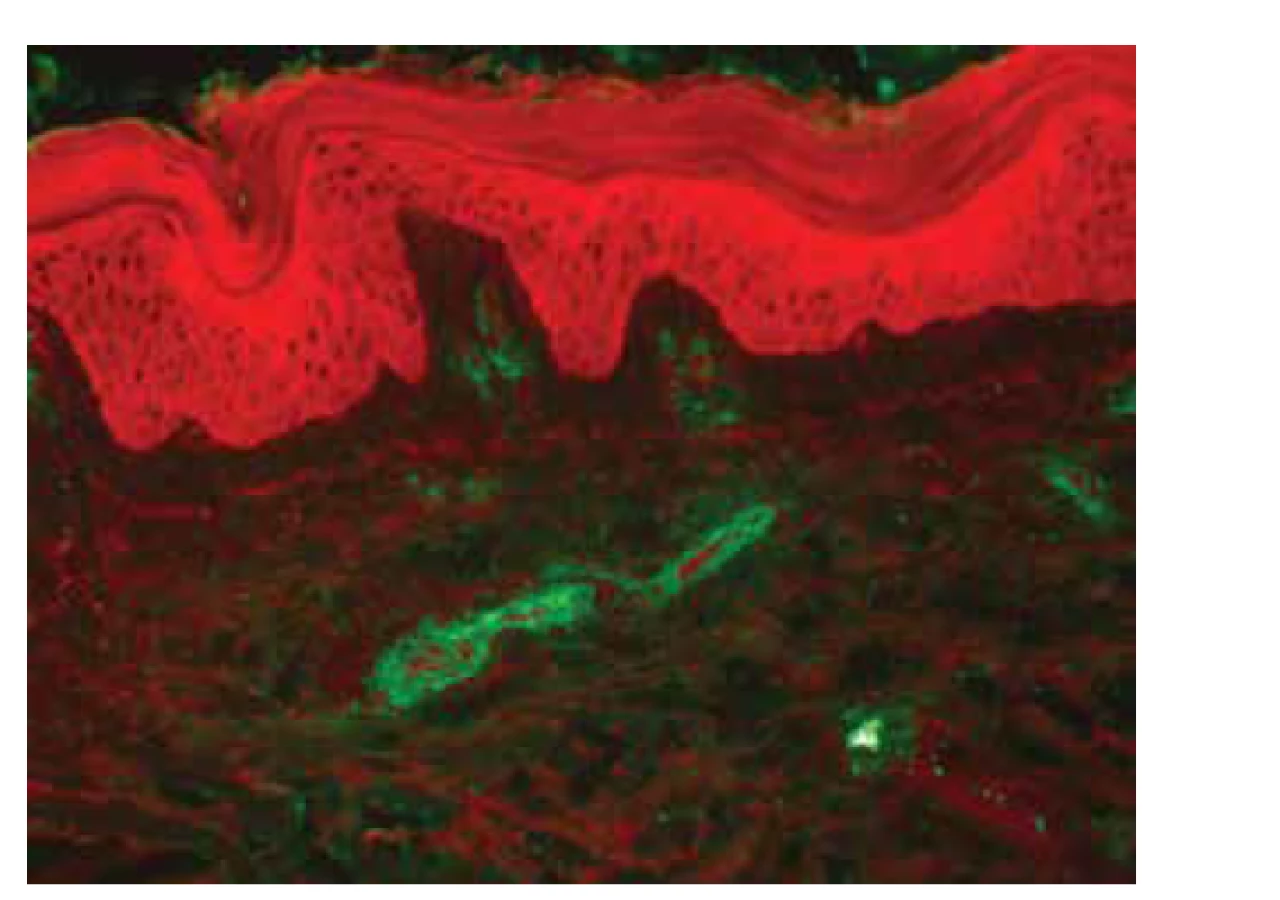
IgA vaskulitida má v typickém klinickém obraze kožní postižení u všech pacientů, s projevy palpovatelné purpury, artritidou, bolestmi břicha, a/nebo nefritidou [128]. Febrilní stav může být přítomen asi u 20 % dospělých pacientů, kdežto u dětí je pozorován prakticky v polovině případů [9]. Projevy purpury začínají nejprve jako erytematózní makuly s postupným rozvojem palpovatelné purpury (obr. 7). Mohou být patrné i urtikariální projevy, vezikuly, buly, nekrotická ložiska (ve středu projevů purpury) nebo terčovité léze. Palpovatelná purpura nejčastěji postihuje symetricky dolní končetiny s maximem na bércích, ale může být patrná i na dolních partiích trupu, horních končetinách nebo v obličeji. Velikost samotných ložisek je od několika milimetrů do splývajících projevů s tvorbou nekróz. Kožní projevy odeznívají postupně podle závažnosti postižení do dvou týdnů až několika měsíců (při tvorbě ne-króz a ulcerací), až u 10 % pacientů však může docházet k recidivám postižení [2]. Artritida postihuje až 75 % pacientů, nejčastěji jsou postiženy klouby dolních končetin (kolena a kotníky). Postižení gastrointestinálního traktu (50–75 % pacientů) může předcházet purpuru a projevuje se bolestmi břicha (65 %), méně často gastrointestinálním krvácením (30 %) a vomitem. Postižení ledvin s mikroskopickou hematurií, nebo i proteinurii, nacházíme u 50 % pacientů. Oproti postižení gastrointestinálního traktu, které kožním projevům může předcházet, se nefritida objevuje až do 3 měsíců od vzniku kožních projevů [128]. V dětském věku je postižení ledvin patrné především u dětí mladších 8 let, a i když průběh onemocnění ledvin může být protrahovaný, k chronickému poškození ledvin dochází pouze asi u 3 % dětských pacientů. U dospělých pacientů je rozvoj chronické renální insuficience pozorován dokonce až u 30 % pacientů. Mezi špatné prognostické faktory patří rozvoj akutní renální insuficience, nefrotický syndrom, sekundární hypertenze a snížená aktivita faktoru XIII [16, 88]. Ze vzácnějších projevů IgA vaskulitidy je možné zmínit postižení plic s projevy difuzního alveolárního krvácení s hemoptýzou, nebo orchitis postihující mladé chlapce, která koreluje s vyšší sérovou hladinou komplementu C3 [31, 53]. IgA vaskulitida může být i jedním z paraneoplastických příznaků, zejména solidních nádorů plic, prsu, prostaty nebo kolorektálního karcinomu [114].

K potvrzení diagnózy IgA vaskulitidy pomocí biopsie vede vedle histopatologického vyšetření zejména vyšetření PIF, ve kterém dominují depozita IgA (obr. 8), o něco méně pak depozita komplementu C3 a fibrinogenu. Mohou být přítomna i depozita IgM a IgG, která však nemusí být plně vyjádřena a jejich průkaz s negativním vyšetřením IgA nebývá spojený s postižením ledvin (může se jednat o samostatnou formu IgM/IgG kožní vaskulitidy). Oproti tomu absence průkazu fibrinogenu ve stěně cév při vyšetření PIF významně koreluje s postižením ledvin [116]. Histopatologicky je patrna leukocytoklastická vaskulitida malých dermálních cév, je častá přítomnost eozinofilů, ale při jejich absenci může být významnější korelace s postižením ledvin (obr. 9) [117]. Přibližně u 6 % pacientů s IgA vaskulitidou lze prokázat pozitivitu anti-MPO (pANCA) protilátek, navíc bylo prokázáno, že tito pacienti mohou mít protrahovaný průběh onemocnění a častější postižení plic či nervového systému [81].
Diferenciální diagnostika IgA vaskulitidy vzhledem k projevům palpovatelné purpury zahrnuje všechny vaskulitidy malých, ale i středních cév. Je nutné se vedle kožní manifestace opírat i o přidružené příznaky artritidy a bolestí břicha. Navíc i u jiných vaskulitid malých cév, nebo i u jiných onemocnění (přecitlivělosti na léky, IgA monoklonální gamapatii, systémového lupus erythematodes, kryoglobulinemie) mohou být přítomna depozita IgA ve stěně cév [91].
Asi nejčastěji užívané léky v léčbě jsou nesteroidní antirevmatika (NSA), systémové glukokortikoidy, které lze využít při terapii artritidy, bolestí břicha, ale i kožních projevů, ale nezabraňují recidivě projevů. Jejich použití je tedy limitované [151]. V dlouhodobé terapii lze zvážit celkovou léčbu dapsonem či kolchicinem, u kterých byl prokázán efekt na rychlejší hojení kožních projevů a snížení frekvence recidiv onemocnění [149]. Při postižení ledvin je použití celkových glukokortikoidů kontroverzní; navzdory prokázanému efektu na snížení chronického poškození ledvin a prokázanému rychlejšímu ústupu proteinurie, glukokortikoidy nezabraňují zcela rozvoji postižení ledvin [123]. Ojedinělé práce prokázaly efekt i dalších alternativních preparátů či postupů jako mykofenolát mofetilu, cyklofosfamidu, rituximabu, IVIg, případně plazmaferézy v případě rezistentního průběhu, převážně při postižení trávicího traktu a ledvin [56, 60, 63, 65, 68, 119, 145].
AKUTNÍ HEMORAGICKÝ EDÉM DĚTÍ
Akutní hemoragický edém dětí je vzácná forma vaskulitidy malých cév postihující kojence ve věku od 4 do 24 měsíců. Toto onemocnění bylo dříve označováno jako varianta IgA vaskulitidy. Samotná etiologie onemocnění není jasná, ale je uváděn sezonní výskyt v zimních měsících s dominantním postižením chlapců (až 70 %). Rozvoj onemocnění je spojován nejčastěji s infekčním prodromem dýchacích cest (virová, bakte-riální), průjmovitým onemocněním či infekcí močových cest a následnou tvorbou imunokomplexů v reakci na antigenní spouštěč [30, 35].
Onemocnění může začínat horečnatým stavem asi 1–2 týdny od spouštějící infekce, ale většinou postižené děti nevypadají nemocně. Kožní projevy začínají jako splývající erytematózní až hemoragická ložiska na tvářích a horních končetinách, může být přítomný edém postižených míst až urtikariální projevy s prstencovitou či terčovitou konfigurací. Postižení trupu je spíše vzácnější, stejně tak tvorba slizničních aft, konjunktivitida, artritida či glomerulonefritida [35]. Léze mohou být asymptomatické, bolestivé nebo svědí. Onemocnění odeznívá bez následků během 2–3 týdnů, ačkoli se mohou objevit exacerbace [30].
V diferenciální diagnostice je nutné odlišit lékové kopřivky, urtikariální vaskulitidu, erythema exsudativum multiforme, Kawasakiho nemoc, či Sweetův syndrom. Laboratorní vyšetření je nespecifické.
Terapie zpravidla není nutná, případně jen symptomatická léčba bolestí. Při možné bakteriální etiologii spouštějící infekce je vhodná cílená antibiotická terapie [89].
URTIKARIÁLNÍ VASKULITIDA
Urtikariální vaskulitida je klinicko-patologická jednotka charakterizovaná perzistujícími urtikariálními projevy, které v histologickém obraze vykazují známky leukocytoklastické vaskulitidy. Onemocnění se vyskytuje samostatně, ale i v souvislosti s autoimunitními onemocněními pojivové tkáně, jako je systémový lupus erythematodes (SLE).
Incidence urtikariální vaskulitidy se odhaduje asi na 0,5 případu na 100 tis. obyvatel za rok. Onemocnění postihuje častěji ženy (60–80 %) v období pátého decennia. Urtikariální vaskulitida vykazuje dvě formy, a to hypokomplementemickou formu, která je podle dodatku klasifikace kožních vaskulitid z roku 2018 řazena mezi kožní formy vaskulitid malých cév a prakticky výlučně postihuje ženy. Normokomplementemická forma je řazena mezi vaskulitidy postihující jeden orgán (kůži) a představuje 70–80 % případů urtikariální vaskulitidy. Je spojena s benigním průběhem onemocnění a průměrnou dobou trvání 3 roky [26, 143].
V patogenezi onemocnění se uplatňují podobné mechanismy jako u jiných kožních vaskulitid malých cév. Aktivace komplementu spouští uvolnění prozánětlivých cytokinů, které aktivují žírné buňky. Dále pak dochází k produkci TNF, který zvyšuje expresi molekul ICAM na žírných buňkách a expresi E-selektinu na endotelových buňkách, kde dochází následně k poškození cévní stěny [26]. Etiologie onemocnění je neznámá a ve většině případů hovoříme o idiopatické urtikariální vaskulitidě. U některých pacientů dochází k výskytu této formy vaskulitidy společně s některými onemocněními pojivové tkáně, jako je zejména Sjögrenův syndrom a SLE. U některých pacientů lze prokázat souvislost s užíváním léků, prodělanou infekcí či hematologickou malignitou. Dále byla popsána souvislost s kryoglobulinemií, sérovou nemocí, či vzácně se solidními nádory, jako je kolorektální či renální karcinom [54].
Projevy urtikariální vaskulitidy představují pomfy, které nemizí do 24 hod. a zanechávají erytémové projevy, sufuze, hematomy, nebo hyperpigmentace (obr. 10). Maximum postižení je na trupu a proximálních částech končetin. Pacienti vnímají projevy spíše jako pálivé či bolestivé než svědivé. Vzácněji jsou přítomny buly, projevy charakteru erythema multiforme a livedo reticularis. Může být přítomen Raynaudův fenomén či otoky sliznic [69].

V případě urtikariálních projevů s histologicky verifikovanou přítomnou leukocytoklastickou vaskulitidou je dalším diagnostickým znakem vyšetření komplementu. U pacientů s normální hladinou komplementu se jedná o onemocnění omezené na kůži. Naproti tomu u pacientů s nízkou hladinou komplementu (hypokomplementemická forma) se může vedle postižení kůže rozvíjet i systémové postižení.
Syndrom hypokomplementemické urtikariální vaskulitidy (HUVS) je závažnější postižení definované specifickými diagnostickými kritérii [69]. Kritéria uplatňující se v diagnostice tohoto syndromu představují dvě hlavní kritéria: 1) kopřivka trvající 6 měsíců; 2) snížená hladina komplementu. K hlavním kritériím patří dvě a více vedlejších kritérií. Ta tvoří:
a) histologicky potvrzená leukocytoklastická vaskulitida,
b) artritida či bolesti kloubů,
c) episkleritida či uveitida,
d) glomerulonefritida,
e) bolesti břicha,
f) nízká hladina C1q, pozitivní precipitovaný test s C1q [69].
Mezi nejčastější extrakutánní projevy patří postižení kloubů, které se projevují až u poloviny pacientů artritidou rukou a nohou, ale i velkých kloubů. Až u 20 % pacientů mohou být plicní příznaky s dominujícím kašlem, otoky sliznic, dušností, exacerbací astma bronchiale a zejména u kuřáků i chronické obstrukční plicní nemoci (CHOPN) [28, 69]. Postižení ledvin se nachází asi u 10 % pacientů ve formě glomerulonefritidy s projevy proteinurie a mikroskopické hematurie [127]. Gastrointestinální projevy s bolestmi břicha, nevolností, zvracením a průjmy se vyskytují až u 30 % pacientů. Časté je i oční postižení (30 %) s projevy konjunktivitidy, episkleritidy či uveitidy. Mohou být přítomny podobné symptomy doprovázející systémová onemocnění, zejména SLE [69].
V laboratorním nálezu u hypokomplementemické formy jsou patrny nízké hladiny komplementu C3 a C4, lze prokázat protilátky anti-C1q a asi u 50 % pacientů i antinukleární protilátky (ANA); protilátky anti-dsDNA nebo anti-Sm jsou však negativní [127].
V histopatologickém obraze u urtikariální vaskulitidy nacházíme leukocytoklazii s nekrózou cévní stěny, nebo fibrinoidními depozity. Dále jsou patrny perivaskulární lymfocytární infiltráty a extravazace erytrocytů. V některých případech může být výraznější neutrofilní infiltrát, a to především u pacientů s hypokomplementemickou formou [26]. V přímé imunoflorescenci lze prokázat depozita IgG, IgM, IgA, C3 nebo fibrinogenu ve stěně cév. U hypokomplementemické formy mohou být granulární depozita imunoreaktantů podél bazální membrány. Tento nález může být spojený s diagnózou SLE [127].
V diferenciální diagnostice je nutné odlišit v první řadě chronickou kopřivku či tlakovou kopřivku. Urtika-riální vaskulitida by měla být také odlišena od neutrofilní kopřivky, která, i když je některými autory považována za součást nálezů urtikariální vaskulitidy, není spojena s hypokomplementemickou formou či autoimunitním onemocněním a je lepší ji považovat za podskupinu klasické urtikarie. Dále je nutné odlišit i některé neutrofilní dermatózy jako Schnitzlerův syndrom, Stillovu chorobu či Sweetův syndrom [80, 127].
V celkové terapii se u mírnějších forem uplatňují antihistaminika a NSA. Pro středně závažné formy jsou vyhrazeny systémové glukokortikoidy, které je vhodné dlouhodobě minimalizovat pro četné nežádoucí účinky, dapson, kolchicin, či hydroxychlorochin. U závažných systémových forem existují ojedinělé údaje o účinnosti pro mykofenolát mofetil, metotrexát, azathioprin, cyklosporin a cyklofosfamid a z řad biologik pak pro anakinru (antagonista pro receptor a interleukinu 1, IL-1ra), canakinumab (anti-IL-1β) či omalizumab (anti-IgE). Rituximab nebo intravenózní imunoglobulin mohou být účinnou terapií u refrakterních hypokomplementických forem [83].
KRYOGLOBULINEMICKÁ VASKULITIDA
Kryoglobuliny jsou imunoglobuliny, které reverzibilně precipitují v chladu a stávají se nerozpustnými. Kryoglobuliny precipitují obvykle při teplotě nižší než 37,0 °C a rozpouštějí se opět při zahřátí krve. Rozlišujeme tři skupiny kryoglobulinů. Kryoglobulinemie I. typu je složena z monoklonálního IgM globulinu (méně pak IgG, vzácně IgA), která vede k mikrovaskulární okluzi bez postižení cévní stěny a spadá mezi tzv. obliterující vaskulopatie. U kryoglobulinemie II. a III. typu dochází již k postižení cévní stěny a rozvoji vaskulitidy. Obecně se tyto dva typy označují jako smíšené kryoglobulinemie, jelikož se precipitáty tvoří jak z monoklonálních (IgM), tak i z polyklonálních (IgM i IgG) složek. Přibližně u 15 % pacientů dochází k rozvoji vaskulitidy postihující kůži, periferní nervový systém a ledviny [92, 131].
Incidence kryoglobulinemické vaskulitidy vykazuje značné geografické rozdíly a je pravděpodobně vázána na prevalenci hepatitidy C (HCV), u které je kryoglobulinemie až u 50 % pacientů a kryoglobulinemická vaskulitida až u 30 % z nich [97]. Smíšená kryoglobulinemie se vyskytuje na pozadí zejména virových onemocnění, autoimunitních onemocnění pojivové tkáně a hematologických malignit. Nejčastější je souvislost s HCV v příbližně 80 % případů, méně pak s HBV či hepatitidou typu E (HEV), či cytomegalovirovou infekcí (CMV). Z autoimunitních onemocnění je spojena s revmatoidní artritidou, Sjögrenovým syndromem a systémovou sklerodermií. Přibližně 5 % pacientů má lymfoproliferativní non-Hodgkinský lymfom z B-buněk, nebo chronickou lymfocytární leukemií. Kryoglobulinemická vaskulitida vzniká při ukládání imunitních komplexů z kryoglobulinů do stěny cév, následně dochází k aktivaci komplementu a indukci zánětlivé odpovědi [137].
Postižení kůže s projevy palpovatelné purpury dolních končetin patří mezi dominantní projevy kryoglobulinemické vaskulitidy. Mezi další kožní nálezy patří erytematózní papuly, noduly, ekchymózy, livedo reticularis, nekrotické léze, ulcerace a buly (obr. 11). Vzácně jsou patrny urtikariální projevy. Kožní projevy nejsou vyvolány chladem na rozdíl od kryoglobulinemie I. typu s obliterující vaskulopatií. Onemocnění má chronický průběh a může být vázáno na úspěšnost antivirové terapie u pacientů s HCV [46].

Mezi nejčastější extrakutánní příznaky patří artritida nebo artralgie (70 % pacientů), periferní (senzorická) neuropatie (40 %), gastrointestinální onemocnění nebo hepatitida (30 %), která může odrážet jak virovou hepatitidu, tak i postižení jater kryoglobuliny. Postižení ledvin s membranoproliferativní glomerulonefritidou postihuje až čtvrtinu pacientů (25 %) [137].
V laboratorním vyšetření je zásadní vyšetření kryoglobulinů, které je vhodné provádět při akutním stadiu onemocnění, v období remise může být vyšetření falešně negativní. Odběr a vyšetření kryoglobulinů je nutné provádět ve specializované laboratoři a během transportu vzorek krve uchovávat při 37 °C, aby nedošlo k precipitaci kryoglobulinů. U většiny pacientů lze prokázat i pozitivitu revmatoidního faktoru, méně pak ANA protilátky. Asi u pětiny pacientů lze imunoelektroforézou detekovat monoklonální gamapatii. Častým nálezem je snížená funkce komplementu, zejména C4. Standardem vyšetření je sérologie hepatitid HCV, HBV a HEV, dála pak CMV a HIV s případným stanovením virové zátěže [97, 137]. Histologicky lze prokázat leukocytoklastickou vaskulitidu, v PIF poté granulární depozita IgM a C3 ve stěně cév.
Základem léčby kryoglobulinemické vaskulitidy je terapie základního onemocnění. V případě HCV infekce vede kombinovaná terapie interferonem-α a ribavirinem ke zhojení kožního, ale i neurologického či renálního postižení. U mírných kožních projevů lze použít glukokortikoidy, kolchicin a dapson. V případě mírné senzorické neuropatie se spíš využívají amytryptylin, duloxetin, gabapentin či pregabalin nežli opioidní analgetika. U mírné formy artralgií či artritid lze využít NSA, nízkou dávku glukokortikoidů či hydroxychlorochin. V případě závažného postižení ledvin lze použit plazmaferézu, či terapii vysokou dávkou systémových glukokortikoidů, optimálně v kombinaci s rituximabem, případně s cyklofosfamidem. Rituximab lze použít i u pacientů s infekcí HCV v kombinaci s antivirovou terapií [109, 137].
Vaskulitidy postihující jeden orgán – kůži
Vaskulitidy omezené na kůži představují skupinu vaskulitid, které nesdílí dostatečné množství klinických, laboratorních a patologických příznaků odpovídajících systémovým vaskulitidám. Tato kategorie vaskulitid vychází z dodatku klasifikace kožních vaskulitid z roku 2018 a není plnohodnotně definována původní revmatologickou klasifikací vaskulitid z roku 2012. Do této skupiny, vedle níže uvedených jednotek, patří i normokomplementemická forma urtikariální vaskulitidy, která je diskutována výše.
Imunokomplexová vaskulitida IgM/IgG
Tato vaskulitida představuje obdobný nález jako IgA vaskulitida s postižením malých cév, vznikem leukocytoklastické vaskulitidy, ale bez rozvoje systémového postižení. Typickým kožním projevem je palpovatelná purpura dolních končetin [143].
Nodulární vaskulitida (erythema induratum Bazin)
Nodulární vaskulitida (NV) je neobvyklá forma panikulitidy charakterizovaná erytematózními noduly typicky postihující bérce, které mohou ulcerovat. NV byla považována za opožděnou hypersenzitivní reakci na antigenní stimul, původně byla spojena čistě s formou kožní tuberkulózy označovanou jako erythema induratum. Souvislost s idovou reakcí tzv. tuberkulidem potvrzují studie prokazující DNA mykobakterií v kožních biopsiích. Jedná se o lobulární panikulitidy s vaskulitidou středních a malých cév v podkoží. Onemocnění postihuje převážně ženy průměrně třetího a čtvrtého decennia, samotná incidence je variabilní podle populace a prevalence tuberkulózy [64]. Díky rutinnímu použití PCR diagnostiky mykobakteriálních infekcí byla prokázána DNA mycobakterium tuberculosis v kožních biopsiích. Byly však zdokumentovány případy, kde byly prokázány i jiné patogeny jako jsou nocardie, HBV nebo HCV. Byl prokázán i spouštěcí mechanismus indukovaný léky (propylthiouracil – léčba hyperfunkce štítné žlázy), nebo autoimunitními onemocněními [154]. Vzácně je nodulární vaskulitida popsána u adenokarcinomu plic [13]. V některých případech byl spouštěcím mechanismem kožní tuberkulinový test [156].
V patogenezi se pravděpodobně uplatňuje proces buněčné reakce IV. typu, i když jsou názory, že samotná vaskulitida je zprostředkována depozity imunokomplexů [64].
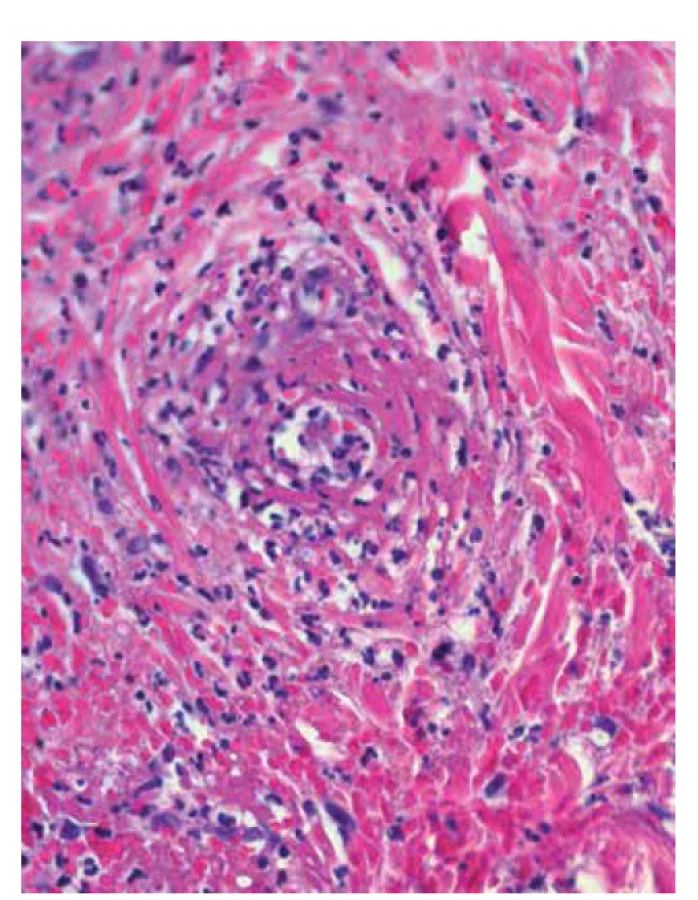
V klinickém obraze dominují bolestivé, erytematózní až lividní noduly a ložiska s typickým postižením dolních končetin, především zadních partií bérců (obr. 12). Projevy mohou ulcerovat až s purulentní sekrecí. Hojí se jizvou s tendencí k recidivám a posunům pigmentu. U erythema induratum spojeného s infekcí mycobacte-rium tuberculosis mohou být přítomny klinické a rentgenové projevy aktivní, ale i latentní tuberkulózy, pozitivní kožní tuberkulinový test, nebo test Quantiferon [115].

V diagnostice se opíráme o provedení hluboké kožní biopsie se zavzetím podkožní tukové tkáně. Erythema induratum v histologii vykazuje lobulární či smíšenou septální a lobulární panikulitidu (obr. 13). Zánětlivá reakce je smíšená a zahrnuje neutrofily, lymfocyty, makrofágy a mnohojaderné buňky. Vaskulitida středně velkých nebo malých cév s granulomatózním zánětem a nekrózou tukové tkáně postihuje v naprosté většině případů žíly nebo tepny septa podkožní tukové tkáně a malé žily tukových lalůčků podkoží. Může být převážně neutrofilní, lymfocytární, nebo i granulomatózní. Lze prokázat nekrózu s koagulačním nebo kazeózním vzhledem, někdy s palisádově uspořádanými granulomy [130, 136].
Přehledový snímek zvětšeno 40x, detail nekrotizující panikulitidy
a obliterované arterie zvětšeno 200x.

V klinicko-patologické diferenciální diagnostice je nutné odlišit projevy při erythema nodosum, kde nenacházíme vaskulitidu, ale výraznější neutrofilní složku infiltrátu. Lupusová panikulitida není granulomatózní, má prominentní lymfoplazmacelulární infiltrát, může vykazovat depozita mucinu a někdy má překrývající se epidermální a dermální změny typické pro lupus erythematosus. Polyarteritis nodosa i tromboflebitida mají tendenci vykazovat zánět omezený na bezprostřední perivaskulární zónu. V některých případech může být obtížné odlišit i např. perniones, kde by však měl být jasný anamnestický údaj o expozici chladu [130].
Léčba by měla být zaměřena na základní příčinu onemocnění, v případě tuberkulózy a po vyšetření pacienta s průkazem aktivní tuberkulózy jsou nutná antituberkulotika. Terapeutické možnosti pro netuberkulózní postižení jsou NSA, systémové glukokortikoidy, tetracyklin, clofazimin, kolchicin, mykofenolát mofetil, či jodid draselný [43].
Erythema elevatum diutinum
Erythema tvorbou červenofialových až červenohnědých papul, nodulů a ložisek, které postihují kůži, elevatum diutinum (EED) je vzácná chronická dermatóza charakterizovaná nad extenzory, ale i např. nad Achillovou šlachou (flexorová skupina). U časných lézí lze prokázat leukocytoklastickou vaskulitidu malých cév.
EED je vzácné až raritní onemocnění, podle dostupné literatury mezi roky 1990–2014 bylo popsáno celosvětově jen 133 případů. Postižení se může vyskytovat v jakémkoliv věku, ale typicky postihuje dospělé pa-cienty středního věku, nevykazuje rasovou ani pohlavní závislost. Časnější nástup onemocnění může souviset s infekcí HIV [32].
Ačkoli etiologie EED není známa, předpokládá se, že cirkulující imunokomplexy s opakovaným ukládáním do stěny cév a indukcí zánětu představují základní patogenezi. Depozita imunokomplexů aktivují komplement a neutrofilní infiltraci. Následně dochází k uvolnění enzymatické degradace a ukládání fibrinu do stěny malých dermálních cév [104]. EED byla popsána v souvislosti s řadou systémových onemocnění, infekcí, autoimunitních onemocnění a maligních hematologických onemocnění, typicky u monoklonální gamapatie typu IgA [32, 112]. Infekční souvislost byla popsána u přidružené infekce zahrnující beta-hemolytické streptokoky, HBV, HIV, tuberkulózu nebo syfilis [32].
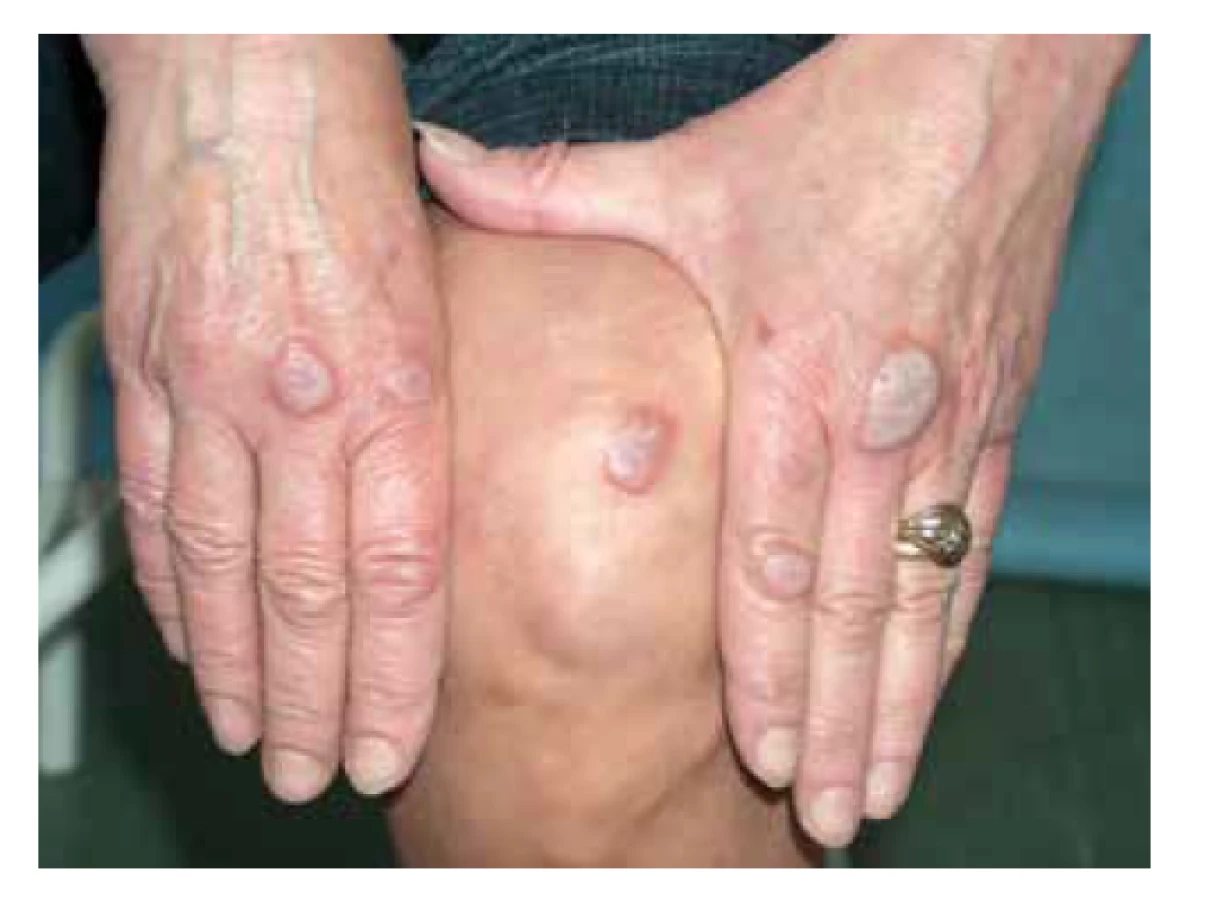
Typickými kožními projevy EED jsou lividní, červenohnědé nebo nažloutlé papuly, noduly a ložiska, symetricky postihující akrální části končetin a oblast nad velkými klouby (lokty, kolena, ruce, kotníky) či nad šlachami (nad Achillovou šlachou) – obr. 14. Z dalších lokalit může být patrné postižení v obličeji, na ušních boltcích, nebo retroaurikulárně. Zpočátku jsou léze erytematózní, ale s vývojem se stávají červenohnědavé až lividní, navíc tuhé v důsledku fibrotizace. Projevy svědí, bolí, až pálí, při lokalizaci nad klouby mohou být přítomné i bolesti kloubů [32]. Mezi extrakutánní projevy patří vzácně oční postižení s keratitidou, nodulární skleritidou až rozvojem slepoty [73]. Průběh onemocnění je chronický s možným spontánním zhojením v horizontu 5–10 let trvání [32].
V histopatologickém obraze čerstvých lézí je patrna leukocytoklastická vaskulitida malých cév s infiltrátem neutrofilů ve střední a horní dermis. V pozdějších fázích mohou být kapiláry fibroticky změněny, či s přítomnou fibrinoidní nekrózou. Onemocnění se řadí mezi heterogenní skupinu neutrofilních dermatóz [32].
Diferenciálně diagnosticky rozlišujeme ostatní neutrofilní dermatózy, jako je Sweetův syndrom anebo neutrofilní revmatoidní dermatóza. Pozdní léze je nutné odlišit od granuloma anulare, revmatoidních nodulů, fibrotických nodulů při borelióze, nebo tuberózních xantomů. Z nádorových onemocnění je projev možné zaměnit např. s dermatofibrosarkoma protuberans [32, 112].
V léčbě se v první řadě uplatňuje dapson, ale exacerbace přes úvodní terapeutické úspěchy jsou časté. Z dalších možností lze zvážit terapii systémovými glukokortikoidy, tetracyklinem (případně claritromycinem, erytromycinem, penicilinem), niacinamidem, chlorochinem, kolchicinem, sulfasalazinem (případně metotrexátem, cyklosporinem, či mykofenolát mofetilem). Intralezionální aplikace glukokortikoidů může pomoci u drobných solitárních projevů a chirurgická excize je rezervovaná pro lokální fibrotické noduly [99].
Rekurentní makulární vaskulitida u hypergamaglobulinemie (Waldenströmova hypergamaglobulinemická purpura)
Recidivující makulární vaskulitida u hypergamaglobulinemie je recidivující epizodická vaskulitida malých cév s tvorbou vaskulárních depozit imunoglobulinů. Nejčastěji je spojena s polyklonální hypergamaglobulinemií. Může být prokázána i pozitivita revmatoidního faktoru, ANA protilátek, anti-SSA (anti-Ro)/anti-SSB (anti-La), nebo přímo asociace např. se Sjögrenovým syndromem, ale již bez tvorby systémové vaskulitidy [39]. Klinicky se tato jednotka projevuje jako drobnější, či splývající erytematózní, či hemoragické makuly postihující dolní končetiny s četnými recidivami. V léčbě se uplatňuje např. dapson [40].
Vaskulitidy s postižením variabilních cév
Behçetova nemoc
Behçetova nemoc je multisystémové onemocnění, postihující prakticky kterýkoliv orgán, s četnými exacerbacemi a s nejistou prognózou. Jedním z projevů jsou vaskulitidy postihující variabilní cévy a orgány.
Incidence onemocnění je celosvětově výrazně varia-bilní od 80 na 100 tis. obyvatel/rok např. v Turecku, k 0,64 na 100 tis. obyvatel/rok ve Spojeném království. V patogenezi se uplatňují faktory genetické (polymorfismus HLA-B51). Dále je diskutována role infekcí, jako je HCV, herpetické viry, zejména herpes simplex, parvovirus B19, ale i streptokokové infekce. Samotná vaskulitida vzniká ukládáním imunokomplexů vzniklých na základně patologické stimulace imunitního systému [5].
Hlavním příznakem je aftózní stomatitida a recidivující afty dutiny ústní, mohou být přítomné i větší ulcerace jazyka. Tyto projevy mohou plnému rozvoji onemocnění předcházet po mnoho let. Následně se mohou objevovat nespecifické horečky, krční lymfadenopatie, faryngitida. Genitální ulcerace jsou větší než v dutině ústní, bolestivější a nepravidelné, postihující skrotum, penis a vulvu. Mezi další primární kožní příznaky patří papulopustulózní či vezikulózní projevy akrálních částí, nebo obličeje. Mohou být přítomny i projevy charakteru erythema nodosum či pyoderma gangrenosum [90]. Mezi systémové příznaky patří postižení oční (konjunktivitida, skleritida, neuritida optického nervu, ale i vaskulitida retinálních cév), kloubů (mono-, či polyartritida), postižení gastrointestinální (ulcerace tenkého střeva, enteroragie, obtížně odlišitelné od nespecifických střevních zánětů), neurologické (meningoencefalitida, neuralgie hlavových nervů), kardiopulmonální postižení (myokarditida, postižení koronárních tepen, aorty, aneurysma plicních žil) [5, 37, 67].
Histologicky lze prokázat vaskulopatie či vaskulitidy arterií a žil v dermis a podkoží. Typický je neutrofilní infiltrát, leukocytoklazie a extravazace erytrocytů. Lze prokázat i kožní leukocytoklastickou vaskulitidu. Může být přítomna ale i trombotizace cév, nebo nekróza stěny cév. V projevech charakteru erythema nodosum lze prokázat neutrofilní lobulární panikulitidu s vaskulitidou podkožních cév.
Diagnostika se vzhledem k množství příznaků opírá na pečlivém zhodnocení jednotlivých projevů. Vedle aftózní stomatitidy recidivující minimálně 3krát během jednoho roku, hlavního diagnostického kritéria, musí být naplněna alespoň dvě vedlejší kritéria (oční postižení, recidivující genitální ulcerace, kožní postižení – léze podobné erythema nodosum, papulopustulózní projevy, akneiformní projevy a pozitivní test patergie do 24–48 hod.) [5, 37, 67].
Vedle NSA a imunosupresivní terapie celkovými glukokortikoidy, kolchicinem, azathioprinem, cyklofosfamidem, cyklosporinem, mykofenolát mofetilem, metotrexátem, thalidomidem nebo interferonem α se v poslední době uplatňují biologika inhibující fos-fodiesterázu 4 (apremilast), TNF (infliximab, adalimumab, etanercept), CD20 (rituximab), či anakinra (IL-1ra), nebo canakinumab (anti-IL-1β). Významná je i podpůrná lokální terapie, např. výrazně bolestivých ulcerací v dutině ústní, nebo na genitálu preparáty s obsahem lokálních anestetik, antibiotik či lokálních glukokortikoidů [3].
Coganův syndrom
Coganův syndrom je vzácná autoimunitní vaskulitida s neznámou etiologií a patogenezí. V patogenezi může být spouštěcím faktorem onemocnění infekce, ale nyní spíše převažuje hypotéza autoimunitního onemocnění. Coganův syndrom je charakterizován očními a audiovestibulárními příznaky podobnými Ménierově nemoci [24, 50]. Přibližně 70 % pacientů má systémové onemocnění, jehož patologickým mechanismem je vaskulitida středně velkých nebo malých arterií i vén. Z kožních příznaků, které jsou velice vzácné, mohou být přítomny léze podobné erythema nodosum s panikulitidou a vaskulitidou podkožních cév [50].
Lékem volby jsou systémové glukokortikoidy, či jiná imunosupresiva, jako je metotrexát, azathioprin, mykofenolát mofetil, cyklofosfamid, cyklosporin, leflunomid, takrolimus. Lze použít i biologika inhibující TNF (zejména infliximab) nebo IL-6r (tocilizumab) [25].
Vaskulitidy spojené se systémovými onemocněními
Kožní vaskulitidy, které jsou spojeny se systémovým onemocněním, mohou být sekundární a často doplňují ostatní (i kožní) příznaky těchto onemocnění. Je vhodné tedy zmínit určitý přehled, který vychází z dodatku klasifikace kožních vaskulitid z roku 2018 [143].
Revmatoidní vaskulitida
Tato vaskulitida postihuje pacienty s revmatoidní artritidou s pozitivním RF, kteří mohou mít erozivní formu artritidy. Vaskulitida postihující kůži vykazuje imunodepozita IgG či IgM, vzácněji i IgA. Jedná se o leukocytoklastickou vaskulitidu postkapilárních venul až arterií na hranici dermis a podkoží nebo jen v podkožní tukové tkáni. Postižení těchto větších cév odlišuje tuto vaskulitidu od imunokoplexových vaskulitid typu IgA či IgG/IgM [135]. Tomu odpovídají i klinické projevy, charakteru tvorby defektů, gangrény prstů, nebo nekrózy nehtových plotének. Často může být přítomna periferní neuropatie v důsledku postižení vasa nervorum, nebo postižení svalů, kde dochází k vaskulitidě svalových postkapilárních venul [62].
Vaskulitidy při systémovém lupus erythematodes a Sjögrenově syndromu
U SLE můžeme pozorovat celou řadu projevů, které vznikají na podkladě kožních vaskulitid postihujících malé, ale i střední cévy. Mohou být přítomny leukocytoklastické vaskulitidy imunokomplexové (postihující dolní končetiny) nebo i hypokomplementemická forma urtikariální vaskulitidy, postihující postkapilární venuly, bez predilekce postižení [127]. Podle nedávné studie byly projevy leukocytoklastické vaskulitidy zaznamenány u 17 % pacientů se SLE a u 35 % docházelo k recidivám postižení. Navíc byla prokázána statistická souvislost s myozitidou a hematologickými změnami při SLE, které byly prediktorem rozvoje leukocytoklastické vaskulitidy a výraznějšího orgánového postižení [75]. Vzácně mohou být leukocytoklastické vaskulitidy projevem dermatomyozitidy nebo systémové sklerodermie, u které jsou popsány i kryoglobulinemické vaskulitidy [157, 118]. U Sjögrenova syndromu se vyskytuje rekurentní makulární vaskulitida s hypergamaglobulinemií nebo kryoglobulinemická vaskulitida s tvorbou smíšených kryoglobulinů II. a III. typu [39, 137].
ZÁVĚR
Vaskulitidy jsou heterogenní skupinou onemocnění, rozmanité etiologie a patogeneze. Kožní vaskulitidy mohou být primární, postihující jen kůži, nebo sekundární, vyskytující se v rámci systémových vaskulitid nebo např. u systémových onemocnění pojiva. Základní klasifikace vaskulitid odráží postižení kalibru cév a nabízí tak jednoduché klinicko-patologické dělení. Typickými příznaky kožních vaskulitid jsou erytematózní makuly, palpovatelná purpura, vezikulózní až bulózní léze, livedo racemosa, noduly, či ulcerace. Mohou být přítomné i urtikariální projevy nebo projevy charakteru erythema nodosum. Pečlivé vyšetření pacienta je nutné k vyloučení systémových vaskulitid a poukazuje na význam mezioborové spolupráce. Jelikož i systémové vaskulitidy mají často kožní projevy, je základní povědomí o jednotlivých projevech vaskulitid nezbytné pro správnou diferenciální diagnostiku. Dermatolog může být navíc prvním lékařem, který stanovuje diagnózu systémové vaskulitidy nebo je vystaven konfrontaci s komplikovanějšími případy s projevy vaskulitid v rámci konziliárních vyšetření.
Poděkování
Grantová podpora: Podpořeno Projektem UK Progress Q28 a projektem CNE (No. CZ.02.1.01/0.0/0.0/16_019/0000785).
Do redakce došlo dne 24. 5. 2021.
Adresa pro korespondenci:
MUDr. Ondřej Kodet, Ph.D.
Dermatovenerologická klinika 1. LF UK a VFN
U Nemocnice 499/2
128 00 Praha 2
e-mail: ondrej.kodet@lf1.cuni.cz
Zdroje
1. AGUR, T., LEVY, Y., PLOTKIN, E. et al. Minocycline-induced polyarteritis nodosa-like vasculitis. Israel Medical Association Journal, 2014, 16(5), p. 322–323.
2. ALFREDO, C. S., NUNES, N. A., LEN, C. A. et al., Henoch-Schönlein purpura: Recurrence and chronicity. J. Pediatr. (Rio. J)., 2007, 83(2), p. 177–180.
3. ALIBAZ-ONER, F., SAWALHA, A. H., DIRESKENELI, H. Management of Behçet’s disease. Current Opinion in Rheumatology, 2018, 30(3), p. 238–242.
4. ALIBAZ-ONER, F., KOSTER, M. J., CROWSON, C. S. et al. Clinical Spectrum of Medium-Sized Ves-sel Vasculitis. Arthritis Care Res., 2017, 69(6), p. 884–891.
5. ALPSOY, E. Behçet’s disease: A comprehensive review with a focus on epidemiology, etiology and clinical features, and management of mucocutaneous lesions. Journal of Dermatology, 2016, 43(6), p. 620–632.
6. ARORA, A., WETTER, D. A., GONZALEZ-SANTIAGO, T. M. et al. Incidence of leukocytoclastic vasculitis, 1996 to 2010: A population-based study in olm-sted county, Minnesota. Mayo Clin. Proc., 2014, 89(11), p. 1515–1524.
7. AWAN, S., MANDHADI, R., ABELES, A. M. Refractory psoriatic arthritis and polyarteritis nodosa: Co-occurrence in a patient with undiagnosed HIV. Conn. Med., 2017, 41(5), p. 285–289.
8. BARRA, L., YANG, G., PAGNOUX, C. Non-glucocorticoid drugs for the treatment of Takayasu’s arteritis: A systematic review and meta-analysis. Autoimmunity Reviews, 2018, 17(7), p. 683–693.
9. BLANCO, R., MARTÍNEZ-TABOADA, V. M., RODRÍGUEZ-VALVERDE, V. et al. Henoch-Schonlein purpura in adulthood and childhood: Two different expressions of the same syndrome. Arthritis Rheum., 1997, 40(5), p. 859–864.
10. BOOTH, A., HARPER, L., HAMMAD, T. et al. Prospective Study of TNFα Blockade with Infliximab in Anti-Neutrophil Cytoplasmic Antibody-Associated Systemic Vasculitis. J. Am. Soc. Nephrol., 2004, 15(3), p. 717–721.
11. BOSCO, L., PERONI, A., SCHENA, D. et al. Cutaneous manifestations of Churg-Strauss syndrome: Report of two cases and review of the literature. Clin. Rheumatol., 2011, 30(4), p. 573–580.
12. BOSCH, X., GUILABERT, A., ESPINOSA, G. et al. Treatment of antineutrophil cytoplasmic antibody-associated vasculitis: A systematic review. JAMA, 2007, 298(6), p. 655–669.
13. BORGES, A. S., BRASILEIRO, A., APETATO, M. Nodular vasculitis associated with lung adenocarcinoma. Anais Brasileiros de Dermatologia, 2018, 93(6), p. 887–889.
14. BORCHERS, A. T., GERSHWIN, M. E. Giant cell arteritis: A review of classification, pathophysiology, geoepidemiology and treatment. Autoimmunity Reviews, 2012, 11(6–7).
15. BREUER, G. S., REINUS, K., NESHER, G. et al. Recur-rent unilateral orchitis as a presenting symptom of polyarteritis nodosa. Isr. Med. Assoc. J., 2015, 17(9), p. 585–586.
16. BROGAN, P., ELEFTHERIOU, D. Vasculitis update: pathogenesis and biomarkers. Pediatric Nephrology, 2018, 33(2), p. 187–198.
17. BRUCATO, A., MAESTRONI, S., MASCIOCCO, G. et al. Il coinvolgimento cardiaco nella sindrome di Churg-Strauss. G. Ital. Cardiol., 2015, 16(9), p. 493–500.
18. CABRAL, D. A., URIBE, A. G., BENSELER, S. et al. Classification, presentation, and initial treatment of Wegener’s granulomatosis in childhood. Arthritis Rheum., 2009, 60(11), p. 3413–3424.
19. CALVO-RÍO, V., LORICERA, J., MATA, C. et al. Henoch-schönlein purpura in Northern Spain: Clinical spectrum of the disease in 417 patients from a single center, Medicine (United States), 2014, 93(2), p. 106–113.
20. CARLSON, J. A., NG, B. T., CHEN, K. R. Cutaneous vasculitis update: Diagnostic criteria, classifica-tion, epidemiology, etiology, pathogenesis, evaluation and prognosis, Am. J. Dermatopathol., 2005, 27(6), p. 504–528.
21. CARUSO, C., GENCARELLI, G., GAETA, F. et al. Efficacy of omalizumab treatment in a man with occupational asthma and eosinophilic granulomatosis with polyangioitis. Ann. Allergy, Asthma Immunol., 2018, 120(2), p. 209–211.
22. COHEN, P., PAGNOUX, C., MAHR, A. et al. Churg-Strauss syndrome with poor-prognosis factors: A prospective multicenter trial comparing glucocorticoids and six or twelve cyclophosphamide pulses in forty-eight patients. Arthritis Care Res., 2007, 57(4), p. 686–693.
23. COMARMOND, C., PAGNOUX, C., KHELLAF, M. et al. Eosinophilic granulomatosis with polyangiitis (Churg-Strauss): Clinical characteristics and long--term followup of the 383 patients enrolled in the French Vasculitis Study Group cohort. Arthritis Rheum., 2013, 65(1), p. 270–281.
24. CUNDIFF, J., KANSAL, S., KUMAR, A. et al. Cogan’s syndrome: A cause of progressive hearing deafness. Am. J. Otolaryngol. – Head Neck Med. Surg., 2006, 27(1), p. 68–70.
25. D’AGUANNO, V., RALLI, M., DE VINCENTIIS, M. et al. Optimal management of Cogan’s syndrome: A multidisciplinary approach. J. Multidisciplin. Healthcare, 2018, 11, p. 1–11.
26. DAVIS, M. D. P., DAOUD, M. S., KIRBY, B. et al. Clinicopathologic correlation of hypocomplementemic and normocomplementemic urticarial vasculitis. J. Am. Acad. Dermatol., 1998, 38(6 I), p. 899–905.
27. DAVIS, M. D. P., DAOUD, M. S., McEVOY, M. T. et al. Cutaneous manifestations of Churg-Strauss syndrome: A clinicopathologic correlation. J. Am. Acad. Dermatol., 1997, 37 (2 I), p. 199–203.
28. DAVID, C., JACHIET, M., PINETON DE CHAMBRUN, M. et al. Chronic obstructive pulmonary disease associated with hypocomplementemic urticarial vasculitis. J. Allergy Clin. Immunol. Pract., 2020, 8(9), p. 3222–3224.e1.
29. DeREMEE, R. A., SPECKS, U., DANIEL SU, W. P. Cutaneous Wegener’s granulomatosis: Clinical, histopathologic, and immunopathologic features of thirty patients. J. Am. Acad. Dermatol., 1994, 31(4), p. 605–612.
30. Di LERNIA, V., LOMBARDI, M., LO SCOCCO, G. Infantile acute hemorrhagic edema and rotavirus infection. Pediatr. Dermatol., 2004, 21(5), p. 548–550.
31. Di PIETRO, G. M., CASTELLAZZI, M. L., MASTRANGELO, A. et al. Henoch-Schönlein Purpura in children: Not only kidney but also lung. Pediatric Rheumatology, 2019, 17(1).
32. DOKTOR, V., HADI, A., HADI, A. et al. Erythema elevatum diutinum: a case report and review of literature. International Journal of Dermatology, 2019, 58(4), p. 408–415.
33. DO, N., RINGOLD, S., BRANDLING-BENNETT, H. Cutaneous polyarteritis nodosa in pediatric patients successfully treated with TNF-α inhibitor and methotrexate: Case series and literature review. Pediatr. Dermatol., 2019, 36(6), p. 932–935.
34. DUMOUCHEL, W., SMITH, E. T., BEASLEY, R. et al. Association of asthma therapy and Churg-Strauss syndrome: An analysis of postmarketing surveillance data. Clin. Ther., 2004, 26(7), p. 1092–1104.
35. E MOURA GARCIA, C. S., SOKOLOVA, A., TORRE, M. L. et al. Acute Hemorrhagic Edema of Infancy. Eur. Ann. Allergy Clin. Immunol., 2016, 48(1), p. 22–26.
36. ERDEN, A., BATU, E. D., SÖNMEZ, H. E. et al. Comparing polyarteritis nodosa in children and adults: a single center study. Int. J. Rheum. Dis., 2017, 20(8), p. 1016–1022.
37. ESATOGLU, S. N., KUTLUBAY, Z., HATEMI, G. Highlights of the 18th International Conference on Beh-çet’s syndrome. Clin. Exp. Rheumatol., 2018, 36(6), p. 3–12.
38. FELICETTI, M., GASPAROTTO, M., FRIGO, A. C. et al. Renal involvement at baseline can predict major renal relapse in anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis. Clin. Exp. Rheumatol., 2020, 38(2), p. 201–206.
39. FERREIRO, J. E., PASARIN, G., QUESADA, R. et al. Benign hypergammaglobulinemic purpura of Waldenström associated with Sjögren’s syndrome. Case report and review of immunologic aspects. Am. J. Med., 1986, 81(4), p. 734–740.
40. FINDER, K. A., McCOLLOUGH, M. L., DIXON, S. L. et al. Hypergammaglobulinemic purpura of Waldenström. J. Am. Acad. Dermatol., 1990, 23(4), p. 669–676.
41. FURUTA, S., IWAMOTO, T., NAKAJIMA, H., Update on eosinophilic granulomatosis with polyangiitis. Allergology International, 2019, 68(4), p. 430–436.
42. GENOVESE, G., TAVECCHIO, S., BERTI, E. et al. Pyoderma gangrenosum-like ulcerations in granulomatosis with polyangiitis: two cases and literature review, Rheumatol. Internat., 2018, 38(6), p. 1139–1151.
43. GILCHRIST, H., PATTERSON, J. W. Erythema nodosum and erythema induratum (nodular vasculitis): Diagnosis and management. Dermatologic Therapy, 2010, 23(4), p. 320–327.
44. GINSBERG, S., ROSNER, I., SLOBODIN, G. et al. Infliximab for the treatment of refractory polyarteritis nodosa. Clin Rheumatol, 2019, 38(10), p. 2825–2833.
45. GIOFFREDI, A., MARITATI, F., OLIVA, E. et al. Eosinophilic granulomatosis with polyangiitis: An overview. Frontiers in Immunology, 2014, 5, (NOV).
46. GIUGGIOLI, D., MANFREDI, A., LUMETTI, F. et al. Cryoglobulinemic vasculitis and skin ulcers. Our therapeutic strategy and review of the literature. Seminars in Arthritis and Rheumatism, 2015, 44(5), p. 518–526.
47. GOLSHEVSKY, D., CHEUNG, M., BURGNER, D. Kawasaki disease: The importance of prompt recognition and early referral. Aust. Fam. Physician, 2013, 42(7), p. 473–476.
48. GRAU, R. G. Churg-Strauss syndrome: 2005–2008 update. Current Rheumatology Reports, 2008, 10(6), p. 453–458.
49. GRECO, A., DE VIRGILIO, A., RIZZO, M. I. et al. Microscopic polyangiitis: Advances in diagnostic and therapeutic approaches. Autoimmunity Reviews, 2015, 14(9), p. 837–844.
50. GRECO, A., GALLO, A., FUSCONI, M. et al. Cogan’s syndrome: An autoimmune inner ear disease. Autoimmunity Reviews, 2013, 12(3), p. 396–400.
51. GRECO, A., MARINELLI, C., FUSCONI, M. et al. Clinic manifestations in granulomatosis with polyangiitis. Intern. J. Immunopathol. Pharmacol., 2016, 29(2), p. 151–159.
52. GROH, M., MASCIOCCO, G., KIRCHNER, E. et al. Heart transplantation in patients with eosinophilic granulomatosis with polyangiitis (Churg-Strauss syndrome). J. Hear. Lung Transplant., 2014, 33(8), p. 842–850.
53. HA, T. S., LEE, J. S. Scrotal involvement in childhood Henoch-Schönlein purpura. Acta Paediatr. Int. J. Paediatr., 2007, 96(4), p. 552–555.
54. HAMAD, A., JITHPRATUCK, W., KRISHNASWAMY, G. Urticarial vasculitis and associated disorders. Ann. Allergy, Asthma Immunol., 2017, 118(4), p. 394–398.
55. HAMADA, H., SUZUKI, H., ONOUCHI, Y. et al. Efficacy of primary treatment with immunoglobulin plus ciclosporin for prevention of coronary artery abnormalities in patients with Kawasaki disease predicted to be at increased risk of non-response to intravenous immunoglobulin (KAICA): a randomised controlled, open-label, blinded-endpoints, phase 3 trial. Lancet, 2019, 393(10176), p. 1128–1137.
56. HERNÁNDEZ-RODRÍGUEZ, J., CARBONELL, C., MIRÓN-CANELO, J. A. et al. Rituximab treatment for IgA vasculitis: A systematic review. Autoim. Rev, 2020, 19, (4).
57. HOFFMAN, G. S., KERR, G. S., LEAVITT, R. Y. et al. Wegener granulomatosis: An analysis of 158 patients. Ann Intern Med, 1992, 116(6), p. 488–498.
58. HOLLE, J. U., DUBRAU, C., HERLYN, K. et al. Rituximab for refractory granulomatosis with polyangiitis (Wegener’s granulomatosis): Comparison of efficacy in granulomatous versus vasculitic manifestations. Ann. Rheum. Dis., 2012, 71(3), p. 327–333.
59. HUNDER, G. G., AREND, W. P., BLOCH, D. A. et al. The American College of Rheumatology 1990 criteria for the classification of vasculitis: Introduc-tion. Arthritis Rheum., 1990, 33(8), p. 1065–1067.
60. CHARTAPISAK, W., OPASTIRAKUL, S., HODSON, E. M. et al. Interventions for preventing and treating kidney disease in Henoch-Schönlein Purpura (HSP). Cochrane Database of Systematic Reviews, 2009, (3).
61. CHASSET, F., FRANCÈS, C. Cutaneous Manifesta-tions of Medium - and Large-Vessel Vasculitis. Clinical Reviews in Allergy and Immunology, 2017, 53(3), p. 452–468.
62. CHEN, K. R., TOYOHARA, A., SUZUKI, A. et al. Clinical and histopathological spectrum of cutaneous vasculitis in rheumatoid arthritis. Br. J. Dermatol., 2002, 147(5), p. 905–913.
63. CHERQAOUI, B., CHAUSSET, A., STEPHAN, J. L. et al. Intravenous immunoglobulins for severe gastrointestinal involvement in pediatric Henoch-Schönlein purpura: A French retrospective study. Arch. Pediatr., 2016, 23(6), p. 584–590.
64. CHO, K. H., LEE, D. Y., KIM, C. W. Erythema induratum of bazin. Int. J. Dermatol., 1996, 35(11), p. 802–808.
65. CHOU, T., LOUISSANT, V. R., ADAMS, A. et al. Suc-cessful treatment of henoch-schönlein purpura with recurrent gastrointestinal involvement with mycophenolate Mofetil: A brief report. Clin. Pediatr. (Phila)., 2015, 54(9), p. 900–903.
66. CHUNG, S. A., SEO, P. Microscopic Polyangiitis. Rheum Dis Clin North Am, 2010, 36(3), p. 545–558.
67. INTERNATIONAL STUDY GROUP FOR BEHÇET’S DISEASE Criteria for diagnosis of Behcet’s disease. Lancet, 1990, 335(8697), p. 1078–1080.
68. ITO, Y., ARITA, M., KUMAGAI, S. et al. Diffuse alveo-lar hemorrhage in iga vasculitis with an atypical presentation. Intern. Med., 2018, 57(1), p. 81–84.
69. JACHIET, M., FLAGEUL, B., DEROUX, A. et al. The clinical spectrum and therapeutic management of hypocomplementemic urticarial vasculitis: Data from a french nationwide study of fifty-seven patients. Arthritis Rheumatol., 2015, 67(2), p. 527–534.
70. JAYNE, D. Challenges in the management of microscopic polyangiitis: Past, present and future. Curr Opin Rheumatol, 2008, 20(1), p. 3–9.
71. JENNETTE, J. C., FALK, R. J. New insight into the pathogenesis of vasculitis associated with antineutrophil cytoplasmic autoantibodies. Curr Opin Rheumatol, 2008, 20(1), p. 55–60.
72. JENNETTE, J. C., FALK, R. J., BACON, P. A. et al. 2012 Revised International Chapel Hill consensus conference nomenclature of vasculitides. Arthritis and Rheumatism, 2013, 65 (1), p. 1–11.
73. JIAO, T., WANG, M., ZHU, X. A case of erythema elevatum diutinum associated with peripheral ulcerative keratitis. Australas. J. Dermatol., 2012, 53(1), p. 78–80.
74. JOHN, K. J., SADIQ, M., THOMAS, M. et al. Henoch-Schonlein purpura associated with HLA-B27 positive axial spondyloarthritis in a young man. BMJ Case Rep., 2019, 12(5).
75. KALLAS, R., GOLDMAN, D., PETRI, M. A. Cutaneous vasculitis in SLE. Lupus Sci. Med., 2020, 7(1), p. e000411.
76. KARADAG, O., JAYNE, D. J. Polyarteritis nodosa revisited: A review of historical approaches, subphenotypes and a research agenda. Clin Exp Rheumatol, 2018, 36, p. S135–S142.
77. KARRAS, A. Microscopic Polyangiitis: New Insights into Pathogenesis, Clinical Features and Therapy. Semin. Respir. Crit. Care Med., 2018, 39(4), p. 459–464.
78. KAWAKAMI, T., KAWANABE, T., SAITO, C. et al. Clinical and histopathologic features of 8 patients with microscopic polyangiitis including two with a slowly progressive clinical course. J. Am. Acad. Dermatol., 2007, 57(5), p. 840–848.
79. KAWAKAMI, T., SOMA, Y. Correlation of livedo racemosa, cutaneous inflammatory plaques, and antiphospholipid antibodies in patients with cutaneous polyarteritis nodosa. Medicine (Baltimore), 2011, 90(2), p. 119–124.
80. KIEFFER, C., CRIBIER, B., LIPSKER, D. Neutrophilic urticarial dermatosis a variant of neutrophilic urticaria strongly associated with systemic disease. report of 9 new cases and review of the literature. Medicine (Baltimore), 2009, 88(1), p. 23–31.
81. KIM, J. Y., CHOI, H., KIM, M. K. et al., Clinical significance of ANCA positivity in patients with IgA vasculitis: a retrospective monocentric study. Rheumatol. Int., 2019, 39 (11), p. 1927–1936.
82. KLUGER, N., PAGNOUX, C., GUILLEVIN, L. et al. Comparison of cutaneous manifestations in systemic polyarteritis nodosa and microscopic polyangiitis. Br. J. Dermatol., 2008, 159(3), p. 615–620.
83. KOLKHIR, P., GRAKHOVA, M., BONNEKOH, H. et al. Treatment of urticarial vasculitis: A systematic review. J. Allergy Clin. Immunol., 2019, 143(2), p. 458–466.
84. KOSSARD, S. Defining lymphocytic vasculitis. Australasian Journal of Dermatology, 2000, 41(3), p. 149–155.
85. KRUSCHE, M., RUFFER, N., SCHNEIDER, U. et al. Tocilizumab treatment for polyarteritis nodosa. Rheumatology (United Kingdom), 2020, 59(10), p. E63–E65.
86. KUO, H. C., LIANG, C. DI, WANG, C. L. et al. Serum albumin level predicts initial intravenous immunoglobulin treatment failure in Kawasaki disease. Acta Paediatr. Int. J. Paediatr., 2010, 99(10), p. 1578–1583.
87. KWAN, Y. W., LEUNG, C. W. Pustulo-vesicular skin eruption in a child with probable Kawasaki disease. Eur. J. Pediatr., 2005, 164(12), p. 770–771.
88. LEUNG, A. K. C., BARANKIN, B., LEONG, K. F. Henoch-Schönlein Purpura in Children: An Updated Review. Curr. Pediatr. Rev., 2020, 16(4), p. 265–276.
89. LEUNG, A. K. C., LEONG, K. F., LAM, J. M. Acute Hemorrhagic Edema of Infancy: A Diagnostic Challenge for the General Pediatrician. Curr. Pediatr. Rev., 2020, 16(4), p. 285–293.
90. LIMTONG, P., CHANPRAPAPH, K., VACHIRAMON, V. et al. Cutaneous and extracutaneous manifesta-tions of behçet’s disease linked to its disease activi-ty and prognosis. Clin. Cosmet. Investig. Dermatol., 2020, 13, p. 639–647.
91. LINSKEY, K. R., KROSHINSKY, D., MIHM, M. C. et al. Immunoglobulin-A – Associated small-vessel vasculitis: A 10-year experience at the Massachusetts General Hospital. J. Am. Acad. Dermatol., 2012, 66(5), p. 813–822.
92. LIPSKER, D. Monoclonal gammopathy of cutaneous significance: review of a relevant concept. Journal of the European Academy of Dermatology and Venereology, 2017, 31(1), p. 45–52.
93. LYNCH, J. P., DERHOVANESSIAN, A., TAZELAAR, H. et al. Granulomatosis with Polyangiitis (Wegener’s Granulomatosis): Evolving Concepts in Treatment. Seminars in Respiratory and Critical Care Medicine, 2018, 39(4), p. 434–458.
94. MAKINO, N., NAKAMURA, Y., YASHIRO, M. et al. Descriptive epidemiology of Kawasaki disease in Japan, 2011–2012: From the results of the 22nd nationwide survey. J. Epidemiol., 2015, 25(3), p. 239–245.
95. MARITATI, F., IANNUZZELLA, F., PAVIA, M. P. et al. Kidney involvement in medium - and large-vessel vasculitis. Journal of Nephrology, 2016, 29(4), p. 495–505.
96. MARZANO, A. V., RAIMONDO, M. G., BERTI, E. et al. Cutaneous Manifestations of ANCA-Associated Small Vessels Vasculitis. Clinical Reviews in Allergy and Immunology, 2017, 53(3), p. 428–438.
97. MAZZARO, C., MAURO, E., ERMACORA, A. et al. Hepatitis C virus - related cryoglobulinemic vasculitis. Minerva Med., 2020, 112(2).
98. MILOSLAVSKY, E. M., SPECKS, U., MERKEL, P. A. et al. Clinical outcomes of remission induction therapy for severe antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheum., 2013, 65(9), p. 2441–2449.
99. MOMEN, S. E., JORIZZO, J., AL-NIAIMI, F. Erythema elevatum diutinum: A review of presentation and treatment. Journal of the European Academy of Dermatology and Venereology, 2014, 28(12), p. 1594–1602.
100. MORGAN, A. J., SCHWARTZ, R. A. Cutaneous polyarteritis nodosa: A comprehensive review. International Journal of Dermatology, 2010, 49(7), p. 750–756.
101. MOTA, A., SAHEBGHADAM LOTFI, A., JAMSHIDI, A. R. et al. Alpha 1-antitrypsin activity is markedly decreased in Wegener’s granulomatosis. Rheumatol. Int., 2014, 34(4), p. 553–558.
102. NAKAMURA, T., KANAZAWA, N., IKEDA, T. et al. Cutaneous polyarteritis nodosa: Revisiting its definition and diagnostic criteria. Archives of Dermatological Research, 2009, 48301(1), p. 117–121.
103. NAKAZAWA, D., MASUDA, S., TOMARU, U. et al. Pathogenesis and therapeutic interventions for ANCA-associated vasculitis. Nature Reviews Rheumatology, 2019, 15(2), p. 91–101.
104. NEWBURGER, J., SCHMIEDER, G. J. Erythema Elevatum Diutinum. 2021.
105. NEWBURGER, J. W., TAKAHASHI, M., BURNS, J. C. Kawasaki Disease. Journal of the American College of Cardiology, 2016, 67(14), p. 1738–1749.
106. NGUYEN, Y., GUILLEVIN, L. Eosinophilic Granulomatosis with Polyangiitis (Churg-Strauss). Semin. Respir. Crit. Care Med., 2018, 39(4), p. 471–481.
107. NIIYAMA, S., AMOH, Y., TOMITA, M. et al. Dermatological manifestations associated with microscopic polyangiitis. Rheumatol. Int., 2008, 28(6), p. 593–595.
108. NINAN, J., LESTER, S., HILL, C. Giant cell arteritis. Best Practice and Research: Clinical Rheumatology, 2016, 30(1), p. 169–188.
109. OSTOJIC, P., JEREMIC, I. R. Managing refractory cryoglobulinemic vasculitis: Challenges and solutions. Journal of Inflammation Research, 2017, p. 49–54.
110. PAGNOUX, C., SEROR, R., HENEGAR, C. et al. Clinical features and outcomes in 348 patients with polyarteritis nodosa: A systematic retrospective study of patients diagnosed between 1963 and 2005 and entered into the French Vasculitis Study Group database. Arthritis Rheum., 2010, 62(2), p. 616–626.
111. PAGNOUX, C., MAHR, A., HAMIDOU, M. A. et al. Azathioprine or Methotrexate Maintenance for ANCA-Associated Vasculitis. N. Engl. J. Med., 2008, 359(26), p. 2790–2803.
112. PATNALA, G. P., SUNANDINI, A. P., RAYAVARAPU, R. et al. Erythema elevatum diutinum in associa-tion with IgA monoclonal gammopathy: A rare case report. Indian Dermatol. Online J., 2016, 7(4), p. 300–303.
113. PIRAM, M. AND MAHR, A. Epidemiology of immunoglobulin A vasculitis (Henoch-Schönlein): Current state of knowledge. Curr Opin Rheumatol, 2013, 25(2), p. 171–178.
114. PODJASEK, J. O., WETTER, D. A., PITTELKOW, M. R. et al. Cutaneous small-vessel vasculitis associated with solid organ malignancies: The Mayo Clinic experience, 1996 to 2009. J. Am. Acad. Dermatol., 2012, 66(2).
115. POLAT, A. K., KARAALI, M. G., AKSU, A. E. K. et al. A rare cutaneous tuberculosis form, erythema induratum of bazin: 6 years’ experience. Acta Dermatovenerologica Alpina, Pannonica Adriat., 2020, 29(3), p. 123–128.
116. POTERUCHA, T. J., WETTER, D. A., GIBSON, L. E. et al. Correlates of systemic disease in adult Henoch-Schönlein purpura: A retrospective study of direct immunofluorescence and skin lesion distribution in 87 patients at Mayo Clinic. J. Am. Acad. Dermatol., 2012, 67(4), p. 612–616.
117. POTERUCHA, T. J., WETTER, D. A., GIBSON, L. E. et al. Histopathology and correlates of systemic disease in adult Henoch-Schönlein purpura: A retrospective study of microscopic and clinical findings in 68 patients at Mayo Clinic. J. Am. Acad. Dermatol., 2013, 68(3).
118. RAINE, C., CANNING, B., MARKS, J. et al. Severe gangrene in a patient with anti-RNP positive limited cutaneous systemic sclerosis/rheumatoid arthritis overlap syndrome caused by vasculopathy and vasculitis. Eur. J. Rheumatol., 2018, 5(4), p. 269–271.
119. RAJAGOPALA, S., SHOBHA, V., DEVARAJ, U. et al. Pulmonary Hemorrhage in Henoch-Schönlein Purpura: Case Report and Systematic Review of the English Literature. Semin. Arthritis Rheum., 2013, 42(4), p. 391–400.
120. REKHTMAN, S., TANNENBAUM, R., STRUNK, A. et al. Mucocutaneous disease and related clinical characteristics in hospitalized children and adolescents with COVID-19 and multisystem inflammatory syndrome in children. J. Am. Acad. Dermatol., 2021, 84(2), p. 408–414.
121. RIMAR, D., ALPERT, A., STAROSVETSKY, E. et al. Tofacitinib for polyarteritis nodosa: A tailored therapy. Annals of the Rheumatic Diseases, 2016, 75(12), p. 2214–2216.
122. RODÓ, X., CURCOLL, R., ROBINSON, M. et al. Tropospheric winds from northeastern China carry the etiologic agent of Kawasaki disease from its source to Japan. Proc. Natl. Acad. Sci. U. S. A., 2014, 111(22), p. 7952–7957.
123. RONKAINEN, J., KOSKIMIES, O., ALA-HOUHALA, M. et al. Early prednisone therapy in Henoch-Schönlein purpura: A randomized, double-blind, placebo-controlled trial. J. Pediatr., 2006, 149(2), p. 241–247.
124. RUSSO, R. A. G., KATSICAS, M. M. Takayasu arteritis. Frontiers in Pediatrics, 6. Frontiers Media S.A., 2018.
125. ROWLEY, A. H., BAKER, S. C., SHULMAN, S. T. et al. Ultrastructural, immunofluorescence, and RNA evidence support the hypothesis of a “new” virus associated with Kawasaki disease. J. Infect. Dis., 2011, 203(7), p. 1021–1030.
126. SAADOUN, D., TERRIER, B., SEMOUN, O. et al. Hepatitis C virus associated polyarteritis nodosa. Arthritis Care Res., 2011, 63(3), p. 427–435.
127. SALIM, S. A., YOUSUF, T., PATEL, A. et al. Hypocomplementemic Urticarial Vasculitis Syndrome With Crescentic Glomerulonephritis. Am. J. Med. Sci., 2018, 355(2), p. 195–200.
128. SAULSBURY, F. T. Clinical update: Henoch-Schönlein purpura. Lancet, 2007, 369 (9566), p. 976–978.
129. SEGELMARK, M., SELGA, D., The challenge of managing patients with polyarteritis nodosa. Curr Opin Rheumatol, 2007, 19(1), p. 33–38.
130. SEGURA, S., PUJOL, R. M., TRINDADE, F. et al. Vasculitis in erythema induratum of Bazin: A histopathologic study of 101 biopsy specimens from 86 patients. J. Am. Acad. Dermatol., 2008, 59(5), p. 839–851.
131. SHAVIT, E., ALAVI, A., SIBBALD, R. G. Vasculitis–What Do We Have to Know? A Review of Literature. International Journal of Lower Extremity Wounds, 2018, 17(4), p. 218–226.
132. SHIMIZU, A., TANAKA, K., TAKAZAWA, S. et al. A large superior mesenteric artery aneurysm and ileal obstruction: A rare presentation of polyarteritis nodosa in an infant. Oxford Med. Case Reports, 2019, 2019, (9), p. 401–404.
133. SHUAI, Z. W., LV, Y. F., ZHANG, M. M. et al. Clinical analysis of patients with myeloperoxidase antineutrophil cytoplasmic antibody-associated vasculitis. Genet. Mol. Res., 2015, 14(2), p. 5296–5303.
134. SHUAI, Z. Q., ZHANG, C. X., SHUAI, Z. W. et al. Efficacy and safety of biological agents in the treatment of patients with takayasu arteritis: A systematic review and meta-analysis. European Review for Medical and Pharmacological Sciences, 2021, 25(1), p. 250–262.
135. SCHROETER, A. L., CONN, D. L., and JORDON, R. E. Immunoglobulin and complement deposition in skin of rheumatoid arthritis and systemic lupus erythematosus patients. Ann. Rheum. Dis., 1976, 35(4), p. 321–326.
136. SCHNEIDER, J. W., JORDAAN, H. F. The histopathologic spectrum of erythema induratum of Bazin. Am. J. Dermatopathol., 1997, 19(4), p. 323–333.
137. SILVA, F., PINTO, C., BARBOSA, A. et al. New in-sights in cryoglobulinemic vasculitis. Journal of Autoimmunity, 2019, 105. Academic Press.
138. SINGH, S., VIGNESH, P., AND BURGNER, D. The epidemiology of Kawasaki disease: A global update. Archives of Disease in Childhood, 2015, 100(11), p. 1084–1088.
139. SMYTH, L., GASKIN, G., PUSEY, C. D. Microscopic polyangiitis. Seminars in Respiratory and Critical Care Medicine, 2004, 25(5), p. 523–533.
140. SONG, G. G., LEE, Y. H. Efficacy and safety of biological agents in patients with giant cell arteritis: A meta-analysis of randomized trials. Int. J. Clin. Pharmacol. Ther., 2020, 58(9), p. 504–510.
141. SÖNMEZ, H. E., ARMAĞAN, B., AYAN, G. et al. Polyarteritis nodosa: lessons from 25 years of experience. Clin. Exp. Rheumatol., 2019, 37(2), p. 52–56.
142. STONE, J. H. Rituximab versus cyclophosphamide for ANCA-associated vasculitis. Journal fur Mineralstoffwechsel, 2010, 17(4), p. 168.
143. SUNDERKÖTTER, C. H., ZELGER, B., CHEN, K. R. et al. Nomenclature of Cutaneous Vasculitis: Der-matologic Addendum to the 2012 Revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheumatol., 2018, 70(2), p. 171–184.
144. UEHARA, R., BELAY, E. D. Epidemiology of Kawasa-ki disease in Asia, Europe, and the United States. J. Epidemiol., 2012, 22(2), p. 79–85.
145. ULUCA, Ü., ECE, A., ŞEN, V. et al. Management of intestinal bleeding with single-dose cyclophosphamide in Henoch-Schönlein purpura. Pediatr. Int., 2015, 57(3), p. 498–500.
146. VAN WIJNGAARDEN, R. A. F. D. L., VAN RIJN, L., HAGEN, E. C. et al. Hypotheses on the etiology of antineutrophil cytoplasmic autoantibody-associated vasculitis: The cause is hidden, but the result is known. Clin J Am Soc Nephrol, 2008, 3(1), p. 237–252.
147. VANKALAKUNTI, M., JOSHI, K., JAIN, S. et al. Polyarteritis nodosa in hairy cell leukaemia: An autopsy report [2]. J Clin Pathol, 2007, 60(10), p. 1181–1182.
148. VIGUIER, M., GUILLEVIN, L., LAROCHE, L. Treat-ment of Parvovirus B19-Associated Polyarteritis Nodosa with Intravenous Immune Globulin. N. Engl. J. Med., 2001, 344(19), p. 1481–1482.
149. VOLEJNIKOVA, J., HORACEK, J., KOPRIVA, F. Dapsone treatment is efficient against persistent cutaneous and gastrointestinal symptoms in children with Henoch-Schönlein purpura. Biomed. Pap., 2018, 162(2), p. 154–158.
150. WATTS, R., LANE, S., HANSLIK, T. et al. Development and validation of a consensus methodology for the classification of the ANCA-associated vasculitides and polyarteritis nodosa for epidemiological studies. Ann. Rheum. Dis., 2007, 66(2), p. 222–227.
151. WEISS, P. F., KLINK, A. J., LOCALIO, R. et al. Corticosteroids may improve clinical outcomes during hospitalization for henoch-schönlein purpura. Pediatrics, 2010, 126(4), p. 674–681.
152. WEYAND, C. M., GORONZY, J. J. Giant-Cell Arteritis and Polymyalgia Rheumatica. N. Engl. J. Med., 2014, 371(1), p. 50–57.
153. WINKLER, A., TRUE, D. Giant Cell Arteritis: 2018 Review. Mo. Med., 2018, 115(5), p. 468–470.
154. WOLF, D., BEN-YEHUDA, A., OKON, E. et al. Nodular vasculitis associated with propylthiouracil therapy. Cutis, 1992, 49(4), p. 253–255.
155. WRIGHT, A. C., GIBSON, L. E., DAVIS, D. M. R. Cutaneous manifestations of pediatric granulomatosis with polyangiitis: A clinicopathologic and immunopathologic analysis. J. Am. Acad. Dermatol., 2015, 72(5), p. 859–867.
156. XU, H., LI, Y. M., MA, H. et al. Mycobacterium tuberculosis found at both skin lesions and Mantoux testing site in a patient with erythema induratum of Bazin. J. Dermatol., 2017, 44(10), p. 1145–1147.
157. XU, J., XU, J., LI, H. et al. Dermatomyositis combined with IgA vasculitis: A case report. Beijing Da Xue Xue Bao., 2019, 51(6), p. 1173–1177.
Štítky
Dermatologie Dětská dermatologieČlánek vyšel v časopise
Česko-slovenská dermatologie

2021 Číslo 3
- Ingenol-mebutát rozšiřuje možnosti léčby aktinické keratózy
- Proces hojení ran krok za krokem a co ho může zkomplikovat
- Léčba chronické blefaritidy vyžaduje dlouhodobou péči
- Léčba zánětů spojivek a mazových žlázek víčka v primární péči
Nejčtenější v tomto čísle
- Vaskulitidy z pohledu dermatologa
- Porovnání hospitalizačních záznamů pacientů se syfilis z let 2007–2009 a 2017–2019
- Atopická dermatitida v registru BIOREP
- Mnohočetné drsné papuly s pozitivním Auspitzovým fenoménem. Stručný přehled
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy