
Klinický případ: Bolestivé noduly na zádech
Autoři:
S. Dudková 1; J. Štork 1; O. Kodet 1,2,3
Působiště autorů:
Dermatovenerologická klinika 1. LF UK a VFN, Praha
přednosta prof. MUDr. Jiří Štork, CSc.
; Anatomický ústav 1. LF UK, Praha
přednosta prof. MUDr. Karel Smetana, DrSc.
2; BIOCEV, Praha
3
Vyšlo v časopise:
Čes-slov Derm, 92, 2017, No. 4, p. 184-188
Kategorie:
Repetitorium
K vyšetření se dostavila 32letá pacientka, která si stěžovala na mnohočetné a silně bolestivé projevy na zádech. V rodinné anamnéze udávala úmrtí otce matky na karcinom prostaty, matka pacientky podstoupila ve 45 letech hysterektomii pro myomatózu. Sama pacientka byla od dětství sledována na plicním pro astma bronchiale na terapii inhalačním budesonidem. Před půl rokem podstoupila enukleaci myomu dělohy, s nálezem hladkosvalového nádoru nejistého maligního potenciálu („smooth muscle tumor of uncertain malignant potential“ – STUMP). Následně podstoupila hysterektomii. Kožní projevy se postupně stále tvoří asi sedm let na zádech, z nichž některé projevy byly silně bolestivé, a proto byly v minulosti ošetřeny CO2 laserem, který zmírnil jejich bolestivost. Při vyšetření byly na zádech mezi lopatkami patrny diseminované erytematózní papuly až noduly velikosti do 15 mm, depigmentované okrouhlé po předchozím laserovém ošetření (obr. 1 a 2). Jeden nodul byl patrný i na pravém rameni. Projevy mezi lopatkami byly silně bolestivé i při šetrné palpaci. Vzhledem k bolestivosti projevů byla provedena excize dvou nejvíce bolestivých projevů a histologické vyšetření (obr. 3 a 4).



HISTOPATOLOGIE
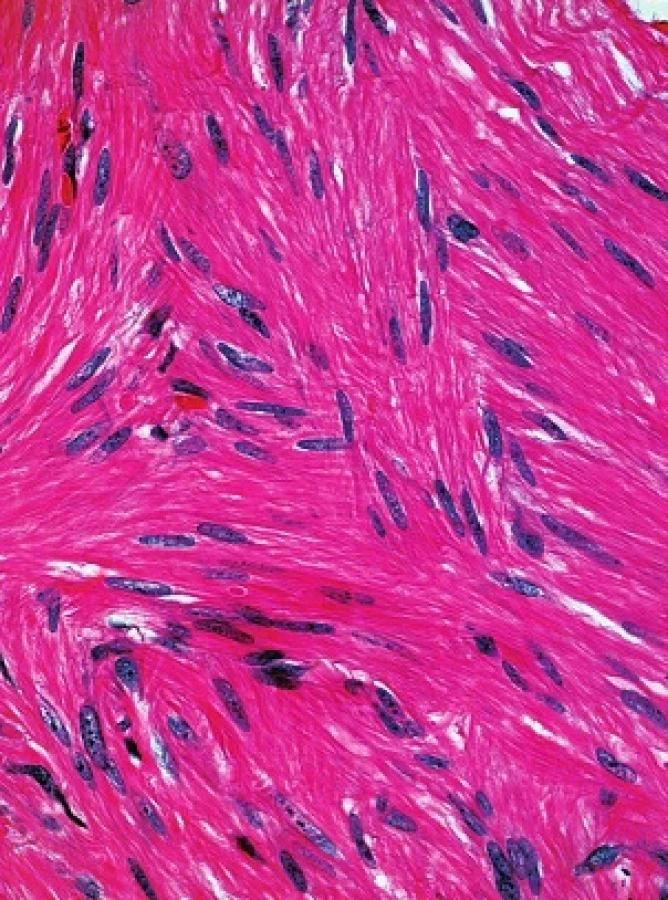
V obou excizích byl zastižen obdobný nález. Epidermis byla beze změn, korium bylo prostoupeno různě se proplétajícími svazky z vřetenitých eozinofilních buněk s protáhlými tupě zakončenými („doutníkovitými“) jádry (viz obr. 3 a 4).
Závěr
Nález v obou excizích svědčil pro piloleiomyom.
PRŮBĚH
Genetické vyšetření prokázalo mutaci genu FH, který kóduje enzym fumarát hydratázu, která je popsána u autozomálně dominantního syndromu hereditární leiomyomatózy a renálního karcinomu. Sonografické vyšetření břicha a základní laboratorní vyšetření včetně moče byla v normě. Pacientka je v naší dispenzární onkologické péči pro možný vznik kožního leiomyosarkomu, zatím bez progrese kožních projevů a vzniku jiných změn.
DISKUSE
Syndrom hereditární leiomyomatózy a renálního karcinomu (Hereditary leiomyomatosis and renal cell cancer, HLRCC) je vzácné autozomálně dominantně dědičné onemocnění s predispozicí ke vzniku mnohočetných kožních a děložních leiomyomů a vysokým rizikem vzniku karcinomu ledvin, nejčastěji agresivního papilárního karcinomu typu II [13]. Onemocnění je známé také jako mnohočetná kožní a děložní leiomyomatóza (multiple cutaneous and uterine leiomyomatosis, MCUL). Syndrom byl poprvé popsán Blumem a Jeanem v roce 1954, ale až v roce 1973 Reedem byla zdokumentována autozomálně dominantní dědičnost, proto je toto postižení nazývané i jako Reedův syndrom [15]. Doposud bylo zdokumentováno celosvětově přibližně 200 rodin, zejména v Severní Americe, Velké Británii, Nizozemsku, Finsku, Španělsku, Indii a Japonsku [5, 13, 17, 19]. Onemocnění nevykazuje pohlavní závislost, ale vzhledem k tvorbě děložních leiomyomů je postižení žen více zatěžující.
Příčinou vzniku je autozomálně dominantně dědičná heterozygotní mutace FH genu na chromozomu 1q42.3-43 kódující enzym fumarát hydratázu. Fumarát hydratáza je tumor supresorový gen, který se vyskytuje v mitochondriích a cytosolu. V cytosolu se účastní metabolismu aminokyselin a nukleotidů. V mitochondriích katalyzuje hydrataci fumarátu na malát jako součást Krebsova cyklu, ale jednoznačná role FH genu v indukci nádorů je nejasná. Předpokládá se role v uplatnění hypoxie a anaerobního metabolismu [6, 11, 18]. Hypoxické mikroprostředí aktivuje řadu proteinů, které se uplatňují v nádorové biologii, jako je GLUT1 či NRF2 [8].
Hlavní a často první klinickou manifestací syndromu je tvorba kožních leiomyomů. Nejčastěji postihují oblast extenzorů končetin, trup, obličej a oblast krku. Je popsána zosteriformní či lineární distribuce, postižení může být ale i symetrické nebo diseminované. Klinicky jsou patrné jako solitární, častěji však mnohočetné papuly a noduly, barvy kůže, světle červené či hnědavé, velikosti od 2 do 20 mm. Počet projevů se v průměru pohybuje okolo 26, ale může dosahovat až 100 a více projevů. Vzhledem k histogenezi lze rozlišit tři podtypy leiomyomů:
- Piloleiomyom – je nejčastějším podtypem, vychází z musculus arrector pilorum a může být výrazně bolestivý.
- Genitální leiomyomy jsou také výrazně bolestivé a vycházejí z tunica dartos, tedy obalů genitálu, případně z oblasti svalů prsního dvorce.
- Vzácnější varianta angioleiomyomů, představuje výrazně bolestivé projevy vycházející z tunica media cévní stěny [8, 13].
K rozvoji kožních leiomyomů dochází asi u 75 % pacientů s HLRCC, zpravidla od adolescence, průměrný věk postižených je 25 let. Bolestivost je přítomna až u 90 % všech nemocných a je nejčastějším příznakem vedoucím k vyšetření [14]. Bolest leiomyomů je ostrá, vystřelující, někdy pálivá. Objevuje se spontánně, může být vyprovokována chladem, stresem, dotykem nebo minimálním traumatem projevu. U čtyřech pacientů s HLRCC byla popsána maligní transformace kožních leiomyomů v leiomyosarkom (riziko se odhaduje na 1–2 %). Klinicky je velice obtížné rozlišit maligní potenciál projevu, proto je vhodné pacienty dlouhodobě dispenzarizovat. Na maligní transformaci projevu nás může upozornit rychlý růst, nepravidelné okraje, povrchová ulcerace, nebo výraznější bolestivost projevu [13].
Mnohočetné děložní leiomyomy postihují pacientky s HLRCC ve více jak 75 %. Mohou dosahovat až 10 cm v průměru a vyskytují se u žen přibližně o 10 let dříve, než je v populaci běžné (již před 30. rokem života) a mohou být důvodem i časné hysterektomie (průměrný věk 30 let oproti v populaci běžnému průměru 40 let) i vzhledem k vyššímu riziku maligní transformace v děložní leiomyosarkom. Nejčastěji se děložní leiomyomy projevují menoragií, nepravidelnou menstruací, bolestivostí nebo poruchami plodnosti vedoucí až k neplodnosti [1, 13].
U nemocných je vysoké riziko vzniku karcinomu ledviny. Jednoznačná souvislost s kožními a děložními leiomyomy byla ustanovena až v roce 2001 [18]. Riziko unilaterálního papilárního karcinomu ledviny typu II se u pacientů s HLRCC odhaduje na 20–34 %, vzácně může být postižení oboustranné. Tento typ karcinomu se vyznačuje agresivním chováním s časným vznikem metastáz, postižení bývá často asymptomatické a k diagnostice dochází až v pokročilém stadiu onemocnění. U více než 70 % pacientů dochází k úmrtí do 5 let od stanovení diagnózy. Mezi příznaky patří hematurie, necharakteristická bolest beder či břicha nebo hmatná rezistence při klinickém vyšetření [5, 6, 13]. Častěji, až ve 36 %, se vyskytují i benigní cysty ledvin oproti nepostižené populaci (cca 8 %) [6, 16]. Z dalších nádorů se při mutaci genu pro FH mohou vyskytovat karcinomy prostaty a močového měchýře, karcinom mammy nebo gastrointestinální stromální tumor [13].
Diagnostická HLRCC se opírá o ustanovení hlavních a vedlejší klinických kritérií (viz tabulka 1) [13]. Definitivní diagnóza je stanovena genetickým potvrzením germinální mutace fumarát hydratázy, genu FH, která však nemusí být pozitivní u všech klinicky manifestovaných případů. Její pozitivita je detekována v 70–90 % případů, přičemž u negativních pacientů, kteří splňují klinická kritéria, nacházíme amplifikaci či delece lokusu pro FH gen, v dalších případech lze doplnit aktivitu enzymu fumarát hydratázy, která může být snížena o více než 60 %, bez přítomné genetické alterace [3, 13, 21].
![Diagnostická kritéria syndromu hereditární leiomyomatózy a renálního karcinomu [13]](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/3871e0cfb61618554212612463f974ca.png)
Histologicky u všech typů kožních leiomyomů nacházíme propletené snopce hladkých svalových buněk, neostře ohraničené vůči okolí s různým množstvím kolagenu v extracelulární matrix. Buňky jsou vřetenité morfologie s eozinofilní cytoplazmou, jádra jsou protáhlá s tupými konci (cigaretový, doutníkovitý tvar). Projevy jsou lokalizovány primárně v dermis, ale mohou zasahovat i do podkožní tukové tkáně. Histologicky lze prokázat i zvýšenou hustotu nervové tkáně oproti okolní zdravé tkáni [8, 13, 14]. Imunohistochemicky prokazujeme silnou pozitivitu hladkého svalového aktinu, kaldesmonu nebo vimentinu [8]. Jako slibný imunohistochemický marker pro diagnostiku HLRCC se zdá být S-(2-succinyl) cysteine (2SC), který je odpadním produktem metabolismu hromadícího se fumarátu. 2SC lze imunohistochemicky prokázat v kožních a děložních leiomyomech, ale i v papilárním karcinomu ledviny u pacientů s HRLCC, senzitivita se pohybuje okolo 95 % [8].
V diferenciální diagnostice je třeba myslet na jiné bolestivé kožní nádory, jako je ekrinní spiradenom, neurinom, neurilemmom, dermatofibrom, angiolipom nebo glomus tumor [7, 8, 13]. Z nebolestivých benigních tumorů zvažujeme trichoepiteliom, lipom, cylindrom či ekrinní porom [7, 13]. Mnohočetné leiomyomy nemusí být výhradně u HLRCC, jsou popisovány i u pacientů s chronickou lymfocytární leukémií [20] a u HIV pozitivních pacientů [10].
Léčba kožních leiomyomů je závislá na jejich počtu, kosmetickém efektu, a především na intenzitě bolestivosti. Při menším postižením je nejúčinnější chirurgická excize, riziko recidivy je však značně vysoké. Na malé případně silně bolestivé léze lze použít CO2 laser, který usnadňuje myolýzu. Na menší projevy lze terapeuticky vyzkoušet efekt elektrodestrukce nebo kryoterapie [13]. Bolestivost lézí může být zmírněna podáním látek blokujících kontrakce hladkého svalstva (blokátory kalciových kanálů, doxazosin, fenoxybenzamin a nitroglycerin), které lze kombinovat s psychofarmaky s analgetickým efektem (gabapentin, pregabalin) [2, 4]. Některé práce poukazují i na účinnost aplikace botulotoxinu ke snížení intenzity bolestivosti leiomyomů [12].
Každý pacient s mnohočetnými kožními leiomyomy by měl podstoupit kompletní vyšetření dermatologické, sonografické vyšetření břicha a u žen i gynekologické vyšetření. Významná je i pečlivě odebraná rodinná anamnéza včetně anamnézy chirurgických výkonů. U pacientů s HLRCC je nutná pravidelná dispenzarizace s multidisciplinárním přístupem. Dermatologické kontroly se doporučují 1krát za rok, gynekologické vyšetření se doporučuje u pacientek také 1krát za rok od věku 18 let, včetně vaginálního ultrazvukového vyšetření dělohy. Doporučuje se i pravidelné samovyšetření prsu a ultrazvuková mamografie od 45 let min. 1krát za 2 roky. Dále se doporučuje provádět ultrazvukové vyšetření břicha se zaměřením na ledviny 1krát za rok, případně u pacientů starších 30 let provádět magnetickou rezonanci břicha 1krát za 2 roky. Při pravidelné kontrole se doporučuje minimálně 1krát za rok provádět vyšetření močového sedimentu [13, 16]. Prognóza pacientů se odvíjí od míry postižení a především možnosti včasně diagnostikovat závažné maligní komplikace tohoto syndromu.
Do redakce došlo dne 20. 7. 2017.
Adresa pro korespondenci:
MUDr. Ondřej Kodet, Ph.D.
Dermatovenerologická klinika 1. LF UK a VFN
U Nemocnice 2
128 00 Praha 2
e-mail: ondrej.kodet@vfn.cz
Zdroje
1. ALAM, N. A., BARCLAY, E., ROWAN, A. J. et al. Clinical features of multiple cutaneous and uterine leiomyomatosis: an underdiagnosed syndrome. Archives of Dermatology, 2005, 141, p. 199–206.
2. ALAM, M., RABINOWITZ, A. D., ENGLER, D. E. Gabapentin treatment of multiple piloleiomyoma-related pain. J. Am. Acad. Dermatol., 2002, 46, 2, p. 27–29.
3. ALAM, N. A., ROWAN, A. J., WORTHAM, N. C. et al. Genetic and functional analyses of FH mutations in multiple cutaneous and uterine leiomyomatosis, hereditary leiomyomatosis and renal cancer, and fumarate hydratase deficiency. Hum. Mol. Genet., 2003, 12, p. 1241–1252.
4. BATCHELOR, R. J., LYON, C. C., HIGHET, A. S. Successful treatment of pain in two patiens with cutaneous leiomyomata with the oral alpha-1 adrenoceptor antagonist, doxazosin. Br. J. Dermatol., 2004, 150, 4, p. 775–776.
5. DE VELASCO, G., MUNOZ, C., SEPUVELDA, J. M., CASTELLANO D. Sequential treatments in hereditary leiomyomatosis and renal cell carcinoma (HLRCC): case report and review of the literature. Can. Urol. Assoc. J., 2015, 9, p. 3–4.
6. GRUBB, R. L. 3rd, FRANKS, M. E., TORO, J. et al. Hereditary leiomyomatosis and renal cell cancer: a syndrome associated with an aggressive form of inherited renal cancer. J. Urol., 2007, 177, p. 2074–2080.
7. HRŇA, Š., KODET, O., BUCIFALOVÁ, M., ŠTORK, J. Klinický případ: bolestivá lividní rezistence. Čes - -slov Derm., 2016, 91, 6, p. 288–290.
8. CHEN, Y. B., BRANNON, A. R., TOUBAJI, A. et al. Hereditary leiomyomatosis and renal cell carcinoma syndrome-associated renal cancer: recognition of the syndrome by pathologic features and the utility of detecting aberrant succination by immunohistochemistry. Am J Surg Pathol., 2014, 38, 5, p. 627–637.
9. JASON, J. E., SHAYNA, S., STEPHEN, E. M. Reeds Syndrome – A Case Of Multiple Cutaneous and Uterine Leiomyomas. J. Clin. Aesthet. Dermatol., 2011, 4, 12, p. 37–42.
10. KANIKATIS, J., CARBONNEL, E., CHOUVET, B. et al. Cutaneous leiomyomas (piloleiomyomas) in adult patient with human immunodeficiency virus infection. Br. J. Dermatol., 2000, 143, p. 1338–1340.
11. LINEHAN, W. M., ROUAULT, T. A. Molecular pathways: fumarate hydratace - deficient kidney cancer - targeting the Warburg effect in Cancer. Clinical Cancer Research, 2013, 19, 13, p. 3345–3352.
12. ONDER, M., ADISEN, E. A new indication of botulinum toxin: leiomyoma - related pain. J. Am. Acad. Dermatol., 2009, 60, 2, p. 325–328
13. PATEL, V. M., HANDLER, M. Z., SCHWARTZ, R. A. et al. Hereditary leiomyomatosis and renal cell cancer syndrome: An update and review. J. Am. Acad. Dermatol., 2017, 77, 1, p. 149–158.
14. RAJ, S., CALONJE, E., KRAUS, M., et al. Cutaneous pilar leiomyoma: Clinicopathologic analysis of 53 lesions in 45 patients. Am. J. Dermatol., 1997, 19, p. 2–9.
15. REED W. B., WALKER, R., HOROWITZ, R. Cutaneous leiomyomata with uterine leiomyomata, Acta Dermato - Venerologica, 1973, 53, 5, p. 409–416.
16. SCHMIDT, L. S., LINEHAN, W. M. Hereditary leiomyomatosis and renal cell carcinoma. Int. J. Nephrol. Renovasc. Dis., 2014, 7, p. 253–260.
17. SMIT, D. L., MENSENKAMP, A. R., BADELOE, S. et al. Hereditary leiomyomatosis and renal cell cancer in families referred for fumarate hydratace germline mutaion analalysis. Clin. Genet., 2011, 79, p. 49–59.
18. TOMLINSON, I. P., ALAM, N. A., ROWAN, A. J. et al. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat. Genet., 2002, 30, p. 406–410.
19. TORO, J. R., NICKERSON, M. L., WEI, M. H. et al. Mutations in the fumarate hydratace gene cause hereditary leiomymatosis and renal cell cancer in families in North America. Am. J. Hum. Genet., 2003, 73, p. 95–106.
20. VERGANI, R., BETTI, R., UZIEL, L. et al. Eruptive multiple Sporady cutaneous piloleiomyomas in a patient with chronic lymphocytic leukemia. Br. J. Dermatol., 2000, 143, p. 907–909.
21. WEI, M. H., TOURE, O., GLENN, G. M. et al. Novel mutations in FH and expansion of the spectrum of phenotypes expressed in families with hereditary leiomyomatosis and renal cell cancer. J. Med. Genet., 2006, 43, 1, p. 18–27.
Štítky
Dermatologie Dětská dermatologieČlánek vyšel v časopise
Česko-slovenská dermatologie

2017 Číslo 4
- Inosin pranobex v léčbě HPV infekcí děložního čípku
- Léčba chronické blefaritidy vyžaduje dlouhodobou péči
- Léčba zánětů spojivek a mazových žlázek víčka v primární péči
- Přidání perorálního acetazolamidu (Diluran) k topické léčbě dorzolamidem může vést k dalšímu snížení nitroočního tlaku u některých dětí s glaukomem
- Nová fixní kombinace pro léčbu akné – 1,2% klindamycinfosfát a 0,025% tretinoin
Nejčtenější v tomto čísle
- Sulfony v dermatologii
- Mnohočetné familiární syringomy
- Rosai-Dorfmanova nemoc – kožní forma
- Klinický případ: Bolestivé noduly na zádech
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy