
CZECANCA: CZEch CAncer paNel for Clinical Application – návrh a příprava cíleného sekvenačního panelu pro identifikaci nádorové predispozice u rizikových osob v České republice
CZECANCA: CZEch CAncer paNel for Clinical Application – Design and Optimization of the Targeted Sequencing Panel for the Identification of Cancer Susceptibility in High-risk Individuals from the Czech Republic
Individuals with hereditary cancer syndromes form a minor but clinically important subgroup of oncology patients, comprising several thousand cases in the Czech Republic annually. In these patients, the identification of pathogenic mutations in cancer susceptibility genes has an important predictive and, in some cases, prognostic value. It also enables rational preventive strategies in asymptomatic carriers from affected families. More than 150 cancer susceptibility genes have been described so far; however, mutations in most of them are very rare, occurring with substantial population variability, and hence their clinical interpretation is very complicated. Diagnostics of mutations in cancer susceptibility genes have benefited from the broad availability of next-generation sequencing analyses using targeted gene panels. In order to rationalize the diagnostics of hereditary cancer syndromes in the Czech Republic, we have prepared the sequence capture panel “CZECANCA”, targeting 219 cancer susceptibility genes. Besides more than 50 clinically important high- and moderate-penetrance susceptibility genes, the panel also targets less common candidate genes with uncertain clinical relevance. Alongside the panel design, we have optimized the analytical and bioinformatics pipeline, which will facilitate establishing a collective nationwide database of genotypes and clinical data from the analyzed individuals. The key objective of this project is to provide diagnostic laboratories in the Czech Republic with a reliable procedure and collective database improving the clinical utility of next-generation sequencing analyses in high-risk patients, which would help improve the interpretation of rare or population-specific variants in cancer susceptibility genes.
Key words:
genetic predisposition testing – hereditary cancer syndromes – high-throughput nucleotide sequencing – genetic information databases – panel sequencing – sequence capture – next-generation sequencing (NGS)
This work was supported by Czech Ministry of Health grants No. NT14054, NV15-28830A, NV15--27695A and The League Against Cancer Prague.
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE recommendation for biomedical papers.
Submitted:
2. 10. 2015
Accepted:
13. 10. 2015
Autoři:
J. Soukupová 1; P. Zemánková 1; P. Kleiblová 1,2; M. Janatová 1; Z. Kleibl 1
Působiště autorů:
Ústav biochemie a experimentální onkologie, 1. LF UK v Praze
1; Ústav biologie a lékařské genetiky, 1. LF UK a VFN v Praze
2
Vyšlo v časopise:
Klin Onkol 2016; 29(Supplementum 1): 46-54
Kategorie:
Původní práce
doi:
https://doi.org/10.14735/amko2016S46
Souhrn
Dědičná nádorová onemocnění tvoří malou, ale klinicky významnou část onkologických onemocnění, v České republice se jedná ročně o několik tisíc osob. Identifikace kauzální mutace v nádorových predispozičních genech má u těchto nemocných zásadní prognostický a v některých případech i prediktivní význam. Mimo to je podmínkou cílené preventivní péče o asymptomatické nosiče mutací v rodinách se zvýšeným rizikem vzniku nádorového onemocnění. Do současné doby bylo charakterizováno více než 150 nádorových predispozičních genů. Mutace většiny z nich se vyskytují vzácně, s výraznou populační specifičností a jejich klinická interpretace je často obtížná. Diagnostiku raritních variant technicky zjednodušují postupy využívající sekvenování nové generace, které umožňují vyšetření rozsáhlých sad genů. Za účelem racionalizace diagnostiky hereditárních nádorových syndromů v České republice jsme navrhli sekvenační panel „CZECANCA“, který cílí na vyšetření 219 genů asociovaných s dědičnými nádorovými onemocněními. Panel obsahuje přes 50 klinicky významných genů vysokého a středního rizika, zbývající geny tvoří málo prozkoumané a kandidátní predispoziční geny, jejichž vrozené mutace mají nejasnou klinickou interpretaci. Společně s návrhem panelu byl optimalizován postup vlastního sekvenování a bioinformatického zpracování sekvenačních dat pro tvorbu jednotné databáze genotypů analyzovaných vzorků. Cílem projektu je nabídnout použití sekvenačního panelu včetně optimalizovaného postupu sekvenování nové generace diagnostickým laboratořím v České republice a zajistit sdílení genotypů a klinických údajů o vyšetřovaných pacientech ve společné databázi za účelem zlepšení možnosti klinické interpretace vzácných mutací u vysoce rizikových osob.
Klíčová slova:
analýza genetické predispozice – dědičné nádorové syndromy – masivní paralelní sekvenování – databáze genetických informací – panelové sekvenování – cílené sekvenování – sekvenování nové generace (NGS)
Úvod
Nemocní s dědičnými nádorovými onemocněními zaujímají malou (obvykle mezi 5 a 10 % všech případů), ale klinicky významnou část onkologických pacientů. S ohledem na celkovou incidenci onkologických onemocnění v ČR se tak jedná o několik tisíc vysoce rizikových pacientů ročně. Plošné testování na přítomnost nádorových predispozičních variant u všech onkologicky nemocných je v současnosti ekonomicky neúnosné, proto je vyšetření nádorové predispozice omezeno na vybrané skupiny pacientů na základě charakteristických znaků, které jsou rozvedeny u jednotlivých diagnóz v tomto supplementu. Hlavním rysem dědičných nádorových onemocnění je zvýšené (a často velmi vysoké) riziko vzniku nádorového onemocnění u nosičů patogenních mutací v postižených rodinách. Z tohoto důvodu je identifikace příčinných mutací v nádorových predispozičních genech u rizikových osob předpokladem účinné strategie léčebné péče, která může zahrnovat širokou škálu terapeutických a preventivních modalit snižujících výskyt či zlepšujících prognózu nádorových onemocnění.
Diagnostika dědičných nádorových onemocnění je jedním ze základních cílů současné onkogenetiky a jejích výstupů do klinické praxe. Nejprostudovanější jsou geny, jejichž mutace způsobují hereditární nádorové syndromy s vysokým rizikem vzniku onkologického onemocnění (např. TP53 u Li-Fraumeni syndromu, BRCA1 a BRCA2 u syndromu hereditárního karcinomu prsu a ovarií či APC u familiární adenomatózní polypózy). Jejich genetickým podkladem jsou převážně monoalelické patogenní mutace ve vysoce penetrantních genech. Mutace v těchto genech však objasňují malou část geneticky podmíněných častých nádorových onemocnění [1]. Zbytek případů připadá na mutace v desítkách až stovkách dalších genů, z nichž jen některé jsou dnes dobře charakterizovány, a případy hereditárního onemocnění na předpokládaném podkladě polygenní dědičnosti [2]. V porovnání s mutacemi v hlavních predispozičních genech se patogenní varianty v těchto dalších predispozičních genech vyznačují nižší penetrancí [3,4], nádorový tropizmus u postižení konkrétního genu je méně vyhraněný [5], vyskytují se s významně nižší populační frekvencí a tato frekvence je mnohdy významně proměnlivá v jednotlivých populacích a etnikách [6]. Identifikace nosičů patogenních variant mimo oblast „klasických“, vysoce penetrantních genů je tak tradičními genetickými postupy analyzujícími jednotlivé geny velmi nákladná a zdlouhavá.
Nástup moderních a výkonných molekulárně biologických technologií posledních let vede k významnému zrychlení identifikace nových predispozičních genů a genetických variant. V současné době jsou známy stovky genů, jejichž dědičné mutace prokazatelně či pravděpodobně zvyšují riziko vzniku nádorových onemocnění. Skutečnou revoluci do preklinického výzkumu, ale i klinické diagnostiky, přinesl nástup sekvenování nové generace (next-generation sequencing – NGS). Flexibilita této metody spočívá v masivním sekvenačním paralelizmu, kde v rámci jednoho sekvenačního běhu je možné analyzovat statisíce až miliony templátových molekul DNA. V rámci konkrétních genetických aplikací je možné tento paralelizmus využít pro sekvenování unikátních DNA templátů, v rámci např. celého genomu, nebo sekvenování vybraných úseků DNA u mnoha různých probandů, jako je tomu u panelového NGS. Pomocí NGS je možné v poměrně krátké době najednou identifikovat genetické varianty ve stovkách genů u desítek probandů s ekonomickými náklady nesrovnatelně nižšími, než by tomu bylo při analýze jednotlivých genů klasickými postupy molekulární biologie zahrnujícími prescreening mutací (DGGE/ HA/ /DHPLC/ HRMA/ RFLP/ PTT) s následnou charakterizací patogenní varianty Sangerovým sekvenováním [7]. Ve srovnání s klasickými analýzami však NGS vyžaduje specifické přístrojové vybavení a laboratorní přístupy, významné zvýšení nároků na bioanalytické zpracování a vysoké nároky na klinické hodnocení identifikovaných genetických variant.
V rámci předchozí studie zahrnující NGS a cílené na panel 581 genů jsme v souboru 325 BRCA1/ BRCA2/ PALB2 negativních pacientek s karcinomem prsu nalezli 127 variant způsobujících zkrácení proteinového produktu v některém ze 73 genů u téměř třetiny vyšetřovaných pacientek [8]. Tato analýza, stejně jako další publikované výstupy NGS, prokázala vysoký výkon, spolehlivost a robustnost cíleného NGS [9]. Proto jsme se rozhodli připravit panel genů – CZECANCA (CZEch CAncer paNel for Clinical Application), který by umožnil komplexní, rentabilní a rychlou analýzu germinálních mutací v hlavních predispozičních genech, ale i kandidátních genech asociovaných se zvýšeným rizikem vzniku nejčastějších solidních nádorů v populaci vysoce rizikových pacientů v ČR.
Koncepce projektu CZECANCA
Sekvenační projekt CZECANCA předpokládá použití panelu CZECANCA pro cílené obohacení (sequence capture) sekvenovaných oblastí DNA zahrnujících především kódující exony a intron-exonové přechody genů, jejichž hereditární varianty byly asociovány se zvýšeným rizikem vzniku nádorových onemocnění u jejich nosičů. Protože se s výjimkou vysoce penetrantních genů jedná obvykle o velmi málo frekventní varianty s předpokládanou výraznou populačně-specifickou variabilitou, je jejich správné klinické zhodnocení a klinická interpretace často velmi obtížná [10]. Zapojení řady klinických pracovišť využívajících jednotný technologický přístup založený na využití panelu CZECANCA umožní získání reprezentativního počtu genotypů u vysoce rizikových osob s různými nádorovými syndromy. Z důvodů minimalizace variability (a tím vznikajících technických chyb ve společné databázi) budou hrubá sekvenační data analyzována na našem pracovišti jednotnou bioinformatickou procedurou (pipeline). Tato data spolu se základními charakteristikami (fenotypem) sekvenovaných osob budou ukládána do jednotné databáze přístupné všem zúčastněným laboratořím. Sdílená databáze nebude obsahovat žádné údaje o vyšetřovaných osobách umožňující jejich identifikaci.
Projekt CZECANCA tak není omezen pouze na sekvenační panel, ale reprezentuje komplexní řešení analýzy nádorové predispozice za účelem zvýšení efektivity klinické interpretace variant nejasného významu a variant genů s nejasným rizikem (schéma 1). O výstupech z projektu CZECANCA bude pravidelně informována odborná veřejnost tak, aby složení sekvenačního panelu i postupy vyšetření odpovídaly aktuálnímu stavu vědeckých poznatků onkogenomiky, NGS a klinických požadavků. Aktuální informace budou dostupné na stránce www.czecanca.cz.

Poznámka k výpočtu sekvenačního výstupu: Při velikosti cílové sekvence panelu CZECANCA (~ 600 kb), cílovém sekvenačním pokrytí 200krát, je pro analýzu jednoho vzorku DNA unikátního pacienta zapotřebí kapacity 120 Mb. S uvažovanou (dolní) mezí sekvenační kapacity chemie V3 (150-cycles), která činí ~ 4 Gb, lze teoreticky analyzovat vzorky 33 unikátních pacientů.
Charakterizace panelu CZECANCA
Sekvenační panel CZECANCA je konstruován na bázi technologie SeqCap EZ choice (Nimblegen/ Roche). Výběr genů zohledňoval četnost různých onkologických diagnóz v ČR, aktuální stav informací o genetické podstatě hereditárních nádorových syndromů, předpoklady pro identifikaci dalších kandidátních genů, ale i technické možnosti pro účinný a spolehlivý způsob cíleného obohacení pro účely panelového NGS. Z technických důvodů, které směřovaly k omezení oblastí genomu s výskytem neunikátních sekvencí (pseudogenů a repetitivních sekvencí), byly z návrhu stávající verze panelu CZECANCA vědomě vynechány některé známé predispoziční geny (např. DIS3L2, DMBT1, PMS2, SBDS, SDHA, SDHC, SDHD) nebo jejich neunikátní části (CHEK2, NF1). V současné podobě (verze 1.0) obsahuje panel CZECANCA sondy cílící na kódující sekvence 219 genů (628 169 b).
Cílené geny jsou, z hlediska klinické významnosti pro účely diagnostiky nádorových predispozičních genů, rozděleny do tří skupin:
Geny skupiny A
- Prokázaná jasná (nezpochybnitelná) asociace s nádorovou hereditou.
- Hlavní predispoziční geny.
- Známé a významně zvýšené relativní riziko pro nosiče (RR > 5).
- Klinicky relevantní alterace genů se referují v plném rozsahu klinickému genetikovi (tzn. patogenní mutace a VUS třídy 3– 5 dle IARC).
- Konsenzuální klinická doporučení pro sledování nosičů mutací jsou jasně definována.
- Vyšetření příbuzných nosičů mutací se provádí z důvodu predikce.
Geny skupiny B
- Prokázána jasná asociace s nádorovou hereditou (evidentní na základě několika publikovaných studií).
- Geny/ alely s (předpokládanou) střední penetrancí.
- Předpokládané významně zvýšené relativní riziko pro nosiče (RR 2– 5).
- Asociace s nádorovou hereditou je evidentní, ale RR není přesně stanoveno (asociace s nádorovými hereditami je zjevná, ale není dostatek studií/ nosičů mutací pro korektní stanovení RR).
- Klinicky relevantní alterace genů se referují v plném rozsahu klinickému genetikovi (tzn. patogenní mutace a VUS třídy 4 nebo 5 dle IARC).
- Klinická doporučení pro sledování nosičů mutací nejsou jasně definována.
- Nosiči mutací jsou požádáni o spolupráci při vyšetření příbuzných pro stanovení segregace.
- V rodinách je provedena segregační analýza identifikované varianty.
- Interpretace výsledků prediktivního testování (pokud je prováděno) je následující:
a) Pozitivně prediktivně testované jedince lze zařadit do preventivních sledovacích programů definovaných pro daný hereditární syndrom (pokud takový program existuje; je nutno vést v patrnosti případné modifikace těchto sledovacích programů). Preventivní chirurgické zákroky nejsou pouze na základě nosičství patogenní mutace v těchto genech indikovány, ale jsou ke zvážení při indikativní rodinné anamnéze.
b) Negativně prediktivně testované jedince dále sledovat, zatím jen dle empirického rizika plynoucího z osobní a rodinné anamnézy.
Geny skupiny C
- Nejasná, avšak předpokládaná asociace s nádorovými hereditami. Infor-mace o nádorové predispozici přinášejí pouze ojedinělé studie nebo preklinická data.
- Informace o klinickém významu genu pro nádorovou predispozici není známa, avšak produkt genu je zapojen v signální dráze, ve které poruchy v jiných genech (kódujících kooperující proteiny) prokazatelně souvisejí s nádorovou predispozicí.
- Klinická doporučení pro sledování nosičů mutací neexistují.
- Alterace genů se nereferují a slouží pro vyhodnocení podílu variant sledovaných genů na vzniku nádorové predispozice u nemocných v ČR.
- Po vyhodnocení kolektovaných údajů mohou být nosiči kandidátních patogenních mutací požádáni o spolupráci při vyšetření příbuzných pro stanovení segregace v případě, že u probanda s indikativní rodinnou anamnézou nebyly nalezeny pravděpodobné patogenní mutace ve skupině A a B.
V tab. 1 jsou uvedeny základní charakteristiky genů ze skupiny A a B, které tvoří geny, jejichž patogenní mutace se prokazatelně podílejí na zvýšeném riziku vzniku nádorů u nosičů. Skupiny A a B odlišuje především existence klinických doporučení pro péči o nosiče mutací (ve skupině A).
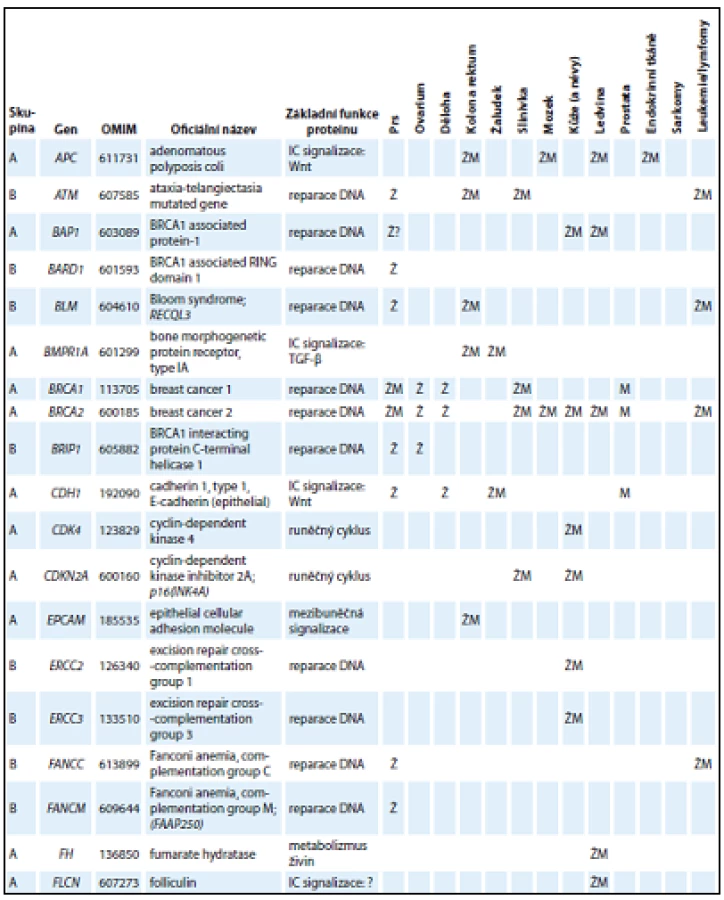


Kromě genů ze skupiny A a B obsahuje panel CZECANCA i skupinu C, která zahrnuje geny, jejichž asociace s nádorovými onemocněními je mnohem méně známá (geny jsou vedeny v abecedním pořadí): AIP, ALK, APEX1, ATMIN, ATR, ATRIP, AURKA, AXIN1, BABAM1, BRAP, BRCC3, BRE, BUB1B, C11ORF30, C19ORF40, CASP8, CCND1, CDC73, CDKN1B, CDKN1C, CEBPA, CEP57, CLSPN, CSNK1D, CSNK1E, CWF19L2, CYLD, DCLRE1C, DDB2, DHFR, DICER1, DMC1, DNAJC21, DPYD, EGFR, EPHX1, ERCC1, ERCC4, ERCC5, ERCC6, ESR1, ESR2, EXO1, EXT1, EXT2, EYA2, EZH2, FAM175A, FAM175B, FAN1, FANCA, FANCB, FANCD2, FANCE, FANCF, FANCG, FANCI, FANCL, FBXW7, GADD45A, GATA2, GPC3, GRB7, HELQ, HNF1A, HOXB13, HRAS, HUS1, CHEK1, KAT5, KCNJ5, LIG1, LIG3, LIG4, LMO1, LRIG1, MAX, MCPH1, MDC1, MDM2, MDM4, MGMT, MMP8, MPL, MRE11A, MSH3, MSH5, MSR1, MUS81, NAT1, NCAM1, NELFB, NFKBIZ, NHEJ1, NSD1, OGG1, PARP1, PCNA, PHB, PHOX2B, PIK3CG, PLA2G2A, PMS1, POLB, PPM1D, PREX2, PRF1, PRKDC, PTTG2, RAD1, RAD17, RAD18, RAD23B, RAD50, RAD51, RAD51AP1, RAD51B, RAD52, RAD54B, RAD54L, RAD9A, RBBP8, RECQL5, RFC1, RFC2, RFC4, RHBDF2, RNF146, RNF168, RNF8, RPA1, RUNX1, SDHAF2, SETBP1, SETX, SHPRH, SMARCA4, SMARCE1, TCL1A, TELO2, TERF2, TERT, TLR2, TLR4, TMEM127, TOPBP1, TP53BP1, TSHR, UBE2A, UBE2B, UBE2I, UBE2V2, UBE4B, UIMC1, XPA, XPC, XRCC1, XRCC2, XRCC3, XRCC4, XRCC5, XRCC6, ZNF350, ZNF365. Vyšetření genů ze skupiny C je nezbytné pro získání informace, zda jejich patogenní mutace mohou vysvětlovat zvýšenou četnost vzniku nádorových onemocnění u jejich nosičů v ČR. Tento význam bude analyzován při dosažení dostatečného počtu vyšetřených osob v databázi projektu.
Sekvenování s panelem CZECANCA
Primární optimalizace sekvenování s panelem CZECANCA probíhala za využití sekvenátoru MiSeq (Illumina), avšak principiálně lze obohacenou knihovnu pravděpodobně použít na libovolné sekvenační platformě současné generace. Použití v současnosti nejrozšířenějšího přístroje firmy Illumina zjednodušuje následné bioinformatické analýzy a snižuje variabilitu výskytu technických sekvenačních artefaktů.
Vstupním materiálem pro přípravu knihovny obohacené o cílové úseky genomové DNA z panelu CZECANCA je fragmentovaná DNA. Fragmentaci lze provádět ultrazvukem (např. systém Covaris) nebo enzymaticky (např. KAPA HyperPlus, Roche) s DNA o doporučeném vstupním množství 0,3– 1 µg. Po úpravě fragmentů DNA (end-repair, A-tailing, ligace adaptorů) provádíme selekci fragmentů o vhodné délce, které pak značíme v průběhu LM-PCR (ligation-mediated PCR) indexy specifickými pro každý vzorek individuální DNA. Takto označené vzorky pak můžeme proporcionálně spojit. Počet spojených vzorků (= společně analyzovaných pacientů) závisí na velikosti panelu, požadovaném pokrytí (= počet čtení každého nukleotidu) a kapacitě sekvenátoru. Za použití panelu CZECANCA lze při cíleném sekvenačním pokrytí 200krát vyšetřit na systému MiSeq (Illumina) bezpečně 30 pacientů v jednom sekvenačním běhu. Fragmenty DNA všech analyzovaných vzorků jsou následně společně hybridizovány se sondami panelu CZECANCA – probíhá obohacování knihovny. Po ukončení hybridizace jsou biotinylované sondy s navázanými fragmenty DNA vychytány magnetickými kuličkami na základě vazby biotin- streptavidin. Vychytané fragmenty DNA jsou následně amplifikovány a po přečištění je obohacená knihovna připravena k sekvenování. Pro sekvenování na MiSeq používáme sekvenační chemii V3 (150-cycle, Illumina).
Zásadní důraz byl kladen na homogenní sekvenační pokrytí (počet čtení jednotlivých nukleotidů sekvenovaného úseku DNA) jednotlivých genů a robustní reprodukovatelnost umožňující minimalizovat sekvenační chyby mezi jednotlivými analýzami a mezi laboratořemi (obr. 1). Otázka sekvenačního pokrytí je dlouhodobě významně diskutované téma. V současné době je za hodnověrné považováno pokrytí 35– 50krát pro jednonukleotidové záměny a malé delece/ inzerce napříč genomem [11].

Bioinformatické zpracování pro účely jednotné databáze (CZECANCA pipeline)
Bioinformatické zpracování sekvenačních dat je založeno na protokolu vypracovaném pracovníky Ústavu dědičných a metabolických poruch (Mgr. Viktor Stránecký, Ph.D. a doc. Ing. Stanislav Kmoch, CSc.) upraveném v Ústavu biochemie a experimentální onkologie (Mgr. Petra Zemánková) 1. LF UK v Praze.
Bioinformatické zpracování pro účely jednotné databáze předpokládá sdílení sekvenačních dat cestou BaseSpace (https:/ / basespace.illumina.com). Soubory jednotlivých vyšetřovaných osob jsou kódovány pracovištěm sdílícím sekvenační data, které jako jediný subjekt má přístup k identifikaci svého konkrétního vzorku. Čtení v podobě fastq souboru jsou namapována pomocí alignovacího softwaru na lidský genom, zároveň vzniká SAM (Sequence Alignment Map) soubor. Pomocí aplikace Picard tools je převeden na BAM soubor, což je binární verze předchozího SAM. K tomuto kroku patří také odstranění duplikátů pomocí stejné aplikace. V této části se také provádí tzv. realignment, který nám umožňuje GATK (The Genome Analysis Toolkit, https:/ / www.broadinstitute.org/ gatk/ ). Soubory BAM slouží pro zobrazení čtení v příslušném prohlížeči, např. Integrative Genomics Viewer (IGV). Dalším důležitým krokem v procesu zpracování dat je převod na VCF (Variant Call File), k čemuž slouží GATK. V tomto souboru se nacházejí varianty nalezené u příslušného pacienta. Tento výstup je zpracován pomocí anotačního softwaru, např. ANNOVAR (http:/ / annovar.openbioinformatics.org/ en/ latest/ ), kde se každé variantě přiřadí její biologická funkce a záznamy, jako je přítomnost varianty v databázích ClinVar (http:/ / www.ncbi.nlm.nih.gov/ clinvar/ ) a HGMD (http:/ / www.hgmd.cf.ac.uk/ ac/ index.php), frekvence varianty v mezinárodních sekvenačních projektech 1 000 genomů (http:/ / www.1000geno-mes.org/ ) nebo ESP6500 (https:/ / esp.gs.washington.edu/ drupal/ ) nebo EXAC (http:/ / exac.broadinstitute.org/ ).
Implementace projektu CZECANCA
Projekt CZECANCA je připraven po technické stránce a v posledním období bylo na základě optimalizovaného protokolu na našem pracovišti analyzováno přes 200 indikovaných osob. V současné době je finalizována podoba společné databáze genotypů a fenotypových charakteristik sekvenovaných pacientů. Údaje o pacientech by měly zahrnovat data, která se vztahují k onkologické diagnóze (věk diagnózy, histologii, imunohistochemická vyšetření, stupeň diferenciace a rozsah onemocnění), osobní anamnéze (věk, pohlaví) a onkologické rodinné anamnéze (reprezentované ideálně rodokmenem).
Vytvoření společné databáze předpokládá rovněž získání genotypů zdravé populace z vyšetření reprezentativního počtu vzorků kontrolního souboru osob bez onkologické diagnózy. Tento důležitý požadavek pro identifikaci populačně specifických genetických variant bez souvislosti s nádorovými onemocněními bude nezbytné řešit formou specifických grantových projektů.
Pro správnou interpretaci získaných sekvenačních dat, zejména v případě nově identifikovaných či kandidátních predispozičních genů, a tím následně pro správnou péči o vyšetřované jedince, je nezbytná úzká spolupráce mezi vyšetřující laboratoří, ambulantním genetikem a ošetřujícím onkologem/ gynekologem. Neexistence této funkční spolupráce vede ke ztrátě řady cenných informací a ve svém důsledku může vést k poškození testovaného probanda, resp. dalších členů jeho rodiny. Na svém významu tak ještě více nabývá kvalitně odebraná rodinná anamnéza včetně informací o zdravých příbuzných a zejména pak její pravidelná aktualizace ošetřujícím lékařem.
Domníváme se, že společné úsilí zainteresovaných pracovišť je racionální cestou k dosažení cíle, kterým je zlepšení klinické diagnostiky dědičných nádorových onemocnění, které by mělo přinést zlepšení péče o nosiče mutací v nádorových predispozičních genech.
Poděkování
Zásadní podíl na provedených bioinformatických analýzách a navržených postupech pro jednotné zpracování dat má Mgr. Viktor Stránecký, Ph.D. a jeho kolegové z Ústavu dědičných metabolických poruch 1. LF UK a VFN v Praze, kterým velmi děkujeme za jejich pomoc a podporu. Za přípravu a kritické poznámky k této práci a složení CZECANCA panelu děkujeme především doc. MU Dr. Lence Foretové, Ph.D. a RNDr. Evě Macháčkové, Ph.D. z Oddělení epidemiologie a genetiky nádorů, MOÚ v Brně a řadě dalších kolegů za přínosné poznámky a doporučení.
Práce byla realizována za podpory Interní grantové agentury MZ ČR (IGA MZ ČR) pod grantovým číslem NT14054, Agentury pro zdravotnický výzkum ČR (AZV ČR) pod čísly NV15-28830A a NV15-27695A a Ligy proti rakovině Praha.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
doc. MUDr. Zdeněk Kleibl, Ph.D.
Ústav biochemie a experimentální onkologie
1. LF UK v Praze
U Nemocnice 5
128 53 Praha 2
e-mail: zdekleje@lf1.cuni.cz
Obdrženo: 2. 10. 2015
Přijato: 13. 10. 2015
Zdroje
1. Foretova L, Petrakova K, Palacova M et al. Genetic testing and prevention of hereditary cancer at the MMCI – over 10 years of experience. Klin Onkol 2010; 23(6): 388– 400.
2. Rahman N. Realizing the promise of cancer predisposition genes. Nature 2014; 505(7483): 302– 308. doi: 10.1038/ nature12981.
3. Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature 2009; 458(7239): 719– 724. doi: 10.1038/ nature07943.
4. Stratton MR, Rahman N. The emerging landscape of breast cancer susceptibility. Nat Genet 2008; 40(1): 17– 22.
5. Saam J, Arnell C, Theisen A et al. Patients tested at a laboratory for hereditary cancer syndromes show an overlap for multiple syndromes in their personal and familial cancer histories. Oncology 2015; 89(5): 288– 293. doi: 10.1159/ 000437307.
6. Kleibl Z, Novotny J, Bezdickova D et al. The CHEK2 c.1100delC germline mutation rarely contributes to breast cancer development in the Czech Republic. Breast Cancer Res Treat 2005; 90(2): 165– 167.
7. Schroeder C, Faust U, Sturm M et al. HBOC multi-gene panel testing: comparison of two sequencing centers. Breast Cancer Res Treat 2015; 152(1): 129– 136. doi: 10.1007/ s10549-015-3429-9.
8. Lhota F, Stranecky V, Boudova P et al. Targeted next-gen sequencing in high-risk BRCA1- and BRCA2-negative breast cancer patients. Curr Oncol 2014; 21(2): e376.
9. Kluska A, Balabas A, Paziewska A et al. New recurrent BRCA1/ 2 mutations in Polish patients with familial breast/ ovarian cancer detected by next generation sequencing. BMC Med Genomics 2015; 8: 19. doi: 10.1186/ s12920-015-0092-2.
10. Cybulski C, Lubiński J, Wokołorczyk D et al. Mutations predisposing to breast cancer in 12 candidate genes in breast cancer patients from Poland. Clin Genet 2014; 88(4): 366– 370. doi: 10.1111/ cge.12524.
11. Sims D, Sudbery I, Ilott NE et al. Sequencing depth and coverage: key considerations in genomic analyses. Nat Rev Genet 2014; 15(2): 121– 132. doi: 10.1038/ nrg3642.
Štítky
Dětská onkologie Chirurgie všeobecná OnkologieČlánek vyšel v časopise
Klinická onkologie

2016 Číslo Supplementum 1
- Metamizol jako analgetikum první volby: kdy, pro koho, jak a proč?
- Incidence asymptomatické hluboké žilní trombózy po totální endoprotéze kyčle a účinnost enoxaparinu v tromboprofylaxi
- Antibiotická rezistence a jiné rizikové faktory prolongovaného hojení ran
- Nejasný stín na plicích – kazuistika
Nejčtenější v tomto čísle
- PALB2 jako další kandidátní gen pro genetické testování u pacientů s hereditárním karcinomem prsu v České republice
- Hepatoblastom, etiologie, kazuistiky
-
Seznam onkologických pracovišť a onkologů zajišťujících prevenci pro osoby (i zdravé) s dědičným rizikem nádorů
Seznam genetických ambulancí při KOC - Genetika tumorigenézy nádorov kolorekta (možnosti testovania a screeningovej predikcie dedičnej formy ochorenia – Lynchovho syndrómu)
Zvyšte si kvalifikaci online z pohodlí domova
Eozinofilní zánět a remodelace
nový kurzVšechny kurzy