
Intersticiální plicní procesy a granulomatózy asociované s běžným variabilním imunodeficitem
Interstitial lung diseases and granulomatoses associated common variable immunodeficiency.
Common variable immunodeficiency disorder belongs to the most common primary human immunodeficiencies and it is characterized by primary defective immunoglobulin production. Hypogammaglobulinemia manifests in every age, usually in adult people. There is no gender predisposition. The prevalence is 1 : 25 000–1 : 50 000. The ethiopathogenesis of the majority of CVIDs is unknown. The main clinical respiratory symptoms include recurrent respiratory infects, especially bacterial etiology, sinusitis, bronchitis, pneumonia, leading to bronchiectasis and lung fibrosis. Interstitial lung fibrosis and granulomatosis often manifest at diagnosis of CVID and they are negative prognostic factors of the disease.
Key words:
common variable immunodeficiency – granulomatosis – interstitial lung fibrosis
Autoři:
Martina Doubková 1; Mojmír Moulis 2; Jana Skřičková 1
Působiště autorů:
Klinika nemocí plicních a tuberkulózy LF MU a FN Brno, pracoviště Bohunice, přednostka prof. MUDr. Jana Skřičková, CSc.
1; Ústav patologie LF MU a FN Brno, pracoviště Bohunice, přednosta doc. MUDr. Leoš Křen, Ph. D.
2
Vyšlo v časopise:
Vnitř Lék 2015; 61(2): 119-124
Kategorie:
Přehledný referát
Souhrn
Běžný variabilní imunodeficit (CVID) patří k nejběžnějším primárním imunitním deficitům a je charakterizován poruchou tvorby imunoglobulinů. Hypogamaglobulinemie se u CVID manifestuje v každém věku, obvykle ale v dospělosti, a není známa závislost výskytu nemoci na pohlaví. Prevalence se odhaduje na 1 : 25 000–1 : 50 000. Patogeneze CVID není plně známa. Základním klinickým respiračním příznakem CVID jsou opakující se infekce dýchacích cest, převážně bakteriální: sinusitidy, bronchitidy, pneumonie, se vznikem bronchiektázií a plicní fibrózy. Intersticiální plicní procesy a granulomatózy se obvykle manifestují již v době diagnózy CVID a jsou většinou špatným prognostickým faktorem onemocnění.
Klíčová slova:
běžný variabilní imunodeficit – granulomatózy – intersticiální plicní procesy
Úvod
Intersticiální plicní procesy (IPP) jsou heterogenní skupina převážně chronických nenádorových a neinfekčních onemocnění alveolů a plicního intersticia. Imunopatologický obraz je charakterizován zánětem se zhojením ad integrum nebo přechodem do nevratné plicní fibrózy s destrukcí plicní tkáně fibrotizujícím procesem. IPP dělíme na IPP známých příčin (pneumokoniózy, polékové postižení a jiné), idiopatické intersticiální pneumonie (běžná intersticiální pneumonie – usual interstitial pneumonia UIP, nespecifická intersticiální pneumonie – NSIP a jiné), granulomatózní IPP (sarkoidóza, granulomatózy jiné etiologie), IPP při systémových onemocněních pojiva a ostatní IPP [1].
Klasifikace imunodeficitů je možná z mnoha hledisek. Pro klinickou praxi je využívané dělení podle důsledku, ke kterému určitá porucha vede, na poruchy protilátkové, buněčné, kombinované, fagocytární a komplementové. Běžný variabilní imunodeficit (commmon variable immunodeficiency – CVID) patří k nejběžnějším primárním imunodeficitům, je charakterizovaný hypogamaglobulinemií a je často provázen bakteriálními infekcemi a bronchiektáziemi. CVID není čistě jen onemocnění B-lymfocytů, ale také zahrnuje abnormality v T-lymfocytech, proto jsou u CVID častěji vidět autoimunitní a lymfoproliferativní onemocnění. IPP jsou přítomny obvykle již v době diagnózy CVID a vyskytují se u 5–22 % pacientů s CVID [2–5]. CVID asociovaný s granulomatózním onemocněním (granulomatous disease – GD) se označuje jako CVID/GD [4,6,7]. Kombinace plicních granulomů a lymfoproliferace se nazývá granulomatous lymphocytic interstitial lung disease (GLILD) [3,8,9]. Pojem lymfoproliferace zahrnuje lymfocytární intersticiální pneumonii LIP, folikulární bronchiolitidu, lymfoidní hyperplazii. GLILD je často doprovázeno splenomegalií a difuzní lymfadenopatií se zvýšeným výskytem vzniku lymfomu [9].
Histologicky nacházíme u pacientů s CVID a IPP sarkoid-like granulomy, organizující se pneumonii, folikulární bronchiolitidu, lymfocytární intersticiální pneumonii a nespecifickou intersticiální pneumonii [10]. Nálezy se mohou vyskytovat samostatně nebo se mohou kombinovat [10].
Primární léčbou je substituce imunoglobuliny, dávka se odvíjí od přítomnosti plicních komplikací. IPP je často dlouhodobě stacionární. Prednison je lékem první volby, u progresivního plicního onemocnění je doporučována kombinovaná imunosuprese. Optimální terapie GLILD není známa [3].
Etiopatogeneze
Genetické, environmentální a další příčiny CVID nejsou dosud dostatečně objasněny. CVID byl v několika případech dán do souvislosti s autozomálně recesivními mutacemi genů pro ICOS (inducible costimulatory receptor expressed on activated T cells) [11], BAFF-R (B-cell activating factor receptor) [12], CD19 (cluster of differentiation, membránové antigeny na povrchu hematopoetických buněk) [13,14], CD20 [15], CD21 [16] a CD81 [17]. U 8–10 % pacientů nacházíme heterozygotní i homozygotní mutace genu pro TACI (B-cell receptor transmembrane activator and calcium-modulating cyclophilin ligand interactor) [18–20]. Selektivní imunodeficit IgA predisponuje ke vzniku CVID, proto by pacienti s tímto defektem měli být pravidelně alespoň 1krát ročně kontrolováni. Vztah CVID a IPP není jasný a u různých druhů IPP a granulomů bude pravděpodobně různý. Zvažuje se vliv infekcí (HHV8 – human herpes virus type 8, EBV – Epstein-Barr virus, HIV – human immunodeficiency virus) [21], porucha B-lymfocytů (snížené zastoupení class-switched memory B cells) [22–24], chronická antigenní stimulace s T imunitní odezvou [25], polymorfizmy TNFα (tumor necrosis factor α) [26].
Diagnostika
CVID patří mezi primární imunodeficity. Prevalence se odhaduje na 1 : 25 000–1 : 50 000 [2]. Podle definice je třeba prokázat poruchu specifické protilátkové odpovědi a vyloučit sekundární příčiny snížení hladin imunoglobulinů (leukemie, lymfomy, léky, infekce, ztráty protilátek gastrointestinálním traktem, ledvinami).
Plicní onemocnění při CVID můžeme rozdělit na obstrukční (bronchiektázie – abnormální rozšíření dýchacích cest způsobené opakovanými záněty, obliterující bronchiolitida) a restrikční (intersticiální plicní fibrózy). Další dělení je možné na postižení respiračního traktu jako důsledku recidivujících infekcí se strukturálním postižením a na neinfekční, kterým je věnována tato práce [2].
Diferenciální diagnostika IPP je široká a než stanovíme diagnózu GLILD, měli bychom vyloučit jiné infekční a neinfekční granulomatózy a IPP. GLILD může být první manifestací CVID a může také předcházet i o několik let diagnózu CVID [7]. Odlišení CVID/GD, GLILD a sarkoidózy bývá mnohdy obtížné, protože u GLILD i sarkoidózy jsou přítomny nenekrotizující granulomy. Na rozdíl od sarkoidózy jsou ale lymfocytární intersticiální pneumonie a folikulární bronchiolitida více pozorovány u GLILD [27,28].
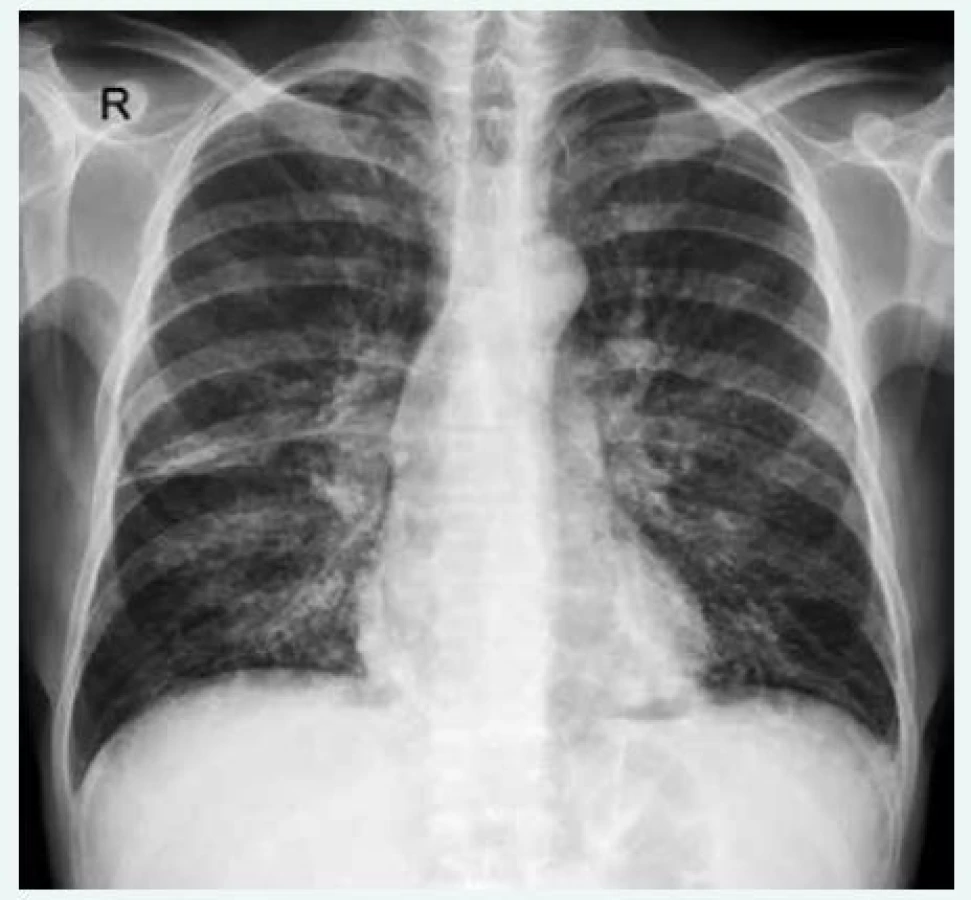
Diagnostika plicních postižení při CVID sestává z klinického a fyzikálního vyšetření, plicního funkčního vyšetření, které může prokázat jak normální ventilační plicní parametry, tak restrikční i obstrukční ventilační poruchu s poruchou plicní difuze (přenos krevních plynů přes alveolokapilární membránu), radiologických metod včetně výpočetní tomografie s vysokou rozlišovací schopností (HRCT hrudníku – high resolution computed tomography) (obr. 1 a 2). Charakteristickými nálezy na HRCT hrudníku jsou difuzní bronchiektázie, ztluštění stěny bronchů, difuzní intersticiální změny, centrilobulární noduly, air traping (mozaiková perfuze způsobená „uvězněním, zadržením“ vzduchu v alveolech) [29,30]. Laboratorní hodnoty SACE (serum angiotensin-converting enzyme) a sIL-2R (soluble interleukine-2 receptor) v krvi využívané při monitoraci aktivity sarkoidózy nevykazují rozdílu mezi sarkoidózou a sarkoid-like (granulomy podobné sarkoidóze) granulomy u CVID a nehodí se k diferenciální diagnostice [2,10]. Další vyšetřovací metodou je bronchoskopie s bronchoalveolární laváží a transbronchiální plicní biopsií. Plicní biopsie cestou transbronchiální nebo chirurgickou cestou torakoskopicky by měla být provedena, aby se vyloučilo jiné lymfoproliferativní onemocnění včetně lymfomu [9].

Histologické plicní nálezy asociované s CVID zahrnují sarkoid-like granuloma, organizující se pneumonie (OP), nespecifické intersticiální pneumonie (NSIP), folikulární bronchiolitidy a lymfocytární intersticiální pneumonie (LIP). Tyto nálezy se mohou u jednoho pacienta kombinovat. GLILD je termín zahrnující granulomy a lymproproliferaci (patří sem lymfocytární intersticiální pneumonie, folikulární bronchiolitida, lymfoidní hyperplazie) [2,31].
U poloviny pacientů s plicními granulomy jsou také přítomny granulomy v mimoplicní lokalizaci (slezina, játra, lymfatické uzliny, ledviny, kůže, gastrointestinální ústrojí) [2,5,7,10]. Jedná se většinou o nenekrotizující (nekaseifikující) granulomy [3], pozorovány ale byly i nekrotizující granulomy [32]. Odlišení od sarkoidózy může být obtížné a je nutné odlišit, zda jde o koincidenci sarkoidózy a CVID nebo o CVID/GD nebo GLILD [33]. Ačkoliv má granulomatózní zánět v obou případech určité podobnosti, u CVID/GD je granulom obvykle složen převážně z vágně organizovaného shluku makrofágů, histiocytů a mnohojaderných obrovskobuněčných buněk v kontrastu od dobře definovaných organizovaných granulomů u sarkoidózy (podrobněji histopatologické rozdíly granulomů sarkoidózy a CVID/GD, obr. 3 a obr. 4). Na rozdíl od sarkoidózy, u níž může být pozorována polyklonální hypergamaglobulinemie bez průkazu monoklonální gamapatie, je CVID/GD, GLILD provázen hypogamaglobulinemií, často s progresivním vývojem plicního postižení bez tendence ke spontánní regresi a s rezistencí na kortikoidní léčbu. Častý je výskyt splenomegalie s trombocytopenií a pancytopenií [34]. Ve studii Bouvryho et al 20 pacientů s průkazem granulomů plic a jiných orgánů nebo jiným typem intersticiálního plicního postižení (organizující se pneumonie, folikulární bronchiolitida, lymfocytární intersticiální pneumonie) bylo porovnáváno s kontrolní skupinou 60 pacientů se sarkoidózou. Bylo zjištěno, že u pacientů s CVID ve srovnání se sarkoidózou jsou fenotypické rozdíly ve vyšší frekvenci autoimunitních onemocnění, ve fyzikálním vyšetření plic (u pacientů s CVID významně přítomen poslechový plicní nález krepitací – auskultačně slyšitelné fenomény připomínající rozepínání suchého zipu), nálezu na HRCT hrudníku (u CVID signifikantně vyšší výskyt bronchiektázií, periferních nodulů s hladkými konturami predilekčně v dolních plicních polích, obklopené opacitami mléčného skla a vzduchový bronchogram, na rozdíl od sarkoidózy s perilymfatickým šířením nodulů lokalizovaných převážně v horních a středních plicních polích), v bronchoalveolární tekutině získané laváží (u obou skupin byla nalezena lymfocytární alveolitida, ale u pacientů s CVID převažoval snížený poměr CD4+/CD8+ 1,6 ± 1,1 oproti 5,3 ± 4 u sarkoidóz, poměr subpopulace pomocných a cytotoxických T-lymfocytů). Histopatologickým vyšetřením byly ve skupině s CVID prokázány vedle granulomatózních lézí i jiné typy postižení jako organizující se pneumonie, folikulární bronchiolitida, lymfocytární intersticiální pneumonie (tab. 1 a tab. 2). Mortalita byla vyšší ve skupině pacientů s CVID oproti pacientům se sarkoidózou bez CVID (30 % oproti 0 % ve skupině sarkoidóz) [10].


![Klinické a laboratorní nálezy u GLILD a sarkoidózy. Upraveno podle Bouvryho [9] a Verbskyho [27]](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/1ac960113f53ca9dd5826cdf8538b4c3.jpg)
![Radiologické nálezy u GLILD a sarkoidózy. Upraveno podle Bouvryho [9] a Verbskyho [27]](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/83875b39e8ddd1428b7206d2be377dda.jpg)
Terapie
Bohužel, chybí kontrolované studie věnující se léčbě CVID a IPP. Optimální terapie není známa, substituce imunoglobuliny není účinná, nezabraňuje progresi. Plicní komplikace CVID vznikají nezávisle na substituční léčbě. Kortikosteroidy jsou nejvíce používané léky, ale nejsou zcela účinné a po jejich vysazení dochází obvykle k návratu nemoci. V retrospektivní studii Boursiquot analyzoval 59 pacientů s CVID/GD [5]. 42 % nemocných s granulomatózním postižením nevyžadovalo léčbu v době diagnózy a bylo stabilních po dobu několika let. Pouze u pacientů s klinickými potížemi, středně těžkým až těžkým plicním funkčním vyšetřením nebo se zhoršováním plicního nálezu byla indikována imunosupresivní léčba spočívající v monoterapii prednisonem nebo v kombinaci s jinými imunosupresivy (cyklofosfamid, cyklosporin, azatioprin, mykofenolát mofetil). Kortikosteroidy se používají v léčbě i u granulomatózního postižení jiných orgánů nežli plic (lymfatické uzliny, játra, slezina) [5].
Vzhledem k tomu, že u GLILD může být přítomen „normální“ skiagram hrudníku a plicní funkční vyšetření bez poruchy, je vhodné provést v čase diagnózy CVID HRCT hrudníku a začít s léčbou včas, abychom zabránili nebo zpomalili vznik nevratné plicní fibrózy [29]. Léčba GLILD je ale svízelná, monoterapie glukokortikoidy je méně účinná a vede ke kompletní remisi jen u některých nemocných [2,5]. Jako steroid šetřící léky jsou přidávána další imunosupresiva, cyklosporin, rituximab, hydroxychlorochin, cyklofosfamid, metotrexát. V práci Chase et al byla prokázána u pacientů s GLILD účinnost kombinované imunosuprese (azatioprin, rituximab) se signifikantním zlepšením HRCT nálezu a plicních funkcí. Kombinovaná imunosuprese byla zvolena proto, že v histologickém vzorku plicní tkáně u GLILD jsou přítomny B - i T-lymfocyty a rituximab ovlivňuje B-lymfocyty (anti-CD20, monoklonální protilátka proti povrchovému antigenu na B-lymfocytech) a azatioprin T-lymfocyty [35]. Kromě glukokortikoidů byly zkoušeny inhibitory anti-TNFα (monoklonální protilátky proti TNFα) s úspěchem pouze v kazuistikách [32,35,36]. Další léčba sestává z plicní rehabilitace, nutriční podpory, řádné léčby infekcí po dobu 2–3 týdnů, preventivní antimikrobiální terapie dle kolonizace bakterií u pacientů s recidivujícími infekcemi, bronchodilatační a mukolytické léčby u pacientů s bronchiektáziemi, preventivní profylaxi Pneumocysty při terapii kortikoidy. V případě plicní progrese je u nemocných s respirační nedostatečností indikována dlouhodobá domácí oxygenoterapie (minimálně 16 hod inhalace kyslíku). S plicními transplantacemi u těchto nemocných mnoho zkušeností není. Diagnóza CVID není kontraindikací k provedení plicní transplantace, i když je provázena infekčními komplikacemi [37].
Prognóza
GLILD je spojována s vyšší mortalitou ve srovnání s pacienty s CVID bez tohoto postižení [4,5,8,10,38–40]. Ve studii Ardenize a Cunninghama ale nebyl zjištěn rozdíl v přežití mezi pacienty s CVID a CVID/GD [7,41]. Ve studii Bouvryho et al bylo přežití pacientů s CVID a IPP ovlivněno výskytem lymfomů, infekčními komplikacemi a nikoliv přítomností IPP [10]. Ve studii Resnicka et al na souboru 473 pacientů s CVID bylo přežití ovlivněno negativně výskytem lymfomu, hepatitidou, chronickým plicním (funkční a strukturální postižení) a gastrointestinálním onemocněním. Přežití nebylo ovlivněno přítomností bronchiektázií, autoimunit, jinými nádory než lymfomy, granulomatózním onemocněním a předešlou splenektomií [38].
Závěr
IPP se manifestují u 5–22 % CVID pacientů a objevují se často již v době diagnózy CVID. Imunologické vyšetření je nedílnou součástí diagnostického a diferenciálně diagnostického algoritmu IPP. Na diagnózu CVID bychom měli pomýšlet u nemocných s IPP, hypogamaglobulinemií a opakujícími se infekcemi zejména respiračního traktu.
MUDr. Martina Doubková
doubkovamartina@seznam.cz
Klinika nemocí plicních a tuberkulózy LF MU a FN Brno
www.fnbrno.cz
Doručeno do redakce 23. 9. 2014
Přijato po recenzi 25. 11. 2014
Zdroje
1. Travis WD, Costabel U, Hansell DM et al. An official American Thoracic Society/European Respiratory Society statement: update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med 2013; 188(6): 733–748.
2. Prasse A, Kayser G, Warnatz K. Common variable immunodeficiency-associated granulomatous and interstitial lung disease. Curr Opin Pulm Med 2013; 19(5): 503–509.
3. Park JH, Levinson AI. Granulomatous-lymphocytic interstitial lung disease (GLILD) in common variable immunodeficiency (CVID). Clin Immunol 2010; 134(2): 97–103.
4. Chapel H, Lucas M, Lee M et al. Common variable immunodeficiency disorders: division into distinct clinical phenotypes. Blood 2008; 112(2): 277–286.
5. Boursiquot JN, Lucas M, Patel S et al. Granulomatous disease in CVID: retrospective analysis of clinical characteristics and treatment efficacy in a cohort of 59 patients. J Clin Immunol 2013; 33(1): 84–95.
6. Cunningham Rundles C. Human B cell defects in perspective. Immunol Res 2012; 54(1–3): 227–232.
7. Ardeniz O, Cunningham-Rundles C. Granulomatous disease in common variable immunodeficiency. Clin Immunol 2009; 133(2): 198–207.
8. Bates CA, Ellison MC, Lynch DA et al. Granulomatous-lymphocytic lung disease shortens survival in common variable immunodeficiency. J Allergy Clin Immunol 2004; 114(2): 415–421.
9. Fernández Pérez ER. Granulomatous lymphocytic interstitial lung disease. Immunol Allergy Clin North Am 2012; 32(4): 621–632.
10. Bouvry D, Mouthon L, Brillet PY et al. Granulomatous-associated common variable immunodeficiency disorder: a case-control study versus sarcoidosis. Eur Respir J 2013; 41(1): 115–122.
11. Grimbacher B, Hutloff A, Schlesier M et al. Homozygous loss of ICOS is associated with adult onset common variable immunodeficiency. Nat Immunol 2003; 4(3): 261–268.
12. Warnatz K, Salzer U, Rizzi M et al. B cell activating factor receptor deficiency is associated with an adult onset antibody deficiency syndrome in humans. Proc Natl Acad Sci USA 2009; 106(33): 13945–13950.
13. van Zelm MC, Reisli I, van der Burg M et al. An antibody deficiency syndrome due to mutations in the CD19 gene. N Engl J Med 2006; 354(18): 1901–1912.
14. Kanegane H, Agematsu K, Futatani T et al. Novel mutations in a Japanese patient with CD19 deficiency. Genes Immun 2007; 8(8): 663–670.
15. Kuijpers TW, Bende RJ, Baars PA et al. CD20 deficiency in humans results in impaired T cell independent antibody responses. J Clin Invest 2010; 120(1): 214–222.
16. Thiel J, Kimmig L, Salzer U et al. Genetic CD21 deficiency is associated with hypogammaglobulinemia. J Allergy Clin Immunol 2012; 129(3): 801–810.
17. van Zelm MC, Smet J, Adams B et al. CD81 gene defect in humans disrupts CD19 complex formation and leads to antibody deficiency. J Clin Invest 2010; 120 : 1265–1274.
18. Salzer U, Chapel HM, Webster AD et al. Mutations in TNFRSF13B encoding TACI are associated with common variable immunodeficiency in humans. Nat Genet 2005; 37(8): 820–828.
19. Castigli E, Wilson SA, Garibyan L et al. TACI is mutant in common variable immunodeficiency and IgA deficiency. Nat Genet 2005; 37(8): 829–834.
20. Pan Hammarstrom Q, Salzer U, Du L et al. Reexamining the role of TACI coding variants in common variable immunodeficiency and selective IgA deficiency. Nat Genet 2007; 39(4): 429–430.
21. Wheat WH, Cool CD, Morimoto Y et al. Possible role of human herpesvirus 8 in the lymphoproliferative disorders in common variable immunodeficiency. J Exp Med 2005; 202(4): 479–484.
22. Detkova D, de Gracia J, Lopes-da-Silva, S et al. Common variable immunodeficiency: association between memory B cells and lung diseases. Chest 2007; 131(6): 1883–1889.
23. Wehr C, Kivioja T, Schmidt C et al. The EUROclass trial: defining subgroups in common variable immunodeficiency. Blood 2008; 111(1): 77–85.
24. Warnatz K, Denz A, Drager R et al. Severe deficiency of switched memory B cells (CD27(+)IgM(-)IgD(-)) in subgroups of patients with common variable immunodeficiency: a new approach to classify a heterogeneous disease. Blood 2002; 99(5): 1544–1551.
25. Kohler PF, Cook RD, Brown WR et al. Common variable hypogammaglobulinemia with T-cell nodular lymphoid interstitial pneumonitis and B-cell nodular lymphoid hyperplasia: different lymphocyte populations with a similar response to prednisone therapy. J Alllergy Clin Immunol 1982; 70(4): 299–305.
26. Mullighan CG, Fanning GC, Chapel HM et al. TNF and lymphotoxin-alpha polymorphisms associated with common variable immunodeficiency: role in the pathogenesis of granulomatous disease. J Immunol 1997; 159(12): 6236–6241.
27. Sutor GCH, Fabel H. Sarcoidosis and common variable immunodeficiency. Respiration 2000; 67(2): 204–208.
28. Verbsky JW, Routes JM. Sarcoidosis and common variable immunodeficiency: similarities and differences. Semin Respir Crit Care Med 2014; 35(3): 330–335.
29. Park JES, Beal I, Dilworth JP et al. The HRCT appearance of granulomatous pulmonary disease in common variable immune deficiency. Eur J Radiol 2005; 54(3): 359–364.
30. Torigian DA, LaRosa DF, Levinson AI et al. Granulomatous-lymphocytic interstitial lung disease associated with common variable immunodeficiency. J Thorac Imaging 2008; 23(3): 162–169.
31. Chua I, Quinti I, Grimbacher H. Lymhoma in common variable immunodeficiency: interplay between immune dysregulation, infection and genetics. Curr Opin Hematol 2008; 15(4): 368–374.
32. Hatab AZ, Ballas ZK. Caseating granulomatous disease in common variable immunodeficiency treated with infliximab. J Allergy Clin Immunol 2005; 116(5): 1161–1162.
33. Drajna M, Kolek V, Kalabusová B. Koincidencia CVID a sarkoidózy. Kazuistiky v alergologii, pneumologii a ORL 2007; 4(1): 20–24.
34. Morimoto Y, Routes JM. Granulomatous disease in common variable immunodeficiency. Curr Allergy Asthma Rep 2005; 5(5): 370–375.
35. Chase NM, Verbsky JW, Hintermeyer MK et al. Use of combination chemotherapy for treatment of granulomatous and lymphocytic interstitial lung disease (GLILD) in patients with common variable immunodeficiency (CVID). J Clin Immunol 2013; 33(1): 30–39.
36. Thatayatikom A, Thatayatikom S, White AJ. Infliximab treatment for severe granulomatous disease in common variable immunodeficiency: a case report and review of the literature. Ann Allergy Asthma Immunol 2005; 95(3): 293–300.
37. Buton CM, Milman N, Andersen CB et al. Common variable immune deficienty and lung transplantation. Scand J Infect Dis 2007; 39(4): 362–367.
38. Resnick ES, Moshier EL, Godbold JH et al. Morbidity and mortality in common variable immune deficiency over 4 decades. Blood 2012; 119(7): 1650–1657.
39. Mechanic LJ, Dikman S, Cunningham-Rundles C. Granulomatous disease in common variable immunodeficiency. Ann Intern Med 1997; 127(8 Pt 1): 613–617.
40. Quinti I, Soresina A, Spadalo G et al. Long-term follow-up and outcome of a large cohort of patients with common variable immunodeficieny. J Clin Immunol 2007; 27(3): 308–316.
41. Cunnigham-Rundles C, Bodian C. Common variable immunodeficiency: clinical and immunological features of 248 patients. Clin Immunol 1999; 92(1): 34–48.
Štítky
Diabetologie Endokrinologie Interní lékařstvíČlánek vyšel v časopise
Vnitřní lékařství

2015 Číslo 2
- Diosmin a hesperidin: Co ukazuje farmakokinetika?
- Omeprazol a mechanismus hojení krvácení z peptického vředu
- Porovnání farmakologických vlastností mikronizovaného diosminu a hesperidinu v terapii chronické žilní insuficience a hemoroidů
- Genetický výzkum potvrdil asociaci mezi autismem a střevními obtížemi
Nejčtenější v tomto čísle
- Spontánní bakteriální peritonitida
- Vyšetření tenkého střeva pomocí magnetické rezonance
- Empagliflozin – nový zástupce inhibitorů transportéru SGLT2 pro léčbu pacientů s diabetem 2. typu
- Význam transkutánneho monitorovania tkanivového kyslíka u pacienta s diabetes mellitus s jeho komplikáciami
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy