
Léčba AL‑ amyloidózy – výsledky jednoho pracoviště a přehled publikovaných zkušeností s novými léky (bortezomibem, thalidomidem a lenalidomidem) u AL‑ amyloidózy
Treatment of AL ‑ amyloidosis – results from one clinic and review of published experience with new agents (bortezomib, thalidomide and lenalidomide) in AL ‑ amyloidosis
Patients:
Fifteen patients with light chain deposits in the form of AL ‑ amyloidosis and 2 patients with light chain deposition as amorphous matter (light chain deposition disease) were treated at our clinic as of 1999. Median age at the diagnosis was 63 (34 – 77) years. The light chain deposition caused: nephrotic syndrome in 12 (70%) patients, renal insufficiency with reduced filtration in 4 (23%) patients, cardiomyopathy in 4 (23%) patients, hepatosplenomegaly in 2 (12%) patients, manifest coagulopathy in 2 (12%) patients, periorbital hematoma in 2 (12%) patients, visceral and somatic neuropathy in 2 (12%) patients. Treatment with high‑dose dexamethasone in combination with adriamycin and vincristine (VAD) or cyclophosphamide (CAD or just CD) was used in 11 patients. In 4 patients, therapy was completed with high‑dose chemotherapy and autologous transplantation; complete haematological and organ treatment response was achieved in all 4 patients with remission lasting 113+, 87+, 50, 45+ months. Of the remaining 7 patients in whom high‑dose dexamethasone therapy was not completed with high‑dose chemotherapy, 3 achieved complete haematological remission (CR) and very good partial remission (VGPR), with 2 patients achieving complete organ treatment response. Organ response in the third patient cannot be assessed due to the short evaluation period. PR with no organ treatment response was achieved in other 2 patients and 2 patients died during the treatment. Therapy with prednisone and alkylating cytostatics was used in 2 patients with serious organ damage, both patients died after a short period of time due to the disease and thus treatment response cannot be evaluated. Combination of thalidomide, dexamethasone and cyclophosphamide (CTD) was used in 4 patients. Two of these patients did not complete full 2 cycles, one for unmanageable thalidomide‑associated constipation, the other died. Two patients underwent a total of 5 and 6 cycles of this treatment with PR effect and plateau after the previous decline of monoclonal immunoglobulin concentrations. Treatment combination of bortezomib (Velcade), cyclophosphamide and dexamethasone (VCD) was used in three patients. In one patient (6 completed CTD cycles with the PR result) this combination led to complete haematological remission, complete remission was also achieved in the second patient and the application of 2 CVD cycles led to CR in the third (5 CTD cycles with PR result). Just one of the 3 female patients has been followed up for more than 12 months and so it is possible to evaluate organ treatment response in this patient; nephrotic syndrome ceased, meaning that organ CR has been achieved.
Conclusion:
Early diagnosis (before severe organ damage occurs) enables administration of aggressive treatment (high‑dose chemotherapy and autologous transplantation) with the outlook of complete haematological and organ treatment response. New drugs thalidomide and bortezomib further expand treatment armamentarium; according to our limited experience and published information, bortezomib may be considered as very effective and well tolerated agent suitable, in combination, for patients with the diagnosis of AL ‑ amyloidosis.
Key words:
AL ‑ amyloidosis – light chain deposition disease – monoclonal gammopathy – thalidomide – bortezomib – high‑dose chemotherapy and autologous transplantation – nephrotic syndrome – cardiomyopathy – neuropathy – coagulopathy
Autoři:
Z. Adam 1; L. Pour 1; M. Krejčí 1; L. Zahradová 1; A. Křivanová 1; J. Šmardová 3; L. Kovářová 1; S. Štěpánková 2; M. Moulis 3; L. Křen 3; K. Veselý 4; I. Svobodová 4; Z. Čermáková 5; M. Nedbálková 6; J. Mayer 1; R. Hájek 1
Působiště autorů:
Interní hematoonkologická klinika Lékařské fakulty MU a FN Brno, pracoviště Bohunice, přednosta prof. MU Dr. Jiří Vorlíček, CSc. 2Interní hepatogastroenterologická klinika Lékařské fakulty MU a FN Brno, pracoviště Bohunice, přednosta prof. MU Dr. Jan Lata
1
Vyšlo v časopise:
Vnitř Lék 2010; 56(3): 190-209
Kategorie:
Původní práce
Souhrn
Pacienti:
Od roku 1999 bylo na našem pracovišti léčeno 15 pacientů s depozity lehkých řetězců ve formě AL ‑ amyloidózy a 2 pacienti s depozicí lehkých řetězců ve formě amorfních hmot (light chain deposition disease). Medián věku při stanovení diagnózy byl 63 (34 – 77) let. Depozita lehkých řetězců způsobovala nefrotický syndrom u 12 (70 %) pacientů, renální insuficienci se sníženou filtrací u 4 (23 %) pacientů, kardiomyopatii u 4 (23 %) pacientů, hepatosplenomegalii u 2 (12 %) pacientů, manifestní koagulopatie u 2 (12 %) pacientů, periorbitální hematomy u 2 (12 %) pacientů, viscerální a somatickou neuropatii u 2 (12 %) pacientů. Léčba založená na vysokých dávkách dexametazonu v kombinaci s adriamycinem a s vinkristinem (VAD) nebo s cyklofosfamidem (CAD nebo jen CD) byla použita u 11 osob. U 4 z nich byla zakončena vysokodávkovanou chemoterapií s autologní transplantací a v těchto 4 případech bylo dosaženo kompletní hematologické a orgánové léčebné odpovědi s trváním remise 113+, 87+, 50, 45+ měsíců. U zbývajících 7 pacientů, u nichž léčba založená na vysokých dávkách dexametazonu nebyla zakončena vysokodávkovanou chemoterapií, dosáhli 3 pacienti kompletní hematologické remise (CR) a velmi dobré parciální remise (VGPR), ve 2 případech s kompletní orgánovou léčebnou odpovědí. Ve 3. případě nelze orgánovou odpověď pro krátké sledování hodnotit. V dalších 2 případech byla dosažena PR bez orgánové léčebné odpovědi a 2 pacienti v průběhu léčby zemřeli. Léčba obsahující prednison a alkylační cytostatika byla použita u 2 nemocných se závažným orgánovým poškozením, oba nemocní v krátké době v důsledku nemoci zemřeli, takže není možné hodnotit léčebnou odpověď. Kombinace thalidomidu, dexametazonu a cyklofosfamidu (CTD) byla použita u 4 pacientů. Dva z těchto pacientů nedokončili ani 2 cykly, jeden pro nezvladatelnou obstipaci po thalidomidu, druhý pro úmrtí. Dva absolvovali celkem 5 a 6 cyklů této léčby s efektem PR s vytvořením plató po předchozím poklesu koncentrace monoklonálního imunoglobulinu. Léčebná kombinace bortezomib (Velcade), cyklofosfamid a dexametazon (VCD) byla použita ve 3 případech. V 1. případě (po ukončených 6 cyklech CTD s výsledkem PR) navodila kompletní hematologickou remisi, ve 2. (intolerancí thalidomidu již v 1. měsíci léčby) navodila také kompletní remisi a ve 3. případě (po 5 cyklech CTD s výsledkem PR) dosáhla aplikace 2 cyklů CVD také CR. Pouze jedna ze 3 pacientek je sledována déle než 12 měsíců a je možné u ní vyhodnotit orgánovou léčebnou odpověď, u této pacientky vymizel nefrotický syndrom, takže byla dosažena orgánová CR.
Závěr:
Časné stanovení diagnózy (bez velkého orgánového poškození) umožní podávat agresivní léčbu (vysokodávkovanou chemoterapii s autologní transplantací) s nadějí na kompletní hematologickou a orgánovou léčebnou odpověď. Nové léky thalidomid a bortezomib dále rozšiřují léčebné možnosti, dle našich limitovaných zkušeností a publikovaných informací lze považovat bortezomib za velmi účinný a dobře tolerovaný lék vhodný v kombinacích pro pacienty s diagnózou AL ‑ amyloidóza.
Klíčová slova:
AL ‑ amyloidosis – ligh chain deposition disease – monoklonální gamapatie – thalidomid – bortezomib – vysokodávkovaná chemoterapie s autologní transplantací – nefrotický syndrom – kardiomyopatie – neuropatie – koagulopatie
Úvod
Amyloidóza z lehkých řetězců (immunoglobulin light chain amyloidosis) – akronymem AL-amyloidóza – je důsledkem tvorby monoklonálních lehkých řetězců se specifickou mutací, která způsobuje odlišnou prostorovou konfiguraci jejich molekul. Díky této odchylné struktuře amyloidogenní lehké řetězce spontánně agregují a vytvářejí oligomery, a ty pak vytvářejí depozita amyloidových fibril ve stabilní lineární struktuře. Podstatně méně často se ukládají ve formě amorfních hmot (light chain deposition disease) [1].
Obecnou histochemickou vlastností depozit patologických proteinů v lineární amyloidové struktuře je charakterické zelené zbarvení v polarizovaném světle po barvení konžskou červení (obr. 1 – 4). V elektronovém mikroskopu jsou zřetelné nevětvené fibrily průměru 7,5 – 10 nm. Ale nejen molekuly monoklonálních lehkých řetězců mohou tvořit amyloidová depozita, existuje více než 11 dalších molekul s amyloidogenní mutací, které také mohou tvořit amyloidová depozita, obtížně odlišitelná od depozit monoklonálních lehkých řetězců [2]. Pro terapeutický plán je zásadní správná typizace amylogenních proteinů, protože omyl může mít tragické následky, např. provedení vysokodávkované chemoterapie s autologní transplantací kostní dřeně u pacienta s amyloidózou tvořenou mutovaným transtyretinem, u něhož je naopak vhodná léčba transplantací jater, a nikoliv kostní dřeně. Proto je nutné provést veškerá dostupná vyšetření a v nejasných případech před zahájením vlastní léčby konzultovat i specializovaná pracoviště [3 – 5].




Průkaz monoklonální gamapatie však neznamená jednoznačný důkaz toho, že se jedná o AL - amyloidózu. Asi 3 % starších osob mají monoklonální gamapatii. Proto je možný současný výskyt hereditárního typu amyloidového depozita a monoklonální gamapatie. V roce 2002 byla uveřejněna analýza 350 pacientů s nálezem amyloidu a monoklonální gamapatie, u nichž se po prvním vyšetření předpokládala souvislost těchto dvou jevů. Vyšetřili u nich mutace genů dalších amyloidogenních bílkovin (transtyretinu, apolipoprotein A - 1, lysozymu a α-řetězce fibrinogenu). Tato vyšetření odhalila hereditární amyloidózu u 9,7 % nemocných, u nichž se pro přítomnost monoklonální gamapatie předpokládala AL-amyloidóza. V 5 % se jednalo o amyloidózu z α-řetězce fibrinogenu, ve 4 % o amyloidózu způsobenou nerozpoznanou mutací transtyretinu. Amyloidóza z aberantního fibrinogenu poškozuje ledviny podobně jako AL-amyloidóza [6].
Amyloidogenní lehké řetězce jsou produkovány monoklonálními plazmatickými buňkami lokalizovanými v kostní dřeni, případně ve slezině, obvykle v nevelkém množství. S pomocí krevního oběhu se pak dostávající k cílovým orgánům, v nichž se usazují díky zatím neznámým interakcím s matrix jednotlivých tkání (s glykosaminoglykany a s molekulami buněčných membrán). Tyto interakce mohou potencovat tvorbu oligomerů klonálních lehkých řetězců, které již mají cytotoxické vlastnosti a konečné formování amyloidového depozita. To, v kterém orgánu dojde k nejmasivnějšímu poškození, je determinováno geny variabilní části lehkého řetězce, a tedy strukturou amyloidogenního lehkého řetězce [7 – 9].
Klinické projevy mohou být velmi pestré a jsou popsány ve vícero českých publikacích [10 – 16] a z didaktických důvodů je připomínáme obr. 5 – 10.



Výskyt amyloidózy není dostatečně zmapován. Autoři z Mayo klinik uvedli 0,5 – 1,2 případů na 100 000 obyvatel [17]. Incidenci dle francouzských autorů uvádí tab. 1.
![Výskyt amyloidóz ve francouzském regionu Magy- Bertrand 2008 [18].](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/488f77b09b31e02a853c2858f20c1ab7.jpeg)
Cílem léčby je rychlá eliminace tvorby amyloidogenních lehkých řetězců, což je podmínkou, aby došlo ke zlepšení funkce amyloidem poškozených orgánů [19,20]. V klinické studii bylo histologicky prokázáno, že po dokončení chemoterapie dochází k postupnému odbourávání a odstraňování amyloidu v místech jeho depozic. Při opakovaném vyšetřování podkožního tuku břišní stěny u pacientů po dokončení léčby AL-amyloidózy bylo zjištěno, že depozita amyloidu vymizela u 80 % pacientů, kteří po 3,2 letech zůstávali v trvalé kompletní remisi, zatímco u 9 pacientů v parciální remisi nedošlo k vymizení amyloidových depozit [21].
Tato práce mění dosavadní pohled na léčbu této nemoci. Jasně dokazuje, že cílem léčby u AL-amyloidózy musí být dosažení kompletní hematologické remise. Dosažení parciální hematologické remise se dnes nepovažuje za dostatečné, protože neumožní reparační pochody v amyloidem poškozených tkáních a orgánech (schéma 1).

Problémem je skutečnost, že s mírou poškození orgánů a tkání se snižuje tolerance nežádoucích účinků léčby. Proto je zásadní, podobně jako u mnohočetného myelomu, stanovit tuto diagnózu v nejranějších fázích nemoci, kdy malé poškození tkání a orgánů umožní podat agresivní léčbu a kdy po dosažení kompletní remise by mělo dojít ke kompletní orgánové léčebné odpovědi.
Cílem následující analýzy je poukázat na skutečnost, že:
- časná diagnostika a účinná léčba mohou vést ke kompletní a dlouhotrvající hematologické i orgánové remisi
- a nové léky v čele s bortezomibem přinášejí prospěch těchto nemocným.
Tato publikace patří k edukační aktivitě České myelomové skupiny zvané CRAB, s cílem přispět k časnějšímu stanovení diagnózy mnohočetného myelomu a dalších monoklonálních gamapatií [22].
Soubor nemocných a léčba
Soubor nemocných a klinické příznaky, které byly indikací k léčbě
V posledních 10 letech (od roku 1999) bylo na našem pracovišti vyšetřeno léčeno celkem 17 nemocných s poškozením organizmu depozity monoklonálních imunoglobulinů. V 15 případech šlo o depozita lehkých řetězců ve formě AL-amyloidu a 2krát v amorfní (neamyloidové) formě. Soubor se skládá z 11 žen a 6 mužů. Medián věku těchto 17 pacientů se systémovou AL - amyloidózou a light chain deposition disease v době diagnózy byl 63 (34 – 77) let.
Mimo uvedených 17 pacientů se systémovou formou depozit monoklonálních lehkých řetězců jsme v průběhu 10 let diagnostikovali 4 případy solitárního izolovaného ložiska amyloidu, 2krát v bronchu, 1krát v laryngu a 1krát v močovém měchýři. Vyjma solitárního ložiska nebylo zřetelné žádné další poškození amyloidem ani monoklonální gamapatie. Ve 2 případech tracheobrochiální amyloidózy jsou tito nemocní sledováni u plicních specialistů. Ložisko v močovém měchýři a v laryngu bylo odstraněno operačně a tito pacienti jsou sledováni mimo naše pracoviště.
Diagnóza AL - amyloidózy byla stanovena na histologickém průkazu amyloidu ve tkáni nejméně jednoho orgánu a současném biochemickém průkazu monoklonální gamapatie. Poté následovalo vždy vyšetření obvyklé u mnohočetného myelomu, jehož cílem bylo zjistit, zda se u konkrétního pacienta s monoklonální gamapatií a s poško-zením organizmu depozity lehkých řetězců má stav klasifikovat jako mnohočetný myelom s přidruženou AL-amyloidózu anebo jako primární systémová AL-amyloidózu bez splnění diagnostických kritérií mnohočetného myelomu. Zásadní pro toto rozlišení je prokázání či neprokázání klonálních plazmocytů v kostí dřeni, jak vyplývá z kritérií International Myeloma Working Group z roku 2003 [23] (tab. 2a a 2b), která pak doplněná o upřesnění léčebných odpovědí vyšla v roce 2009 [24]. Z hlediska biologie AL-amyloidózy není toto členění zásadní, neboť jde o pohledy z různých úhlů na jeden biologický jev s kontinuálním přechodem od nulového po vysoký počet klonálních plazmocytů v kostní dřeni. Nicméně z hlediska formálního je toto rozdělení důležité, protože pro mnohočetný myelom jsou registrovány nové léky (bortezomib, thalidomid a lenalidomid), zatímco pro primární systémovou amyloidózu registrovány nejsou.
![Tab. 2a. Kritéria symptomatického myelomu [23].](https://pl-master.mdcdn.cz/media/image/b6241a38ac13d117f94dbc61056d2f5b.jpeg?version=1537790298)
![Tab. 2b. Kritéria poškození orgánů či tkání myelomem [23].](https://pl-master.mdcdn.cz/media/image/fce3db42b93f55bc6dd32a4a922ba164.jpeg?version=1537790298)
Z popisovaných 15 pacientů s depozity lehkých řetězců ve formě AL-amyloidu nebyla naplněna kritéria mnohočetného myelomu v 8 případech, takže diagnóza byla uzavřena jako primární systémová AL-amyloidóza. U 7 pacientů ve vzorku kostní dřeně, získaném trepanobiopsií, byly imunohistochemicky v plazmocytech prokázány lehké řetězce λ, méně často κ, a tím prokázána jejich klonalita. U těchto 7 pacientů byla diagnóza uzavřena jako symptomatický mnohočetný myelom provázený AL-amyloidózou, přesně dle kritérií IMWG 2003. Zajímavé je, že u pacientů s diagnózou uzavřenou jako mnohočetný myelom s přidruženou amyloidózou nebyla stejně jako u pacientů s diagnózou primární systémová AL-amyloidóza detekována osteolytická ložiska ani difuzní osteoporóza.
Přínos vyšetření vzorku kostní dřeně průtokovou cytometrií
Pomocí průtokové cytometrie (FC) byla vyšetřena kostní dřeň u 7 pacientů ze 17 (3 s mnohočetným myelomem provázeným amyloidózou a 4 s primární systémovou AL-amyloidózou). Plazmocyty (PC) byly identifikovány jako CD38+CD138+ buňky s mediánem zastoupení 0,2 % (0,2 – 2,2). Dle mediánu počtu CD45+PC 40,4 % (12,2 – 76,0) lze usoudit, že u všech pacientů bylyv různé míře nalézány klonální CD45 - PC,což bylo dále potvrzeno sníženým za-stoupením CD19+PC 29,0 % (2,5 – 69,3)a zvýšeným zastoupením CD56+PC 41,3 % (2,7 – 96,3). U jedné pacientky došlo ke ztrátě CD56 na patologických PC a u další pacienty byly naopak nalezeny CD19+ l + klonální PC. Překvapivě PC téměř všech hodnocených pacientů exprimovaly též znak CD27, nicméně k významu jeho exprese se prozatím nelze vyjádřit (obr. 11). Je zřejmé, že analýza PC pomocí senzitivní průtokové cytometrie může napomoci orientaci a stanovení diagnózy tím, že prokáže patologickou populaci plazmocytů v kostní dřeni, jinými metodami nedetekovanou. Průkaz klonální plazmocytární populace v kostní dřeni při histologickém nálezu amyloidu, jehož složení se ne vždy podaří pojmenovat, umožní uzavřít diagnózu jako AL-amyloidóza.

Současně pak FC může sloužit k monitorování účinnosti léčby.
Depozita lehkých řetězců v neamyloidové podobě
U 2 pacientů s nefrotickým syndromem a sníženou filtrací (zvýšená hodnota kreatininu) odhalila biopsie ledvin depozita lehkých řetězců ve formě amorfních hmot, takže u nich byla diagnóza uzavřena jako light chain deposition disease. Plazmocelulární proliferace v kostní dřeni měla u jednoho charakter monoklonální gamapatie nejistého významu a u druhého pak mnohočetného myelomu. Charakter souboru dokumentuje tab. 3.

Spektrum orgánového poškození
V uvedeném sedmnáctičlenném souboru pacientů s poškozením organizmu depozity lehkých řetězců byl nejčastějším příznakem nemoci nefrotický syndrom 12 pacientů (70 %). Snížená filtrace se zvýšenou hodnotou kreatininu v séru byla nalezena jen u 4 nemocných (23 %). Poškození srdce bylo echokardiograficky prokázáno u 4 (23 %) nemocných, v jednom z těchto případů byla amyloidová kardiomyopatie prokázána biopticky. Všichni pacienti s kardiomyopatií měli sníženou fyzickou zdatnost (NYHA II – III).
Hepatosplenomegalie byla palpačně a sonograficky prokázána u 2 (12 %) nemocných, biopsie jater však nebyla u žádného z pacientů provedena, na souvislost s AL-amyloidózou jsme pouze usuzovali, protože nebyl znám žádný další důvod pro tyto změny.
Klinicky manifestní koagulopatie byla přítomna ve 2 případech (12,0 %). V jednom případě byla zřejmě kombinované etiologie (trombocytopatie i alterace koagulačních faktorů), ve druhém případě šlo o izolovaný deficit faktoru X, jeho nejnižší hodnoty oscilovaly kolem 10 %. Snížená hladina faktoru X u této pacientky způsobovala metrorrhagii. Ta byla důvodem pro kyretáž, a když se krvácení pro kyretáže zhoršilo, následovala hysterektomie. Operace byla komplikována hemoragickým šokovým stavem, který naštěstí pacientka přežila. V resekátu dělohy byl prokázán amyloid.
Závažná somatická a viscerální neuropatie s klinicky závažnou ortostatickou hypotenzí byla diagnostikována u 2 (12 %) nemocných, syndrom karpálního kanálu měli 4 (23 %) nemocní. Pacienty s typickými velkými periorbitální hematomy v našem souboru nemáme. U 2 (12,0 %) pacientů z našeho souboru však byly přítomny pouze malé, diskrétní hematomy na víčkách. Stejně tak se v našem souboru nevyskytla makroglosie (typický příznak AL - amyloidózu, který se u jiných typů amyloidózy nevyskytuje). Příznaky poškození organizmu depozity lehkých řetězců a základní charakteristiku souboru shrnuje tab. 4.
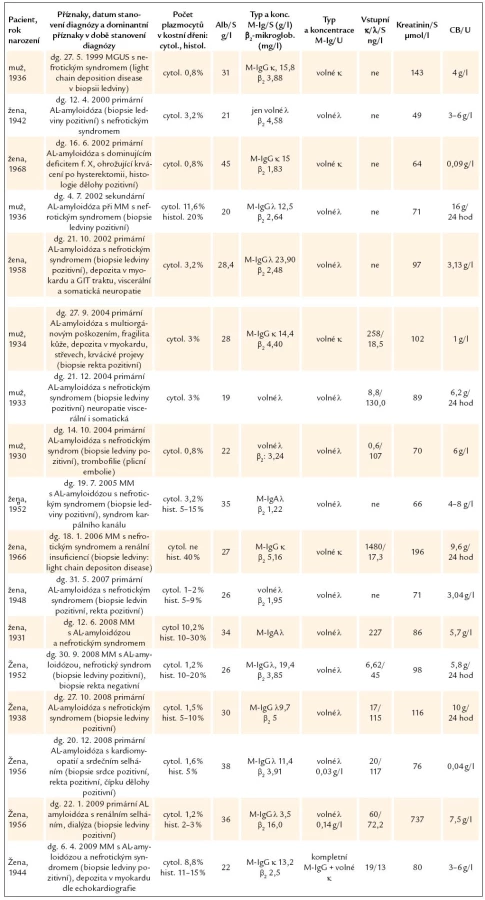
Léčba
Léčba pacientů této skupiny nebyla homogenní, a to jak pro odlišný biologický stav jednotlivých nemocných, tak pro měnící se socioekonomické podmínky a dostupnost použitých protinádorových léčiv v posledních 9 letech. Při volbě léčebného režimu bylo přihlíženo k celkové fyzické a mentální zdatnosti, věku a k hodnotám krevního obrazu před léčbou.
Klasickou léčbou, používanou jak u mnohočetného myelomu, tak u primární AL-amyloidózy i light chain deposition disease při MGUS, je kombinace vinkristinu, adriamycinu a dexametazonu (VAD). Tuto léčbu jsme použili u 6 (35 %) pacientů.
Vinkristin je však možné vyměnit za cyklofosfamid. Kombinací CAD byli léčeni 2 (12 %) pacienti.
Dalším léčebným schématem, které jsme použili v našem souboru, je kombinace cyklofosfamidu (buď perorálně 50 mg denně dlouhodobě, nebo 500 mg/ m2 i.v. infuze ve 14denních intervalech) a podávání dexametazonu (20 – 40 mg) 1. – 4. den a 15. – 18. den ve 28denních intervalech. Toto léčebné schéma bylo použito u 2 (12 %) pacientů.
Kombinace cyklofosfamidu, thalidomidu a dexametazonu byla použita ve 4 (23 %) případech a u 3 z nich jsme po 6 cyklech změnili léčbu na kombinaci bortezomibu (Velcade), cyklofosfamidu a dexametazonu (VCD).
Léčebné režimy, které mají prednison místo dexametazonu, jsme použili pouze ve 2 (12 %) případech, jednou to byla kombinace vinkristin, melfalan, cyklofosfamid a prednison (VMCP) a jednou to byla kombinace prednisonu a cyklofosfamidu.
Vysokodávkovanou chemoterapii (melfalan 100 mg/ m2, maximálně 140 mg/ m2) jsme použili u 4 (23 %) nemocných.
Nežádoucí účinky léčby
Léčebné režimy založené na dexametazonu jsou sice účinné, ale mají pravidelné nežádoucí účinky. Běžnými nežádoucími účinky uvedených dávek dexametazonu jsou psychické problémy, které jsou u někoho největší právě ve dnech užívání dexametazonu (poruchy spánku, agresivita, či naopak deprese), u jiného naopak jsou nejintenzivnější těsně po vysazení dexametazonu (detrakční syndrom s výraznými projevy fatigue). Tyto účinky postihnou více či méně každého léčeného. Běžným nežádoucím účinkem je také manifestace latentního diabetes mellitus či dekompenzace stávajícího diabetes mellitus. S dekompenzací diabetu jsme se setkali u 4 nemocných, u 2 z nich to bylo důvodem k hospitalizaci, 2 to zvládali s navýšením inzulinu dle aktuálních hodnot glykemií, měřených doma glukometrem. Dalším problémem jsou infekce. V našem souboru 17 léčených bylo celkem 10 hospitalizací pro infekční komplice v průběhu léčby. V jednom případě šlo o závažný pásový opar s dlouhodobě trvajícími neuralgickými bolestmi.
U pacientů s nefrotickým syndromem, závažnou hypoalbuminemií, se objevuje další problém, který u pacientů s mnohočetným myelomem se v té míře nevyskytuje. Retence tekutin v době užívání dexametazonu působila ve 3 případech závažné problémy a u jedné pacientky retence tekutin při dexametazonu vyústila v počínající plicní edém. Proto rizikové osoby s nefrotickým syndromem jsme hospitalizovali po dobu podávání dexametazonu a dle vývoje hmotnosti upravovali dávky furosemidu.
Myelosuprese není pro používané režimy typická. Problémem bylo však neurotoxické působení vinkristinu, zvláště pokud nasedlo na viscerální neuropatii související s amyloidózou. Proto jsme ve 2 případech museli pro subileózní stav po 1. cyklu chemoterapie VAD vysadit vinkristin a pokračovat pouze kombinací AD.
U 4 pacientů s AL-amyloidózou při mnohočetném myelomu byl použit režim s thalidomidem. V jednom případně byl režim CTD opuštěn ihned po 1. cyklu pro nezvladatelnou obstipaci, jedna pacientka zemřela po 2 cyklech CTD a 2 pacientky absolvovali 5 a 6 cyklů CTD. Typickými nežádoucími účinky, které tento režim provázely, byla zácpa, fatigue a postupně nastupující příznaky neuropatie.
Režim založený na bortezomibu dostali celkem 4 pacienti, obvykle následoval při netoleranci thalidomidového režimu anebo při nedosažení CR thalidomidovým režimem. Při této léčbě dominovaly nežádoucí účinky dexametazonu (retence tekutin, psychické problémy). Neuropatie, které provází léčbu Velcadem, byla jen mírná, pouze I. a II. stupně, srovnatelná jako u pacientů s mnohočetným myelomem.
Výsledky léčby
V případě AL-amyloidózy a ligh chain deposition disease je nutné hodnotit jak hematologickou léčebnou odpověď (změny koncentrace monoklonálního imunoglobulinu a volných lehkých řetězců a změny v počtu plazmocytů v kostní dřeni), tak také orgánovou odpověď. Hematologickou léčebnou odpověď a orgánovou léčebnou odpověď hodnotíme dle kritérií z roku 2005 [25], jejich český překlad byl zveřejněn v časopise Vnitřní lékařství [10].
Kompletní hematologické léčebné odpovědi bylo dosaženo celkem 8krát (47 %).
Velmi dobré parciální léčebné odpovědi (koncentrace M-Ig je poklesla na tak nízké hodnoty, že není možné kvantitativní stanovení denzitometrií, ale je stále prokazatelné pomocí imunofixační elektroforézy) bylo dosaženo u 2 (12 %) pacientů.
Parciální léčebné odpovědi bylo dosaženo u 2 (12 %) pacientů.
U 5 pacientů nebylo dosaženo žádné léčebné odpovědi.
Orgánovou léčebnou odpověď bylo možné hodnotit jen u 13 pacientů, u 4 ji pro krátký interval ji hodnotit.
K vymizení příznaků poškození orgánu monoklonálním imunoglobulinem (neboli k orgánové CR) došlo celkem u 5 (38 %) pacientů. U jedné nemocné byla orgánová léčebná odpověď hodnocena jako parciální.
U 6 pacientů nedošlo k žádným změnám orgánového poškození, v 1 případě se míra orgánového poškození v průběhu léčby zhoršovala.
Jednoznačným předpokladem k orgánové léčebné odpovědi bylo dosažení kompletní hematologické remise. Léčbu, léčebné odpovědi a dobu sledování celého souboru uvádí tab. 5.

Doba sledování a délka přežití
Z uvedeného souboru 17 pacientů zemřelo 5 pacientů, takže není možné stanovit medián přežití. Medián sledování celého souboru je 18,5 (0,3 – 124) měsíců. Průměrná doba sledování je 39,6 měsíců. Tento rozdíl mezi medián a průměrem je způsobem relativně vyšším počtem nemocných s touto diagnózou v posledních letech.
Nicméně z údajů uvedených v tab. 5 je zřetelné, že dosažení kompletní hematologické remise je velmi často provázeno dosažením kompletní orgánové léčebné odpovědi a dlouhodobými remisemi.
Diskuze
Prognostické členění pacientů s AL-amyloidózou
Tolerance, a tím i výsledky léčby u pacientů s AL-amyloidózou závisí na míře poškození orgánů a tkání nemocného depozity amyloidu. Volba léčebného re-žimu je výsledek odhadu poměru pravděpodobnosti závažných nežádoucích účinků léčby při zjištěné míře a rozsahu poškození organizmu amyloidem a přínosu léčby pro konkrétního pacienta.
V poslední době se ukazuje, že prognosticky nejzávažnější je míra poškození srdce amyloidem [26,27]. Proto prognostická klasifikace zohledňuje míru poškození srdce. Je založen na 2 parametrech, na hodnotě troponinu a hodnotě NT - (N-terminální) proBNP (proBrain Natriuretic Peptide).
ProBNP je peptid o délce 108 aminokyselin, tvořený myocyty dominantně v levé komoře v důsledku napětí srdeční stěny. Po uvolnění je štěpen na 2 fragmenty, na aktivní BNP obsahují 77. až 108. aminokyselinu a na NT-proBNP obsahující 1. – 76. aminokyselinu. BNP i NT - proBNP jsou markery srdečního selhání.
Autoři z Mayo kliniky navrhli a ověřili jednoduchý klasifikační systém, založení na troponinu a NT-proBNP. Pacienti, kteří měli obě hodnoty ve fyziologickém rozmezí, tvořili skupinu s nízkým rizikem, pacienti s oběma parametry nad fyziologickou hranicí tvořili skupinu s vysokými rizikem. Střední skupiny tvořili pacienti s jednou patologickou a jednou fyziologickou hodnotou. Uvedené hodnoty byly zvýšené také u několika pacientů s normálními echokardiografickými nálezy, kteří ale při dalším sledování měli nepříznivý průběh a krátkou dobu přežití, podobně jako ostatní pacienti se zvýšenými hodnotami. Proto se autoři domnívají, že tato laboratorní klasifikace svou přesností předčí echokardiografické vyhodnocení [28]. Italští autoři k této klasifikaci připojili ještě další parametry, tak jak je uvádíme v tab. 6 [29].
![Prognostické skupiny pacientů s AL‑ amyloidózou [29].](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/78053e433d106296260304236ca638fe.jpeg)
Za pacienty s nízkým rizikem považuje Palladini nemocné mladší 65 let, s postižením maximálně 2 orgánů, s hodnotami troponinu a NT-proBNP ve fyziologickém rozmezí, clearencí kreatininu nejméně 50 ml/ min a plicní difuzní kapacitě nad 50 % a systolickým tlakem nad 90 mm Hg. Tito nemocní mohou být považováni za kandidáty pro vysokodávkovanou chemoterapii. Počet těchto pacientů však obvykle nepřesahuje 15 % všech pacientů s AL-amyloidózou.
Pacienti s vysokým rizikem mají krátký medián přežití asi kolem 3,5 měsíce a představují přibližně 20 % všech pacientů s AL-amyloidózou Tito nemocní jsou velmi křehcí a špatně tolerují nežádoucí účinky chemoterapie. Nicméně i tito nemocní mohou profitovat z léčby, pokud jim jejich zdravotní stav umožní, aby ještě žili tak dlouho, aby mohli dokončit chemoterapii a mít prospěch z případné kompletní remise. Ze skupiny 22 těchto pacientů zemřelo 6 nemocných před dokončením 3. cyklu chemoterapie CTD a pouze 8 pacientů dosáhlo hematologické léčebné odpovědi a z nich 4 dosáhli dlouhodobého zlepšení funkce srdce [29]. Jsou však popsány i další případy, kdy kombinovaná léčba obsahující thalidomid přinesla nejen hematologickou léčebnou odpověď, ale i zlepšení srdeční funkce [30].
Podobně jako u mnohočetného mye-lomu důležitým prognostickým faktorem je také masa patologických plazmocytů, a tedy produkce volných lehkých řetězců. Výše jejich koncentrace v době stanovení diagnózy má prognostickou výpověď. O míře léčebné odpovědi svědčí více změny v koncentraci volných lehkých řetězců než změny koncentraci kompletní molekuly monoklonálního imunoglobulinu [31 – 38].
Léčebné alternativy
Režimy kombinující cytostatikum s vysokou dávkou dexametazonu
Klasickým léčebným postupem v předchozích desetiletích byla léčba melfalanem a prednisonem [39]. Tato léčba přinášela však nevelký počet léčebných odpovědí, nevedla tedy k zásadnímu prodloužení přežití [40]. Léčba kombinací více alkylačních cytostatik také nevedla k zásadnímu zlepšení [41]. Zvýšení počtu léčebných odpovědí přineslo až přidání vysokých dávek dexametazonu do léčebných schémat, počet léčebných odpovědí byl vyšší než v předchozích studiích. Monoterapie dexametazonem a udržovací léčba dexametazon + interferon dosáhla 24 % kompletních remisí [42].
Podobně i další klinické studie, v nichžbyly použity léčebné režimy obsahující vysoké dávky dexametazonu, prokázaly vyšší počet léčebných odpovědí než dříve používaný alkeran a prednison (alkeran a dexametazon) [43], VAD a jemu podobné režimy [44,45], střední dávky melfalanu a dexametazon [42,46]. Ve studii zveřejněné roku 2007 dosáhla kombinace melfalan + dexametazon léčebné odpovědi v 67 % případů (33 % CR) a orgánové léčebné odpovědi ve 48 %. Dosažené léčebné odpovědi trvaly v 70 % nejméně 3 roky [47]. Poslední vyhodnocení dat z italského centra uvádí v souboru 126 pacientů 26 % CR, 36 % PR a 33 % orgánových léčebných odpovědí [48]. Menší počet léčebných odpovědí uvádí studie se 70 pacienty nevhodnými k transplantační léčbě, u 22 z nich pro časné úmrtí nebylo možné hodnotit léčbu a ze zbytku 48 hodnotitelných nemocných bylo dosaženo jen 15 % hematologických léčebných odpovědí [49]. Je zřejmé, že výsledky léčby zásadně závisí od složení léčebné skupiny nemocných.
V našem souboru jsem chemoterapii VAD, CAD či jen AD použili v 9 případech, ve 4 z nich pak zakončili vysokodávkovaným melfalanem. Z 5 léčených klasickými režimy VAD, CAD a AD bylo v 1 případě dosaženo CR a poději i orgánové CR, v jednom případě VGPR s kompletní orgánovou CR, v jednom případě PR bez orgánové léčebné odpovědi a ve 2 případech nebyla dosažena žádná léčebná odpověď a tito pacienti po 5 a 6 měsících od zahájení léčby zemřeli.
Thalidomid v léčbě AL-amyloidózy
Po roce 2000 se do klinického použití dostaly nové léky pro mnohočetný myelom, thalidomid, bortezomib a lenalidomid. Registrační klinické studie, na jejichž základě jsou tyto léky používány u mnohočetného myelomu, byly opakovaně zveřejňovány. Naproti tomu mnohem méně pozornosti bylo věnováno přínosu těchto léků pro AL-amyloidózu, neboť toto onemocnění je méně časté, a tak se farmaceutickým firmám nevyplatí provádět velké registrační studie. Práce, které vyhodnocují jejich podání, musí tedy vždy uvést, že byly použity of lable.
Nicméně díky akademickým studiím, v rámci nichž byly tyto nové léky u AL-amyloidózy použity, máme již k dispozici informace, které potvrzují, že tyto léky přinášejí minimálně stejně velký prospěch pacientům s AL-amyloidózou jako pacientům s mnohočetným myelomem.
Thalidomid v monoterapii je v případě AL-amyloidózy málo účinný při použití v nízkých dávkách a špatně tolerovatelný při použití dávek vyšších [50,51].
Palladini, který používal maximální tolerovatelné dávky, až 400 mg, uvádí symptomatickou bradykardii u 26 % léčených [52].
Při kombinaci s dexametazonem se však účinnost léčby zvyšuje a počet léčebných odpovědí dosáhl 48 % [52]. Kombinovaná léčba (cyklofosfamid, thalidomid a dexametazon) v dávkách upravovaných dle předpokládané tolerance dosáhla celkem 74 % léčebných odpovědí [53]. Kombinace melfalanu, dexametazonu a thalidomidu pomohla i v případech závažné kardiální formy amyloidózy, pokud srdeční postižení umožnilo absolvovat alespoň 3 cykly [29]. Největší popsaný soubor vyšel z britských ostrovů, celkem 202 pacientů bylo léčeno tímto režimem. Hematologickou léčebnou odpověď popisují o 62 % hodnocených pacientů, z toho bylo 25 % CR a 3 % nCR. Léčebná odpověď byla dosažena u 67 % nově léčených nemocných a u 54 % nemocných s relapsem nemocí. Celkový počet orgánových léčebných odpovědí dosáhl 42 %. Frekvence dosažení orgánové léčebné odpovědi však závisí od toho, který orgán je poškozen.
Orgánová léčebná odpověď byla dosažena při poškození ledvin u 46 % léčených, při poškození jater u 29 % léčených, při poškození nervů u 13 % léčených, při poškození srdce u 8 % léčených, při poškození gastrointestinálního traktu u 25 % [54]. Nicméně jsme v literatuře našli 2 zveřejněné srovnávací studie klasické léčby melfalanem a dexametazonem (MD) s kombinací thalidomidu, dexametazonu a cyklofosfamidu (CTD). Po 12 měsících léčby byla orgánová léčebná odpověď prokázána u 44/ 113, tedy 39 % pacientů léčených režimem CTD a u 12/ 56, tedy 21 % pacientů léčených MD (p = 0,03). Autoři uvádějí, že mezi oběma režimy nebyl rozdíl v počtu hematologických kompletních remisí, celkovém počtu hematologických odpovědí, toxicitě a celkovém přežití, pouze medián nástupu léčebné odpovědi (dle FLC) byl 3 měsíce po MD a 2 měsíce po CTD [55]. Shodné závěry získané na podstatně menším souboru pacientů (12 MD + 12 CTD) uvádějí i další autoři [56]. Zkušenosti s thalidomidem shrnuje tab. 7.

V našem souboru nemocných dostali celkem 4 pacienti režim CTD v rámci první linie léčby. Pouze 2 z nich absolvovali více než 2 cykly (5 a 6 cyklů, ale pro nedosažení CR byla léčbě nakonec změněna za bortezomibový režim). V jednom případně pro intoleranci (neřešitelná zácpa) muselo být podávání thalidomidu po měsíci ukončeno a léčba byla změněna na bortezomibový režim a jedna pacientka zemřela v průběhu druhého léčebného cyklu na infekci.
Bortezomib v léčbě AL-amyloidózy
První klinické zkušenosti s bortezomibem přinesly pozitivní překvapení. Kastritis aplikoval bortezomib a dexametazon skupině 18 pacientům (z toho 7 bylo relabujících) a dosáhl 94 % léčebných odpovědí, z toho 44 % kompletních remisí, přičemž medián intervalu do dosažení léčebné odpovědi bylo jen 0,9 měsíce. Při mediánu sledování 11,2 měsíce zaznamenali již 28 % orgánových léčebných odpovědí [59,60]. Tak vysoký počet léčebných odpovědí po klasické léčbě zatím nebyl zaznamenán. Hypotetickým vysvětlením je velmi vysoká citlivost amyloidotvorných plazmocytů na bortezomib. Úlohou proteasomu je regulovaná degradace proteinů. Degradovány jsou nesprávně složené proteiny, aberantní proteiny nebo proteiny, které regulují např. buněčný cyklus nebo apoptózu. Proteasom hraje např. klíčovou roli v regulaci aktivity NF-κB (nukleární faktor κ B), a to degradací jeho inhibitoru I - κB (inhibitor κ B).
Účinek inhibitorů proteasomu (včetně bortezomibu) na myelomové buňky souvisí především s jejich dopadem na funkci faktoru NF-κB, který, je li aktivní, zprostředkovává přežití MM buněk a jejich rezistenci k chemoterapii a radioterapii. V důsledku inhibice degradace proteinů nedochází k degradaci I-κB, a tím k aktivaci NF-κB. Působením bortezomibu je tedy I-κB stabilizován, tím je NF-κB inhibován, a nemůže tak zajistit přežití MM buněk.
Mechanizmus účinku bortezomibu na amyloidogenní plazmatické buňky zahrnuje stejný mechanizmus jako u myelomových buněk, tedy inhibici NF-κB, ale navíc ještě další. Degradace proteasomem je spřažena s uvolňováním proteinů určených k degradaci z lumen endoplazmatického retikula (ER). Pokud je tento proces zastaven, v ER se proteiny hromadí a to způsobuje stres ER. Stres ER představuje stav, při kterém je porušena metabolická a redoxní rovnováha a buňka se stává citlivější k apoptóze, např. v důsledku stabilizace některých proapoptotických proteinů. Dlouhodobý stres ER pak vede k apoptóze.
V případě amyloidózy je v ER postižených buněk velké množství aberantních imunoglobulinů. Inhibice proteasomu působením bortezomibu vede k další akumulaci proteinů v ER, způsobí tak stres ER, který následně indukuje apoptózu [61].
Dávky bortezomibu byly u AL amyloidózy testovány v eskalační studii 0,7 – 1,6 mg 1krát týdně a 0,7 – 1,3 mg 2krát týdně. Celkem 31 pacientů bylo rozděleno do 7 skupiny. Celkem bylo dosaženo 20 % kompletních remisí opět s krátkým mediánen pro dosažení léčebné odpovědi 1,2 měsíce. Autoři konstatují, že při dávce bortezomibu 1,3 mg/ m2 při klasickém dávkování 1, 4, 8, 11 a nebo 1,6 mg/ m2 při podávání ve dnech 1, 8, 15, 22 v 35denních cyklu se neliší tolerance od pacientů s myelomem a dosahuje vysokého počtu léčebných odpovědí. V dalších studiích budou tedy používat stejné dávky [62].
Zkušenosti z Velké Británie sumarizuji Wachelaker. V souboru 20 intenzivně předléčených pacientů (medián předchozích léčebných linií = 3), kteří dostávali bortezomib v monoterapii nebo v kombinaci s dexametazonem, dosáhli 15 % kompletních remisi a 65 % parciálním remisi a orgánová léčebná odpověď byla dosažena u 38 % léčených [63.]
Doposud největší soubor pacientů je obsažen souhrnném hodnocení italských, britských a řeckých center, věnujících se léčbě amyloidózy – obsahující 94 pacientů. Tito pacienti byli léčení bortezomibem v kombinaci s dexametazonem. Celkem bylo dosaženo 71 % léčebných odpovědí, z toho 25 % kompletních. V podskupině, která tuto léčbu dostala jako iniciální, bylo dokonce dosaženo 47 % CR, zatímco ve skupině, jejíž pacienti byli refrakterní či relabovali po jiné iniciální léčbě, to bylo jenom 20 %. Orgánové léčebné odpovědi bylo dosaženo u 30 % pacientů (22 % kardiálních, 17 % renálních a 19 % hepatálních léčebných odpovědí). Opět byl potvrzen krátký interval pro dosažní léčebné odpovědi (medián 1,2 měsíce) a medián intervalu pro dosažení kompletní remise 2,3 měsíce. Autoři svoje pozorování uzavírají slovy: kombinovaná léčba amyloidózy obsahující bortezomib a dexametazon je velmi účinná a má zvladatelné nežádoucí účinky [64]. Pro srovnání u dříve používaných léčebných postupů to bylo 3,5 a 6,2 měsíce. Zkušenosti s bortezomibem v léčbě AL - amyloidózy shrnujeme v tab. 8.

V našem souboru jsme kombinaci obsahující bortezomib použili celkem 3krát při intoleranci thalidomidu anebo při nedosažení kompletní remise thalidomovým režimem. Ve 2 případech tato léčba navolila CD a v jednom VGPR. Naše zkušenosti tedy potvrzují vysoký účinek bortezomibu u AL-amyloidózy.
Lanalidomid v léčbě AL - amyloidózy
Lenalidomid, lék s ověřenou účinností u mnohočetného myelomu, byl použit také v léčbě AL-amyloidózy. První studie ověřovaly dávku. Standardní dávka 25 mg/ den se ale v iniciální léčbě amyloidózy ukázala velmi toxická, a proto byla snížena na 15 mg/ den, podobně jako i v pozdějších studiích (tab. 9). Při použití této dávky bylo dosaženo 29 % kompletních hematologických léčebných odpovědí, 38 dosáhlo PR [69]. Další zkušenosti byly získány na Mayo klinice v Rochesteru. První 3 cykly podávali lenalidomid v monoterapii. Z těchto 23 nemocných nedokončilo 10 plánované 3 cykly a z těch, kteří tyto plánované 3 cykly dokončili, pouze u jednoho došlo k hematologické léčebné odpovědi. Pak přidali k lenalidomidu dexametazon a v průběhu dalších měsíců léčby se zvýšil počet hematologických léčebných odpovědí na 9 z 22 (45 %). Tato studie tedy prokázala nízkou účinnost v lenalidomidu v monoterapii, poukázala na jeho toxicitu a prokázala jeho účinnost v kombinaci s dexametazonem [70]. V analýze souboru z roku 2008 tito autoři uvádějí, že lenalidomid v monoterapii není vhodný pro léčbu AL amyloidózy, ale v kombinaci může být pro tyto nemocné užitečný [71].

Další klinické studie proto začínají testovat revlimid v trojkombinaci (lenalidomid, dexametazon a cyklofosfamid), zatím však bez jasného doporučení pro praxi [72]. Nicméně aplikace revlimidu může být spojena se zhoršováním ledvinných i srdečních funkcí [73]. Současné zkušenosti s touto léčbou sumarizuje tab. 9.
Vysokodávkovaná chemoterapie
Léčba vysokodávkovaným melfalanem s transplantací autologních krvetvorných buněk dosahuje vysokého počtu kompletních hematologických remisí, což sebou přináší zlepšení kvality života a prodloužení přežití [79]. Nicméně tento postup je spojen s určitou toxicitou, a to při zvýšené fragilitě pacientů, způsobené depozity amyloidu, je spojeno se zvýšenou mortalitou, která se pohybuje od 11 % [80] do 27 % [81]. Tuto vysokou mortalitu je možné zmenšit pečlivou selekcí nemocných vhodných pro tuto léčbu, založenou pečlivým vyhodnocením míry srdeční dysfunkce [82 – 89].
Další možností, jak snížit mortalitu, je snížení dávky melfalanu na 100 – 140 mg/ m2. Redukce dávky však snížila počet hematologických odpovědí (53 %) a přitom pořád zůstala nezanedbatelná mortalita [90].
V retrospektivní studii měli pacienti s vysokodávkovanou chemoterapií delší přežití než pacienti léčení melfalanem a prednisonem [91].
První multicentrickou studií, která měla za cíl srovnat klasickou chemoterapii melfalan a dexametazon s vysokodávkovanou chemoterapií, byla studie francouzské myelomové skupiny. V této studii nebyl zjištěn signifikantní rozdíl v počtu hematologických a orgánových léčebných odpovědí v obou skupinách [92].
Kritické hlasy sice vyčítají této studii suboptimální výběr pacientů pro vysokodávkovanou chemoterapii, nicméně vyplývá z ní, že konvenční režim založený na vysokodávkovaném dexametazonu a alkylačním cytostatiku má pořád své místo v léčbě těchto nemocných.
Tato provokativní francouzská studie iniciovala další analýzy této otázky. Randomizovaná studie z Mayo kliniky prokázala, že vysokodávkovaná chemoterapie je jednoznačně lepší než kombinace melfalanu a dexametazu pro pacienty s počtem plazmocytů v kostní dřeni nad 20 % [93].
Důležitost dosažení kompletní remise pro prognózu nemocných zdůrazňuje analýza 80 pacientů, kteří podstoupili vysokodávkovanou chemoterapii s au-tologní transplantací před 10 lety. Sice 22 % léčených umřelo v průběhu 1. roku (14 % důsledkem toxicity léčby a 8 % důsledkem progrese). Z 63 pacientů, kteří přežili 1. rok po transplantaci, se 51 dostalo do kompletní remise nemoci. Pro celou skupinu 80 pacientů byl medián přežití 4,75 roku. Medián přežití pacientů, kteří se dostali do kompletní remise, přesahoval 10 roků, zatímco medián přežití těch, kteří se nedostali do kompletní remise, byl pouze 50 měsíců. Tato studie prokázala, že dosažení kompletní remise navodí u AL - amyloidózy dlouhodobé přežití [94]. A ani postižení srdce není kontraindikací, v souboru 194 pacientů s kardiálním poškozením a vysokodávkovanou chemoterapií byla dosažena hematologická léčebná odpověď u 69 % nemocných a orgánová léčebná odpověď u 47 %, a to za cenu 16,5 % mortality [95].
Z našeho souboru byla vysokodávkovaná chemoterapie podána celkem 4 pacientům a všichni jsou v dlouhodobé remisi trvající 113+, 87+, 50+ a 45+ měsíců. Nikdo z našich pacientů po vysokodávkované chemoterapii s autologní transplantací neumřel na komplikaci ani na progresi nemoci. Vysokodávkovanou chemoterapii považujeme stále za vhodnou volbu pro nemocné bez závažného orgánového poškození.
Vysokodávkovaná chemoterapie následovaná konsolidační chemoterapií
Pro pacienty, kteří po vyskodávkované chemoterapii nedosáhnou kompletní remise, jsou vhodné další léčebné linie. Kombinace thalidomidu a dexametazonu byla v této indikaci zkoušena u 31 nemocných, u 42 % z nich prohlou-bila léčebnou odpověď, ale tolerance této léčby nebyla bezproblémová [58].
V následující klinické studii (27 pacientů) zaměnili thalidomid za bortezomib a u 7/ 8 pacientů došlo ke zlepšení léčebné odpovědi [96].
Časnou konsolidační léčbu navazující na vysokodávkovanou chemoterapii s autologní transplantací, která může prohloubit dosaženou léčebnou odpověď u těch pacientů, kteří nejsou v CR, podporují i výsledky dalších studií [98]. Výsledky nejnovější studie shrnujeme do tab. 10.

Závěry pro praxi
- AL-amyloidóza může mít projevy zře-telné na první pohled – periorbitálníhematomy, lehká tvorba hematomů nebo makroglosie, anebo může způsobovat poškození organizmu prostému oku lékaře skryté (nefropaties nefrotickým syndromem, kardio-myopatie, neuropatie, syndrom karpálního kanálu, poškození zažívacího traktu, získanou poruchu koagulace).
- Na poškození orgánu amyloidem je nutno pomýšlet při zjištění některého z výše uvedených problémů.
- Prokázat lze amyloid jedině histologickým vyšetřením s cíleným barvením na amyloidózu a typizací bílkovin, z nichž jsou vytvořena amyloidová depozita. Histologie však není vždy schopna prokázat, že se jedná o amyloid z lehkých řetězců. K závěru, že se jedná o amyloid vytvořený z depozit lehkých řetězců, lze sice přijít proteomickou analýzou, tak však není v ČR dostupná. Proto k této diagnóze dospíváme nepřímo, metodou vyloučení jiných příčin amyloidózy a průkazem monoklonální gamapatie na základě biochemických metoda a na základě vyšetření kostní dřeně histologicky, ale i flowcytometricky.
- Úspěch léčby závisí na míře poškození organizmu amyloidem, při malém poškození organizmu a věku do 65 let lze použít vysokodávkovanou chemoterapii s vysokou pravděpodobností dosažení úspěchu. V případě pozdní diagnózy a velkého poškození orgánů, zvláště pak srdce, obvykle pacient již léčbu netoleruje a obvykle zmírá dříve, než by mohl nastoupit případný léčebný efekt.
- V případech, kdy nelze použít vysokodávkovanou chemoterapii, se považuje za vhodné podat léčebnou kombinaci obsahující dexametazon, alkylační cytostatikum a případně některý z nových léků.
- Z nových léků lze pro tuto nemoc použít bortezomib, ale také thalidomid či lenalidomid.
- Při léčbě AL-amyloidózy je nutno mít vždy na paměti, že cílem léčby musí být vždy dosažení kompletní hematologické remise (vymizení amyloidotvorných lehkých řetězců, potvrzeno negativní imunofixací a normálním nálezem volných lehkých řetězců v séru).
- Řádově měsíce od dosažení hematologické kompletní remise může dojít k orgánové léčebné odpovědi (vymizení depozit amyloidu v orgánech), a tím ke zlepšení funkce poškozených orgánů.
- Vyšetřování volných lehkých řetězců v séru umožňuje monitorovat účinnost léčby AL-amyloidózy, a 90 % pokles jejich koncentrace je spojen se signifikantně delším přežitím.
Text byl připraven v rámci projektu CRAB – „Časnou diagnostikou za lepší kvalitu života“, který byl aktivován v roce 2007, a dále v rámci následujících aktivit: MŠMT LC 06027, MSM0021622434, GACR 301/ 09/ P457, IGA NS10408 - 3a IGA NS10406 - 3.
prof. MUDr. Zdeněk Adam, CSc.
www.fnbrno.cz
e-mail: z.adam@fnbrno.cz
Doručeno do redakce: 26. 11. 2009
Přijato po recenzi: 4. 1. 2010
Zdroje
1. Merlini G, Bellotti V. Molecular mechanisms of amyloidosis. N Engl J Med 2003; 349 : 583 – 596.
2. Sideras K, Gertz MA. Amyloidosis. Adv Clin Chem 2009; 47 : 1 – 44.
3. Lavatelli F, Perlman DH, Spencer B. Amyloidogenic and associated proteins in systemic amyloidosis proteome of adipose tissue. Mol Cell Proteomics 2008; 7 : 1570 – 1583.
4. Anesi E, Palladini G, Perfetti V et al. Therapeutic advances demand accurate typing of amyloid deposits. Am J Med 2001; 111 : 243 – 244.
5. Vrana JA, Zeldenrust SR, Theis JD et al. Diagnosis and classification of amyloidosis in abdominal subcutaneous fat specimens using mass spectrometry based proteomics. Blood 2008; 112 : 937. Abstract 2710.
6. Lachmann HJ, Booth DR, Booth SE et al. Misdiagnosis of hereditary amyloidosis as AL (primary) amyloidosis. N Engl J Med 2000; 346 : 1786 – 1791.
7. Abraham RS, Geyer SM, Price ‑ Troska TL et al. Immunoglobulin light chain variable (V) region genes influence clinical presentation and outcome of light chain‑associated amyloidosis (AL). Blood 2003; 101 : 3801 – 3808.
8. Comenzo RL, Zhang Y, Martinez C et al. The tropism of organ involvement in primary systemic amyloidosis: contributions of Ig V(L) germ line gene use and clonal plasma cell burden. Blood 2001; 98 : 714 – 720.
9. Bellotti V, Chiti F. Amyloidogenesis in its biological environment: challenging a fundamental issue in protein misfolding diseases. Curr Opin Struct Biol 2008; 18 : 771 – 779.
10. Ščudla V, Pika T. Současné možnosti léčby systémové AL amyloidózy. Vnitř Lék 2009; 55 (Suppl 1): S77 – S87.
11. Adam Z, Ščudla V. Klinické projevy a diagnostika AL ‑ amyloidózy a některých dalších typů amyloidóz. Vnitř Lék 2001; 47 : 36 – 45.
12. Tichý M. Primární amyloidóza. Lék Zpr LF UK Hradec Králové 1999; 44 : 99 – 107.
13. Kroupa R, Dastych M, Šenkyřík M et al. Systémová amyloidóza s dominující klinickou manifestací v trávicím traktu. Vnitř Lék 2005; 51 : 588 – 592.
14. Ryšavá R. Amyloidóza ledvin. Postgrad Med 2006; 8 : 207 – 212.
15. Ryšavá R. Léčba paraproteinemické nefropatie a primární amyloidózy ledvin. Aktual v Nefrol 2005; 11 : 62 – 65.
16. Linhartová K, Daum O. Srdeční amyloidóza. Cor Vasa 2005; 47 : 328.
17. Kyle RA, Linos A, Beard CM et al. Incidence and natural history of primary systemic amyloidosis in Olmsted County Minnesota, 1950 through 1989. Blood 1992; 79 : 1817 – 1822.
18. Magy ‑ Bertrand N, Dupond JL, Mauny F et al. Incidence of amyloidosis over 3 years. The AMYPRO study. Clin Exp Rhematol 2008; 26 : 1074 – 1078.
19. Merlini G, Palladini G. Amyloidosis: is a cure possible? Ann Oncol 2008; 19 (Suppl 4): iv63 – iv66.
20. Merlini G, Stone MJ. Dangerous small B ‑ cell clones. Blood 2006; 108 : 2520– – 2530.
21. van Gameren II, van Rijswijk MH, Bijzet Jet al. Histological regression of amyloid in AL amyloidosis is exclusively seen after normalization of serum free light chain. Haematologica 2009; 94 : 1094 – 1100.
22. Štraub J, Adam, Z, Gregora E et al. Mnohočetný myelom – projekt časné diagnostiky CRAB. Zdravotnické noviny – Lékařské listy 2007; 16 : 4 – 5.
23. International Myeloma Working Group: Criteria for the classification of monoclonal gammopathies, multiple myelom and releated disorders: a report of the International Myeloma Working Group. Brit J Haematol 2003; 121 : 749 – 757.
24. Kyle RA, Rajkumar SV. Criteria for diagnosis, staging, risk stratification and response assessment of multiple myeloma. Leukemia 2009; 23 : 3 – 9.
25. Gertz MA, Comenzo R, Falk RH. Definition of organ involvement and treatment response in immunoglobulin light chain amyloidosis (AL): a consensus opinion from the 10th International Symposium on Amyloid and Amyloidosis, Tours, France, 18 – 22 April 2004. Am J Hematol 2005; 79 : 319 – 328.
26. Obici L, Perfetti V, Palladini G et al. Clinical aspects of systemic amyloid diseases. Biochim Biophys Acta 2005; 1753 : 11 – 22.
27. Palladini G, Campana C, Klersy C et al. Serum N‑terminal pro‑brain natriuretic peptide is a sensitive marker of myocardial dysfunction in AL amyloidosis. Circulation 2003; 107 : 2440 – 2445.
28. Dispenzieri A, Gertz MA, Kyle RA et al. Serum cardiac troponins and N‑terminal pro‑brain natriuretic peptide: a staging system for primary systemic amyloidosis. J Clin Oncol 2004; 22 : 3751 – 3757.
29. Palladini G, Russo P, Lavatelli F. Treatment of patients with advanced cardiac AL amyloidosis with oral melphalan, dexamethasone, and thalidomide. Ann Hematol 2009; 88 : 347 – 350.
30. Oh IY, Kim HK, Kim YJ et al. An intriguing case of primary amyloidosis with cardiac involvement: symptomatic and echocardiographic improvement with thalidomide treatment. Int J Cardiol 2006; 113 : 141 – 143.
31. Bird J. Guidelines in the diagnosis and management of AL amyloidosis. Brit J Haematol 2004; 125 : 681 – 700.
32. Dispenzieri A, Lacy MQ, Katzmann JA et al Absolute values of immunoglobulin free light chains are prognostic in patients with primary systemic amyloidosis undergoing peripheral blood stem cell transplantation. Blood 2006; 107 : 3378 – 3383.
33. Kumar S, Gertz MA, Lacy MQ et al. Changes in serum free light chain rather then intact monoclonal imunoglobin levels predict outcome with therapyin patients with light chain amyloidosis. Blood 2009; 114 (Suppl N22): 311. Abstract 747.
34. Lachmann HJ, Gallimore R, Gillmore JD et al. Outcome in systemic AL amyloidosis in relation to changes in concentration of circulating free immunoglobulin light chains following chemotherapy. Br J Haematol 2003; 122 : 78 – 84.
35. Palladini G, Lavatelli F, Russo P et al. Circulating amyloidogenic free light chains and serum N‑terminal natriuretic peptide type B decrease simultaneously in association with improvement of survival in AL amyloidosis. Blood 2006; 107 : 3854 – 3858.
36. Ščudla V, Minařík J, Schneiderka P et al. Význam sérových hladin volných lehkých řetězců imunoglobulinu v diagnostice a hodnocení aktivity mnohočetného myelomu a vybraných monoklonálních gamapatií. Vnitř Lék 2005; 51 : 1249 – 1259.
37. Waldenström H. On the formation and disappearance of amyloid in man. Acta Chir Scand 1928; 63 : 479 – 530.
38. Wechalekar AD, Merlini G, Gillmore JD et al. Role of NT ‑ ProBNP to assess the adequacy of treatment response in AL amyloidosis. Blood 2008; 112 : 596 – 759.
39. Kyle RA, Gertz MA, Greipp PR et al. A trial of three regimens for primary amyloidosis: colchicine alone, melphalan and prednisone, and melphalan, prednisone, and colchicine. N Engl J Med 1997; 336 : 1202 – 1207.
40. Gertz MA, Kyla RA, Greip PP. Response rates, and survival in primary systemic amyloidosis. Blood 1991; 77 : 257 – 262.
41. Gertz MA, Lacy MQ, Lust JA et al. Prospective randomized trial of melphalan and prednisone versus vincristine, carmustine, melphalan, cyclophosphamide and prednisone in the treatment of primary systemic amyloidosis. J Clin Oncol 1999; 17 : 262 – 267.
42. Dhodapkar MV, Hussein MA, Rasmussen E et al. Clinical efficacy of high‑dose dexamethasone with maintenance dexamethasone/ alpha interferon in patients with primary systemic amyloidosis: results of United States Intergroup Trial Southwest Oncology Group (SWOG) S9628. Blood 2004; 104 : 3520 – 3526.
43. Palladini G, Perfetti V, Obici L et al. Association of melphalan and high‑dose dexamethasone is effective and well tolerated in patients with AL (primary) amyloidosis who are ineligible for stem cell transplantation. Blood 2004; 103 : 2936 – 2938.
44. Goodmann H, Lachmann H, Bradsell AR et al. Intermediate dose melpahalan and dexamethasone treatment in 144 patients with systemic AL ‑ amyloidosis. Blood 2004; 104 : 755 – 759.
45. Goodman H, Wechalekar A, Lachmann H et al. Clonal disease response and clinical outcome in 229 patients with AL ‑ amyloidosis treated with VAD‑like chemotherapy. Hematologica 2008; 90 : 201 – 203.
46. Palladini G, Anesi E, Perfetti V et al. A modified high‑dose dexamethasone regimen for primary systemic (AL) amyloidosis. Br J Haematol 2001; 113 : 1044 – 1046.
47. Palladini G, Russo P, Nuvolone M et al. Treatment with oral melphalan plus dexamethasone produces long‑term remissions in AL amyloidosis. Blood 2007; 110 : 787 – 788.
48. Palladini G, Russo P, Foli A et al. Treatment with oral melphalan and dexamethasone in an extended population with AL amyloidosis. Blood 2009; 114 (Suppl N22): 1496. Abstract 3889.
49. Sanchorawala V, Seldin DC, Sloan JM et al. Oral cyclic melphalan and dexametasone treatment in the treatment of patients with high dose amyloidosis, ineligible for high dose melphalan and stem cell transplantation. Blood 2009; 114 (Suppl N22): 748. Abstract 1883.
50. Dispenzieri A, Lacy MQ, Rajkumar SV et al. Poor tolerance to high dose thalidomide in patients with light chain associated (AL) amyloidosis. Amyloid 2003; 10 : 257 – 261.
51. Dispenzieri A, Lacy MQ, Geyer SM et al. Low dose single agent thalidomide is tolerated in patients with primary amyloidosis, but responses are limited. Blood 2004; 104. Abstract 4920.
52. Palladinini G, Perfetti V, Perlini S et al. The combination of thalidomide and intermediate‑dose dexamethasone is an effective but toxic treatment for patients with primary amyloidosis (AL). Blood 2005; 105 : 2949 – 2951.
53. Wechalekar AD, Goodman HJ, Lachmann HJ et al. Safety and efficacy of risk‑adapted cyclophosphamide, thalidomide, and dexamethasone in systemic AL amyloidosis. Blood 2007; 109 : 457 – 464.
54. Gibbs SDJ, Sattianayagam PT, Lachmann H et al. Risk adapted cyclophosphamide, thalidomide and dexamethasone (CTD) for treatment of systemic AL ‑ amyloidosis: long term outcome of 202 Patients. Blood 2008; 112 : 611. Abstract 1733.
55. Gibbs SD, Gillmore J, Satttianayagam PT et al. AL ‑ amyloidosis both oral melphalan and dexametzasone and risk adapted cyclophosphamide, thalidomide, and dexamethasone (CTD) have similar efficacy as upfront treatment. Blood 2009; 114 (Suppl N22): 310. Abstract 745.
56. Gillmore J, Cocks K, Gibbs DJB et al. Cyclophosphamide, thalidomide and dexamethasone (CTD) versus melphalan and dexamethasone (MD) for newly diagnosed systemic AL amyloidosis. Result from the UK amyloidosis treatment trial. Blood 2009; 114 (Suppl N22): 1120. Abstract 2869.
57. Seldin DC, Choufani EB, Dember LM et al. Tolerability and efficacy of thalidomide for the treatment of light chain associated (AL) amyloidosis. Clin Lymphoma 2003; 3 : 241 – 246.
58. Cohen AD, Zhou P, Chou J et al. Risk‑adapted autologous stem cell transplantation with adjuvant dexamethasone ± thalidomide for systemic light‑chain amyloidosis: results of a phase II trial. Br J Haematol 2007; 139 : 224 – 233.
59. Kastritis E, Anagnostopoulos A, Rousnou M et al. Treatment of light chain deposition disease with the combination of bortezomibe and dexamethasone. Blood 2007; 110 (Suppl N11). Abstract 64.
60. Kastritis E, Anagnostopoulos A, Roussou M et al. Treatment of light chain (AL) amyloidosis with the combination of bortezomib and dexamethasone. Haematologica 2007; 92 : 1351 – 1358.
61. Sitia R, Palladini G, Merlini G. Bortezomib in the treatment of AL amyloidosis: targeted therapy? Haematologica 2007; 92 : 1302 – 1307.
62. Reece D, Hegenbart U, Merlini G et al. Weekly and twice‑weekly bortezomib in patients with systemic AL ‑ amyloidosis: results of a phase 1 dose‑escalation study. Blood 2009; 114 : 1489 – 1497.
63. Wechalekar AD, Lachmann HJ, Offer M et al. Efficacy of bortezomib in systemic AL amyloidosis with relapsed/ refractory clonal disease. Haematologica 2008; 93 : 295 – 298.
64. Kastritis E, Wechalekar AD, Dimopoulos C et al. Significant activity of bortezomib‑based therapy in patients with primary systemic AL ‑ amyloidosis. Blood 2008; 112 : 321. Abstract 869.
65. Zonder JA, Sanchorawala V, Snyder RM et al. Melphalan, dexamethosone plus bortezomib induces hematologic and organ response in AL ‑ amylolidosis with tolerable neurotoxicity. Blood 2009; 114 (Suppl N22): 310. Abstract 746.
66. Jimenez ‑ Zepeda VH, Reeder CB, Mikhael JR et al. Cyclophophamide, bortezomib and dexamethasone induces rapid and complete response in patients with amyloidosis non eligible for peripheral stem cell transplant. Blood 2009; 114 (Suppl N22): 737. Abstract 1857.
67. Singh V, Saad A, Palmer J et al. Response to bortezomib based induction therapy in newly diagnosed light chain (AL) amyloidosis. Blood 2009; 114 (Suppl N22): 740. Abstract 1867.
68. Lamm W, Willenbacher W, Zojer N et al. Efficacy of the combination of bortezomib and dexamethasone in systemic AL amyloidosis. Blood 2009; 114 (Suppl N22): 1121. Abstract 2871.
69. Sanchorawala V, Wright DG, Rosenzweig M et al. Lenalidomide and dexamethasone in the treatment of AL amyloidosis: results of a phase 2 trial. Blood 2007; 109 : 492 – 496.
70. Dispenzieri A, Lacy MQ, Zeldenrust Set al. The activity of lenalidomide with or without dexamethasone in patients with primary systemic amyloidosis. Blood 2007; 109 : 465 – 470.
71. Dispenzieri L, Lacy M, Zeldenrust SR et al. Long term follow up of patients with immunoglobulin light chain amyloidosis treated with lenalidomide and dexamethasone. Blood 2008; 112 : 612. Abstract 1737.
72. Kastritis E, Zahoura F, Rousnou M et al. Phase I/ II study of lenalidomid, intermediate dose of dexamethasone and low dose cyclophosphamide for the treatment of AL ‑ amyloidosis. Blood 2008; 112 : 611. Abstract 1734.
73. Schoenland SO, Bochler T, Ditrich S et al. Single center experience of lenalidomide/ dexametazone treatment in 40 patients with light chain amyloidosis. High toxicity in patients with impaired renal and cardiac function. Blood 2008; 112 : 612. Abstract 1736.
74. Moreau P, Jaccard A, Benboubker L et al. Lenalidomide with melphalan and dexamethasone in patients with newly diagnosed light chain amyloidosis. A multicenter phase I/ II dose escalation study. Blood 2009; 114 (Suppl N22): 177. Abstract 427.
75. Kastritis E, Roussou M, Migkou M et al. A phase I/ II study of lenalidomide with low dose dexamethasone and cyclophosphamide for patients with primary systemic light chain amyloidosis. Blood 2009; 114 (Suppl N22): 177. Abstract 428.
76. Sloan JM, Sanchowarala V, Girnius S et al. Melphalan, lenalidomide and dexamethasone combination therapy in patients with AL amyloidosis. Blood 2009; 114 (Suppl N22): 737. Abstract 1859.
77. Palladini G, Russo P, Bragotti LZ et al. A phase II trial of cyclophosphamide, lenalidomide and dexamethasone in previously treated patients with AL ‑ amyloidosis. Blood 2009; 114 (Suppl N22): 1117. Abstract 2863.
78. Kumar S, Hayman SR, Buaudi F et al. A phase II trial of lenalidomide, cyclophosphamide and dexamethasone in patients with light chain amyloidosis. Blood 2009; 114 (Suppl N22): 1482. Abstract 3853.
79. Sanchorawala V, Skinner M, Quillen K et al. Long‑term outcome of patients with AL amyloidosis treated with melphalan and stem cell transplantation. Blood 2007; 110 : 3561 – 3563.
80. Gertz MA, Lacy MQ, Dispenzieri A et al. Effect of hematologic response on outcome of patients undergoing transplantation for primary amyloidosis: importance of achieving a complete response. Haematologica 2007; 92 : 1415 – 1418.
81. Vesole DH, Perez WS, Akasheh M et al. High‑dose therapy and autologous hematopoietic stem cell transplantation for patients with primary systemic amyloidosis: A Center for International Blood and Marrow Transplant Research Study. Mayo Clin Proc 2006; 81 : 880 – 888.
82. Dispenzieri A, Gertz MA, Kyle RA et al. Prognostication of survival using cardiac troponins and N‑terminal pro‑brain natriuretic peptide in patients with primary systemic amyloidosis undergoing peripheral blood stem cell transplantation. Blood 2004; 104 : 1881 – 1887.
83. Dispenzieri A, Kyle RA, Gertz MA et al. Survival in patients with primary systemic amyloidosis and raised serum cardiac troponins. Lancet 2003; 361 : 1787 – 1789.
84. Gertz MA, Lacy M, Dispenzieri A et al. Troponin T level as an exclusion criterion for stem cell transplantation in light‑chain amyloidosis. Leuk Lymphoma 2008; 49 : 36 – 41.
85. Skinner M, Sanchorawala V, Seldin DC et al. High‑dose melphalan and autologous stem ‑ cell transplantation in patients with AL amyloidosis: an 8‑year study. Ann Intern Med 2004; 140 : 85 – 93.
86. Yagi S, Akaike M, Ozaki S et al. Improvement of cardiac diastolic function and prognosis after autologous peripheral blood stem cell transplantation in AL ‑ cardiac amyloidosis. Intern Med 2007; 46 : 1705 – 1710.
87. Perfetti V, Siena S, Palladini G et al. Long‑term results of risk adapted approach to melphalan conditioning in autologous peripheral blood stem cell transplantation for primary AL amyloidosis. Hematologica 2006; 91 : 1635 – 1643.
88. Dispenzieri A, Kyle RA, Lacy MQ et al. Superior survival in primary systemic amyloidosis patients undergoing peripheral stem cell transplantation. A case control study. Blood 2004; 103 : 2949 – 2951.
89. Goldsmith YB, Liu J, Chou J et al. Frequencies and types of arrhythmias in patients with systemic light-chain amyloidosis with cardiac involvement undergoing stem cell transplantation on telemetry monitoring Am J Cardiol 2009; 104 : 990–994.
90. Gertz MA, Lacy MQ, Dispenzieri A et al. Risk‑adjusted manipulation of melphalan dose before stem cell transplantation in patients with amyloidosis is associated with a lower response rate. Bone Marrow Transplant 2004; 34 : 1025 – 1031.
91. Mhaskar R, Kumar A, Behera M et al. Role of high-dose chemotherapy and autologous hematopoietic cell transplantation in primary systemic amyloidosis: a systematic review. Biol Blood Marrow Transplant 2009; 15 : 893–902.
92. Jaccard A, Moreau P, Leblond V et al. High‑dose melphalan versus melphalan plus dexamethasone for AL amyloidosis. N Engl J Med 2007; 357 : 1083 – 1093.
93. Leung N, Gunderson H, Tan TS et al. Malphalan and dexamethason is less effective for patients with immunoglobulin light chain amyloidosis with high bone marrow plasmocytosis. Blood 2008; 112 : 612. Abstract 1735.
94. Sanchorawala V, Seldin DC, Magnani Bet al. Serum free light‑chain responses after high‑dose intravenous melphalan and autologous stem cell transplantation for AL (primary) amyloidosis. Bone Marrow Transplant 2005; 36 : 597 – 600.
95. Madan S, Kumar S, Dispenzieri A et al. Outcomes with high dose therapy and peripheral blood stem cell transplantation for light chain amyloidosis with cardiac involvement. Blood 2009; 114 (Suppl N22): 223. Abstract 534.
96. Landau H, Hassoun H, Cohen AD et al. Adjuvant bortezomib and dexamethasone following risk adapted melphalan and stem cell transplant in systemic light chain amyloidosis. Blood 2009; 114 (Suppl N22): 222. Abstract 533.
97. Landau HJ, Hoffman J, Hassoun H et al. Adjuvant bortezomib and dexamethasone following risk‑adapted melphalan and stem cell transplant in patients with light‑chain amyloidosis (AL). J Clin Oncol 2009; 27 : 15. Abstract 8540.
98. Hoffman JE, Hassoun H, Landau H et al. Early adjuvant treatment after risk adapted autologou stem cell transplant for systemic light chain amyloidosis. Increased hospitalization and impaired immune recovery but improved responses and overall survival. Blood 2008; 112 : 1143. Abstract 3329.
Štítky
Diabetologie Endokrinologie Interní lékařstvíČlánek vyšel v časopise
Vnitřní lékařství

2010 Číslo 3
- Diosmin a hesperidin: Co ukazuje farmakokinetika?
- Omeprazol a mechanismus hojení krvácení z peptického vředu
- Porovnání farmakologických vlastností mikronizovaného diosminu a hesperidinu v terapii chronické žilní insuficience a hemoroidů
- Srovnání vlivu omeprazolu a pantoprazolu na antiagregační účinek klopidogrelu
Nejčtenější v tomto čísle
- Příspěvek k diferenciální diagnostice chronických břišních bolestí
- Pretrvávajúce symptómy, diastolická dysfunkcia a nízka koronárna rezerva u pacientky po úspešnej korekcii rekoarktácie aorty
- Léčba dospělých pacientů s akutní lymfoblastickou leukemií dle protokolu GMALL 07/ 2003 a její výsledky – první zkušenosti v České republice
- Nealkoholová steatóza a steatohepatitida – editorial
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy