
Poškození ledvin při mnohočetném myelomu a dalších monoklonálních gamapatiích
Kidney damage in multiple myeloma and other monoclonal gammopathies
Multiple myeloma typically damages the skeleton in the form of osteolytic lesions or diffuse osteoporosis and causes a decrease in blood production. Renal insufficiency is diagnosed immediately at the onset of illness when establishing diagnosis in up to 20% of patients. Where patients suffer from an advanced form of the illness, it occurs in up to 40%. The predominant cause of damage to the kidneys is the monoclonal light chains. Most frequently, nephropathy is caused by the precipitation of light chains with the Tamm-Horsfall protein in the distal part of the loop of Henle and subsequent tubular ruptures and the creation of fibrous changes in the interstitium. Less frequently, there is clinically serious damage to tubular functions without indication of renal insufficiency. In some patients monoclonal immunoglobulin induces changes in the glomeruli. A rare type of damage is deposits of light chains in the form of AL-amyloid and subsequent nephritic syndrome. A very rare form is the deposition of monoclonal immunoglobulin in the form of amorphous matter (light-chain deposition disease) or in the form of crystals within tissue histiocytes (crystal storing histiocytosis). Both of these disorders cause renal insufficiency and less frequently nephritic syndrome such as AL amyloidosis. With timely and intensive treatment of multiple myeloma, which quickly suppresses the creation of light chains, a significant proportion of patients experience reparative changes and improved kidney function. The benefit of plasmapheresis for patients with severe kidney damage has not been confirmed by randomised studies. At the present time the first positive results are becoming available from tests of the use of pre-emptive haemodialysis with special columns that are permeable for light chains. The aim of the text is to provide information on the various forms of nephropathy whose closer analysis can reveal multiple myeloma and contribute to the timely diagnosis of the cause of the nephropathy.
Key words:
multiple myeloma – myeloma kidney – cast nephropathy – AL-amyloidosis – light-chain deposition disease – crystal storing histiocytosis – bortezomib – thalidomid – plazmaferesis
Autoři:
Z. Adam 1; L. Pour 1; M. Krejčí 1; S. Štěpánková 2; I. Svobodová 3; K. Veselý 3; R. Hájek 1
Působiště autorů:
Interní hematoonkologická klinika Lékařské fakulty MU a FN Brno, pracoviště Bohunice, přednosta prof. MUDr. Jiří Vorlíček, CSc.
1; Interní gastroenterologická klinika Lékařské fakulty MU a FN Brno, pracoviště Bohunice, přednosta prof. MUDr. Jan Lata, CSc.
2; I. patologicko‑anatomický ústav Lékařské fakulty MU a FN u sv. Anny Brno, přednostka doc. MUDr. Markéta Hermanová, CSc.
3
Vyšlo v časopise:
Vnitř Lék 2008; 54(9): 847-861
Kategorie:
Přehledný referát
Souhrn
Mnohočetný myelom typicky poškozuje skelet ve formě osteolytických ložisek či difuzní osteoporózy a způsobuje útlum krvetvorby. Renální insuficience je diagnostikována ihned v počátku nemoci, při stanovení diagnózy, až u 20 % nemocných. U pacientů s pokročilou chorobou se vyskytuje až u 40 %. Příčinou poškození ledvin jsou dominantně monoklonální lehké řetězce. Nejčastěji je nefropatie způsobena precipitací lehkých řetězců s Tammovým-Horsfalovým proteinem v distální části Henleovy kličky s následnou rupturou tubulů a vzniku fibrózních změn v intersticiu. Méně časté je klinicky závažné poškození tubulárních funkcí bez známek renální insuficience. U ně-kte-rých nemocných indukuje monoklonální imunoglobulin změny v glomerulech. Vzácným typem poškození jsou depozita lehkých řetězců ve formě AL-amyloidu s následným nefrotickým syndromem. Výjimečnou formou je ukládání monoklonálního imunoglobulinu ve formě amorfních hmot (light-chain deposition disease), případně ve formě krystalů střádaných ve tkáňových histiocytech (crystal storing histiocytosis). Obě tyto poruchy způsobují renální insuficienci a již ne tak často nefrotický syndrom jako AL-amyloidóza. Při časné a intenzivní léčbě mnohočetného myelomu, která rychle utlumí tvorbu lehkých řetězců, dochází u značné části nemocných k reparativním změnám a ke zlepšení funkce ledvin. Přínos plazmaferézy pro nemocné se závažným poškozením ledvin nebyl potvrzen randomizovanou studií. V současnosti jsou k dispozici první pozitivní výsledky testování preemptivní hemodialýzy na speciálních kolonách, které propouštějí lehké řetězce. Cílem textu je upozornit na jednotlivé formy nefropatií, při jejichž bližší analýze lze odhalit mnohočetný myelom, a přispět tak k časné diagnóze příčiny nefropatie.
Klíčová slova:
mnohočetný myelom – myelomová ledvina – odlitková nefropatie – AL‑amyloidosis – nemoc z depozit lehkých řetězců – histiocytóza se střádáním krystalů – bortezomib – thalidomid – plazmaferéza
Úvod
Poškození ledvin s renální insuficiencí anebo s nefrotickým syndromem je důsledkem chorob, které poškozují specificky ledviny (glomerulonefritis, intersticiální nefritis, jiné formy autoimunitních chorob s poškozením ledvin) anebo důsledkem jiných interních nemocí (cukrovka, hypertenze). Po těchto nemocech se standardně pátrá, zjistí‑li lékař u pacienta stoupající hodnoty urey a kreatininu nebo proteinurie. Nicméně poškození ledvin může být i prvním symptomem hematologického onemocnění produkujícího monoklonální imunoglobulin, nejčastěji mnohočetného myelomu.
Mnohočetný myelom je choroba, při níž v kostní dřeni dochází k nekontrolované proliferaci a akumulaci maligních plazmocytů, které způsobují osteolýzu, útlum fyziologické krvetvorby a které nejčastěji produkují kompletní molekulu monoklonálního imunoglobulinu, méně často produkují pouze lehké řetězce (v moči prokazatelné jako Bence-Jonesova bílkovina). Zcela výjimečně jsou plazmocyty tak nediferencované, že netvoří žádné monoklonální imunoglobuliny. Incidence mnohočetného myelomu v ČR je 4/100 000 obyvatel. Nemoc se vyskytuje hlavně u osob starších 60 let. Poškození ledvin s retencí dusíkatých látek v počátku nemoci není časté, řádově do 10 % všech nově diagnostikovaných nemocných [1].
S pokročilostí nemoci narůstá výskyt renální insuficience, u pokročilých forem, opakovaně relabujících po léčbě, postihuje až 40 % nemocných.
Při hodnocení velkého souboru pacientů v M. D. Anderson Cancer Center nalezli tříprocentní incidenci renální insuficience (kreatinin nad 2 mg/ml čili 176 μmol/l) mezi pacienty v počáteční fázi nemoci, a tedy s nevelkou masou myelomových buněk, zatímco ve skupině s pokročilým onemocněním mělo koncentraci kreatininu nad 176 μmol/l již 40 % pacientů [2].
Dle statistických dat z Oxfordu 2 % ze všech pacientů v chronickém dialyzačním programu představují nemocní s poškozením ledvin monoklonální gamapatií [3–5].
V rámci diferenciální diagnostiky renální insuficience či v rámci diferenciální diagnostiky proteinurie se obvykle pátrá po častějších příčinách. Souvislost s monoklonální gamapatií zůstává často zpočátku neodhalena. Diagnóza mnohočetného myelomu je proto mnohdy stanovována relativně pozdě, až v době těžkého orgánového poškození, patologické fraktury skeletu anebo těžkého poškození ledvin s nutností pravidelné dialýzy.
Proto Česká myelomová skupina zorganizovala edukační akci nazvanou CRAB s cílem edukačními akcemi přispět k časnějšímu stanovení diagnózy mnohočetného myelomu a dalších monoklonálních gamapatií. Toto přehledové sdělení je součástí edukačního projektu CRAB České myelomové skupiny.
Definice monoklonální gamapatie
Termín monoklonální gamapatie lze použít jak ve smyslu čistě biochemickém, kdy se tímto termínem označuje přítomnost monoklonálního imuno-globulinu v séru, tak ve smyslu klinickém, kdy se tímto termínem označuje skupina benigních a maligních nemocí spojených s plazmocelulární proliferací.
Benigní proliferace plazmatických buněk je podstatou asyptomatického stavu, označovaného jako monoklonální gamapatie nejistého významu – monoclonal gammopathy of unknown (undetermined) significance. Slovem unknown nebo undetermined je vyjádřeno, že prognózu a vývoj benigní plazmocelulární proliferace nelze odhadnout, u části pacientů dojde k přechodu do maligní plazmocelulární proliferace. Pokud je monoklonální imunoglobulin tvořen benigní plazmocelulární proliferací a přitom vyvolává specifické symptomy, označují se tyto stavy dle přítomných symptomů (kryoglobulinemie, nemoc chladových aglutininů).
Z maligních jednotek je nejčastější příčinou tvorby monoklonálního imunoglobulinu mnohočetný myelom. Přibližně 10krát nižší incidenci mají další nemoci, které jsou pravidelně spojené s tvorbou monoklonálního imunoglobulinu – Waldenströmova makroglobulinemie a primární systémová AL‑amyloidóza. Vzácně a v nízkých koncentracích provází monoklonální imunoglobulin i jiné maligní lymfoproliferativní nemoci [6].
Monoklonální imunoglobulin může poškozovat organizmus následujícími způsoby:
- v závislosti na jeho koncentraci a na fyzikálních a chemických vlastnostech vznikají příznaky hyperviskozity
- vlivem strukturálních aberací molekul jsou narušeny cesty jeho metabolizmu, molekula přestává být jednoduše rozložitelnou hydrolytickými enzymy a dochází ke vzniku depozit různé morfologie v orgánech, a tím k jejich poškození
- mnohé nežádoucí účinky monoklonálního imunoglobulinu jsou však způsobeny jeho vazbou na ně-kte-ré struktury (antigeny) vlastního těla.
Vedle nefropatie jsou nejčastějšími nežádoucími účinky monoklonálního imunoglobulinu:
- hypo-, ale i hyperkoagulopatie vlivem vazby na trombocyty, ale i na základě ovlivnění koagulačních faktorů monoklonálním imunoglobulinem
- neuropatie, vznikající na základně vazby monoklonálního imunoglobulinu na struktury nervové tkáně
- kryoglobulinemie, nemoc chladových aglutininů
- hyperviskozita (krvácení ze sliznic, neurologické poruchy)
- AL‑amyloidóza a jiné formy poškození organizmu depozity monoklonálního imu-noglobulinu
Principy vzniku nefropatie u mnohočetného myelomu
Monoklonální imunoglobulin nejčastěji poškozuje tubuly nefronu, vzácně může poškodit ledviny na podkladě vaskulitidy anebo poškodit glomeruly podobně jako některé formy glomerulo-nefritid.
Patofyziologie poškození ledvin monoklonálním imunoglobulinem závisí zcela na vlastnostech monoklonálního lehkého řetězce. Tato souvislost byla prokázána na pokusech na myších, jimž byly podány nitrožilně monoklonální lehké řetězce, izolované od pacienta s monoklonální gamapatií a nefropatií. Následně byly histologicky vyšetřeny ledviny těchto myší a výsledky srovnány s bioptickými nálezy od nemocných osob. Histologické typy poškození ledvin u myší a u lidí se shodovaly [7].
Poškození ledvin u mnohočetného myelomu lze rozdělit zhruba do šesti kategorií:
- postižení proximálního a distálního tubulu se vznikem odlitkové nefropatie – cast nephropathy
- depozita lehkých řetězců ve formě amyloidu
- depozita lehkých řetězců v neamyloidové podobě (light chain deposition diesease)
- proliferativní glomerulonefritida, způsobená depozicí kompletní mo-lekuly monoklonálního imunoglobulinu v glomerulech
- kryoglobulinemická glomerulonefritida
- pyelonefritis při zvýšené vnímavosti pacientů k infekcím
Protože je velká variabilita ve vlastnostech monoklonálního imunoglobulinu, existuje také variabilita v rámci každé z uvedených skupin [8].
Nejčastějším typem renálního poškození je akutní tubulární nekróza podmíněná odlitkovou nefropatií, zatímco depozita monoklonálního imunoglobulinu v amyloidové či neamyloidové podobě jsou podstatně vzácnější.
Údaje o incidenci jednotlivých typů poškození se liší dle definice studovaných sekčních souborů a dle indikací k biopsii ledvin, pokud šlo o bioptické studie.
Autoptické studie popisují u osob zemřelých v důsledku mnohočetnéhomyelomu odlitkovou nefropatii u 30 až50 %, depozita lehkých řetězců v neamyloidové podobě u 2–3 %, depozita lehkých řetězců v amyloidové podobě ve 4–5 % [9].
Bioptické studie uvádějí u pacientů s monoklonální gamapatií a nefropatií odlitkovou nefropatii u 40–63 % vyšetřených, depozita lehkých řetězců ve formě amyloidu u 19–26 %, depozita lehkých řetězců v neamyloidové podobě u 7–30 % a méně než 1 % nález kryoglobulinemické glomerulonefritidy [10,11]. Údaje o četnosti však v jednotlivých pracích kolísají (tab. 1), charakteristiku jednotlivých typů nefropatií uvádí tab. 2.
![Časté a méně časté formy nefropatií při monoklonálních gamapatiích a jejich sekční frekvence [49].](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/878c80c7c9f1f6fec0e7b9d87a39a032.png)
![Poruchy spojené s depozity monoklonálního imunoglobulinu [50].](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/1b663d5794e6bc2b54b23a33de4b43c6.png)
Klinický obraz postižení ledvin při monoklonální gamapatii
Akutní selhání ledvin
Akutní selhání ledvin (obvykle s normálním objemem moče anebo oligurií a výjimečně anurií) může být vůbec první manifestací mnohočetného myelomu. Udávané počty případů se v řadě studií liší (5–10 %, někteří autoři až 30 %). Symptomy provázející akutní renální selhání jsou často důvodem, proč nemocný člověk přijde k lékaři a následná diferenciálně diagnostická vyšetření odhalí mnohočetný myelom.
Příčina rozvoje akutního selhání ledvin u pacienta, u něhož jsou lehké řetězce vylučovány po delší dobu, má obvykle svoje zevní či vnitřní příčiny:
- náhlá progrese nemoci se zvětšující se náloží lehkých řetězců
- hyperkalcemie, která sama o sobě poškozuje tubuly a která dále zvyšuje nefrotoxicitu lehkých řetězců
- dehydratace v souvislosti s jinými zdravotními komplikacemi či operací
- excesivní podávání kličkových diuretik s následujícím zmenšením extracelulárního objemu s poklesem glomerulární filtrace
- hyperurikemie
- sepse – zvýšená nálož katabolitů akceleruje tvorbu odlitkových válců
- nesteroidní antirevmatika, tvořící základ analgetické léčby pacientů s kostními bolestmi
- podání nefrotoxických léků – aminoglykozidová antibiotika
- přímá i nepřímá nefrotoxicita cytostatické léčby
- močová infekce, zejména při současné dehydrataci [2,4–6,12–17]
Chronická renální insuficience
Tato forma může nezřídka předcházet stanovení diagnózy mnohočetného myelomu [18,19]. Pacienti opět mívají různě intenzivní proteinurii a laboratorní známky již výraznější retence dusíkatých látek.
Podrobnějším vyšetřením typu proteinurie zjistíme přímo Bence-Jonesovu bílkovinu. Pokud jsou monoklonálním imunoglobulinem poškozeny glomeruly, mívají tito pacienti často neselektivní proteinurii [15,20–23].
Proteinurie bez klinicky významné renální insuficience a bez nefrotického syndromu
Tato forma postižení je charakterizována především proteinurií, která je u 70 % pacientů asymptomatická. Nejčastěji prokážeme Bence-Jonesovu bílkovinu. Tíže proteinurie je různá a může dosáhnout až hodnot několika gramů lehkých řetězců [24]. Po provedení odpovídajících vyšetření byly popsány i u těchto nemocných poruchy tubulárních funkcí – koncentrační, acidifikační až do obrazu renální tubulární acidózy nebo kompletního Fanconiho syndromu. Ve většině případů však tubulární poruchy nebývají klinicky významné. Tito nemocní mívají po řadu let jen nepatrné snížení hodnot clearence kreatininu a žádné či nepatrné zvýšení hodnot kreatininu v séru. Podle některých autorů je postupný pokles renálních funkcí v přímé souvislosti s množstvím vylučovaných lehkých řetězců [25].
Nefrotický syndrom
Typickým klinickým projevem jsou symetrické otoky nohou. Typickými laboratorními nálezy je výrazná proteinurie, hypoalbuminemie a hypoproteinemie, snížený onkotický tlak. Hodnota kreatininu v séru může, ale nemusí být zvýšená.
Příčinou nefrotického syndromu bývá nejčastěji diabetická nefropatie nebo některé formy glomerulonefritidy, nesmí se však zapomínat na to, že nefrotický syndrom může také být důsledkem AL‑amyloidózy či mnohočetného myelomu. Klinickým obrazem ani biochemickými nálezy nelze stanovit etiologickou příčinu, tu je možné rozpoznat jedině biopsií ledviny.
Patofyziologie a klinické projevy tubulárního poškození monoklonálním imunoglobulinem
Ledviny jsou hlavním místem katabolizmu lehkých řetězců imunoglobulinů. Za fyziologických okolností je v krvi prokazatelné malé množství volných polyklonálních lehkých řetězců imunoglobulinů ve formě monomerů s molekulární hmotností kolem 22 kDa nebo ve formě dimerů.
Metabolizmus těchto lehkých řetězců závisí zcela na glomerulární filtraci, snížení glomerulární filtrace způsobí zvýšení koncentrace volných lehkých řetězců v krvi. Úbytek počtu nefronů vede ke kompenzačnímu zvýšení filtrace, a tím i ke zvýšení nálože volných řetězců v ostatních nefronech, což zvyšuje nefrotoxický potenciál lehkých řetězců.
Lehké řetězce díky malé molekule volně procházejí přes glomerulus do proximálního tubulu. Zde se dostávají do kontaktu s membránou tubulárních buněk, na které jsou umístěny receptory, skládající se z tzv. megalinu a cubilinu [26–29]. Po navázání na tyto receptory se dostávají volné lehké řetězce do nitra proximálních tubulárních buněk mechanizmem endocytózy. Snížení pH (okyselení) způsobí oddělení ligandu a receptoru uvnitř buňky. Receptory recyklují zpět na buněčnou membránu, kde čekají na další náklad, aby jej dopravily znovu do nitra buňky. Lysozymální enzymy pak hydrolyzují lehké řetězce na aminokyseliny, které se pak dostávají do oběhu. V tomto malém, fyziologickém množství, fyziologické volné lehké řetězce funkci ledvin neovlivňují. Fyziologický stav znázorňuje obr. 1.
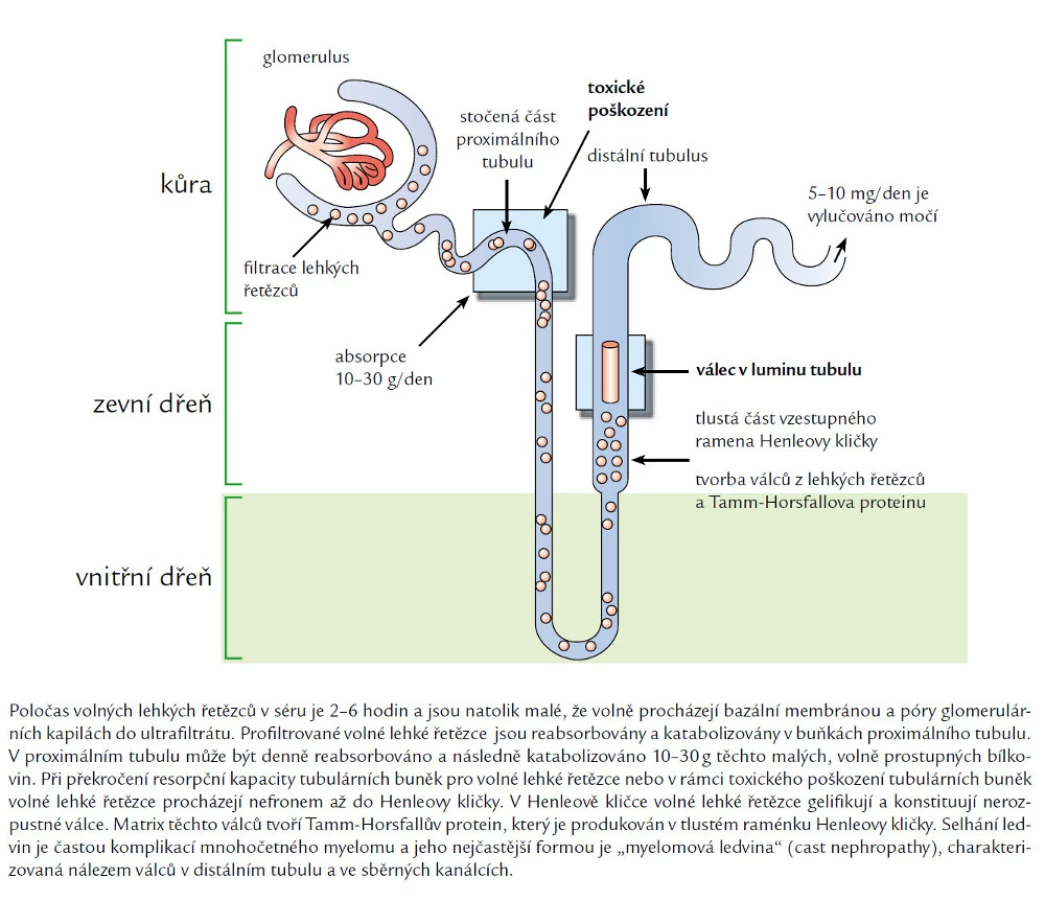
Nicméně transportní mechanizmus má svoje limity a saturace multiligandového endocytického receptorového komplexu vede k tomu, že se volné lehké řetězce dostávají do distálnějších částí nefronu a do moče.
V případě monoklonální gamapatie, kdy je produkováno větší množství monoklonálních lehkých řetězců, může docházet k poškození funkce proximálních tubulů v různém rozsahu. Za patofyziologickou podstatu poškození epitelií proximálních tubulů se považují fyzikálně‑chemické vlastnosti monoklonálních lehkých řetězců, díky nimž dochází k agregaci lehkých řetězců po endocytóze a tvorbě takových prostorových forem lehkých řetězců, které jsou obtížně hydrolyzovatelné. Proto do-chází k distenzi endolysozomálního systému, k akumulaci nehydrolyzovatelných monoklonálních lehkých řetězců v buňkách, což způsobuje funkční a morfologické změny těchto buněk. Důvod, proč fyziologické lehké řetězce nepoškozují proximální tubuly, zatímco monoklonální ano, je zřejmě dán mutací genu lehkého řetězce, která vede k diskrétním změnám v molekule těchto řetězců [30].
Pokud se u tohoto typu poškození provede histologické vyšetření, je patrné poškození proximálních tubulů způsobené depozity lehkých řetězců v tubulárních buňkách, charakteristickou tubulární změnou je homogenní rozšíření bazálních membrán neatrofických tubulů. Dále bývají pomocí imunofluorescenčních nebo imunope-roxidázových metod prokazována depozita lehkých řetězců podél bazálních membrán a v intersticiu. Dále jsou popisovány různě těžké změny: atrofie kartáčkového lemu, vakuolizace buněk, nekrózy a deskvamace tubulárních buněk apod.
U minority případů lze při bioptickém vyšetření prokázat v cytoplazmě proximálních tubulárních epitelií krystalické inkluze při vyšetření konvenční optickou mikroskopií. Pomocí imuno-fluorescence lze prokázat restrikci lehkých řetězců v proximálních tubulech, elektronově mikroskopickým vyšetřením lze opět u části postižených prokázat krystaloidní intracytoplazmatické struktury [31].
Endocytóza strukturálně změněných volných lehkých řetězců, které nepodléhají tak snadno enzymatické hydrolýze jako fyziologické lehké řetězce, je hlavním mechanizmem vzniku tubulárních poruch. Ve většině případů však tyto funkční tubulární poruchy, defekty v acidifikaci a koncentraci, nejsou klinicky manifestní a jen zcela výjimečně se vyvine kompletní forma získaného Fanconiho syndromu.
Fanconiho syndrom
Je charakterizován renální tubulární acidózou typu II, defektními transportními procesy spřaženými s transportem sodíku, které vedou k aminoacidurii, glykozurii, hyperchloremické metabolické acidóze, hypokalemii, tubulární proteinurii a hypourikemii. Hlavní klinickou manifestací je pak osteomalacie, polyurie, chronická acidóza, epizody dehydrace a přechod do renální insuficience. Biopsie ledvin v těchto případech popisuje typické krystalické či fibrilární inkluze v cytoplazmě buněk proximálních tubulů, tvořené lehkými řetězci, i když u části pacientů mohou jakékoli krystalické formace chybět, dokonce i při užití elektronové mikroskopie. U části pacientů s plně vyvinutým Fanconiho syndromem může být při bioptickém vyšetření přítomna typická odlitková nefropatie [32].
Fanconiho syndrom může ohlašovat mnohočetný myelom. Lehké řetězce, které způsobují tento syndrom, jsou obvykle typu κ a podrodiny κ-1 a mají další aberace, které vedou k jejich krystalizaci. Vznik Fanconiho syndromu záleží nejen na kvalitě těchto řetězců, ale také na jejich kvantitě. Předpokládá se, že při větší kvantitě těchto lehkých řetězců by docházelo k závažnějšímu typu poškození ledvin [33]. Uvádí se, že při plně vyjádřeném Fanconiho syndromu obvykle není přítomna nefropatie z odlitkových válců, z čehož se dedukuje, že lehké řetězce způsobující Fanconiho syndrom mají jiné chemicko‑fyzikální vlastnosti a zřejmě se nevážou na Tammův-Horsfallův protein [34].
Z pohledu hematologa musíme říci, že stanovení diagnózy získaného Fanconiho syndromu u mnohočetného myelomu je zcela výjimečné a je obtížné říci, zda proto, že incidence plně vyjádřeného syndromu je velmi malá, nebo proto, že u mnohočetného myelomu se standardně screeningově neprovádějí odpovídající diagnostické kroky pro stanovení této diagnózy.
Patofyziologie a klinické projevy odlitkové nefropatie s následnou tubulární nekrózou
Patofyziologie a morfologie odlitkové nefropatie
Původní studie dávaly odlitkovou nefro-patii do souvislosti s lehkými řetězci λ, následující práce však nepotvrdily predominanci některého z typu lehkých řetězců u odlitkové nefropatie [10].
Patofyziologickým podkladem vzniku odlitkové nefropatie a akutní tubulární nekrózy je tvorba monoklonálních lehkých řetězců v množství, které vysoce přesahuje vstřebávací vlastnosti proximálního tubulu, takže volné lehké řetězce se volně dostávající do distálních tubulů a sběrných kanálků. Zde se setkávají s Tammovým-Horsfallovým proteinem, vážou se na něj a precipitují ve formě odlitkových válců.
Tammův-Horsfallův protein tvoří nejvýznamnější podíl fyziologicky se vyskytujících bílkovin v moči. Tammův-Horsfallův protein je tvořen v ascendentní části Henleovy kličky a v solubilní formě je detekován v tekutině distálního tubulu a posléze v moči. V pokusech na krysách bylo prokázáno, že zvýšený perorální příjem soli zvyšuje množství Tammova-Horsfallova proteinu v moči [35,36].
Rychlost, s jakou se tvoří odlitkové válce, záleží nejen na koncentraci lehkých řetězců v distálním tubulu, ale také na jejich fyzikálně‑chemických vlast-nostech. Lehké řetězce, vytvářející odlitkovou nefropatii, mají obvykle vyšší izoelektrický bod a častěji jsou typu λ [24]. Nejdůležitější pro vytvoření odlitkového válce je však afinita lehkých řetězců k Tammovu-Horsfallovu proteinu. Bylo zjištěno, že důležitá doména pro vazbu lehkých řetězců na Tammův-Horsfallův protein je CDR3 region imunoglobulinu [36].
Při studiu pomocí in vivo izolované mikroperfuze bylo zjištěno, že odlitkové válce vznikají pouze v distální části nefronu, počínaje silnou částí ascendentního raménka Henleovy kličky. Izolovaná obstrukce nefronu způsobí zvýšení tlaku v proximálních částech nefronu, jejímž důsledkem je atrofie nefronu proximálně od místa obstrukce. Současně dojde ke snížení průtoku krve glomerulem. Zvýšený tlak v nefronu způsobí tubulární rupturu, což pak indukuje vznik intersticiální nefritidy. Četnost odlitkových válců v tubulech popisovaná při vyšetření však není přímo úměrná míře intersticiální fibrózy a tubulární atrofii, ačkoliv vznik odlitkového válce má zásadní roli v patogenezi tohoto typu poškození. Odlitkové válce lze zachytit také při mikroskopickém vyšetření moče (obr. 2) a je možné je prokázat pomocí biopsie ledviny s imunofluorescenčním nebo imunoperoxidázovým průkazem lehkých řetězců, které myelom produkuje (obr. 3). Odlitková nefropatie je ve světelné mikroskopii charakterizována dilatovanými atrofickými renálními tubuly s eozinofilními válci v distálních tubulech. Tyto válce jsou tvořené především monoklonálními lehkými řetězci a Tammovým-Horsfallovým proteinem a dále pak fibrinogenem a albuminem. Makrofágy vytvářejí kolem těchto vícevrstevných válců mnohojaderné buňky (giant cells). Válce, makrofagická a obrovskobuněčná reakce jsou prakticky diagnostické. Intersticiální nefritida s fibrózou obvykle komplikuje tubulární formy poškození ledviny monoklonálním imunoglobulinem. Při imunofluorescenčním vyšetření mohou válce obsahovat oba lehké řetězce z důvodu zachytávání normálních lehkých řetězců, imuno-fluorescenční vyšetření tedy nemusí být přínosné. Tento typ postižení, odliková nefropatie (cast nephropathy), je nejčastější forma poškození ledviny u myelomu neboli „myelomové ledviny“. Míra nevratného poškození funkce ledviny koreluje hlavně s mírou intersticiální fibrózy a tubulární atrofie [10].


Klinický obraz
Pacienti s odlitkovou nefropatií mají obvykle závažnou renální insuficienci. Asi v 50 % se rozvine akutní renální selhání v souvislosti s vlivy, které akcelerují precipitaci (dehydratace, infekce, hyperkalcemie, kontrastní jodové látky či vyšší dávky nesteroidních antiflogistik). A ačkoliv je proteinurie přítomna u všech nemocných, méně než 10 % má nefrotický syndrom. Riziko akutního selhání souvisí s množstvím vylučovaných lehkých řetězců. Renální insuficience byla nalezena u 16 % nemocných s odpadem lehkých řetězců menším než 1 g/24 hod, u 47 % s odpadem mezi 1 a 10 g/24 hod a 63 % s odpadem nad 10 g/24 hod [37]. Diagnózu odlitkové nefropatie lze stanovit pouze histologicky.
Léčba renální insuficience u mnohočetného myelomu, která nejčastěji vzniká právě na základě odlitkové nefropatie
Akutní renální selhání u mnohočetného myelomu má s vysokou pravděpodobností patofyziologický podklad ve výše popsané odlitkové nefropatii. Prvním akutním léčebným opatřením by měla být agresivní hydratace, dosahující diurézy 2–3 litrů denně, alkalizace moče, přerušení podání nesteroidních antiflogistik, pokud byly podávány.
Protimyelomová léčba s co nejrychlejším nástupem léčebné odpovědi
Dále je nutné zahájit co nejdříve antimyelomovou léčbu a preferovat léčbu s rychlým nástupem léčebného účinku, neboli s rychlým poklesem tvorby mo-noklonálního imunoglobulinu. Z hle-diska rychlosti nástupu léčebného účinku jsou mezi léčebnými režimy značné rozdíly. Cílem léčby je co nejrychleji utlumit tvorbu lehkých řetězců, aby mohly nastoupit reparativní změny v ledvině, a tím došlo i ke zlepšení funkce.
Při volbě protimyelomových léků je třeba znát jejich farmakokinetická data při renální insuficienci. Dříve se používal léčebný režim obsahující doxorubicin, vinkristin a dexametazon, tyto léky není nutné redukovat při renálním selhání. V současnosti se jako optimální jeví kombinované léčebné režimy, obsahující bortezomib nebo thalidomid, neboť léčebný efekt – pokles tvorby monoklonálního imunoglobulinu – nastupuje velmi rychle. Bortezomib se také nemusí při renálním selhání redukovat. Možné je zvolit i léčebný režim obsahující thalidomid, jehož dávky se rovněž nemusí redukovat při selhání ledvin [38,39].
Přínos plazmaferézy pro nemocné s myelomovou ledvinou
Otázkou, která byla řešena formou popisů případů [40] a dále 2 menšími studiemi a 1 větší randomizovanou studií, je, zda je přínosné provedení akutní plazmaferézy s cílem snížit nálož monoklonálního imunoglobulinu, a tedy i volných lehkých řetězců. První 2 malé studie obsahovaly nevelký počet nemocných, první studie obsahovala 29 nemocných [41], druhá celkem pouze 21 nemocných [42]. Obě tyto studie popsaly přínos plazmaferézy, leč pro malý počet nemocných neměly výsledky Johnsonovy studie statistickou významnost. Od těchto 2 malých studií se však dříve odvozovalo doporučení plazmaferéz u osob s monoklonální gamapatií a závažným renálním selháním. Teoretickou slabostí této léčebné metody je však skutečnost, že patofyziologicky jsou významné volné lehké řetězce, a nikoliv kompletní molekula monoklonálního imunoglobulinu a že přítomnost volných lehkých řetězců není omezena na intravaskulární objem.
Doposud největší klinickou studii hledající odpověď na otázku, jak dalece pomáhá plazmaferéza k obnovení funkce ledvin, publikoval Clark v roce 2005. Do této studie bylo zařazeno 104 nemocných s nově diagnostikovaným mnohočetným myelomem a s akutním selháním ledvin ze 14 center. Všichni byli léčeni konvenční chemoterapií a jedna skupina měla k této standardní léčbě ještě navíc 5–7 plazmaferéz. Celkem 30 % nemocných vyžadovalo v úvodu dialýzu. V průběhu 6 měsíců byla dialýza ukončena u 36,8 % nemocných v kontrolní skupině a u 41,6 % pacientů podstoupivších opakované plazmaferézy. Nově byla dialýza v průběhu 6 měsíců zahájena u 20 % nemocných v každé skupině. Mezi skupinou s plazmaferézou a skupinou kontrolní nebyl nakonec signifikantní rozdíl v počtu úmrtí, nutnosti chronické dialýzy ani v počtu případů s přetrvávající těžkou renální insuficiencí (58 vs 69 %). Tato studie tedy neprokázala přínos plazmaferézy při současné chemoterapii a případné dialyzační léčbě [43,44].
Odstraňování lehkých řetězců pomocí hemodialýzy
Nová technologie hemodialýzy umožnila odstraňovat z plazmy volné lehké řetězce. Podařilo se to pomocí dialyzační kolony Gambro HCO 1100, která se ze všech testovaných kolon jevila v tomto ohledu nejúčinnější. Podstatou metody je membrána, které propouští přes svoji stěnu i molekuly volných lehkých řetězců. Klinické testy pro-kázaly, že koncentrace volných lehkých řetězců poklesla o 35–70 % v průběhu prvních 2 hod dialýzy, po přerušení dialýzy však docházelo k vyrovnávání koncentrací mezi intravaskulárním a extravaskulárním prostorem, koncentrace lehkých řetězců se opět navýšily, ale již nedosáhly původních hodnot. A tak se při opakovaných dialýzách zároveň snižovala hladina volných lehkých řetězců. U jednoho nemocného se touto metodou podařilo v průběhu 6 týdnů odstranit 1,7 kg volných lehkých řetězců. Hemodialýza s pomocí dialyzační kolony Gambro HCO 1100 umožnila kontinuální bezpečné odstraňování velkých kvant lehkých řetězců. Zda bude mít tento postup klinický přínos, ukážou jedině větší klinické studie [45].
Transplantace ledviny
Transplantace ledvin není standardním postupem pro obvykle nepříznivou prognózu nemocných. Nicméně Korbet [10] uvádí, že v případě kompletní remise trvající déle než 1 rok a nutnosti chronické dialýzy je možné zvažovat transplantaci ledviny. Zásadní však je, že musí být kompletní remise s negativní imunofixací a normálním výsledkem vyšetření volných lehkých řetězců, jinak dojde k odlitkové nefropatii v transplantované ledvině [46]. V ČR se u myelomu s renálním selháním trans---plantace neprovádí, přítomnost ma-ligní nemoci je obecnou kontraindikací, a to nejen pro nedostatek vhodných ledvin. Dlouhodobá imunosupresivní léčba cyklosporinem A a případně kor-tikosteroidy výrazně snižuje T bu-něč-nou imunitu, a tím i protinádoro-vou aktivitu imunitního systému. To může způsobit akceleraci průběhu již přítomného maligního onemocnění a souvisí to se zvýšenou četností maligních onemocnění u osob dlouhodobě užívajících imunosupresivní léky. Nicméně v dnešní době, kdy jsou díky novým lékům dosahovány kompletní remise, někdy i na molekulární úrovni s dlouhodobým trváním, bude možná tato otázka znovu diskutována.
Amyloidóza a amyloidová nefropatie
Amyloidóza není jednotnou nemocí. Jde o název skupiny chorobných stavů, které mají jednu společnou vlastnost, extracelulární ukládání nerozpustných fibrilárních proteinů. Termínem amyloid nazval Virchow v roce 1854 patologický extracelulární materiál v játrech. V následujících letech bylo zjiš-těno, že amyloid lze barvit konžskou červení. V normálním světelném mikroskopu jsou takto obarvené amyloidové hmoty červené, v polarizovaném světle jsou však jasně zelené. Téměř za 100 let po objevení amyloidu byla pomocí elektronového mikroskopu popsána speciální fibrilární struktura amyloidu, která souvisí s jeho histochemickými vlastnostmi.
Všechny druhy amyloidu jsou složeny z lineárních, nerozvětvených fibrilárních proteinů v β konfiguraci. Amyloidová hmota však obsahuje ještě nefibrilární glykoprotein (amyloidovou komponentu P) a glukózaminoglykany.
To, že podstatu některých typů amyloidu tvoří fragmenty lehkých řetězců imunoglobulinů, bylo objeveno v roce 1971. Později se zjistilo, že i jiné proteiny neimunoglobulinové povahy mo-hou vytvářet amyloidové hmoty. To je podstatou reaktivní (sekundární) amyloidózy a familiárních amyloidóz.
Moderní klasifikace amyloidóz je za-ložena na identifikaci proteinů, které tvoří amyloidové fibrily (WHO–IUIS Nomenclature subcomitee, 1993). Plazmatické proteiny vytvářející amyloid jsou velmi různorodé a chemicky nepříbuzné, přesto všechny tvoří amyloidová depozita s typickou β fibrilární strukturou. Chemickou klasifikaci amyloidóz uvádí tab. 3.
![Chemická klasifikace amyloidóz. WHO- IUIS nomenclature subcomittee:
Nomenclature of amyloid and amyloidoses [48].](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/79a0d509f4e54ff524d60d64ed9590aa.png)
Amyloid je v podstatě definován svojí afinitou ke konžské červeni a jakýkoli proteinový materiál s touto afinitou a charakteristickým zeleným dvojlomem v polarizovaném světle je definován jako amyloid. Stejné vlastnosti mají i ně-kte-ré intracelulární bílkovinné agregáty, a proto je pojetí pouze extracelulárního výskytu amyloidu již překonané [47].
Ze všech typů amyloidóz je nejčastější AL‑amyloidóza (AL‑amyloid light chain). AL‑amyloidóza může provázet mnohočetný myelom a méně často i Waldenströmovu makroglobulinemii, pak jde o sekundární AL‑amyloi-dózu. V případě, že maligní lymfocy-tární či plazmocytární proliferace není prokázána, používá se termín primární AL‑amyloidóza. V těchto případech se jedná o benigní klonální proliferaci jako u monoklonální gamapatie nejistého významu, benigní klon plazmocytů však produkuje toxické lehké řetězce, které mají potenciál tak závažně poškodit orgány a tkáně, že nemocný na toto poškození umírá.
Pro nedostupnost údajů z České republiky demonstrujeme incidenci amy-loi-dózy čísly z USA. Incidence pri-mární systémové AL‑amyloidózy je 0,8/100 000 obyvatel. Incidence trans-thyretinové amyloidózy není přesně známa ani v USA, ví se pouze, že se vyskytuje méně často než AL‑amyloidóza. Amyloidóza způsobená ukládáním β-2 mikroglobulinu provází pouze dialyzované pacienty. Objevuje se po 5–10 letech dialyzační léčby a velmi častou se stává po 15 letech.
Patofyziologie amyloidóz
AL‑amyloidóza
Záměna aminokyselin v lehkém řetězci nebo chybění ně-kte-rých aminokyselin destabilizuje lehký řetězec a je příčinou ukládání těchto řetězců ve formě amyloidu. Fibrily AL‑amyloidu jsou tvořeny N‑terminálním fragmentem lehkého řetězce, obsahují jeho variabilní část a zlomek konstantní části. Specifické vlastnosti aberantního lehkého řetězce jsou příčinou, proč je u jednoho pacienta postižen dominantně ten či onen orgán.
AL‑amyloid je většinou tvořen λ ře-tězci, méně často κ řetězci.
Zcela výjimečné jsou popisy amyloidu tvořené fragmenty variabilní části těžkého řetězce, které tvoří tzv. amyloidózu z těžkých řetězců (haevy chain amyloidosis).
AA-amyloidózy
AA-amyloid je tvořen sérovým amyloidovým A-proteinem (SAA), což je protein akutní fáze tvořený jako odpověď na zánět. Amyloidový A-protein je tvořen v játrech jako důsledek vyšších hladin interleukinů tvořených při zánětu (Il-1, Il-6 a TNF). Existuje více typů sérových amyloidových A-proteinů, lidský AA-amyloid je tvořen nejméně 5 molekulárními formami sérových amyloidových A-proteinů.
S částečným potlačením mnoha chronických infekcí, např. tuberkulózy a osteomyelitidy, se velmi podstatně snížil i výskyt AA-amyloidózy. Stále je však dosti pacientů s revmatickým kloubním onemocněním, zejména s revmatickou artritidou a Bechtěrevovou chorobou, a také s nespecifickými chronickými záněty střeva, např. Crohnovou chorobou, u nichž se s AA-amyloidózou můžeme stále ještě setkat.
Stanovení diagnózy
Základem stanovení diagnózy je odběr histologie a požádání patologů o průkaz amyloidu. Dle definice amyloid může být diagnostikován pouze morfologicky, žádné klinické, radiologické nebo sérologické vyšetření nenahrazuje vyšetření tkáňového vzorku pro definitivní diagnostický průkaz. Běžně se provádí necílený odběr sliznice rekta. V literatuře se popisuje průkaz amyloidu v aspirovaném podkožním tuku, tato metoda však není standardně prováděna na všech pracovištích a my s ní vlastní zkušenosti nemáme. Literatura uvádí, že biopsie sliznice rekta je senzitivnější než aspirační biopsie tuku a umožňuje širší imunohistochemické vyšetření. Senzitivita rektálních biopsií kolísá v rozsahu 70–97 % a závisí především na množství vyšetřované tkáně [47]. Falešnou negativitu lze očekávat až v 60 % případů, pokud bioptický vzorek nezahrnuje submukózu. V případě negativního výsledku a podezření na orgánové postižení nezbývá nic jiného než získat histologii ze suspektního místa depozice amyloidu.
Pro průkaz se používá dvojlom polarizovaného světla a dichroizmus indukovaný vazbou konžské červeně na fibrily amyloidu. Reakce je velmi specifická. Falešně pozitivní nebo falešně negativní výsledky jsou obvykle způsobeny defektní technikou barvení, neadekvátní mikroskopickou výbavou nebo vznikají v důsledku velmi malých množství amyloidu. Velmi citlivá, ale méně specifická je vazba fluorogenu – tioflavinu-T a -S na amyloid. Všechny druhy amyloidu obsahují P-komponentu, kterou lze příslušnými protilátkami prokázat.
Fibrilární struktura amyloidu je dobře patrná elektronovým mikroskopem, jímž lze prokázat přítomnost 8–10 nm silných fibril. U mnoha chorob se nicméně mohou vyskytnout biochemicky různá depozita mikrofibril, některá ultrastrukturálně velmi podobná fibrilám amyloidu. Průkaz konžskou červení a imunohistochemická detekce amyloidové P-komponenty je tak metodou volby při odlišení od jiných fibrilárních depozit.
Pokud se prokáže obecně amyloid, je dalším úkolem stanovit jeho etiologii. Protože nejpravděpodobnější je AL‑amyloidóza, je cílem dalších vyšetření prokázání lymfocytární či plazmocytární klonální proliferaci.
Logické by bylo v případě průkazu amyloidu dalšími vyšetřeními prokázat jeho typ. Neexistují žádné histologické znaky, které by umožnily rozlišit jednotlivé formy amyloidu. Historicky byla užívána reakce s manganistanem draselným k odlišení AL ‑ a AA-amyloidu, v současnosti jsou používány specifičtější metody, např. imunohistochemické, dvojitá imunodifuze, Western blottingu a další. Typizace amyloidu však zvláště u AL‑amyloidózy není tak lehce proveditelná, protože lehké řetězce, z nichž je amyloid tvořen, se obvykle nedaří prokázat imunohistochemickou reakcí, neboť komerčně dostupné protilátky proti lehkým řetězcům jsou zaměřeny proti jejich konstantní části. Amyloidová depozita jsou tvořena dominantně variabilní částí lehkých řetězců a mimo to v průběhu amyloidogeneze dochází ke změnám prostorové struktury. Proto imunohistochemické reakce, prokazující antigeny lehkých řetězců v amyloidu, mohou být falešně negativní; udává se pouze 40% senzitivita při užití komerčně dostupných protilátek. Negativní výsledek proto nevylučuje AL‑amyloid. Naopak falešná pozitivita může být způsobena běžnou přítomností imunoglobulinů v amyloidu ne-imunoglobulinového původu. Z tohoto důvodu se snažíme podezření, že se jedná o AL‑amyloid, podepřít průkazem monoklonální gamapatie a provádíme následující vyšetření:
- trepanobiopsie kostní dřeně a vyšetření klonality plazmocytů + průkaz amyloidu v kostní dřeni
- průkaz monoklonální gamapatie s po-mocí imunofixačního vyšetření krve a moče
- kvantitativní stanovení lehkých ře-tězců v séru (free light chain assay)
U pacientů s AL‑amyloidózou bývá v kostní dřeni přítomno jen mírné zmnožení plazmocytů, pod 10 %, někdy však zmnožení plazmocytů nemusí být zachyceno vůbec. Morfologicky a biologicky odpovídá populace plazmocytů při primární AL‑amyloidóze populaci plazmocytů u MGUS. Transformace této primární AL‑amyloidózy do mnohočetného myelomu je výjimečná. Na druhé straně bioptický nález v kostní dřeni odhalí plazmocytární proliferaci počtem a dalšími znaky odpovídající mnohočetnému myelomu. V tomto případě pak mluvíme o sekundární AL‑amyloidóze při mnohočetném myelomu.
Renální manifestace AL‑amyloidózy
AL‑amyloidóza má nejširší spektrum orgánového postižení ze všech amyloidóz. Potíže nemocného odrážejí postižení jednoho či více orgánů, kde je deponováno nejvíce amyloidu a kde dochází k nejvýraznějšímu poškození tkáně či orgánu.
Amyloidová depozita v ledvinách bývají jak u mnohočetného myelomu, tak u primární systémové AL‑amyloidózy. Amyloid je lokalizován obvykle v bazálních membránách kapilár a glomerulů, prvotní depozita jsou detekována obvykle v mezangiu glomerulu. Tubuly a in-tersticium jsou depozity méně často postiženy. Průkaz těchto změn je možný ve vzorku získaném renální biopsií.
Monoklonální lehké řetězce jsou detekovatelné v moči u 70 % případů a ve většině případů jsou typu λ [51].
Postižení ledvin je nejčastějším projevem AL‑amyloidózy (až 74 %). Prokázaná amyloidóza ledvin se v 50 % případů projeví nefrotickým syndromem (masivní proteinurií, edémy a hypoalbuminemií, hypercholesterolemií). Dříve se za příčiny otoků jednoznačně považoval snížený onkotický tlak, dnes je za hlavní příčinu otoků považována retence sodíku poškozenými ledvinami. Nejzávažnějším projevem je anasarka. Koncentrace kreatininu a urey v séru může být zpočátku normální či jen nepatrně zvýšená, teprve postupně vzniká renální insuficience s retencí dusíkatých látek, která má většinou chronický charakter a je detekována asi u 50 % pacientů s amyloidózou ledvin. Závažná renální insuficience s retencí dusíkatých látek je přítomna u 18 % případů AL‑amyloidózy ledvin. Pokud se analyzují příčiny nefrotického syndromu u osob nemajících cukrovku, tak AL‑amyloidóza ledvin je přítomna u 10 % z nich.
Léčba primární systémové AL‑amyloidózy
Cílem léčby je zcela zastavit tvorbu toxických lehkých řetězců maligním či benigním klonem plazmocytů. Pokud se podaří léčbou dosáhnout vymizení lehkých řetězců z moče a krve (hemato-logická léčebná odpověď), pak obvykle ještě několik měsíců trvá, než dojde ke zlepšení funkce amyloidem poškozených orgánů (orgánová léčebná odpověď).
Pro léčbu AL amyloidózy se doporučuje intenzivní léčba jako u mnohočetného myelomu.
Vysokodávkovaná chemoterapie s au--tologní transplantací má však podstatně vyšší mortalitu (10–30 % dle vý-běru nemocných) než u mnohočetného myelomu, a proto je tato léčbě proveditelná jen u osob mladších 65 let, které nemají závažné poškození tkání či orgánů.
Nověji byly z ně-kte-rých zahraničních pracovišť popsány úspěchy kombinovaných léčebných postupů, které obsahují jedno čí více klasických cytostatik, vysoké dávky dexametazonu a bortezo-mib nebo thalidomid. Bortezomib a tha-lidomid však zatím nemají registraci pro léčbu primární AL‑amyloidózy. Lze je však použít, pokud se jedná o AL‑amyloidózu provázející mnohočetný myelom.
Léčbu AL‑amyloidózy monitorujeme dle odpadu lehkých řetězců v moči za 24 hod a dle výsledků analýz volných lehkých řetězců v plazmě.
U pacientů do 65 let v dobrém stavu je indikována vysokodávkovaná chemoterapie a autologní transplantace krvetvorných buněk, pokud není přítomno závažné poškození orgánů. Nutno však zdůraznit, že orgánové po-škození amyloidovými depozity zhoršuje toleranci léčby.
Symptomatická léčba někdy vyžaduje kličková diuretika, v případě hypertenze jsou léčbou první volby ACE inhibitory. Častější je však těžká hypotenze způsobená autonomní neuropatií. Nefrotický syndrom je spojen s hyperkoagulačním stavem [52–60].
Klinické projevy a léčba AA-amyloidózy
AA‑amyloidóza neboli sekundární (též reaktivní) amyloidóza provází chronické zánětlivé nemoci, dříve to bývaly infekční nemoci, dnes bývá příčinou AA‑amyloidózy nejčastěji revmatoidní artritida, Bechtěrevova nemoc či Crohnova choroba. Tato AA-amyloidóza se projevuje nejčastěji poškozením ledvin, případně hepatomegalií a splenomegalií. Na Mayo Clinic tvořil tento typ amyloidózy jen 10 % všech amyloidóz. Poškození srdce je u AA‑amyloidózy vzácné. Ale i pokud je echokardiograficky patrné, nevede k srdečnímu selhání jako u AL‑amyloidózy srdce. Makroglosie se u AA‑amyloidózy nevyskytuje, podobně jako se nevyskytuje u transtyretinové amyloidózy.
Podstatou léčby je velmi aktivní léčba chronické zánětlivé nemoci, která způsobila tuto AA‑amyloidózu.
Nemoc způsobená depozity monoklonálních lehkých řetězců v neamyloidové podobě (light chain deposition disease)
V anglické literatuře existuje termín light chain deposition disease, který je standardně používán po mnoho let. Do češtiny jej přeložíme jako „nemoc z ukládání lehkých řetězců“, přičemž budeme respektovat, že je tím míněna nemoc z ukládání lehkých řetězců v neamyloidové podobě. I AL‑amyloidóza je patofyziologické nemocnění z ukládání lehkých řetězců, nicméně drtivá většina anglických publikací označuje pod pojmem light chain deposition di-sease právě patologická depozita amorfních neamyloidových forem lehkých řetězců, většinou typu κ. Jedná se o velmi vzácné onemocnění.
První popis této nemoci u pacientů s renální insuficiencí, způsobenou gra-nulárními neamyloidovámi depozity v ledvině, v nichž byly prokázány lehké řetězce, zveřejnil Randall [61] v roce 1976. Tato depozita nereagovala s konž--skou červení, což znamenalo, že lehké ře-tězce nejsou uloženy ve formě amyloidu.
Nemoc z ukládání těžkých řetězců je velmi výjimečná, provází výjimečně lymfoproliferativní choroby. Podstatou této nemoci je tvorba abnormálního těžkého řetězce (truncated haevy chain), který tvoří depozita v tkáních [62,63].
Podstatou light chain deposition disease nemoci je maligní (65 %) či nemaligní plazmocelulární proliferace (35 %), produkující monoklonální imunoglobulin. Ten je s pomocí imunofixace detekovatelný u 94 % nemocných se zřetelnou dominancí lehkých řetězců κ [64,65]. Nicméně jsou popsány i případy, kdy jsou depozita lehkých ře-tězců κ diagnostikována morfologicky a přitom v moči a v krvi nebyl detekován monoklonální imunoglobulin.
Postižení ledvin je nejčastějším projevem light chain deposition disease. Symptomatická extrarenální depozita se vyskytují jen zřídka. U všech pacientů s light chain deposition disease byla extrarenální depozita prokázána v srdci (21 %), v játrech (19 %) a na periferních nervech (8 %), ale i v CNS.
Klinicky se light chain deposition di-sease projevuje selháváním funkce ledvin. Proteinurie nad 1 g/24 hod byla popsána u 84 % pacientů a 40 % nemocných mělo proteinurii odpovídající nefrotickému syndromu.
Podezřelé z této formy nefropatie jsou všechny formy renální insuficience u mnohočetného myelomu, u nichž dochází k rychlému zhoršování funkce ledvin. Do klinického obrazu mimo proteinurii patří mikrohematurie a hypertenze.
Retence dusíkatých látek bývá ob-vykle závažnější než v případě depozice amyloidových hmot v ledvině.
Nemocní, u nichž je přítomen mnohočetný myelom, mají vyšší pravděpodobnost akutního renálního selhání, pokud je jejich nemoc neléčená, než osoby, u nichž je podkladem nemoci nemaligní plazmocelulární proliferace typu MGUS. V případě mnohočetného myelomu při vyšším kvantu vylučovaných lehkých řetězců může být ledvina poškozována současně amorfními depozity lehkých řetězců a odlitkovými válci. Koexistence těchto dvou patofyziologických podkladů nefropatie byla popsána asi v 1/3 případů [66–73].
Diagnostikovat lze tuto jednotku pouze pomocí biopsie ledviny. V op-tické mikroskopii je charakteristická přítomnost mezangiálních uzlů, zesílení bazálních membrán glomerulárních kapilár a tubulů. Obraz nodulární sklerózy v základním barvení (H&E, PAS) je histologicky těžko odlišitelný od diabetické glomerulosklerózy. V zásadě u LCDD se noduly vyskytují difuzněji než u diabetické glomerulosklerózy. Histologický obraz LCDD je však velice variabilní a nepřítomnost mezangiálních uzlů nevylučuje toto onemocnění. Při nepřítomnosti nodulárních formací bývá fokální nebo difuzní mezangiální expanze někdy se zvýšenou buněčností. Pomocí imunofluorescence lze imunohistochemickým barvením prokázat depozici monoklonálního imunoglobulinu (řetězce κ) v glomerulární bazální membráně, v mezangiu a v tubulární bazální membráně. V elektronomikroskopickém obraze vidíme depozita nefibrilárního materiálu ve zmíněných lokalizacích.
Diferenciální diagnostika je velice široká. Zahrnuje diabetickou glomerulosklerózu, idiopatickou nodulární glomerulosklerózu, membranoproliferativní glomerulonefritidu, amyloidózu a fibrilární glomerulonefritidu.
Faktory, které způsobily to, že se lehké řetězce ukládají v oblasti bazální membrány glomerulů a tubulů, nejsou známy.
Celkové přežití pacientů s light chain deposition disease se uvádí kolem 49 měsíců, 31 % nemocných však žije déle než 8 let.
Pro prognózu je zásadní biologický charakter plazmocelulární proliferace.
Nepříznivými prognostickými faktory je vyšší věk, agresivní průběh myelomu a depozita lehkých řetězců i v dalších orgánech [74–79].
Histiocytóza s ukládáním krystalů (crystal storing histiocytosis)
Tato forma poškozování organizmu monoklonálním imunoglobulinem je ještě vzácnější než light chain deposition disease. Poprvé byla popsána v roce 1978 Terashimou a od té doby se objevují zprávy o asociaci této jednotky s plazmatickými a lymfoidními klonálními proliferacemi [76].
Crystal storing histiocytosis je porucha, při níž dochází k intralysozomální kumulaci monoklonálního imunoglobulinu, který agreguje do krystalické struktury. Krystaly jsou tvořeny povětšinou lehkými řetězci κ.
Krystalické inkluze bývají nalézány v tkáňových histiocytech (někdy i ve fibroblastech) měkkých tkáních či pa-renchymatózních orgánech. V kostní dřeni byla popsána tato depozita jak v histiocytárních, tak v plazmatických buňkách kostní dřeně. Dále byly krystalické inkluze popsány v lymfatických uzlinách, ve slezině, játrech, žaludku, štítné žláze, v nadledvinách, ledvinách, ale také v rohovce, v kůži a ve varlatech [80,81].
Klinické příznaky této nemoci se liší dle toho, ve které tkáni dochází k de-pozicím. Popsané případy nefropatie vzniklé na principu crystal‑storing histiocytózy shrnuje Stokes [82]. Klinicky se tento typ postižení nejčastěji manifestoval akutní renální insuficiencí, která byla zřejmě výsledkem tkáňové infiltrace histiocytárními buňkami s deponovanými krystaly a akutním tubulárním poškozením. U 8 z 10 popsaných případů nefropatie byly nalezeny krystalické inkluze nejen v ledvinách, ale také v buňkách kostní dřeně. Ve všech případech se jednalo o κ řetězce, většinou (u 9 případů z 10) byly tyto κ řetězce detekovatelné i v moči. Krystalické inkluze byly nalezeny v bioptických preparátech nejen v intersticiálních histiocytech, ale také v tubulárních epiteliích, v mezangiálních buňkách a v glomerulárních endoteliálních buňkách [83].
Základem léčby je eliminace patologického klonu, který produkuje tyto toxické lehké řetězce, čili dosažení kompletní remise nemoci. Je proto pochopitelné, proč je agresivní chemoterapie zahrnující vysokodávkovanou chemoterapii s autologní transplantací a léčebné postupy obsahující nové léky (thalidomid, bortezomib) účinnější než dřívější klasická léčba melfalanen a prednisonen [8,77,84].
Proliferativní glomerulonefritis způsobená monoklonálním imunoglobulinem
Monoklonální imunoglobulin ve vzácných případech vytváří depozita v glomerulech. Ve světelném mikroskopu se tento typ poškození ledvin projevuje difuzní endokapilární proliferativní glo-merulonefritidou, charakterizovanou en--dokapilární hypercelularitou a leukocytární infiltrací. Monoklonální imunoglobulin vytváří elektron-denzní depozita zřetelná primárně v mezangiu a subendoteliálně. Při použití imunofluorescence bylo prokázáno, že tato depozita mají monoklonální původ. V těchto případech nebyla depozita detekována v jiných částech ledvin, v tubulární membráně či intersticiu. Komplementová depozita byla přítomna u 90 % a u 40 % pacientů byly snížené plazmatické koncentrace komplementu [77,85–87].
Kryoglobulinemie
Kryoglobulinemie je termín pro přítomnost bílkovin, které v cévním řečišti při poklesu teploty pod fyziologické rozmezí kryoprecipitují.
Kryoglobulinemie se klasifikuje dle komponenty, která precipituje. Klinické příznaky závisí na teplotě, při níž kryoprotein gelifikuje.
Kryoglobulin I. typu je tvořen monoklonálním imunoglobulinem IgM nebo případně jiným typem monoklonálního imunoglobulinu, který samostatně precipituje při ochlazení, aniž by se specificky vázal na jiné bílkoviny. Patofyziologickým podkladem klinických příznaků je intravaskulární precipitace kryoglobulinu a narušení cirkulace vlivem gelifikace. Kryoglobulinemie I. typu může mít těžké projevy, ale může být také pouhým laboratorním fenoménem bez klinických projevů. Závisí to na teplotě, při níž nastává kryoprecipitace. V případě makroglobulinemie se vyskytuje nejčastěji právě tento typ.
Kryoglobulin II. typu je definován jako monoklonální imunoglobulin vázající se v chladu na Fc fragmenty polyklonálních imunoglobulinů jiných tříd. Typicky je to monoklonální imunoglobulin typu IgM, vázající se na Fc fragmenty polyklonálních imunoglobulinů typu IgG. Monoklonální IgM má v tomto případě charakter revmatoidního faktoru. Tato nemoc je nazývána smíšená kryoglobulinemie (mixed cryoglobulinaemia).
Kryoglobulin III. typu (synonymem polyklonální kryoglobulin) je tvořen pouze polyklonálními imunoglobuliny s tepelnou charakteristikou kryoglobulinů. Je pozorována nejčastěji v souvislosti s hepatitidou C.
Typickými příznaky kryoglobulinemie I. typu je zbělení akrálních částí končetin nebo lividní zbarvení těchto okrsků pro prochlazení. V ně-kte-rých případech způsobuje chladovou urtiku a také purpuru. Porucha prokrvení tkání na pokladě kryoglobulinu může vést i k trvalému poškození, nejtěžšími projevy jsou kožní ulcerace a nekrózy a případně poškození ledvin.
Kryoglobulinemie II. typu je imunokomplexová choroba. Způsobuje vaskulitidy dominantně malých cév kůže, ledvin, jater a periferních nervů. Typickým klinickým projevem je purpura.
U Waldenströmovy makroglobulinemie se frekvence výskytu kryoglobulinemie pohybuje kolem 10 %. Dimopoulos popisuje nález monoklonálního kryoglobulinu u 20 %, ale klinicky významné příznaky kryoglobulinemie pouze u 5 % [88].
Klinicky se poškození ledvin při kryo-globulinemii projevuje asi u 20 % nemocných výraznou proteinurií až nefrotického charakteru a hematurií. U 1/4 pacientů dominuje v obraze akutní nefritický obraz s hematurií, hypertenzí a proteinurií až s rozvojem akutní renální insuficience [89].
Při histologickém vyšetření nalézáme nejčastěji obraz membranoproliferativní glomerulonefrity typ 1 a typ 3. V ně-kte-rých případech může být obraz difuzní endokapilární proliferativní glomerulonefritidy. Glomerulární depozita kryoglobulinů jsou v subendoteliální a intraluminální lokalizaci, tvořící intrakapilární „proteinové tromby“, méně nápadně v mezangiu. Imunofluorescence prokáže glomerulární depozita komplementu a imunoglobulinů podle typu kryoglobulinu. V elektronmikroskopickém obraze se v typických případech nacházejí depozita se substrukturou (mikrotubulární) především v subendoteliální lokalizaci. Do diferenciální diagnózy patří membranoproliferativní glomerulonefritida a systémový lupus erythematodes.
Závěr
Z výše uvedených odstavců vyplývá, že monoklonální imunoglobulin může poškodit ledviny a způsobit různě intenzivní proteinurii, od nepatrného zvýšení nad fyziologickou mez až po hodnoty odpovídající těžkému nefrotického syndromu. Nezávisle na míře proteinurie může dojít k poškození funkce ledvin a k retenci dusíkatých látek.
Proto u každého nemocného s proteinurií či s retencí dusíkatých látek by mělo být zváženo provedení průkazu monoklonálního imunoglobulinu v moči a v séru metodou imunofixace. Pokud je výrazné podezření na souvislost nefropatie s monoklonální gamapatií, považuje se za užitečné provést kvantifikaci volných lehkých řetězců v séru a stanovení jejich poměru [90–98] a případně doplnění zobrazovacích vyšetření [99].
Pokud je potvrzena přítomnost monoklonálního imunoglobulinu, je nutné ihned zahájit diferenciální diagnostiku monoklonální gamapatie. Včasné stanovení diagnózy mnohočetného myelomu a zahájení léčby vede k udržení lepší kvality života než v případě pozdního zahájení léčby, kdy poškození organizmu monoklonálním imunoglobulinem je již nevratné.
Práce vznikla v rámci VZ MSM 0021622434 a grantu LC06027 Masarykovy univerzity, Česká republika a je součástí edukačního programu CRAB České myelomové skupiny.
Doručeno do redakce: 7. 4. 2008
Přijato po recenzi: 26. 5. 2008
prof. MUDr. Zdeněk Adam, CSc.
www.fnbrno.cz
e‑mail: z.adam@fnbrno.cz
Zdroje
1. Adam Z, Krejčí M, Vorlíček J. Přehled maligních krevních chorob. Praha: Grada 2008.
2. Alexanian R, Barlogie B, Dixon D. Renal failure in multiple myeloma. Pathogenesis and prognostic implication. Arch Intern Med 1990; 150 : 1693–1695.
3. Iggo N, Palmer ABD, Severn A et al. Chronic dialysis in patients with multiple myeloma and renal failure: a worthwhile treatment. Q J Med 1989; 73 : 903–910.
4. Iggo N, Parsons V. Renal disease in multiple myeloma: current perspectives. Nephron 1990; 56 : 229–233.
5. Iggo N, Winearls CG. The development of cast nephropathy in multiple myeloma. Q J Med 1998; 90 : 653–656.
6. International Myeloma Working Group: Criteria for the classification of monoclonal gammopathies multiple myeloma and related disorders. A report of International Myeloma Working Group. Br J Haematol 2003; 121 : 749–757.
7. Solomon A, Weiss DT, Kattine AA. Nephrotoxic potential of Bence Jones proteins. N Engl J Med 1991; 324 : 1845–1851.
8. Markowitz GS. Dysproteinemia and the kidney. Adv Anat Pathol 2004; 11 : 46–63.
9. Herrera GA, Joseph L, Hough A et al. Renal pathologic spectrum in an autopsy series of patients with plasma cell dyscrasia. Arch Path Lab Med 2004; 128 : 875–879.
10. Korbet SM, Schwartz M. Multiple myeloma. J Am Soc Nephrol 2006; 17 : 2533–2545.
11. Montseny JJ, Kleinknecht D, Meyrier A et al. Long term outcome according to renal histological lesions in 118 patients with monoclonal gammopathies. Nephrol Dial Transplant 1998; 13 : 1438–1445.
12. Sanders PW, Booker BB. Pathobiology of cast nephropathy from human Bence Jones proteins. J Clin Invest 1992; 89 : 630–639.
13. Ehrenfeld M. Acute renal failure precipitated by non‑steroidal anti‑inflammatory drugs in multiple myeloma. Am J Hematol 1998; 58 : 142–144.
14. Pasquali S, Casanova S, Zucchelli A et al. Long term survival patients with acute and severe renal failure due to myeloma. Clin Nephrol 1990; 34 : 247–254.
15. Pasquali S, Zucchelli P, Casanova S et al. Renal histological lesions and clinical syndromes in multiple myeloma. Clin Nephrol 1987; 27 : 222–228.
16. Shpilberg O, Douer D, Ehrenfeld M et al. Naproxen associated fatal acute renal failure in multiple myeloma. Nephron 1990; 55 : 448–449.
17. Waught DA, Ibels LS. Multiple myeloma presenting as recurrent obstructive uropathy. Aust N Z J Med 1980; 10 : 555–568.
18. Border WA, Cohen AH. Renal biopsy diagnosis of clinically silent multiple myeloma. Ann Intern Med 1980; 93 : 43–46.
19. Rivera F. Renal insufficiency and proteinuria: glomerulonefritis or myeloma? Nefrologia 1998; 18 : 253–254.
20. Kleinknecht D. Long‑term outcome according to renal histological lesions in 118 patients with monoclonal gammopathies. Nephrol Dial Transplant 1998; 13 : 1438–1445.
21. Meyrier A, Simon P, Mignon F et al. Rapidly progressive glomerulonephritis and monoclonal gammapathies. Nephron 1984; 38 : 156–162.
22. Esnault VLM, Jayne DRW, Keogan MT. Anti‑neutrophil cytoplasm antibodies in patients with monoclonal gammopathies. J Clin Lab Immunol 1990; 32 : 153–159.
23. Vigil A, Oliet A, Gallar P et al. Rapidly progressive immunotactoid glomerulonephritis and multiple myeloma. Nephron 1998; 79 : 238–240.
24. Penfield JG. Multiple myeloma and end‑stage renal disease. Semin Dial 2006; 19 : 329–334.
25. Kyle RA. Monoclonal gammopathies and the kidney. Annu Rev Med 1989; 40 : 53–60.
26. Batuman V, Verroust PHJ, Navar GL. Myeloma light chains are ligand for cubilin. Am J Physiol 1998; 275: F246–F254.
27. Batuman V, Guan S. Receptor mediated endocytosis of Bence Jones proteins by proximal tubular cells. Am J Physiol 1997; 272: F521–F530.
28. Klassen RB, Allen PL, Batuman V et al. Light chains are a ligand for megalit. J Appl Physiol 2005; 98 : 257–263.
29. Bradley JR, Thiru S, Evans DB. Light chains and the kidney. J Clin Pathol 1987; 40 : 53–60.
30. Guillermo A, Herrera A, Sanders PW. Paraproteinemic renals disease that involve the tubulo-interstitium. Contrib Nephrol 2007; 153 : 105–115.
31. Kapur U, Barton K, Fresco R et al. Expanding the pathologic spectrum of immunoglobulin light chain proximal tubulopathy. Arch Pathol Lab Med 2007; 131 : 1368–1372.
32. Messiaen T, Deret S, Mougenot B et al. Adult Fanconi syndrome secondary to light chain gammopathy. Clinicopathologic heterogeneity and unusual features in 11 patients. Medicine (Baltimore) 2000; 79 : 135–154.
33. Decourt C, Bridoux F, Touchard G et al. A monoclonal V kappa λ light chain responsible for incomplete proximal tubulopathy. Am J Kidney Dis 2003 : 41 : 497–504.
34. Ronco P, Aucouturier P, Mougenot B. Plasma cell dyscrasias-releated glomerulopathies and Fanconi’s syndrome: a molecular approach. J Nephrol 2000; 13 (Suppl 3): S34–S44.
35. Ying WZ, Sander PW. Mapping the binding domain of imunoglobulin light chain for Tammm Horsfall protein. Am J Pathol 2001; 158 : 1589–1866.
36. Ying WZ, Sanders PW. Expression of Tamm Horfall glycoprotein is regulated by dietary salt in rats. Kidney Int 1998; 20 : 198–210.
37. Knudsen LM, Hippe E, Hjort M et al. Renal function in newly diagnosed multiple myeloma: A demographic study of 1353 patients. The Nordic Myeloma Study Group. Eur J Haematol 1994; 53 : 207–212.
38. Chanan-Khan AA, Kaufman JL, Mehta J et al. Activity and safety of bortezomib in multiple myeloma with advanced renal failure. Blood 2007; 109 : 2604–2606.
39. Fakhouri F, Guerraoui H, Presne C et al. Thalidomide in patients with multiple myeloma and renal failure. Br J Haematol 2004; 125 : 96–97.
40. Solling K, Solling J. Clearence of Bence Jones proteins during peritoneal dialysis of plasmapheresis in myelomatosis associated with renal failure. Contrib Nephrol 1988; 68 : 259–262.
41. Zucchelli P, Pasqualli S, Cognoli L et al. Controlled plasma exchange trial in acute renal failure due to multiple myeloma. Kidney Int 1988; 33 : 1175–1180.
42. Johnson WJ, Kyle RA, Pineda AA et al. Treatment of renal failure associated with multiple myeloma. Arch Intern Med 1990; 150 : 863–869.
43. Clark, WF, Steward AK, Rock GA et al. Plasma exchange when myeloma presents as acute renal failure: a randomized controlled trial. Ann Intern Med 2005; 143 : 777–784.
44. Gerz MA. Managing myeloma kidney. Ann Intern Med 2005; 143 : 835–837.
45. Hutchison CA, Cockwell P, Reid S et al. Efficient removal of immunoglobulin free light chains by hemodialysis for multiple myeloma. In vitro and in vivo studies. J Amer Soc Nephrol 2007; 18 : 886–895.
46. Alpers CE, Marchioro TL, Johnson RJ. Monoclonal immunoglobulin deposition disease in renal allograft recipients: probable recurrent disease in a patient with myeloma. Am J Kidney Dis 1989; 13 : 418–423.
47. Röcken C, Sletten K. Amyloid in surgical pathology. Virchows Arch 2003; 443 : 3–16.
48. Merlini M. WHO-IUIS nomenclature subcomittee: Nomenclature of amyloid and amyloidoses. N Engl J Med 2003; 349 : 583–596.
49. Leung N, Rajkumar V. Renal manifestation of plasma cell disorders. Amer J Kidney Disease 2007; 50 : 155–165.
50. Merlini M. Monoclonal gammopathy and nephropathy. Blood 2006; 108 : 2520–2530.
51. Falk RH, Comenzo RL, Skinner M. The systemic amyloidoses. N Engl J Med 1997; 337 : 898–909.
52. Adam Z, Ščudla V, Tomíška M. Léčba AL‑amyloidózy a ně-kte-rých dalších typů amyloidóz. Vnitř Lék 2001; 47 : 46–52.
53. Adam Z, Ščudla V. Klinické projevy a diagnostika AL‑amyloidózy a ně-kte-rých dalších typů amyloidóz. Vnitř Lék 2001; 47 : 36–45.
54. Kroupa R, Dastych M, Šenkyřík M et al. Systémová amyloidóza s dominující klinickou manifestací v trávicím traktu. Vnitř Lék 2005; 51 : 588–592.
55. Linhartová K, Daum O. Srdeční amyloidóza. Cor Vasa 2005; 47 : 328.
56. Ryšavá R. Amyloidóza ledvin. Postgrad Med 2006; 8 : 207–212.
57. Ryšavá R. Léčba paraproteinemické nefropatie a primární amyloidózy ledvin. Aktual v nefrol 2005; 11 : 62–65.
58. Bird JM, Fuge R, Sirohy B et al. The clinical outcome and toxicity of high‑dose chemotherapy and autologous stem cell transplantation in patients with myeloma or amyloid and severe renal impairment: a British Society of Blood and Marrow Transplantation study. Br J Haematol 2006; 134 : 385–390.
59. Pirani CL, Silva F, D’Agati V et al. Renal lesions in plasma cell dyscrasias: ultrastructural observations. Am J Kidney Dis 1987; 10 : 208–221.
60. Ramos R, Poveda R, Sarra J et al. Renal involvement in non malignant IgM gammopathy. Nephrol Dial Transplant 2007; 22 : 627–630.
61. Randall RE, Williamson WC, Mulinax F et al. Manifestation of systemic light chain deposition. Am J Med 1976; 60 : 293–299.
62. Wahner-Roedler DL, Kyle R. Heavy chain disease. Best Pract Res Clin Haematol 2005; 18 : 729–746.
63. Tichý M, Maisnar V, Stulík J et al. Nemoc z těžkých řetězců µ. Klin Biochem Metab 2007; 15 : 78–81.
64. Pozzi C, D’Amico M, Fogazzi GB et al. Light chain deposition disease with renal involvement: clinical characteristics and prognostic factors. Am J Kidney Dis 2003; 42 : 1154–1163.
65. Cogne M, Preudhomme JL, Bauwens M. Structure of monoclonal kappa chain of the V and IV subgroup in the kidney and plasma cells in light chain deposition disease. J Clin Invest 1991; 87 : 2186–2192.
66. Gu X, Herrera AG. Light chain mediated acute tubular interstitial nephritis. Arch Pathol Lab Med 2006; 130 : 165–169.
67. Gallo GR, Lazowski P, Kumar A et al. Renal and cardiac manifestations of B-cell dyscrasias with nonamyloidotic monoclonal light chain and light and heavy chain deposition disease. Adv Nephrol Necker Hosp 1998; 28 : 355–382.
68. Gallo G, Picken M, Buxbaum J et al. The spectrum of monoclonal immunoglobulin deposition disease associated with immunocytic dyscrasias. Semin Hematol 1989; 26 : 234–238.
69. Ganeval D, Noel LH, Preud’homme JL et al. Light chain deposition disease: Its relation with AL type amyloidosis. Kidney International 1984; 26 : 1–9.
70. Ganeval D, Rabian C, Guerin V et al. Treatment of multiple myeloma with renal involvement. Adv Nephrol Necker Hosp 1992; 21 : 347–370.
71. Girelli CM. Kappa light chain deposition of the liver. Eur J Gastroenterol Hepatol 1998; 10 : 429–430.
72. Steuhl KP, Knorr C, Rohrbach JM et al. Paraproteinemic corneal deposits in plasma cell myeloma. Am J Ophthalmol 1991; 111 : 312–318.
73. Lin J, Markowitz GS, Valeri AM et al. Renal monoclonal immunoglobulin deposition disease. The disease spectrum. J Am Soc Nephrol 2001; 12 : 1482–1487.
74. Confalonieri R, Barbiano di Belgiojoso G. Light chain nephropathy: histological and clinical aspects in 15 cases. Nephrol Dial Transplant 1988; 3 : 150–156.
75. Bladé J, Rosinol L. Renal, hematologic and infectious complication in multiple myeloma. Best Pract Res Clin Haematol 2005; 18 : 635–652.
76. Heilman RL, Velosa JA, Holley JJ et al. Long‑term follow up and response to chemotherapy in patients with light chain deposition disease. Amer J Kidney Dis 1992; 20 : 34–41.
77. Phillips AO, O’Donnel PJ, Nelson SR et al. Light chain nephropathy and anti‑neutrofil cytoplasmatic antibody associated vasculitis. Nephrol Dial Transplant 1993; 8 : 1178–1180.
78. Pozzi C, D’Amico M, Fogazzi GB et al. Light chain deposition disease with renal involvement: clinical characteristics and prognostic factors. Am J Kidney Dis 2003; 42 : 1154–1163.
79. Terashima K, Takahashi K, Kojima M et al. Kappa‑type light chain crystal storage histiocytosis. Acta Pathol Jpn 1978; 28 : 111–138.
80. Pock L, Stuchlík D, Herzogová J et al. Crystal storing histiocytosis of the skin associated with multiple myeloma. Int J Dermatol 2006; 45 : 1408–1411.
81. Preddie CD, Markowitz GS et al. Multiple myeloma, nephrotic syndrome and crystaloid inclusions in podocytes. Kidney Int 2006; 69 : 616–620.
82. Stokes MB, Aronoff B, Siegel D et al. Dysproteinemia releated nephropathy associated with crystal storing histiocytosis. Kidney International 2006; 70 : 597–602.
83. Lebeau A, Zeindl-Ebergart E, Müller ECh et al. Generalized crystal storing histiocytosis with monoclonal gammopathy: molecular analysis of a disorder with rapid clinical course and review of the literature. Blood 2002; 100 : 1817–1827.
84. Nasr SH, Nasr SH, Markowitz GS et al. Proliferative glomeruloneftitis with monoclonal IgG deposits: a distinct entity mimicking with immune-complex glomerulonephritis. Kidney Int 2004; 65 : 85–96.
85. Rosenstock JL, Markowitz GS, Valeri AM et al. Fibrillary and immunotactoid glomerulonephritis: Distinct entities with different clinical and pathological features. Kidney Int 2003; 63 : 1450–1461.
86. Soares SM, Lager DJ, Leung N et al. A proliferative flomerulonefritis secondary to a monoclonal IgA. Amer J Kidney Dis 2006; 47 : 342–349.
87. Silva FG, Meyrier A, More L et al. Proliferative glomerulopathy in mutliple myeloma. J Pathol 1980; 130 : 229–235.
88. Dimopoulos MA, Kyle RA, Anagnastopoulos A et al. Diagnosis and management of Wadenström’s macroglobulinemia. J Clin Oncol 2005; 23 : 1564–1577.
89. Shihabi ZK. Cryoglobulins: An important but neglected clinical test. Ann Clin Lab Science 2006; 36 : 395–408.
90. Ščudla V, Minařík J, Schneiderka P et al. Význam sérových hladin volných lehkých řetězců imunoglobulinu v diagnostice a hodnocení aktivity mnohočetného myelomu a vybraných monoklonálních gamapatií. Vnitř Lék 2005; 51 : 1249–1259.
91. Tichý M, Hrnčíř Z, Urban P et al. Monoklonální imunoglobuliny. Klin Biochem Metabol 2004; 12 : 84–87.
92. Tichý M. Monoklonální gamapatie. Labor Aktuel CS 2000; 5 : 7–10.
93. Tichý M, Urban P, Matěja P et al. Laboratorní analýza souboru 3049 monoklonálních imunoglobulinů. Klin Biochem Metabol 2002; 10 : 257–261.
94. Tichý M. Viscosity of paraproteinemic séra. Acta Med 1996; 39 : 41–43.
95. Tichý M. Laboratorní analýza monoklonálních imunoglobulinů (paraproteinů). Český Těšín: Finidr 1997.
96. Špička I, Mašek Z, Jirsa M et al. Interleukin‑6, tumor-nekrotizující faktor a jejich solubilní receptory u Bence-Jonesovy nefropatie – možná úloha v patogenezi a význam stanovení pro prognózu renální insuficience. Klin Onkol 1996; 9 : 126–129.
97. Špička I, Merta M, Cieslar P et al. Postižení ledvin u monoklonálních gamapatií. Čas Lék Čes 1996; 135 : 374–377.
98. Špička I, Merta M, Cieslar P et al. Postižení ledvin u monoklonálních gamapatií. Klinická studie. Čas Lék Čes 1995; 134 : 478–481.
99. Ščudla V, Nekula J, Bačovský J et al. Nukleární magnetická rezonance v hodnocení páteře u mnohočetného myelomu. Čes Revmatol 1997; 5 : 51–52.
Štítky
Diabetologie Endokrinologie Interní lékařstvíČlánek vyšel v časopise
Vnitřní lékařství

2008 Číslo 9
- Inovace v hojení ran: krytí Zetuvit Plus Silicone Border pro optimální management exsudátu z ran
- Ivabradin zlepšuje kvalitu života starších pacientů se srdečním selháním
- Fixní kombinace kandesartan/amlodipin v terapii arteriální hypertenze
- Clopidogrel je v prevenci kardiovaskulárních příhod přínosnější než kyselina acetylsalicylová
Nejčtenější v tomto čísle
- Poškození ledvin při mnohočetném myelomu a dalších monoklonálních gamapatiích
- BK virová infekce po transplantaci ledvin
- Hypertenze u dialyzovaných pacientů
- Ischemická choroba srdeční s předčasnou manifestací u mladých pacientů
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy