
Význam Downova syndromu v hematologii
Importance of Down syndrome in haematology
Down syndrome (DS) is one of the most common congenital syndromes in humans. The cause of DS is complete or partial trisomy of chromosome 21. The disease is associated with a typical phenotypic manifestation and a higher risk of health complications, including haemato-oncological diseases. The most serious haematological complications include transient myeloproliferative disease (TMD), myelodysplastic syndrome and acute leukaemia. TMD affects up to 10% of new-borns with DS. While spontaneous remission occurs in most cases, 20–30% of cases develop into acute leukaemia. Acute leukaemia associated with DS is most often of myeloid origin, rarely of lymphoid origin. TMD is not defined by unambiguous morphological criteria and characterised by the transient presence of megakaryocyte lineage blasts in the peripheral blood of children with chromosome 21 trisomy.
Keywords:
leukaemia – Down syndrome – peripheral blood – blastic elements – GATA1 mutation – transient myeloproliferative disease – chromosome 21
Autoři:
L. Kolařík 1,2
![]() ; H. Zůnová 3; H. Kollárová 2
; H. Zůnová 3; H. Kollárová 2
![]()
Působiště autorů:
Oddělení klinické hematologie, FN Motol, Praha
1; Ústav veřejného zdravotnictví, LF UP, Olomouc
2; Oddělení Lékařské cytogenetiky, Ústav biologie a lékařské genetiky, 2. LF a FN Motol, Praha
3
Vyšlo v časopise:
Transfuze Hematol. dnes,28, 2022, No. 2, p. 101-111.
Kategorie:
Původní práce
doi:
https://doi.org/10.48095/cctahd2022prolekare.cz7
Souhrn
Downův syndrom (DS) patří mezi nejčastější vrozené syndromy vyskytující se v lidské populaci. Příčinou DS je úplná nebo částečná trizomie chromozomu 21. Onemocnění je provázeno typickým fenotypovým projevem a vyšším rizikem zdravotních komplikací, vč. hemato-onkologických onemocnění. Mezi nejzávažnější hematologické komplikace patří tranzientní myeloproliferativní onemocnění (TMD), myelodysplastický syndrom a akutní leukemie. TMD postihuje až 10 % novorozenců s DS. Ve většině případů dochází ke spontánní remisi onemocnění, avšak ve 20–30 % případů dojde k rozvoji akutní leukemie. Akutní leukemie spojená s DS bývá nejčastěji myeloidního původu a vzácně lymfoidního původu. TMD není definováno jednoznačnými morfologickými kritérii. Jedná se o tranzientní stav přítomnosti blastů megakaryocytární linie v periferní krvi u dětí s trizomií chromozmomu 21.
Klíčová slova:
leukemie – Downův syndrom – periferní krev – blastické elementy – GATA1 mutace – tranzietní myeloproliferativní onemocnění – chromozom 21
ÚVOD
Downův syndrom (DS) patří mezi nejčastější vrozené syndromy vyskytující se v lidské populaci. Příčinou DS je v 95 % úplná trizomie chromozomu 21, zbývající případy jsou způsobeny částečnou trizomií nebo mozaikou chromozomu 21 [1]. Onemocnění je charakterizováno komplexním fenotypovým projevem. Ten je kromě typické faciální dysmorfie provázen vyšším rizikem výskytu zdravotních komplikací, postihující více orgánových systémů: centrální nervový systém, kardiovaskulární systém, muskulo-skeletární systém, senzorický systém, respirační a imunitní systém [2]. Významnou zdravotní komplikací DS z oblasti hematologie je vyšší riziko výskytu hemato-onkologických onemocnění zahrnujících tranzientní myeloproliferativní onemocnění, myelodysplastický syndrom a akutní leukemie.
GENETICKÁ PODSTATA DS
DS patří mezi jeden z nejčastějších a nejznámějších syndromů. Syndrom byl poprvé definován anglickým lékařem Johnem Langdonem Downem v roce 1866 [3]. Genetická podstata onemocnění, spočívající v přítomnosti nadbytečného genetického materiálu, a sice nadpočetného chromozomu 21, však byla odhalena o téměř 100 let později (1959).
DS spadá mezi tzv. numerické chromozomové aberace (aneuploidie). Vyskytovat se může v několika podobách karyotypu, a to buď jako volná (prostá) trizomie nebo ve formě translokace. Volná trizomie je důsledkem chybného rozchodu chromozomů při I. či II. meiotickém buněčném dělení, tzv. nondisjunkce [4]. K nondisjunkci nejčastěji dochází ve fázi oogeneze, tedy na straně matky. Při translokační formě DS je nadbytečný chromozom 21 translokován a fúzován s jiným, zpravidla akrocentrickým chromozomem (tzv. robertsonská translokace). Nejčastěji je popisována translokace mezi chromozomy 21 a 14 [5]. Robertsonská translokace jako taková však představuje balancovanou formu aberace a nositel je zcela bez klinických projevů. Problémem je následný přenos derivovaného chromozomu 21 na potomky, kdy je předán společně derivovaný chromozom i normální chromozom 21 od nosiče translokace a další chromozom 21 od druhého rodiče. Výsledkem je plod, jehož chromozomová konstituce je představována dvěma normálními chromozomy 21 a třetím, derivovaným chromozomem 21. Vzácněji pak může být popsána také mozaiková forma DS. V tomto případě je nadbytečný chromozom 21 přítomen pouze v některých buňkách, zatímco ostatní buňky mají fyziologický počet chromozomů [6]. Celkový fenotyp těchto jedinců je pak odrazem procentuálního zastoupení aberantní, tedy trizomické buněčné linie.
PRENATÁLNÍ SCREENING V ČR
Prenatální diagnostika neboli vyšetření tkáně plodu v průběhu těhotenství představuje významný bod v záchytu těhotenství s DS. Právě DS patřil mezi první sledovanou chromozomovou abnormalitu v oblasti prenatální diagnostiky. Existují dva základní přístupy – invazivní prenatální testování a neinvazivní prenatální testování, které zpravidla předchází invazivnímu testování.
Neinvazivní prenatální testování je primárně založeno na hodnocení biochemických a ultrazvukových markerů (UZ). Základy tohoto testování položili vědci Brock a Sutcliffe [7]. Mezi sledované biochemické markery patří lidský choriový gonadotropin (cg, resp. free bhCG), volný estriol (uE3), specifický těhotenský protein A (PAPP-A), inhibin A a placentární růstový faktor (PIGF). V rámci UZ se primárně hodnotí temeno-kostrční délka (CRL), nuchální translucence (NT) a přítomnost/nepřítomnost nosní kůstky (NB). Změna koncentrace biochemických ukazatelů a UZ markerů jsou pro přehlednost shrnuty v tab. 1. [8–12]. Na kombinaci markerů jsou založeny screeningové testy v těhotenství. V základní rovině rozlišujeme screening prvotrimestrální a druhotrimestrální. Prvotrimestrální screening je prováděn mezi 10. a 14. týdnem těhotenství (tt) a kombinuje vyhodnocení hodnot PAPP-A, free bhCG a NT. Pokud je hodnocení rozšířeno o další UZ parametry, hovoříme o tzv. kombinovaném, kontingenčním testu [13]. Druhotrimestrální screening je založen čistě na hodnocení biochemických markerů a je prováděn v 15.–22. tt. Výsledkem screeningu je pak vyjádření míry rizika narození plodu s DS. V případě pozitivity screeningu je těhotné doporučeno podstoupit invazivní prenatální testování. Cílem je odběr tkáně plodu a jeho následné genetické testování za účelem potvrzení diagnózy. Mezi metody invazivního testování patří odběr choriových klků (choriocentéza), plodové vody (amniocentéza), případně odběr pupečníkové krve (kordocentéza).

FENOTYPOVÉ PROJEVY DS
U jedinců s DS je popsán charakteristický komplexní fenotypový projev. Pacienti s DS jsou nejčastěji diagnostikováni na základě velmi specifické faciální dysmorfie zahrnující mongoloidní směr očních štěrbin, níže posazené uši, makroglosii, kratší nos a široký kořen nosu. Na duhovce mohou být patrné skvrny označované jako Brushfieldovy linie. Další morfologické změny lze pozorovat na rukou a nohou. Ruce bývají široké se zkrácenými prsty, charakteristická je také tzv. čtyřprstá rýha na dlaních. V oblasti chodidel bývá širší mezera mezi palcem a ukazovákem, označovaná jako sandálová mezera [14]. U jedinců s DS jsou kromě nápadných fenotypových znaků pozorovány také různé orgánové vady, postihující nejčastěji kardiovaskulární a nervový systém. Změny v oblasti kardiovaskulárního systému jsou pozorovány u 40 % jedinců s DS. Mezi vadu s největší prevalencí patří prolaps mitrální chlopně [15,16]. U jedinců s DS jsou popsány také různé strukturní a funkční abnormality gastrointestinálního traktu či endokrinního systému [17,18]. Postižení nervového systému má spíše funkční než morfologický charakter. Psychomotorický vývoj těchto dětí bývá opožděný, provázený také poklesem intelektových funkcí. U dětí je často pozorována porucha motoriky a koordinace. U některých jedinců byla popsána epilepsie, s nástupem prvních záchvatů před ukončeným 1. rokem života [19,20]. Po 35. roce života vzrůstá riziko rozvoje neuropatických změn a zvýšené riziko rozvoje Alzheimerovy choroby [21].
EPIDEMIOLOGIE
K nejčastěji sledovaným ukazatelům u DS patří incidence. Na incidenci můžeme nahlížet dvěma způsoby: Celková incidence DS a Incidence DS u živě narozených dětí. Incidence může být vyjádřena relativně nebo absolutně, kdy je hodnota nejčastěji vztažena na 10 000 živě narozených dětí.
V celkové incidenci jsou zahrnuti jedinci narození s DS i případy, kdy byl DS prokázán prenatálně s následným ukončením těhotenství [22]. V současné době pozorujeme rostoucí trend u celkové incidence DS. Vysvětlení nalezneme v metodách prenatálního screeningu, kdy vývoj těchto metod zvýšil jejich účinnost a diagnostika onemocnění byla možná v časnější fázi těhotenství. Dalším důležitým faktorem zvyšujícím celkovou incidenci je rostoucí věk rodiček (advanced maternal age – AMA), definovaný jako (věk ≥ 35 let v době porodu). S rostoucím věkem ženy pozorujeme zvyšující se riziko výskytu vrozených vad. Primární příčinou je snižující se kvalita oocytů [23]. Kromě DS je v souvislosti s AMA popisováno zvýšené riziko rozvoje trizomie chromozomu 18 (Edwardsův syndrom) a trizomie chromozomu 13 (Patauův syndrom) [23]. Problematika věkového složení rodiček není omezena jen na Českou republiku, ale jedná se o celosvětový fenomén [24]. Počty narozených novorozenců dle věkového složení rodiček v České republice znázorňuje graf 1 [25].

Další epidemiologickou charakteristikou je pohlaví. Incidence DS u jednotlivých pohlaví je přibližně shodná. Incidenci DS dle pohlaví v podmínkách České republiky znázorňuje graf 2 [26–37]. V České republice byl v letech 2004–2015 poměr zastoupení chlapců a dívek s DS průměrně 1 : 1,1.

Opačný trend pozorujeme u incidence DS u narozených dětí, kdy je zásluhou prenatální diagnostiky trend klesající viz. graf 3.

HEMATOLOGICKÉ KOMPLIKACE
DS je spojován s řadou hematologických onemocnění, a to zejména v dětském věku. Nespecifické hematologické abnormality u novorozenců s DS zahrnují neutrofílii (80 %), trombocytopenii (66 %), polycytémii (33 %) a koagulopatii (9 %) [38,39]. Mezi specifické hematologické abnormality u DS patří tranzientní myeloproliferativní onemocnění (TMD/TAM tranzientní abnormální myelopoéza), vyšší riziko rozvoje myelodysplastického syndromu (MDS) a akutní leukemie (AL) [40]. Klasifikace Světové zdravotnické organizace (WHO) pro myeloidní novotvary a akutní leukemie obsahuje jednotku „Myeloidní proliferace související s Downovým syndromem“, která se následně dělí na dvě podjednotky: Přechodná abnormální myelopoéza a Myeloidní leukemie spojená s Downovým syndromem [41].
Tranzientní myeloproliferativní onemocnění můžeme podle WHO definovat jako stav, při kterém dochází k přítomnosti myeloidních blastických elementů v nátěru periferní krve u novorozenců s DS [42–44]. Na základě provedených studií byla kritéria různými autory doplňována o podmínku pozitivity blastických elementů na mutaci GATA-vazebného proteinu 1 (GATA1) u dětí s DS mladších 3 měsíců [18,45].
První zmínku o tomto onemocnění přinesli Schunk a Lehman v roce 1954 [46]. Patogeneze TMD začíná již in utero (v hepatolienálním období) [44,47], jako následek nadbytečného genetického materiálu chromozomu 21. Pro rozvoj TMD je u naprosté většiny případů nezbytné získání somatické mutace v genu kódujícím hematopoetický transkripční faktor GATA1 (OMIM *305371) [48,49].
Trizomie chromozomu 21 při prenatální hematopoéze v játrech vede k expanzi megakaryocytárních a erytroidních prekurzorů (MEP) a snížení prekurzorů pro granulocyty a makrofágy. Populace MEP vyznačující se zvýšenou proliferační aktivitou je citlivá na získání GATA1 mutace. Získání GATA1 mutace má za následek hyperproliferaci megakaryocytární linie [47]. Hyperproliferace blastů megakaryocytární linie může být jednou z příčin vedoucí ke klinickým obtížím, provázející TMD. Při hyperproliferaci megakaryoblastů může docházet k infiltraci tkání megakaryoblasty, zejména k infiltraci jater anebo kůže. Následkem infiltrace jater dochází k hepatomegálii a tvorbě výpotků v pleurálních a perikardiálních prostorech. Postižení jater je provázeno zvýšením transamináz s konjugovanou hyperbilirubinémií. Při infiltraci kůže se u pacienta objevuje papulární nebo vezikopustulózní vyrážka [48]. Žádné ze zmíněných příznaků nejsou zcela specifické pro TMD, ale u pacientů s TMD se vyskytují s vyšší frekvencí než u DS bez GATA1 mutace. Tranzientní myeloproliferativní onemocnění postihuje přibližně 5–10 % dětí s DS [2,44]. Nález v periferní krvi (PK) je variabilní – od přítomnosti/nepřítomnosti leukocytózy, anémie a trombocytopenie. V nátěru PK pozorujeme výskyt blastických elementů myeloidního původu, morfologicky vzhledu megakaryoblastů. Počet blastických elementů v periferní krvi ve většině případů dosahuje vyššího procentuálního zastoupení než v kostní dřeni [44,50]. Nález v kostní dřeni je též variabilní, pozorujeme normální či snížený počet megakaryocytů. Megakaryocyty mohou vykazovat poruchu zrání s přítomností dysplastických změn, může být přítomna i dyserytropoéza [50]. Postižení kostní dřeně nekoreluje se závažností onemocnění [48]. Pro stanovení diagnózy TMD není nutné provádět vyšetření kostní dřeně. Tranzientní myeloproliferativní onemocnění ve většině případů spontánně ustoupí, avšak u 20–30 % případů dojde k rozvoji AL, obvykle do 5. roku života [45]. I přes spontánní remisi onemocnění provází TMD v 15–23 % předčasné úmrtí pacienta [48]. Mezi nejčastější příčiny vedoucí k předčasnému úmrtí pacienta patří: progresivní hepatopatie vedoucí k fulminální jaterní firbóze, diseminovaná intravaskulární koagulopatie a multiorgánové selhání [48]. Z důvodu rizika rozvoje MDS a AL se doporučuje sledování v ordinaci dětského hematologa [44,51].
Jedinci s DS mají 10–20x vyšší riziko rozvoje AL [52]. Akutní leukemie spojená s DS bývá nejčastěji myeloidního, vzácněji lymfoidního původu. Subtyp akutní myeloidní leukemie (AML) vyskytující se u DS je ve většině případů akutní megakaryoblastická leukemie [53,54]. Akutní megakaryoblastická leukemie představuje relativně vzácný subtyp AML. U dětí s DS je riziko rozvoje tohoto subtypu AML až 500x vyšší v porovnání se zdravou populací bez DS [40,45]. Akutní lymfoblastické leukemie u dětí s DS jsou téměř vždy B-prekurzorové [55]. Retrospektivní studie provedená autory Buitenkmp et al ukazuje, že ze souboru 708 ALL u DS pouze 5 případu tvořilo akutní lymfoblastické leukemie z T buněk [56]. Riziko rozvoje akutní lymfoblastické leukemie u dětí s DS je přibližně 20× vyšší než u dětí bez DS [40,52].
Odpověď na léčbu akutní megakaryoblastické leukemie u dětí s DS dosahuje lepších výsledků než léčba u dětí bez DS. Vyšší úspěšnost léčby je částečně způsobena zvýšenou in vitro citlivostí blastických elementů na cytosin arabinosid a daunorubicin a vyšší generací ara-C trifosfátu [57]. Míra relapsu AML u dětí s DS je o 20 % nižší než u dětí bez DS [57]. Opačně je tomu u akutní lymfoblastické leukemie, kde je prognóza u dětí s DS horší než u dětí bez DS [52]. Důvodem tohoto trendu je vyšší mortalita související s léčbou a vyšší počet relapsů onemocnění [56].
METODIKA
Přiložené kazuistiky demonstrují možnosti průběhu a vývoje tranzientního myeloproliferativního onemocnění. Kazuistiky doplňují laboratorní výsledky popisující danou fázi onemocnění (TMD/AML). Kazuistiky 1 a 2 demonstrují novorozence s TMD, u kterých došlo k samovolnému odeznění onemocnění. V kazuistice číslo 3 je zachycena progrese TMD do akutní megakaryoblastické leukemie.
Data obsažená v kazuistikách byla zpracována a anonymizována dle zásad GDPR se souhlasem etické komise FN Motol v rámci výzkumu incidence hemato-onkologických onemocnění v ČR EK-1278/21.
Hematologická vyšetření byla provedena na oddělení klinické hematologie Fakultní nemocnice Motol, analýza krevních obrazu byla provedena na analyzátorech Sysmex XE-5000 (Japonsko) a Sysmex XN-1000 a XN-3000 (Japonsko). Morfologické hodnocení nátěru periferní krve bylo provedeno pomocí digitální morfologie na přístroji CellaVisionTM DM96 Sysmex (Japonsko).
Sběr dat o incidenci Downova syndromu v ČR byl proveden z reportu Vrozené vady u narozených v roce 2004–2015 pod správou Ústavu zdravotnických informací a statistiky ČR, dostupné na: https: //www.uzis.cz/.
Sběr dat o počtu narozených dětí v ČR byl proveden z reportu Rodička a novorozenec v letech 1985–2015 pod správou Ústavu zdravotnických informací a statistiky ČR, dostupné na: https: //www.uzis.cz/.
Stanovení karyotypu bylo provedeno na Ústavu biologie a lékařské genetiky 2. LF UK a FN Motol pomocí metody G-pruhování.
KAZUISTIKY
Kazuistika 1
Pacient ročník 2015 přijat 8. 6. 2015 z novorozeneckého oddělení s diagnózou jiné předčasně narozené dítě. V krevním obraze významná leukocytóza (WBC 41,1×109/l), nátěr periferní krve bez přítomnosti blastických buněk. Dne 11. 6. 2015 přetrvávající mírná leukocytóza, v nátěru periferní krve přítomno 13,1 % blastických buněk (obr. 1–3).



Vyšetřením karyotypu byla zjištěna volná trizomie chromozomu 21 : 47,XY,+21 (obr. 4). Dne 27. 7. 2015 byl proveden kontrolní odběr, s normálními hodnotami krevního obrazu a bez přítomnosti blastických buněk. Hodnoty krevního obrazu a výsledky hodnocení nátěru periferní krve viz v tab. 2. Pacient byl následně předán do pozorování dětskému hematologovi.


Kazuistika 2
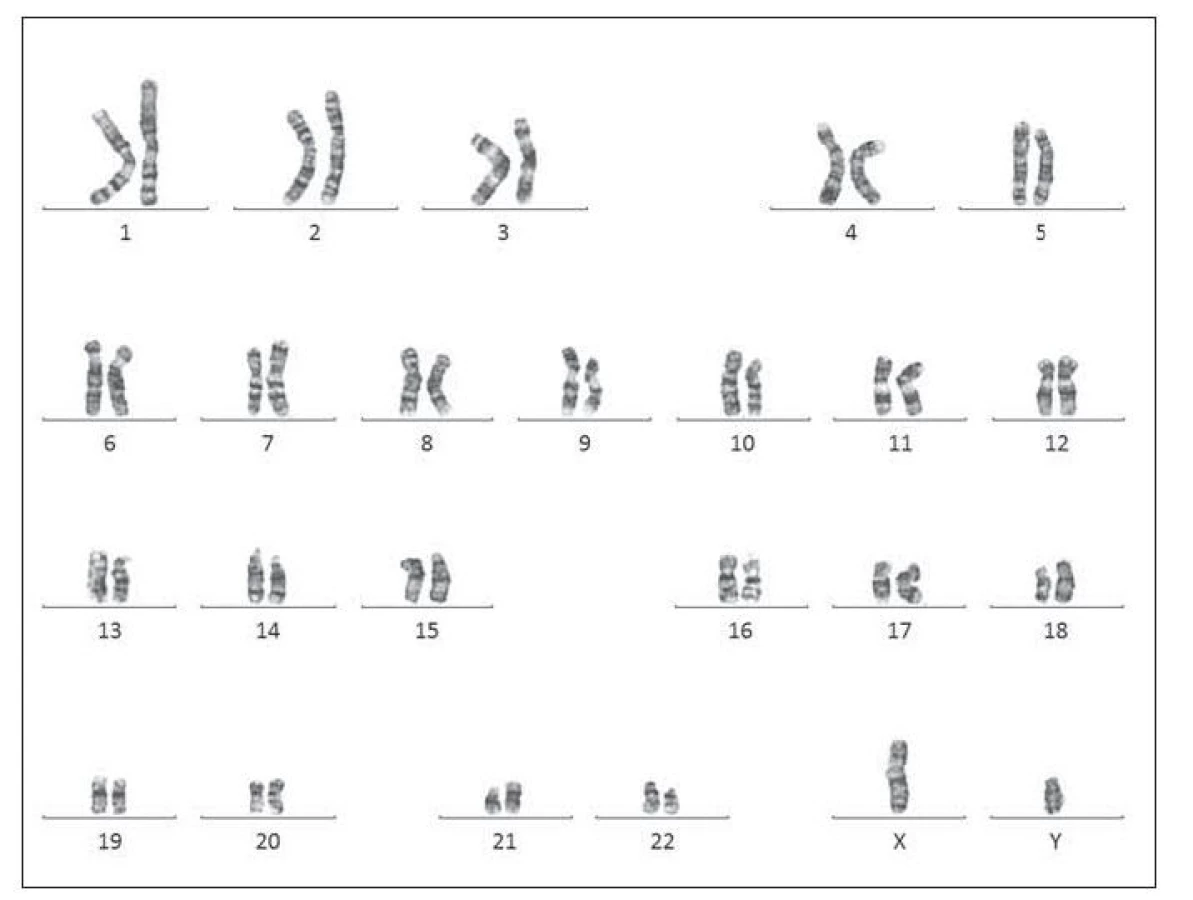
Pacient ročník 2011 přijat 20. 11. 2011 z Kliniky dětské hematologie a onkologie 2. LF UK a FN Motol s diagnózou kvalitativní poruchy trombocytů. Ve vstupním krevním obraze výrazná leukocytóza (WBC 59,6×109/l), mírná anémie (Hgb 132 g/l) a výrazná trombocytóza (PLT 1464×109/l), v nátěru periferní krve přítomno 71 % blastických buněk vzhledu megakaryoblastů (obr. 5–7). Vyšetření karyotypu vedlo k průkazu translokační formy DS 46,XY,t (21; 21) (q10; q10),+21 viz (obr. 8). V průběhu kontrolních odběrů postupný pokles počtu blastických buněk v nátěru periferní krve viz tab. 3, dne 2. 2. 20218 již bez nálezu blastických buněk. Pacient byl následně předán k pozorování dětskému hematologovi.




Kazuistika 3
Pacient ročník 2018 přijat 1. 2. 2018 (postnatální věk 2 dny) z Kliniky dětské hematologie a onkologie 2. LF UK a FN Motol s diagnózou DS. Ve vstupním krevním obraze výrazná leukocytóza (WBC 52,2×109/l), mírná anémie (Hgb 129 g/l) a trombocytóza (PLT 613×109/l), v nátěru periferní krve 46,4 % blastických buněk vzhledu megakaryoblastů (obr. 9–11). Dne 25. 2. 2018 (postnatální věk 26 dní) již bez nálezu blastických buněk v nátěru periferní krve. Dne 27. 11. 2019 pacient přijat na urgentní příjem pro děti a dorost ve FN Motol s diagnózou horečka, v krevním obraze leukocyty 2,8×109/l, hemoglobin 66 g/l, trombocyty 13×109/l, v nátěru periferní krve 0,6 % blastických buněk. Následující den provedeno vyšetření kostní dřeně z pravé kyčle – 22 % atypických buněk, stanovení dg. akutní megakaryoblastické leukemie při DS. Kontrolní vyšetření KD: D+28, D+30 a D+84 bez nálezu blastických buněk. Vývoj absolutního počtu blastických buněk viz. graf 4.




DISKUZE
Stanovení diagnózy tranzientního myeloproliferativního onemocnění je dle WHO je založeno na výskytu blastických elementů v nátěru periferní krve u novorozenců s DS. V současné době však neexistuje doporučení, které by určovalo minimální procentuální zastoupení blastických elementů potřebných pro stanovení diagnózy TMD [42]. Z tohoto důvodu je ve světě napříč odborníky používána hranice nejméně 1–10 % blastických elementů potřebných ke stanovení TMD u DS [58]. Goemans et al. [58] doporučují hodnotu 5 % blastických elementů v nátěru periferní krve (pomocí morfologického vyšetření nebo průtokové cytometrie), pro stanovení diagnózy TMD anebo přítomnost GATA1 mutace. Pětiprocentní hranice v jejich sledovaném souboru vedla k 58% senzitivitě a 100% specificitě pro imunofenotypizaci. Výsledky studie Goemans et al. dále zmiňují roli screeningového vyšetření GATA1 mutace v diagnostice TMD a ML-DS. Vyšetření GATA1 mutace vykazuje 95% senzitivitu a 100% specificitu při kombinaci metod klasického Sangerova skenování (Ss) a cíleného hlubokého sekvenování (NGS). Výsledky studie uvádí dva benefity plynoucí ze screeningového stanovení GATA1 mutace u novorozenců s DS. Prvním benefitem je možnost identifikace rizikové skupiny pacientů s DS, u které hrozí rozvoj akutní myeloidní leukemie. Všichni pacienti, u kterých došlo k rozvoji akutní myeloidní leukemie při DS, měli přítomnou GATA1 mutaci. Druhý benefit screeningového stanovení se zaměřuje na nepřítomnost GATA1 mutace, kdy pacienti bez GATA1 mutace mají riziko rozvoje akutní myeloidní leukemie velmi nízké [58].
Přínos screeningového vyšetření GATA1 mutace potvrzují Roberts et al. [42]. Z důvodu klasifikace TMD dle WHO založené pouze na přítomnosti neurčitého množství blastických elementů v periferní krvi u novorozenců s Downovým syndromem autoři využívají přítomnost GATA1 mutace jako druhé kritérium pro stanovení diagnózy. Pro stanovení diagnózy TMD navrhují přítomnost ≥ 10 % blastických elementů a zároveň pozitivní vyšetření na GATA1 mutaci. Detekce GATA1 mutace při využití metod cíleného hlubokého sekvenování vede k vyšší záchytnosti a pomáhá identifikovat pacienty s nízkým počtem buněčných klonů s GATA1 mutací. Pomocí této metody byli identifikováni novorozenci s původně negativním nálezem GATA1 mutace, vyšetření pomocí méně citlivých metod, a dále novorozenci bez klinických a hematologických známek TMD [42].
Závěry obou studií však poukazují především na informativní či diagnostickou hodnotu screeningového vyšetření GATA1 mutace u novorozenců s Downovým syndromem. Klinická hodnota screeningového testování je ovlivněna absencí doporučení zahrnující péči o pacienty s „pre-leukemickým“ stavem. Jediný klinický benefit vycházející ze znalosti GATA1 mutace spočívá v častějších kontrolách u dětského hematologa, s možným včasným záchytem rozvoje akutní myeloidní leukemie a zahájením příslušné léčby [42,58].
U všech pacientů v přiložených kazuistikách probíhalo klinicky a hematologicky zjevné TMD. Pacienti zároveň splňovali doporučení obou autorů na hodnotu procentuálního zastoupení blastických elementů v nátěru periferní krve a prokázání GATA1 mutace. V případě kazuistiky č. 1 byla vstupní hodnota blastických elementů 13,1 %, kazuistika 2 – 71 % a kazuistika 3 – 46,6 % blastických elementů. Kazuistika 1 a 2 demonstrují průběh TMD s postupným samovolným vymizením onemocnění. Pacienti jsou i nadále v péči dětského hematologa s pravidelnou observací. Kazuistika číslo 3 popisuje TMD s progresí do akutní megakaryoblastické leukemie. U pacienta došlo k progresi do akutní megakaryoblastické leukemie za 22 měsíců od stanovení diagnózy TMD. Při automatickém morfologickém hodnocení nátěru periferní krve (pomocí digitální morfologie CellaVision DM96 Sysmex) bylo u pacienta zastiženo pouze 0,6 % „podezřelých buněk“ tj. 1 buňka na 179 hodnocených leukocytů. Tento případ též poukazuje, že hodnocení nátěru periferní krve u pacientů s DS by měl provádět laboratorní pracovník se zkušenostmi s hodnocením nátěrů periferních krví u novorozenců a dětí, vč. infekčních a septických stavů. Morfologické odlišení mladších forem leukocytů při infekčním či septickém stavu od mladších forem leukocytů při patologickém procesu může být v řadě případů složité, obzvláště v případech, kdy je přítomnost patologických buněk v nátěru periferní krve jen ojedinělá.
V České republice se vyšetření GATA1 mutace provádí až v případě, kdy je pacient s Downovým syndromem v péči dětského hematologa. Přínosnost screeningového stanovení této mutace u všech novorozenců s Downovým syndromem se mezi oslovenými odborníky v České republice liší. Je potřeba se zamyslet, zda má vyšetření GATA1 mutace pozitivní přínos pro pacienta. V případě klinicky a hematologicky „zjevného“ TMD dojde k diagnostice tohoto stavu v rámci několika dní po porodu. Otázkou je, bude-li u pacienta probíhat klinicky a hematologicky „tichý“ TMD, bude znalost této informace využitelná pro následnou lékařskou péči? V současné době neexistují klinické postupy, které by upravovaly péči u „pre-leukemických stavů“. I v případě nepřítomnosti GATA1 mutace jsou děti s DS nadále ohroženy vyšším rizikem rozvoje akutní lymfoblastické leukemie [58].
ZÁVĚR
Z důvodu vyššího výskytu MDS a AL u pacientů s DS představuje tento syndrom jak důležitou součást práce dětského hematologa, tak výzvu v oblasti laboratorní hematologie. V případě podezření/prokázání DS u novorozence by mělo být provedeno hodnocení nátěru periferní krve maximálně do tří až sedmi dnů po porodu. Hodnocení by měl provádět zkušený laboratorní pracovník se znalostmi nátěrů periferních krví od novorozenců, vč. předčasně narozených novorozenců či novorozenců v septickém stavu.
Definice WHO je založena pouze na průkazu přítomnosti neurčitéhomnožství blastických elementů v PK u novorozence s DS. v PK u novorozence s DS. Průkaz výskytu blastických elementů může být zatížen schopnostmi hodnotitele či jiným stavem nesouvisejícím s DS. Řada případů TMD proběhne bez zjevných klinických projevů nebo detekovatelných odchylek hematologických parametrů. Z tohoto důvodu vyvstává otázka budoucí aktualizace a revidace klasifikace dle WHO. Nová klasifikace by měla jasně stanovovat kritéria pro diagnózu TMD a měla by být rozšířena o další kritéria, než je pouze přítomnost neurčitého množství blastických elementů u pacientů s DS. Mezi další rozšiřující kritéria by mohla být zařazena přítomnost, či nepřítomnost GATA1 mutace. Otázka screeningového stanovení GATA1 mutace u všech novorozenců s DS je otázkou, která by mohla být předmětem dalšího zkoumání, zvláště ve spojení s klinickým benefitem pro pacienta. V tuto chvíli je screeningové stanovení GATA1 mutace spojeno spíše s diagnostickým nežli klinickým benefitem.
SEZNAM ZKRATEK
AL – akutní leukemie
AMA – pokročilý věk matky (advanced maternal age)
CRL – temeno-kostrční délka (crown rump lenght)
DS – Downův syndrom
Hgb – hemoglobin
MDS – myelodysplastický syndrom
MEP – megakaryocytární a erytroidní prekurzory
ML-DS – myeloidní leukemie při Downově syndromu
NB – vyšetření přítomnosti/nepřítomnosti nosní kůstky
NGS – cílené hluboké skenování (next generation sequencing)
NRBC – normoblast
NT – nuchální translucence
PAPP-A – specifický těhotenský protein A (pregnancy associated plasma protein-A)
PIGF – placentární růstový faktor (placental growth factor)
PK – periferní krev
PLT – trombocyty (platelets)
Ss – Sangerovo skenování
TMD – tranzientní myeloproliferativní onemocnění (transient myeloproliferative disease)
tt – týden těhotenství
uE3 – volný estradiol
UZ – ultrazvukové markery/metody
WBC – leukocyty (white blood cells)
WHO – Světová zdravotnická organizace (World Health Organization)
PODÍL AUTORŮ NA PŘÍPRAVĚ RUKOPISU
KL – koncept, kazuistiky a příprava rukopisu
ZH – příprava rukopisu
KH – revize rukopisu, konečné schválení
ČESTNÉ PROHLÁŠENÍ
Autoři práce prohlašují, že v souvislosti s tématem, vznikem a publikací tohoto článku nejsou ve střetu zájmů a vznik ani publikace článku nebyly podpořeny žádnou farmaceutickou firmou.
PODĚKOVÁNÍ
Rádi bychom poděkovali vážené paní prim. MUDr. Jitce Segethové z OKH FN Motol, za umožnění přístupu k datům potřebných pro zpracování jednotlivých kazuistik a za podporu během zpracovávání článku.
Doručeno do redakce dne: 3. 2. 2022.
Přijato po recenzi dne: 14. 3. 2022.
Mgr. Lukáš Kolařík, DiS.
Oddělení klinické hematologie
FN Motol
V Úvalu 84, 150 06 Praha 5
e-mail: lukas.kolarik@fnmotol.cz
Zdroje
1. Coppedé F. Risk factors for Down syndrome. Arch Toxicol. 2016; 90 (12): 2917–2929.
2. Antonarakis SE, Skotko BG, Rafii MS, et al. Down syndrome. Nature Rev Dis Primers. 2020; (6): 1–49.
3. Down JLH. Observations on an Ethnic Classification of Idiots. In: London Hospital Reports, 3 : 1866 : 259–262.
4. Antonarakis SE, Petersen MB, McInnis MG, et al. The meiotic stage of nondisjunction in trisomy 21: determination by using DNA polymorphisms. Am J Hum Genet. 1992; 50 (3): 544–550.
5. Muntau AC. Autozomální aberace autozomů. In: Muntau AC. Pediatrie. 6. vyd. Praha, Grada Publishing, 2009 : 39–42.
6. Niikawa N, Kajii T. The origin of mosaic Down syndrome: four cases with chromosome markers. Am J Hum Genet. 1984; 36 (1): 123–130.
7. Brock DJ, Sutcliffe RG. Alpha-fetoprotein in the antenatal diagnosis of anencephaly and spina bifida. Lancet. 1972; 2 (7770): 197–199.
8. Wald N, Stone R, Cuckle HS, et al. First trimester concentrations of pregnancy associated plasma protein A and placental protein 14 in Down‘s syndrome. BMJ. 1992; 305 (6844): 28.
9. Cuckle HS, Wald NJ, Lindenbaum RH. Maternal serum alpha-fetoprotein measurement: a screening test for Down syndrome. Lancet. 1984; 1 (8383): 926–929.
10. Bashore RA, Westlake JR. Plasma unconjugated estriol values in high-risk pregnancy. Am J Obstet Gynecol. 1977; 128 (4): 371–380.
11. David M, Merksamer R, Israel N, et al. Unconjugated estriol as maternal serum marker for the detection of Down syndrome pregnancies. Fetal Diagn Ther. 1996; 11 (2): 99–105.
12. Aitken DA, Wallace EM, Crossley JA, et al. Dimeric inhibin A as a marker for Down‘s syndrome in early pregnancy. N Engl J Med. 1996; 334 (19): 1231–1236.
13. Wald NJ, Rodeck C, Hackshaw AK, et al. First and second trimester antenatal screening for Down‘s syndrome: the results of the Serum, Urine and Ultrasound Screening Study (SURUSS). Health Technol Assess. 2003; 7 (11): 1–77.
14. Stark A. Down syndrome: Advances in biomedicine and behavioral science. In: Püschel SM, Rynders JE. Dentistry. Cambridge,1982 : 198–203.
15. Goodman RM., Gortin JR. Down Syndrome (mongolism). In: Goodman RM., Gortin JR. The malformed infant and child: an illustrated guide, New York, Oxford University, 1983 : 122–123.
16. Barnett ML, Friedman D, Kastner T. The prevalence of mitral valve prolapse in patients with Down‘s syndrome: implications for dental management. Oral Surg Oral Med Oral Pathol. 1988; 66 (4): 445–447.
17. Hawli Y, Nasrallah M, El-Hajj Fuleihan G. Endocrine and musculoskeletal abnormalities in patients with Down syndrome. Nat Rev Endocrinol. 2009; 5 (6): 327–334.
18. Akhtar F, Bokhari SRA. Down Syndrome. StatPearls, publikováno elektronicky 12. prosince 2021. PMID 30252272.
19. Arya R, Kabra M, Gulati S. Epilepsy in children with Down syndrome. Epileptic Disord. 2011; 13 (1): 1–7.
20. Pueschel SM, Louis S, McKnight P. Seizure disorders in Down syndrome. Arch Neurol. 1991; 48 (3): 318–320.
21. Janicki MP, Dalton AJ. Prevalence of dementia and impact on intellectual disability services. Ment Retard. 2000; 38 (3): 276–288.
22. Downův syndrom, Vrozené vady [Online]. http: //www.vrozene-vady.cz/vrozene-vady/index.php?co=downuv_syndrom. Citováno dne: 20.11.2021
23. Šídlo L, Štastná A, Kocourková J, et al. Vliv věku matky na zdravotní stav novorozenců v Česku. Demografie. 2019; 61 : 154–174.
24. Šípek A., Gate2Biotech. Proč se zvyšuje četnost Downova syndromu? [Online] http: //www.gate2biotech.cz/proc-se-zvysuje-cetnost-downova-syndromu/. Citováno dne: 20.11.2021
25. Ústav zdravotnických informací a statistiky ČR. Vývoj počtu živě narozených podle věku matky. Rodička a novorozenec 2014-2015. 2017 : 36-37.
26. Ústav zdravotnických informací a statistiky ČR. Vrozené vady rok 2004. Zdravotnické ročenka ČR 2005. 2006 : 74.
27. Ústav zdravotnických informací a statistiky ČR. Vrozené vady rok 2005. Zdravotnické ročenka ČR 2006. 2007 : 74.
28. Ústav zdravotnických informací a statistiky ČR. Vrozené vady rok 2006. Zdravotnické ročenka ČR 2007. 2008 : 74.
29. Ústav zdravotnických informací a statistiky ČR. Vrozené vady rok 2007. Zdravotnické ročenka ČR 2008. 2009 : 74.
30. Ústav zdravotnických informací a statistiky ČR. Vrozené vady rok 2008. Zdravotnické ročenka ČR 2009. 2010 : 74.
31. Ústav zdravotnických informací a statistiky ČR. Vrozené vady rok 2009. Zdravotnické ročenka ČR 2010. 2011 : 76.
32. Ústav zdravotnických informací a statistiky ČR. Vrozené vady rok 2010. Zdravotnické ročenka ČR 2011. 2012 : 76.
33. Ústav zdravotnických informací a statistiky ČR. Vrozené vady rok 2011. Zdravotnické ročenka ČR 2012. 2013 : 76.
34. Ústav zdravotnických informací a statistiky ČR. Vrozené vady rok 2012. Zdravotnické ročenka ČR 2013. 2014 : 76.
35. Ústav zdravotnických informací a statistiky ČR. Vrozené vady rok 2013. Zdravotnické ročenka ČR 2014. 2016 : 63.
36. Ústav zdravotnických informací a statistiky ČR. Vrozené vady rok 2014. Zdravotnické ročenka ČR 2015. 2016 : 63.
37. Ústav zdravotnických informací a statistiky ČR. Vrozené vady rok 2015. Zdravotnické ročenka ČR 2016. 2017 : 60.
38. Henry E, Walker D, Wiedmeier SE, et al. Hematological abnormalities during the first week of life among neonates with Down syndrome: Data from a multihospital healthcare system. Am J Med Gen Part A. 2007; 143A (1): 42–50.
39. Gamis AS, Smith FO. Transient myeloproliferative disorder in children with Down syndrome: clarity to this enigmatic disorder. Br J Haematol. 2012; 159 (3): 277–287.
40. Mateos MK, Barbaric D, Byatt S-A, Sutton R, Marshall GM. Down syndrome and leukemia: insights into leukemogenesis and translational targets. Translat Pediatr. 2015; 4 (2): 76–92.
41. Vardiman JW, Thiele J, Arber AD, et al. The 2008 revision of the World Health Or - ganization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009; 114 (5): 937–951.
42. Roberts I, Alford K, Hall G, et al. GATA1-mutant clones are frequent and often unsuspected in babies with Down syndrome: identification of a population at risk of leukemia. Blood. 2013; 122 (24): 3908–3917.
43. Swerdlow SH, Campo E, Harris NL, et al. Myeloid proliferations associated with Down syndrome. In: Arber DA, Beumann I, Niemyer CM. WHO classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. Lyon, IARC, 2017 : 169–170.
44. Brink DS. Transient leukemia (transient myeloproliferative disorder, transient abnormal myelopoiesis) of Down syndrome. Adv Anat Pathol. 2006; 13 (5): 256–262.
45. Hasaart KAL, Bertrums EJM, Goemans BF, et al. Increased risk of leukaemia in children with Down syndrome: a somatic evolutionary view. Exp Rev Mol Med. 2021; 23: e5.
46. Massey GV, Zipursky A, Chang MN, et al. A prospective study of the natural history of transient leukemia (TL) in neonates with Down syndrome (DS): Children‘s Oncology Group (COG) study POG-9481. Blood. 2006; 107 (12): 4606–4613.
47. Grimm J, Heckl D, Klusmann J-H. Molecular mechanisms of the genetic predisposition to acute megakaryoblastic leukemia in infants with Down syndrome. Front. Oncology. 2021; 11 : 1-14
48. Tunstall O, Bhatnagar N, Beki J, et al. Guidelines for the investigation and management of transient leukaemia of Down syndrome. Br J Haematol. 2018; 182 (2): 200–211.
49. Caldwell JT, Yubin GE, Taub JW. Prognosis and management of acute myeloid leukemia in patients with Down syndrome. Exp Rev Hematol. 2014; 7 (6): 831–840.
50. Hayashi Y, Eguchi M, Sugita K, et al. Cytogenetic findings and clinical features in acute leukemia and transient myeloproliferative disorder in Down‘s syndrome. Blood. 1988; 72 (1): 15–23.
51. Ropper AH, Bull MJ. Down Syndrome. New Engl J Med. 2020; 382 (24): 2344–2352.
52. Brown AL, Smith AJ, Gant V, et al. Inherited genetic susceptibility to acute lymphoblastic leukemia in Down syndrome. Blood. 2019; 134 (15): 1227–1237.
53. Mast KJ, Taub JW, Alonzo TA, et al. Pathologic features of Down syndrome myelodysplastic syndrome and acute myeloid leukemia: a report from the Children‘s Oncology Group Protocol AAML0431. Arch Pathol Lab Med. 2020; 144 (4): 466–472.
54. Lange B. The management of neoplastic disorders of haematopoeisis in children with Down‘s syndrome. Br J Haematol. 2000; 110 (3): 512–524.
55. Roberts I, Izraeli S. Haematopoietic development and leukaemia in Down syndrome. Br J Haematol. 2014; 167 (5): 587–599.
56. Buitenkamp TD, Izraeli S, Zimmermann M, et al. Acute lymphoblastic leukemia in children with Down syndrome: a retrospective analysis from the Ponte di Legno study group. Blood. 2014; 123 (1): 70–77.
57. Taub JW, Huang X, Matherly LH, et al. Expression of chromosome 21-localized genes in acute myeloid leukemia: Differences between Down syndrome and non-Down syndrome blast cells and relationship to in vitro sensitivity to cytosine arabinoside and daunorubicin Blood. 1999; 94 (4): 1393–1400.
58. Goemans BF, Noort S, Blink M, et al. Sensitive GATA1 mutation screening reliably identifies neonates with Down syndrome at risk for myeloid leukemia. Leukemia. 2021; 35 (8): 2403–2406
Štítky
Hematologie a transfuzní lékařství Interní lékařství OnkologieČlánek vyšel v časopise
Transfuze a hematologie dnes

2022 Číslo 2
- Diosmin a hesperidin: Co ukazuje farmakokinetika?
- Evaluace skóre m7-FLIPI u pacientů s folikulárním lymfomem odhalila slibný prediktivní biomarker k určení vhodné chemoterapie
- Neuropatie výrazně zpomaluje proces hojení u povrchových ran
- Diagnostika von Willebrandovy choroby krok za krokem
- Těhotenství a porod u ženy s VWD − kazuistika
Nejčtenější v tomto čísle
- Význam Downova syndromu v hematologii
- Mutace v epigenetických regulátorech – potenciální prognostické markery a terapeutické cíle u akutní myeloidní leukemie
- Novinky v translačním výzkumu akutní lymfoblastické leukemie – výběr z konference Evropské hematologické školy
- Real-world dáta ohľadom účinnosti a bezpečnosti ibrutinibu a venetoklaxu u pacientov s chronickou lymfocytovou leukémiou, skúsenosti jedného centra
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy