
The Immune Adaptor ADAP Regulates Reciprocal TGF-β1-Integrin Crosstalk to Protect from Influenza Virus Infection
Infection of avian influenza virus, especially the highly pathogenic strain H5N1, is a serious threat to public health worldwide, which causes severe fatal respiratory disease and excessive levels of inflammation. It has been reported that both transforming growth factor-beta 1 (TGF-β1) and the integrin CD103 induced by TGF-β1 play protective roles in influenza virus infections. We aimed to find which protein regulates the TGF-β1-integrin cross-talk to protect against H5N1 virus infection. This study provides the first evidence that the intracellular signaling protein ADAP (adhesion and degranulation-promoting adapter protein) up-regulates TGF-β1 production and TGF-β1 induced CD103 expression in CD8+ T cells via the TβRI-TRAF6-TAK1-SMAD3 pathway. Importantly, in response to H5N1 and H1N1 virus infection, ADAP deficiency decreases TGF-β1 production and CD103 expression in lung infiltrating CD8+ T cells with the enhanced mortality in mice. Since various SNPs or mutations in key molecules of TGF-β1 pathway, including polymorphisms located in ADAP/FYB gene, are associated with inflammatory diseases, future work should investigate whether these SNPs or mutations enhance disease susceptibility or clinical manifestations in response to acute influenza virus infection.
Published in the journal:
. PLoS Pathog 11(4): e32767. doi:10.1371/journal.ppat.1004824
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004824
Summary
Infection of avian influenza virus, especially the highly pathogenic strain H5N1, is a serious threat to public health worldwide, which causes severe fatal respiratory disease and excessive levels of inflammation. It has been reported that both transforming growth factor-beta 1 (TGF-β1) and the integrin CD103 induced by TGF-β1 play protective roles in influenza virus infections. We aimed to find which protein regulates the TGF-β1-integrin cross-talk to protect against H5N1 virus infection. This study provides the first evidence that the intracellular signaling protein ADAP (adhesion and degranulation-promoting adapter protein) up-regulates TGF-β1 production and TGF-β1 induced CD103 expression in CD8+ T cells via the TβRI-TRAF6-TAK1-SMAD3 pathway. Importantly, in response to H5N1 and H1N1 virus infection, ADAP deficiency decreases TGF-β1 production and CD103 expression in lung infiltrating CD8+ T cells with the enhanced mortality in mice. Since various SNPs or mutations in key molecules of TGF-β1 pathway, including polymorphisms located in ADAP/FYB gene, are associated with inflammatory diseases, future work should investigate whether these SNPs or mutations enhance disease susceptibility or clinical manifestations in response to acute influenza virus infection.
Introduction
H5N1 influenza viruses are highly pathogenic avian influenza (HPAI) virus, which also infect humans and cause fatal human respiratory diseases [1, 2]. Numerous animal or clinical studies have indicated that virus-induced cytokine dysregulation is one central reason for H5N1 pathogenesis and disease severity [2–4]. Compared with the influenza virus subtype H1N1, H5N1-infected patients showed unusually high serum levels of chemokines and inflammatory cytokines. And, H5N1-infected patients who died had higher serum levels of these mediators than those who survived [1, 2]. Mouse studies also suggest that sustained induction of the inflammatory response after H5N1 virus infection is correlated with the ability of H5N1 virus to disseminate to extrapulmonary organs [5]. Although the appearance of cytokine storm (also termed hypercytokinemia) is one of the most important features of H5N1 and H1N1 immunopathogenesis, mouse models deficient of cytokines IL-6 or MIP-1α show comparable mortality as influenza-challenged wild type controls [4, 6]. This might be due to the redundancy between cytokines or chemokines. Therefore, identification of other strategies to prevent cytokine storm or damage of respiratory tract is crucial and should shed new light on effective clinical anti-H5N1 or H1N1 treatment.
Transforming growth factor β1 (TGF-β1) is the predominant member of the TGF-β isoforms in the immune system, and controls initiation and resolution of inflammation during multiple pathogenic progress, including influenza virus infection [7, 8]. TGF-β is secreted as an inactive precursor, which cannot bind to its receptor in its latent form (i.e. LTGF-β) [9]. The neuraminidase glycoprotein (NA) of most influenza strains including H1N1 could activate latent TGF-β (LTGF-β), while H5N1 cannot activate latent TGF-β [10, 11]. In agreement with the protective role of TGF-β during influenza virus infection, exogenous TGF-β delays mortality in H5N1-infected mice, and depletion of TGF-β increases mortality of H1N1 infection [10]. We aimed to dissect new mechanism of TGF-β production in host cells to ameliorate H5N1 or H1N1 virus infection.
Integrins have been identified to play critical roles in the regulation of TGF-β1 activation. Furthermore, once activated TGF-β1 binds to its heterodimeric receptor complex TβRI/II to induce SMAD-dependent or independent pathways, expression of multiple integrins is enhanced, including CD103 (also termed αEβ7) and VLA-1 (very late antigen-1, also termed α1β1) [12]. CD103 is preferentially expressed on CD8+ T cells specific for influenza but not for EBV or CMV, and maintains CD8+ T cells in lungs by binding to its ligand epithelial cadherin (E-Cadherin), which enhances immune surveillance to clear influenza infection [13, 14]. Despite of this, it remains to be elucidated which cytoplasmic effector(s) plays an indispensable role in the control of reciprocal TGF-β1-integrin crosstalk.
We and others have demonstrated that upon infection or chemokine stimulation, T cells use the immune adaptor protein ADAP (adhesion and degranulation-promoting adapter protein, also known as Fyn-binding protein [Fyb] or SLP-76 associated protein of 130 kD [SLAP-130]) to increase β2 integrin activation and adhesion [15–17]. However, it remains unclear whether ADAP regulates the expression of TGF-β1, CD103 and VLA-1 or controls the reciprocal TGF-β1-integrin crosstalk during influenza infection. In this study, we provided the first evidence that ADAP was indispensable for autocrine TGF-β1 production and CD103 expression via the TβR-TRAF6-ADAP-TAK1-SMAD3 pathway. Importantly, ADAP knock-out (KO) mice as well as Rag1 KO mice receiving ADAP KO T cells enhanced mortality in response to H5N1 or H1N1 infection, which displayed severe cytokine storm and lung damages with reduced levels of TGF-β1, CD103 and VLA-1 in CD8+ T cells.
Results
ADAP regulates TGF-β1 production in T cells via an autocrine manner
ADAP is an intracellular hematopoietic-specific adaptor protein that is constitutively and mainly expressed in T lymphocytes. Multiple studies have demonstrated that ADAP plays a crucial role in enhancing β2 integrin activation in response to antigen or chemokine stimulation [15, 16, 18, 19]. Since TGF-β1 and integrins have close relationship and function on each other, we asked whether ADAP regulated the crosstalk between integrins and TGF-β1. We first examined whether ADAP regulated TGF-β1 production in CD8+ cytotoxic T lymphocytes (CTLs). ADAP-/- mice were crossed with OT1 TCR transgenic (OT1 Tg) mice to obtain CD8+ Tg cells, which express specific TCR for recognizing OVA257-264 peptide. Splenocytes from wild type or ADAP-/- OT1 mice were incubated with OVA257-264 peptide for 7 days to generate OVA257-264-specific CD8+ CTLs. LAP (TGF-β1) is TGF-β1 precursor containing LAP (latency-associated peptide) and anti-LAP (TGF-β1) antibody is used to detect the latent form of TGF-β1. Compared to that of wild type cells, we observed that OVA-stimulated ADAP-/- CD8+ CTLs significantly reduced TGF-β1 production at precursor levels by anti-LAP (TGF-β1) staining (7.64% vs. 24.61%) or at mRNA levels by RT-PCR analysis (Fig 1A and 1B). Next, ADAP was overexpressed in human C8166 T cell line, followed by stimulation with anti-CD3/CD28 antibodies. ADAP overexpression enhanced LAP (TGF-β1) surface expression compared to the GFP controls (Fig 1C, 74% vs. 43.7%). In agreement with this observation, wild type splenocytes enhanced LAP (TGF-β1) production after anti-CD3/CD28 stimulation, while ADAP-/- splenocytes failed to increase LAP (TGF-β1) production (1.29% vs. 13.8%, Fig 1D). Next, we measured the mature/active form and the inactive form of TGF-β1 by ELISA assay in wild type and ADAP-/- CD8+ CTLs after stimulation with OVA257-264 peptide and exogenous TGF-β1. According to the manufactory’s instruction, the mouse ELISA kit recognizes the mature/active form of TGF-β1. After samples received acid treatment and neutralization to remove LAP from TGF-β1 precursor, both active and inactive TGF-β1 in the supernatants could be detected by ELISA assay. We found that in response to OVA257-264 peptide and exogenous TGF-β1 stimulation, ADAP deficient CD8+ CTLs indeed decreased both active and inactive TGF-β1 concentrations (Fig 1E).
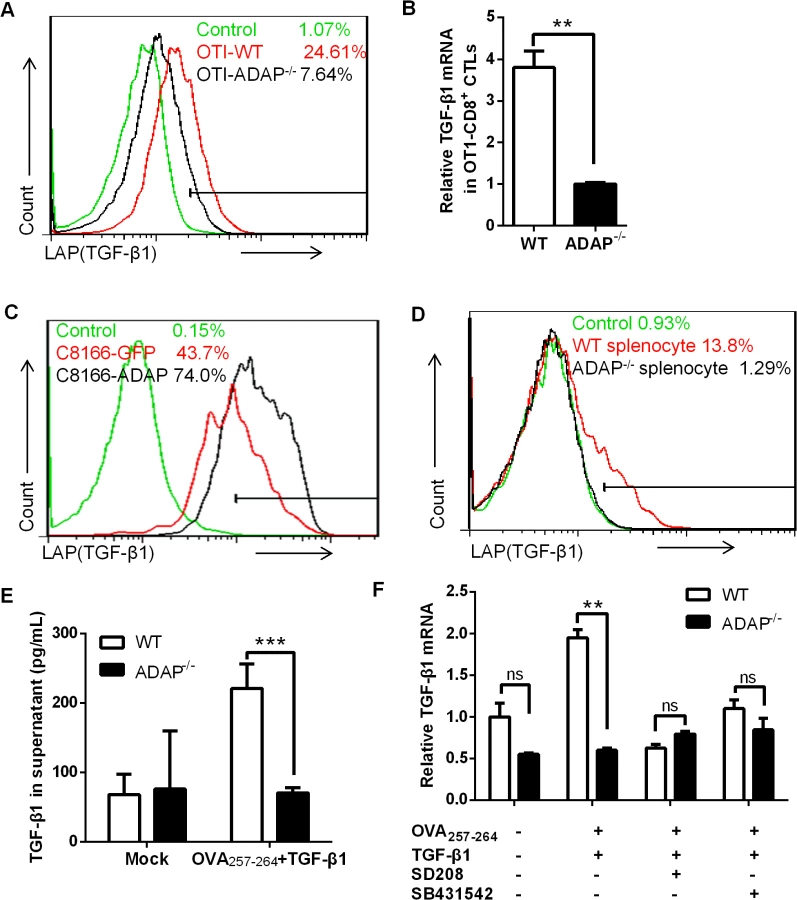
It has been demonstrated that TGF-β1 production is regulated by an auto-feedback loop via TGF-β receptor I/II (TβRI/II) [20, 21]. TGF-β1 binds to a type II receptor, which recruits and phosphorylates the type I receptor TβRI. We observed that treatment with exogenous TGF-β1 enhanced the mRNA levels of TGF-β1 in OVA257-264-stimulated wild-type CD8+ OT1 Tg cells, while ADAP-/- CD8+ OT1 Tg cells did not show this enhancement (Fig 1E). Next, the TGF-β receptor I kinase inhibitor SD208 or SB431542 was used to inhibit TGF-β signaling. In the presence of SD208 or SB431542, exogenous TGF-β1 failed to effectively induce TGF-β1 production in wild type cells, which was reduced to similar levels as that in ADAP-/- OT1 Tg cells (Fig 1F). Together, we suggest ADAP as a critical intracellular effector for autocrine TGF-β1 production in T cells, which is dependent on TβRI kinase activity.
ADAP enhances TGF-β1/TβRI signaling via TRAF6-TAK1-SMAD3
Upon TGF-β1 stimulation, TβRI recruits and phosphorylates R-SMADs (receptor-regulated Smads, i.e. SMAD2 or SMAD3). R-SMADs then bind the common SMAD4 and enter the nucleus as transcription factors to induce expression of various genes, including TGF-β1 itself. Given that ADAP was involved in TGF-β1/TβRI signaling for the induction of TGF-β1 (Fig 1), we asked whether ADAP regulated the TβRI/II-induced SMAD activity. Jurkat T cells were transfected with GFP control or ADAP-GFP together with the (CAGA)12-luciferase or (SBE)4-luciferase reporters respectively, which contain SMAD binding sequences [22, 23]. Exogenous TGF-β1 treatment increased the luciferase activity compared to untreated samples; and compared to the GFP control, ADAP overexpression further enhanced this by about 30% (Fig 2A, left two panels). Next, wild type or ADAP deficient Jurkat cells (i.e. termed JDAP) were transfected with the (CAGA)12-luciferase or (SBE)4-luciferase reporters respectively. In response to exogenous TGF-β1 treatment, wild type Jurkat cells increased the luciferase activity around two-folds compared to that in JDAP cells (Fig 2A, right two panels).

To further confirm whether ADAP regulated the transcriptional activity of R-SMAD, we measured the phosphorylation levels of R-SMAD. Compared to OVA257-264-stimulated wild type CD8+ OT1 Tg cells, ADAP-/- CD8+ OT1 Tg cells induced lower levels of phosphorylated SMAD3 (p-SMAD3 ser 423/425) in response to exogenous TGF-β1 stimulation (Fig 2B, the relative p-SMAD3/actin ratio was shown in the right panel). ADAP deficiency did not affect the total amount of SMAD3 expression. In addition, nuclear and cytoplasmic extracts from wild type and ADAP-/- CD8+ OT1 Tg cells were used to quantify the amount of nuclear localized SMAD3. TGF-β1 treatment induced SMAD3 nuclear translocation in wild type CD8+ CTLs, while ADAP deficiency decreased SMAD3 nuclear translocation compared to wild type cells (Fig 2C).
As an immune adaptor protein, ADAP does not have kinase or enzyme activity, but contains several domains (i.e. proline-rich domain, SH3 domain and multiple tyrosines) for recruitment of other signaling proteins. To explore how ADAP was involved in TGF-β1 signaling, we overexpressed ADAP with SMAD2 or SMAD3 in 293T cells respectively, but did not find direct interaction between ADAP and SMAD2 or SMAD3 (S1A Fig). We next examined whether ADAP regulated the non-canonical SMAD signaling that participate in TGF-β1 production. Previous studies have shown interesting feature of the PI3K/AKT pathway, the MAPKs (mitogen-activated protein kinases) including p38 and JNK (c-Jun N-terminal kinase) to modulate SMAD activity [24–28]. In response to TGF-β1 and OVA peptide stimulation, our representative data showed that wild type and ADAP-/- CD8+ OT1 Tg cells displayed similar levels of p-p38, p-JNK, the PI3K subunit p-p85 (Tyr458) and p-AKT (Ser473) (Fig 2D, the relative ratio compared to β-actin was shown in S1B Fig).
TAK1 (TGF-β activated kinase 1) is another non-canonical SMAD signaling protein to regulate SMAD3 activation [29]. The TβRI-TRAF6 interaction was previously identified for TGF-β-induced autoubiquitylation of TRAF6 and subsequent activation of TAK1 [30]. Interestingly, when GFP-tagged ADAP was co-transfected with Flag-tagged TRAF6 or HA-tagged TAK1 respectively in 293T cells, we observed direct binding of ADAP to TRAF6 (S1C Fig, left panel). Although no direct interaction between ADAP and TAK1, co-transfection of TRAF6 could bring ADAP and TAK1 into the same complex (S1C Fig, right panel). We next examined whether endogenous ADAP interacted with TRAF6 in primary CD8+ CTLs. Anti-TRAF6 immunorecipitation was performed followed by immunoblotting with antibodies against ADAP and TAK1. We confirmed that endogenous TRAF6 indeed forms a complex with ADAP and TAK1 in primary CD8+ T cells (Fig 2E).
To test whether TAK1 played a central role together with ADAP in TGF-β1 production, wild type and ADAP-/- CD8+ OT1 CTLs were stimulated with OVA peptide and exogenous TGF-β1 in the presence of the TAK1 inhibitor 5Z-7-oxozeaenol. The mRNA levels of TGF-β1 were profoundly decreased by 5Z-7-oxozeaenol treatment, and no difference was detected between wile type and ADAP-/- CD8+ OT1 CTLs (Fig 2F). Together, these data suggest that ADAP forms a signaling module with TRAF6 and TAK1 to enhance TGF-β1-induced SMAD3 activation and TGF-β1 production via an autocrine manner.
ADAP-/- mice enhanced mortality with reduced TGF-1 production in response to H5N1 virus infection
Since TGF-β has been identified to play a protective role during the influenza H5N1 virus infection [10], we asked whether and how ADAP regulated H5N1 virus infection. Previously reports showed that ADAP deficient mice have normal T cell development in thymus and normal percentages or numbers of mature peripheral T cells [15–17]. Wild type and ADAP-/- mice were challenged with the H5N1 strain A/DK/GuangXi/12/03 that was isolated from ducks in China (i.e. termed GX/12 in this paper) [31]. As reported earlier [31], the infected wild type mice started to recover from day 8 and remained survival. In contrast, the infected ADAP-/- mice markedly lost bodyweight and 100% died around day 10 post infection (Fig 3A). In the PBS-treated mock control groups, ADAP-/- mice and wild type mice showed normal lung structure without immune cell infiltration. At day 10 post infection, lungs from ADAP-/- mice were swollen with increased weight compared to those from wild type mice (Fig 3B). Substantially, lung structure was disrupted with massive infiltration of immune cells in the H5N1-infected ADAP-/- mice by H&E staining (Fig 3C). At day 10 post infection, the number of infiltrated CD8+ T cells into BAL (bronchoalveolar lavage) of lungs was significantly enhanced in the infected ADAP-/- mice examined by FACS analysis or by IHC staining (Fig 3D). The number of infiltrating CD8+ T into BAL was also examined at different time points after infection. At day 3–4 post infection, detectable number of infiltrating CD8+ T cells was noticed in lungs of wild type or ADAP-/- mice. At day 8–9, infiltration of wild type CD8+ T cells reached maximal levels, then declined with recovery of wild type mice from infection. By contrast, the number of infiltrating ADAP-/- CD8+ T cells was continuously and substantially increased around day 10 post-infection and ADAP-/- mice were unable to recover from infection (S2A Fig).

Despite of enhanced ADAP-/- CD8+ T lymphocytes in the infected lungs, concentrations and the mRNA levels of TGF-β1 in lungs were severely decreased in H5N1-infected ADAP-/- mice compared to those from wild type mice (Fig 3E). Serum TGF-β1 concentrations were also reduced from the infected ADAP-/- mice (Fig 3F). Since TGF-β1 plays a key role in the development of Foxp3+CD4+ Treg cells and Treg cells also inhibit excessive inflammatory responses, we examined the percentage of Treg cells in H5N1-infected ADAP-/- mice. ADAP deficiency did not affect the percentage of lung infiltrating Treg cells (S2B Fig). Previous studies suggested that ADAP deficient Treg cells mediated suppressive capacity at wild type levels when co-transferred with effector T cells into NOD/SCID mice to monitor recipients for diabetes; and ADAP deficient Treg cells inhibited TCR-stimulated effector T cell proliferation in vitro as that by wild type Treg cells [32]. Together, our findings show that after H5N1 virus infection, ADAP deficiency impairs TGF-β1 production with enhanced mortality and lung infiltrating CD8+ T cells.
Since TGF-β and ADAP have been demonstrated to regulate cell cycle and apoptosis [16–18, 33, 34], we examined whether T cell proliferation and apoptosis were affected in H5N1-infected ADAP-/- mice. The BrdU incorporation assay confirmed no significant difference of CD8+ T cell proliferation in lungs, MLN or spleens between the infected ADAP-/- and wild type mice (S3A, S3B and S3C Fig; left panels). Also, ADAP deficiency did not change CD8+ T cell apoptosis in lungs, MLN (mediastinal lymph-node), and spleens as examined by PI/Annexin V staining (S3A, S3B and S3C Fig; right panels). Interestingly, the number of CD8+ T cells was reduced in spleens of the infected ADAP-/- mice (S3D Fig).
Severe inflammation and lung damage of H5N1-infected ADAP-/- mice
In agreement with the impaired TGF-β1 production in CD8+ T cells and ADAP-/- lungs, we detected cytokine storm phenotype in H5N1-infected ADAP deficient mice. The relative mRNA levels of IL-1β, IL-6, TNFα and IFNγ were markedly increased in lungs from H5N1-infected ADAP-/- mice with no significant changes of IL-4 (Fig 4A). Besides TGF-β1, IL-10 could also inhibit cytokine production. However, we found no significant difference of IL-10 mRNA levels in lungs between H5N1-infected wild type and ADAP-/- mice (Fig 4A, right panel). Importantly, we also observed much higher levels of chemokines in lungs of H5N1-infected ADAP-/- mice, including CCL2/MCP-1 (Monocyte Chemotactic Protein-1), CCL3/MIP-1α (Macrophage Inflammatory Protein-1α), CCL5 and CXCL10/IP-10 (Interferon-γ-inducible Protein) (Fig 4B). This might attract more immune cells to migrate from spleen to lung (S3D Fig) and enhance the number of lung infiltrating ADAP-/- CD8+ T cells (Fig 3D).

Uncontrolled lung inflammation was account for the destructive structure of respiratory tract in H5N1-infected ADAP-/- mice, including alveoli, bronchioles and bronchus of lungs (Fig 4C). This suggests that ADAP deficiency induces a possible link between reduced TGF-β1 production and declined lung function with acute lung inflammation.
ADAP deficiency reduces TGF-1-induced CD103 expression in CD8+ T cells
TGF-β1 signaling has been reported to induce and maintain integrin CD103 expression preferentially on influenza-specific CD8+ T cells, and CD103+CD8+ T cells plays a protective role in the control of influenza infection [13]. We therefore examined CD103 expression on ADAP deficient CD8+ T cells after H5N1 infection. Compared to that of the H5N1-infected wild type mice, surface CD103 expression on ADAP-/- CD8+ T cells from BAL and MLNs was decreased (Fig 5A and 5B, percentages and MFI/Mean Fluorescence Intensity of CD103+ staining). The reduced CD103 mRNA levels were also observed in lungs of the infected ADAP-/- mice (Fig 5C, left panel). The expression of the CD103 ligand E-cadherin was not significantly affected by ADAP deficiency, which is mainly expressed in lung endothelial cells (Fig 5C, right panel).

Next, we used exogenous TGF-β1 and anti-CD3/CD28 treatment to induce CD103 expression in wild type or ADAP deficient CD8+ CTLs in vitro. The mRNA levels of CD103 were profoundly reduced in ADAP-/- CD8+ CTLs compared to those in wild type cells (Fig 5D). After treated with different concentrations of exogenous TGF-β1, surface CD103 expression was increased in wild type CD8+ T cells, while ADAP-/- CD8+ T cells failed to significantly increase TGF-β1-induced CD103 expression (Fig 5E, percentages and MFI of CD103+ staining). We then asked whether ADAP co-operated with the TβRI-TRAF6-TAK1 pathway for TGF-β1-induced CD103 expression. The TAK1 inhibitors 5Z-7-oxozeaenol and the TβRI kinase inhibitor SB431542 significantly reduced surface CD103 expression in wild type CD8+ T cells, which reached to the same levels as that in the ADAP-/- cells (Fig 5F). Since TRAF6-/- mice are embryonic lethal, we isolated CD8+ T cells from TRAF6 heterozygous mice and the wild type control mice followed by stimulation with anti-CD3/CD28 in the presence of different concentrations of TGF-β1. Compared to wild type cells, TRAF6 heterozygous CD8+ T cells reduced surface CD103 expression by about 15–20% (Fig 5G). Together, we propose that TβRI, TAK1, TRAF6 and ADAP are indispensable for TGF-β1-induced CD103 expression.
ADAP deficiency decreases VLA-1 expression in CD8+ T cells with increased amount of H5N1 virus
As another critical integrin, VLA-1 (very late antigen-1, also termed α1β1) was reported to regulate CD8+ T cell-mediated immune protection against influenza infection [14]. In addition, β1 integrin has been suggested for LAP-TGF-β1 activation [35]. Interestingly, at day 10 post H5N1 virus infection, ADAP-/- CD8+ CTLs in BAL reduced VLA-1 expression on cell surface (Fig 6A, percentages and MFI of VLA-1+ staining). The mRNA levels of the VLA-1 ligand VCAM-1 in the infected lungs from ADAP-/- mice were also decreased (Fig 6B).

In agreement with the protective function of CD103 and VLA-1 in CD8+ T cells, wild type mice controlled H5N1 virus replication; while higher amount of H5N1 was detected in lungs of the infected ADAP-/- mice at day 10 (Fig 6C, IHC staining with anti-H5N1 NP mAb). This was correlated with the reduced level of anti-viral cytokine IFN-β in lungs of the infected ADAP-/- mice (Fig 6D). The copy numbers of H5N1 NP gene were measured by RT-PCR at different time points after infection. The amount of H5N1 virus reached the maximal level around day 4, then significantly declined at day 6 in lungs of H5N1-infected wild type mice. Although ADAP-/- mice reduced the amount of H5N1 virus at day 6 compared to day 4, they were unable to clear virus effectively as wild type mice (S4 Fig).
Rag1-/- mice receiving ADAP-/- T cells reduces protection and CD103 expression in response to H5N1 virus infection
To confirm that ADAP deficiency in T cells represented the main reason to cause more severe H5N1 virus infection, we transferred wild type or ADAP-/- T cells into Rag1-/- recipient mice. The comparable percentages of CD8+ T cells were detected in MLN and spleens in these Rag1-/- mice, confirming a success of reconstitution of wild type or ADAP-/- T cells (S5 Fig). Four days after adaptive T-cell transfer, these mice were infected with the H5N1 virus strain GX/12. Rag1-/- mice received ADAP-/- T cells significantly reduced body weight, compared to those receiving wild type T cells (Fig 7A). HE staining showed that more immune cells were infiltrated into lungs of the Rag1-/- recipient mice that were adoptively transferred with ADAP-/- T cells (Fig 7B). Furthermore, the lung infiltrating ADAP-/- CD8+ T cells significantly reduced surface CD103 expression (Fig 7C). These findings have demonstrated that ADAP deficiency impairs T cell-mediated protection against H5N1 virus infection.

ADAP-/- mice enhance mortality with defective expression of TGF-1 and CD103 in response to H1N1 infection
To assess whether ADAP regulates infection of other influenza virus subtype (i.e. not HPAI), wild type and ADAP-/- mice were intranasally infected with the H1N1 virus strain A/PR8. Compared to wild type mice, ADAP-/- mice showed heightened mortality within shorter periods after A/PR8 infection (Fig 8A). We observed that the A/PR8-infected ADAP-/- mice had edematous lungs with enhanced weight and numbers of lung infiltrating immune cells (Fig 8B and 8C). At day 8 post infection, the mRNA level of viral PR8 in the lungs of ADAP deficient mice was significantly higher than that of wild type mice (Fig 8D). Moreover, the number of CD8+ T cells in BAL of lungs was significantly enhanced in the A/PR8-infected ADAP-/- mice examined by FACS or real-time PCR (Fig 8E). Similar to the results of H5N1 infection, concentrations of active and total TGF-β1 in lungs were severely decreased in H1N1-infected ADAP-/- mice compared to those from wild type mice (Fig 8F).

Previous studies have demonstrated that highly elevated IL-6, IL-1, TNF-α, IFN-γ, MIP, CXCL10 are major causes of ARDS (acute respiratory distress syndrome) and mortality [4, 6, 36, 37], and TGF-β1 could restrict inflammatory response. In agreement with the reduced TGF-β1 production, we detected markedly increased mRNA levels of IL-6, TNFα and IFNγ (Fig 8G) or CCL2, CCL3, CCL5 and CXCL10 (Fig 8H) in lungs from A/PR8-infected ADAP-/- mice. By contrast, IL-4 or IL10 expression was not substantially changed (Fig 8G). Similar to H5N1 infection, we also detected reduced surface CD103 levels in lung infiltrating ADAP deficient CD8+ T cells (Fig 8I). Taken together, ADAP enhances TGF-β1 production and signaling, and is indispensable for protection against H1N1 and H5N1 virus infection.
Discussion
As the highly pathogenic avian influenza (HPAI), H5N1 virus infection induces unbalanced host immune responses with highly elevated cytokines and significant lung pathology [36–38]. TGF-β1 is suggested to suppress cytokine storm during H5N1 virus infection [10]. Since HPAI loads and replicates in the respiratory tract for much longer time than seasonal influenza [2], viral antigen activated T cells become one of the major sources to produce TGF-β1. In this study, we observed the enhanced mortality in the H5N1 - or H1N1-infected ADAP KO mice and this could be possibly explained by three main findings including (1) defective TGF-β production and TGF-β signaling; (2) cytokine storm; (3) reduced CD103 expression. Previous studies suggest the protective role of TGF-β to control initiation and resolution of inflammation, while CD103+CD8+ T cell-mediated immune responses play vital roles in recovery from H5N1 virus infection [39]. Our study has provided the first evidence that CD8+ T cells secreted TGF-β1 in an autocrine manner via the TβRI-TRAF6/ADAP/TAK1-SMAD3 pathway (Fig 1). Further, ADAP forms a complex with TRAF6/TAK1 in CD8+ T cells to regulate SMAD3 activation and TGF-β-induced CD103 expression (Figs 2 and 4). Consistent with the in vitro findings, H5N1-infected ADAP KO mice reduce TGF-β production and CD103 expression (Figs 3 and 5). Moreover, the reduced TGF-β production was correlated to the elevated levels of inflammatory cytokines and chemokines in H5N1 - or H1N1-infected ADAP KO mice (Figs 3 and 8). We further used Rag1 KO mice that were adoptively transferred with ADAP KO T cells to demonstrate that ADAP deficient T cells play a direct role in protection against virus infection (Fig 7). These observations together suggest the enhanced mortality in ADAP KO mice was closely related to the defective TGF-β production and signaling. To demonstrate a direct link of the ADAP KO mortality phenotype to TGF-β, it is better to set a ‘rescue’ experiment provided by TGF-β1 or TGF-β1 receptor, which protects ADAP KO mice effectively as the wild type control mice. Since ADAP deficient T cells not only decreased TGF-β1 production, but also poorly responded to TGF-β1 signaling, we were unable to expect a miraculous reversal of lung pathology or reduction of mortality in ADAP-/- mice after exogenous injection of TGF-β1. Together, this study suggests that ADAP is indispensable for TGF-β1 production and TGF-β1-triggered CD103 expression via the TβRI-, TRAF6-, TAK1-dependent manner and protects from influenza virus infection.
Although mature TGF-β1 plays a critical role in the suppression of cytokine storm, H5N1 virus fails to activate the latent form of TGF-β1 (LAP-TGF-β1) [10]. We have observed that ADAP controls surface expression levels of VLA-1 (i.e. α1β1) in lung infiltrating CD8+ T cells during H5N1 virus infection (Fig 6). β1 integrin has been suggested for LAP-TGF-β1 activation [35], and TGF-β1 regulates expression of various β1 integrins. It is likely that ADAP is indispensable for CD8+ T cells to (1) promote VLA-1 expression, which activates LAP-TGF-β1 on neighboring immune cells to release active TGF-β1; (2) produce autocrine or paracrine TGF-β1, which then binds TβRI on CD8+ T cells to amplify the reciprocal TGF-β1-integrin crosstalk.
Interestingly, the combination of TGF-β1 and TCR stimulation could induce CD103 expression as well as FoxP3 expression, which are both dependent on SMAD2/3 and NFAT1 [40]. In agreement with this line, alloantigen-induced CD103+CD8+ T cells display strong suppressive activity in a mixed lymphocyte culture [41]. In addition, our recent findings suggest that ADAP deficient CD8+ T cells enhance cytotoxicity to kill tumor cells. Since ADAP deficient CD8+ T cells reduce TGF-β1/CD103 expression and decrease the ability to protect from H5N1 virus infection, further investigation should ask whether ADAP regulates suppressive function of CD103+CD8+ T cells, whether the TGF-β1/TβRI-ADAP-TRAF6-TAK1 pathway represents a general mechanism for reciprocal TGF-β1-integrin crosstalk to mediate inflammation-associated autoimmune diseases or graft transplantation.
Past efforts have identified various SNPs (Single Nucleotide Polymorphisms) or mutations in key molecules of TGF-β1 pathway, which are associated with inflammatory, infections or human cancer including TGFB1, TβRI/II, SMADs, Foxo3A [42–44]. Interestingly, a recent study has found an association between polymorphisms located in ADAP/FYB gene and Systemic Lupus Erythematosus (SLE) [45]. Considering high mortality of humans in H5N1 virus infection, it is critical to investigate whether these human SNPs or mutations in TGF-β1-regulated pathway (including the ADAP gene) could possibly enhance disease susceptibility or clinical manifestations in response to acute H5N1 virus infection.
Methods
Mice and ethics statement
ADAP-/- mice and OT-1 TCR transgenic mice were kindly provided by Drs. E. Peterson (University of Minnesota, USA) and CE Rudd (University of Cambridge, UK) respectively. ADAP-/- mice were crossed with OT1 TCR transgenic mice to generate ADAP-/- OT1 mice. RAG1-/- mice and wild type C57BL/6 mice were respectively purchased from Model Animal Research Center of Nanjing University or Shanghai Slac Laboratory Animal co. Ltd. Mice were bred under specific pathogen-free conditions at the Animal Care Facility of Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences. Sex and age-matched mice (8–12 weeks old) were used and animal infection with the H5N1 avian influenza virus A/Duck/Guangxi/12/2003 (GX12) were conducted in BSL3+ approved by the Chinese Ministry of Agriculture. This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the Ministry of Science and Technology of the People's Republic of China. The protocols for animal studies were approved by the Committee on the Ethics of Animal Experiments of the Harbin Veterinary Research Institute (HVRI) of the Chinese Academy of Agricultural Sciences (CAAS) (approval numbers BRDW-XBS–12).
Viral infection
The H5N1 virus A/Duck/Guangxi/12/2003 (termed GX/12) was used which was isolated from ducks in China. The viruses were propagated in 10-day-old specific-pathogen-free embryonated eggs and were stored at -70°C. Mice were lightly anesthetized with CO2 and were challenged intranasally with 106 50% egg infectious doses (EID50) of GX/12 in a volume of 50 μl. A/PR8 virus, kindly provided by Prof. Dongming Zhou (Institut Pasteur of Shanghai, Chinese Academy of Sciences, Shanghai, China), was grown in embryonated chicken eggs, and titrated for mean lethal dose (LD50) in adult C57BL/6 mice. C57BL/6 mice were inoculated intranasally with 10 LD50 of A/PR8 virus. At the indicated time points, samples from the infected mice were collected for RNA extraction or preparation of tissue lymphocytes. The remaining mice were monitored for 14 days for weight loss and mortality. Animal experiments were performed at least two independent times.
T cell adoptive transfer
Splenocytes were collected from ADAP-/- or wild type control mice, then CD4+ and CD8+ T cells were purified by positive selection with BD IMag™ CD4+ and CD8+ Magnetic Particles respectively. 4×106 CD4+ and 2.5×106 CD8+ T cells were transferred into Rag1-/- mice by tail vein injection. After 4 days, the recipient mice were challenged with the H5N1 virus (GX/12).
Preparation of tissue lymphocytes
Briefly, BAL fluids from the infected mice were collected by lavage with 1ml PBS for three to five times. Or, MLN and spleen were homogenized, filtered through 70 μm nylon mesh; and red blood cell were lysed and removed. Immune cells were collected after extensive wash with PBS.
ELISA assay
Lung was harvested and homogenized in 1mL 1×PBS on ice. Serum was gained in blood after clots were formed at 4°C. TGF-β1 concentrations in lung homogenate, serum, and supernatant from cell cultures were quantified by ELISA according to the manufacture’s instruction (Westong Biotechnology, Shanghai). The TGF-β ELISA kit recognizes the mature/active form of TGF-β1 and does not detect the latent form of TGF-β1. To measure the total concentration of TGF-β1 (both active and latent form), samples required acid-treatment and neutralization to remove LAP from the latent TGF-β1. Active TGF-β1 in samples was assayed without acid treatment.
Histology and immunohistochemistry
Lungs from the H5N1 virus infected mice were collected at the indicated time points, fixed in formalin and embedded in paraffin. Sections were cut and stained with hematoxylin and eosin (H&E) or immunohistochemistry (IHC). The relative degree of lung inflammation and tissue damage were examined and scored using the method described previously [46, 47]. Briefly, lung infiltration of inflammatory cells was scored as follows: 0, no inflammation; 1, mild peribronchial and peribronchiolar infiltrates, extending throughout, 10% of the lung; 2, moderate inflammation covering 10–50% of the lung; 3, severe inflammation involving over one-half of the lung. Degeneration of bronchi and bronchiolar epithelium, alveoli degeneration and collapse was scored as follows: 0, no degeneration; 1, little vacuolar degeneration of bronchi and bronchial epithelium cells, normal pulmonary alveoli; 2, mild necrosis of bronchi and bronchial epithelium, mild alveoli damage; 3, severe degeneration. Necrosis was scored as follows: 0, no necrosis; 1, mild necrosis with scant exudate; 2, marked necrosis with abundant exudate; 3, severe interstitial edema around blood vessels, apparent injured parenchyma and alveolar epithelial cells with greater infiltration of inflammatory cells.
TGF-β1 induction in vitro
To check whether ADAP regulated TGF-β1 production in vitro, naïve splenocytes from WT or ADAP-/- OT1 Tg mice were stimulated with 10nM OVA257-264 and 20U/ml human recombinant IL-2 for 3 days to generate CD8+ CTLs. Alternatively, splenocytes from WT or ADAP-/- mice or C8166 T cell lines overexpressing GFP or ADAP-GFP were stimulated with 2ug/mL plate bound anti-CD3/CD28 antibody for 3 days. Cells were harvested to examine surface expression by anti-LAP (TGF-β1) staining or TGF-β1 mRNA levels by RT-PCR. To block TGF-β signaling, splenocytes from WT or ADAP-/- OT1 Tg mice were stimulated with 10nM OVA257-264 and 5ng/mL exogenous TGF-β1 in presence or absence of the TGF-β receptor I kinase inhibitor SD208 or SB431542 (10uM). TGF-β1 mRNA levels were then measured by RT-PCR.
Flow cytometry
Lymphocytes from each organ was stained with various combinations of mAbs to CD4-FITC/APC, CD8α-PE/APC, CD3α-PE, and B220-FITC (eBiosciences), CD103-APC, LAP (TGF-β1)-PE (Clone TW7-20B9, Biolegend). Fixation/Permeabilization Concentrate and Diluent Kit (eBiosciences) were used for the intracellular staining of FoxP3-PE/APC according to the user’s handbook. After stained with surface markers, cells were stained with Annexin V and PI (BD Pharmingen) to examine apoptosis. For in vivo BrdU incorporation assay, mice were administered of BrdU (16 mg/ml; 50 μl) via i.n. route [48]; cells were harvested after 24hours and stained with BD FastImmune BrdU kit (BD Biosciences). Samples were run on BD Biosciences FACSCalibur, FACSAria or Accuri C6 instruments, and analyzed with FlowJo software (Tree Star, Ashland, OR).
Luciferase reporter assay
Wild type Jurkat, ADAP deficient Jurkat (JDAP cells) or ADAP overexpressing Jurkat cells were transfected with (CAGA)12-luciferase or (SBE)4-luciferase reporter plasmids respectively. Cells were then stimulated with 5ng/mL exogenous TGF-β1 for 6–8 hrs, followed by measurement of luciferase activity using Dual-Glo luciferase system (Gibco).
Western blot assay
CD8+ CTL cells generated with 10nM OVA257-264 treatment from WT and ADAP-/- OT1 Tg mice were left unstimulated or stimulated with 5ug/mL TGF-β1 for different time points. Cell lysates were prepared for immunoblotting with the indicated antibodies. To quantify the amount of nuclear SMAD3, WT and ADAP KO CD8+ CTLs were treated with 5ng/mL TGF-β1 for 30min. The extraction of cytoplasmic and nuclear protein was performed according to the product manufacturer instructions. Equal amounts of cytoplasmic and nuclear extract were loaded for western blotting assay with antibodies against SMAD3, SP1 and tubulin.
RNA extraction and real-time PCR analysis
Total RNA was extracted from tissues or cells using Trizol reagent (Invitrogen) following the supplier’s protocol and first-strand cDNA was synthesized using M-MLV Reverse Transcriptase RNase H Minus (Promega). Relative quantitative Real-Time PCR was carried out on BIO-RAD CFX96 Touch TM Real-Time PCR Detection System (BIO RAD) using Bestar TM Real-Time PCR Master Mix (SYBR Green methods, DBI Bioscience) with 2-△△CT method. Actin was used as the reference gene. All primer sequences are listed in Table 1.

CD103 induction in vitro
Naïve splenocytes from WT and ADAP-/- OT1 Tg mice were stimulated with 10nM OVA257-264 in presence of different concentrations of TGF-β1 for 3 days, CD103+CD8+ CTLs were identified by staining with anti-mouse CD8-FITC and anti-mouse CD103-APC. Alternatively, to examine surface CD103 expression, CD8+ T cells were purified with BD IMag™ CD8+ Magnetic Particles; WT and ADAP-/- CD8+ T cells were stimulated with 2ug/mL plate bound anti-CD3/CD28 and 5ng/mL exogenous TGF-β1 in the presence of the TAK1 inhibitor 5Z-7-oxo (2uM) or the TGF-β receptor I kinase inhibitor SB431542 (10uM); WT and TRAF6+/- CD8+ T cells were stimulated with 2ug/mL plate bound anti-CD3/CD28 and different concentrations of exogenous TGF-β1 for 3 days.
Statistical analysis
Data were analyzed using unpaired two tailed Student’s t test. Survival rates were analyzed by log-rank (Mantel-Cox) test. p value <0.05 or <0.01 was marked as * or ** respectively.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Peiris JS, Yu WC, Leung CW, Cheung CY, Ng WF, Nicholls JM, et al. Re-emergence of fatal human influenza A subtype H5N1 disease. Lancet. 2004;363(9409):617–9. 14987888
2. de Jong MD, Simmons CP, Thanh TT, Hien VM, Smith GJ, Chau TN, et al. Fatal outcome of human influenza A (H5N1) is associated with high viral load and hypercytokinemia. Nature medicine. 2006;12(10):1203–7. 16964257
3. Tumpey TM, Lu X, Morken T, Zaki SR, Katz JM. Depletion of lymphocytes and diminished cytokine production in mice infected with a highly virulent influenza A (H5N1) virus isolated from humans. Journal of virology. 2000;74(13):6105–16. 10846094
4. Szretter KJ, Gangappa S, Lu X, Smith C, Shieh WJ, Zaki SR, et al. Role of host cytokine responses in the pathogenesis of avian H5N1 influenza viruses in mice. Journal of virology. 2007;81(6):2736–44. 17182684
5. Cilloniz C, Pantin-Jackwood MJ, Ni C, Goodman AG, Peng X, Proll SC, et al. Lethal dissemination of H5N1 influenza virus is associated with dysregulation of inflammation and lipoxin signaling in a mouse model of infection. Journal of virology. 2010;84(15):7613–24. doi: 10.1128/JVI.00553-10 20504916
6. Salomon R, Hoffmann E, Webster RG. Inhibition of the cytokine response does not protect against lethal H5N1 influenza infection. Proc Natl Acad Sci U S A. 2007;104(30):12479–81. 17640882
7. Massague J. TGF-beta signal transduction. Annual Review of Biochemistry. 1998;67 : 753–91. 9759503
8. Li MO, Wan YY, Sanjabi S, Robertson A - KL, Flavell RA. Transforming growth factor-beta regulation of immune responses. Annu Rev Immunol. 2006;24 : 99–146. 16551245
9. Annes JP, Munger JS, Rifkin DB. Making sense of latent TGFbeta activation. Journal of cell science. 2003;116(Pt 2):217–24.
10. Carlson CM, Turpin EA, Moser LA, O'Brien KB, Cline TD, Jones JC, et al. Transforming growth factor-beta: activation by neuraminidase and role in highly pathogenic H5N1 influenza pathogenesis. PLoS pathogens. 2010;6(10):e1001136. doi: 10.1371/journal.ppat.1001136 20949074
11. Schultz-Cherry S, Hinshaw VS. Influenza virus neuraminidase activates latent transforming growth factor beta. Journal of virology. 1996;70(12):8624–9. 8970987
12. Kang SG, Park J, Cho JY, Ulrich B, Kim CH. Complementary roles of retinoic acid and TGF-[beta]1 in coordinated expression of mucosal integrins by T cells. Mucosal immunology. 2011;4(1):66–82. doi: 10.1038/mi.2010.42 20664575
13. Piet B, de Bree GJ, Smids-Dierdorp BS, van der Loos CM, Remmerswaal EB, von der Thusen JH, et al. CD8(+) T cells with an intraepithelial phenotype upregulate cytotoxic function upon influenza infection in human lung. The Journal of clinical investigation. 2011;121(6):2254–63. doi: 10.1172/JCI44675 21537083
14. Ray SJ, Franki SN, Pierce RH, Dimitrova S, Koteliansky V, Sprague AG, et al. The collagen binding alpha1beta1 integrin VLA-1 regulates CD8 T cell-mediated immune protection against heterologous influenza infection. Immunity. 2004;20(2):167–79. 14975239
15. Wang H, Wei B, Bismuth G, Rudd CE. SLP-76-ADAP adaptor module regulates LFA-1 mediated costimulation and T cell motility. Proceedings of the National Academy of Sciences. 2009;106(30):12436–41. doi: 10.1073/pnas.0900510106 19617540
16. Griffiths EK, Krawczyk C, Kong YY, Raab M, Hyduk SJ, Bouchard D, et al. Positive regulation of T cell activation and integrin adhesion by the adapter Fyb/Slap. Science. 2001;293(5538):2260–3. 11567140
17. Peterson EJ, Woods ML, Dmowski SA, Derimanov G, Jordan MS, Wu JN, et al. Coupling of the TCR to integrin activation by Slap-130/Fyb. Science. 2001;293(5538):2263–5. 11567141
18. Medeiros RB, Burbach BJ, Mueller KL, Srivastava R, Moon JJ, Highfill S, et al. Regulation of NF-kappaB activation in T cells via association of the adapter proteins ADAP and CARMA1. Science. 2007;316(5825):754–8. 17478723
19. Suzuki J, Yamasaki S, Wu J, Koretzky GA, Saito T. The actin cloud induced by LFA-1-mediated outside-in signals lowers the threshold for T-cell activation. Blood. 2007;109(1):168–75. 16973965
20. Liu G, Ding W, Neiman J, Mulder KM. Requirement of Smad3 and CREB-1 in mediating transforming growth factor-beta (TGF beta) induction of TGF beta 3 secretion. The Journal of biological chemistry. 2006;281(40):29479–90. 16891311
21. Tang B, Bottinger EP, Jakowlew SB, Bagnall KM, Mariano J, Anver MR, et al. Transforming growth factor-beta1 is a new form of tumor suppressor with true haploid insufficiency. Nature medicine. 1998;4(7):802–7. 9662371
22. Dennler S, Itoh S, Vivien D, ten Dijke P, Huet S, Gauthier JM. Direct binding of Smad3 and Smad4 to critical TGF beta-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. The EMBO journal. 1998;17(11):3091–100. 9606191
23. Jonk LJ, Itoh S, Heldin CH, ten Dijke P, Kruijer W. Identification and functional characterization of a Smad binding element (SBE) in the JunB promoter that acts as a transforming growth factor-beta, activin, and bone morphogenetic protein-inducible enhancer. The Journal of biological chemistry. 1998;273(33):21145–52. 9694870
24. Conery AR, Cao Y, Thompson EA, Townsend CM Jr., Ko TC, Luo K. Akt interacts directly with Smad3 to regulate the sensitivity to TGF-beta induced apoptosis. Nature cell biology. 2004;6(4):366–72. 15104092
25. Remy I, Montmarquette A, Michnick SW. PKB/Akt modulates TGF-beta signalling through a direct interaction with Smad3. Nature cell biology. 2004;6(4):358–65. 15048128
26. Runyan CE, Schnaper HW, Poncelet AC. The phosphatidylinositol 3-kinase/Akt pathway enhances Smad3-stimulated mesangial cell collagen I expression in response to transforming growth factor-beta1. The Journal of biological chemistry. 2004;279(4):2632–9. 14610066
27. Zhang YE. Non-Smad pathways in TGF-beta signaling. Cell research. 2009;19(1):128–39. doi: 10.1038/cr.2008.328 19114990
28. Kretzschmar M, Doody J, Timokhina I, Massague J. A mechanism of repression of TGFbeta/ Smad signaling by oncogenic Ras. Genes & development. 1999;13(7):804–16.
29. Moustakas A, Heldin CH. Non-Smad TGF-beta signals. Journal of cell science. 2005;118(Pt 16):3573–84. 16105881
30. Sorrentino A, Thakur N, Grimsby S, Marcusson A, von Bulow V, Schuster N, et al. The type I TGF-beta receptor engages TRAF6 to activate TAK1 in a receptor kinase-independent manner. Nature cell biology. 2008;10(10):1199–207. doi: 10.1038/ncb1780 18758450
31. Jiao P, Tian G, Li Y, Deng G, Jiang Y, Liu C, et al. A single-amino-acid substitution in the NS1 protein changes the pathogenicity of H5N1 avian influenza viruses in mice. Journal of virology. 2008;82(3):1146–54. 18032512
32. Zou L, Mendez F, Martin-Orozco N, Peterson EJ. Defective positive selection results in T cell lymphopenia and increased autoimmune diabetes in ADAP-deficient BDC2.5-C57BL/6 mice. European journal of immunology. 2008;38(4):986–94. doi: 10.1002/eji.200737881 18383041
33. Srivastava R, Burbach BJ, Mitchell JS, Pagan AJ, Shimizu Y. ADAP regulates cell cycle progression of T cells via control of cyclin E and Cdk2 expression through two distinct CARMA1-dependent signaling pathways. Molecular and cellular biology. 2012;32(10):1908–17. doi: 10.1128/MCB.06541-11 22411628
34. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57–70. 10647931
35. Wipff PJ, Rifkin DB, Meister JJ, Hinz B. Myofibroblast contraction activates latent TGF-beta1 from the extracellular matrix. The Journal of cell biology. 2007;179(6):1311–23. 18086923
36. Li Q, Zhou L, Zhou M, Chen Z, Li F, Wu H, et al. Epidemiology of Human Infections with Avian Influenza A(H7N9) Virus in China. New England Journal of Medicine. 2014;370(6):520–32. doi: 10.1056/NEJMoa1304617 23614499
37. Chen Y, Liang W, Yang S, Wu N, Gao H, Sheng J, et al. Human infections with the emerging avian influenza A H7N9 virus from wet market poultry: clinical analysis and characterisation of viral genome. Lancet. 2013;381(9881):1916–25. doi: 10.1016/S0140-6736(13)60903-4 23623390
38. Laidlaw BJ, Decman V, Ali MA, Abt MC, Wolf AI, Monticelli LA, et al. Cooperativity Between CD8+ T Cells, Non-Neutralizing Antibodies, and Alveolar Macrophages Is Important for Heterosubtypic Influenza Virus Immunity. PLoS pathogens. 2013;9(3):e1003207. doi: 10.1371/journal.ppat.1003207 23516357
39. Wu H, Kumar A, Miao H, Holden-Wiltse J, Mosmann TR, Livingstone AM, et al. Modeling of influenza-specific CD8+ T cells during the primary response indicates that the spleen is a major source of effectors. Journal of immunology. 2011;187(9):4474–82. doi: 10.4049/jimmunol.1101443 21948988
40. Mokrani M, Klibi J, Bluteau D, Bismuth G, Mami-Chouaib F. Smad and NFAT Pathways Cooperate To Induce CD103 Expression in Human CD8 T Lymphocytes. J Immunol. 2014;192(5):2471–9. doi: 10.4049/jimmunol.1302192 24477908
41. Uss E, Rowshani AT, Hooibrink B, Lardy NM, van Lier RA, ten Berge IJ. CD103 is a marker for alloantigen-induced regulatory CD8+ T cells. Journal of immunology. 2006;177(5):2775–83. 16920912
42. Lee JC, Espeli M, Anderson CA, Linterman MA, Pocock JM, Williams NJ, et al. Human SNP links differential outcomes in inflammatory and infectious disease to a FOXO3-regulated pathway. Cell. 2013;155(1):57–69. doi: 10.1016/j.cell.2013.08.034 24035192
43. Chen T, Triplett J, Dehner B, Hurst B, Colligan B, Pemberton J, et al. Transforming Growth Factor-β Receptor Type I Gene Is Frequently Mutated in Ovarian Carcinomas. Cancer research. 2001;61(12):4679–82. 11406536
44. Antony ML, Nair R, Sebastian P, Karunagaran D. Changes in expression, and/or mutations in TGF-beta receptors (TGF-beta RI and TGF-beta RII) and Smad 4 in human ovarian tumors. Journal of cancer research and clinical oncology. 2010;136(3):351–61. doi: 10.1007/s00432-009-0703-4 19916025
45. Addobbati C, Brandao LA, Guimaraes RL, Pancotto JA, Donadi EA, Crovella S, et al. FYB gene polymorphisms are associated with susceptibility for systemic lupus erythemathosus (SLE). Human immunology. 2013;74(8):1009–14. doi: 10.1016/j.humimm.2013.04.026 23628395
46. Crowe CR, Chen K, Pociask DA, Alcorn JF, Krivich C, Enelow RI, et al. Critical Role of IL-17RA in Immunopathology of Influenza Infection. Journal of immunology. 2009;183(8):5301–10. doi: 10.4049/jimmunol.0900995 19783685
47. Wang X, Chan CCS, Yang M, Deng J, Poon VKM, Leung VHC, et al. A critical role of IL-17 in modulating the B-cell response during H5N1 influenza virus infection. Cellular & molecular immunology. 2011;8(6):462–8.
48. Lawrence CW, Ream RM, Braciale TJ. Frequency, specificity, and sites of expansion of CD8(+) T cells during primary pulmonary influenza virus infection. Journal of immunology. 2005;174(9):5332–40. 15843530
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2015 Číslo 4
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Toxin-Induced Necroptosis Is a Major Mechanism of Lung Damage
- Transgenic Fatal Familial Insomnia Mice Indicate Prion Infectivity-Independent Mechanisms of Pathogenesis and Phenotypic Expression of Disease
- Role of Hypoxia Inducible Factor-1α (HIF-1α) in Innate Defense against Uropathogenic Infection
- A Temporal Gate for Viral Enhancers to Co-opt Toll-Like-Receptor Transcriptional Activation Pathways upon Acute Infection
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy