
Quadruple Quorum-Sensing Inputs Control Virulence and Maintain System Robustness
Quorum-sensing (QS) is a microbial cell-cell communication process that allows bacteria to function as a collective group. Many pathogens, including Vibrio cholerae, the causative agent of cholera, depend on QS to regulate important cellular processes that are essential for survival and adaptation inside and outside of their hosts. Since its discovery, the V. cholerae QS system has served as a model to understand how bacterial pathogens employ QS for temporal control of virulence factor production. Yet, after a decade of research, our understanding of the V. cholerae QS system is still incomplete. Here we re-define the QS network architecture of this important pathogen. We show that two novel sensory inputs function in parallel with the two canonical QS pathways to regulate V. cholerae virulence gene expression. Moreover, our study illustrates a strategy that bacteria employ to maintain QS system robustness. By perceiving multiple parallel sensory inputs, the V. cholerae QS network is structured to be highly resistant to signal perturbations, therefore preventing premature commitment to QS. Our study provides new insights into how bacterial pathogens integrate multiple sensory signals to elicit robust and coordinated QS responses.
Published in the journal:
. PLoS Pathog 11(4): e32767. doi:10.1371/journal.ppat.1004837
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004837
Summary
Quorum-sensing (QS) is a microbial cell-cell communication process that allows bacteria to function as a collective group. Many pathogens, including Vibrio cholerae, the causative agent of cholera, depend on QS to regulate important cellular processes that are essential for survival and adaptation inside and outside of their hosts. Since its discovery, the V. cholerae QS system has served as a model to understand how bacterial pathogens employ QS for temporal control of virulence factor production. Yet, after a decade of research, our understanding of the V. cholerae QS system is still incomplete. Here we re-define the QS network architecture of this important pathogen. We show that two novel sensory inputs function in parallel with the two canonical QS pathways to regulate V. cholerae virulence gene expression. Moreover, our study illustrates a strategy that bacteria employ to maintain QS system robustness. By perceiving multiple parallel sensory inputs, the V. cholerae QS network is structured to be highly resistant to signal perturbations, therefore preventing premature commitment to QS. Our study provides new insights into how bacterial pathogens integrate multiple sensory signals to elicit robust and coordinated QS responses.
Introduction
Bacteria produce and detect multiple classes of chemical signals called autoinducers to monitor local population density and species complexity. This cell-to-cell communication process, called Quorum Sensing (QS), allows groups of bacteria to synchronize population-wide gene expression and effectively carry out collective behaviors that are presumably ineffective if performed by a single bacterial cell acting alone. Disruption of the QS signal transduction cascade leads to uncoordinated gene expression and renders many pathogenic bacteria avirulent [1–5].
Vibrio cholerae, the etiological agent of the diarrheal disease cholera, uses QS to regulate virulence factor production, biofilm formation, Type VI secretion, and competence development, all of which are important for survival and adaptation inside and outside of its human hosts [6–17]. Two parallel QS signaling systems that function via phosphorelay-type regulatory pathways have been identified in V. cholerae [6]. The CqsA/CqsS system, which produces and detects CAI-1 (S-3-hydroxytridecan-4-one) as a QS signal, is present in many Vibrio species and is believed to be used for intra-genus communication [18–23]. The LuxS/LuxPQ system, which produces and detects AI-2 (S-TMHF-borate) as a QS signal, is present in many bacterial species and is believed to be used for inter-species signaling [6, 24–27]. In environments where the concentrations of these two autoinducers are below their detection threshold, such as at low cell density (LCD), CqsS and LuxQ function as kinases. They hydrolyze ATP and shuttle the phosphoryl group, via a histidine-phosphotransfer protein LuxU, to the key response regulator, LuxO. Phosphorylated LuxO (LuxO~P) activates transcription of the genes encoding four regulatory sRNAs, called Qrr1-4 [28]. Aided by the RNA chaperone Hfq, Qrr1-4 activate the translation of the AphA regulator and inhibit the translation of the HapR regulator (Fig 1A) [6, 28–30].
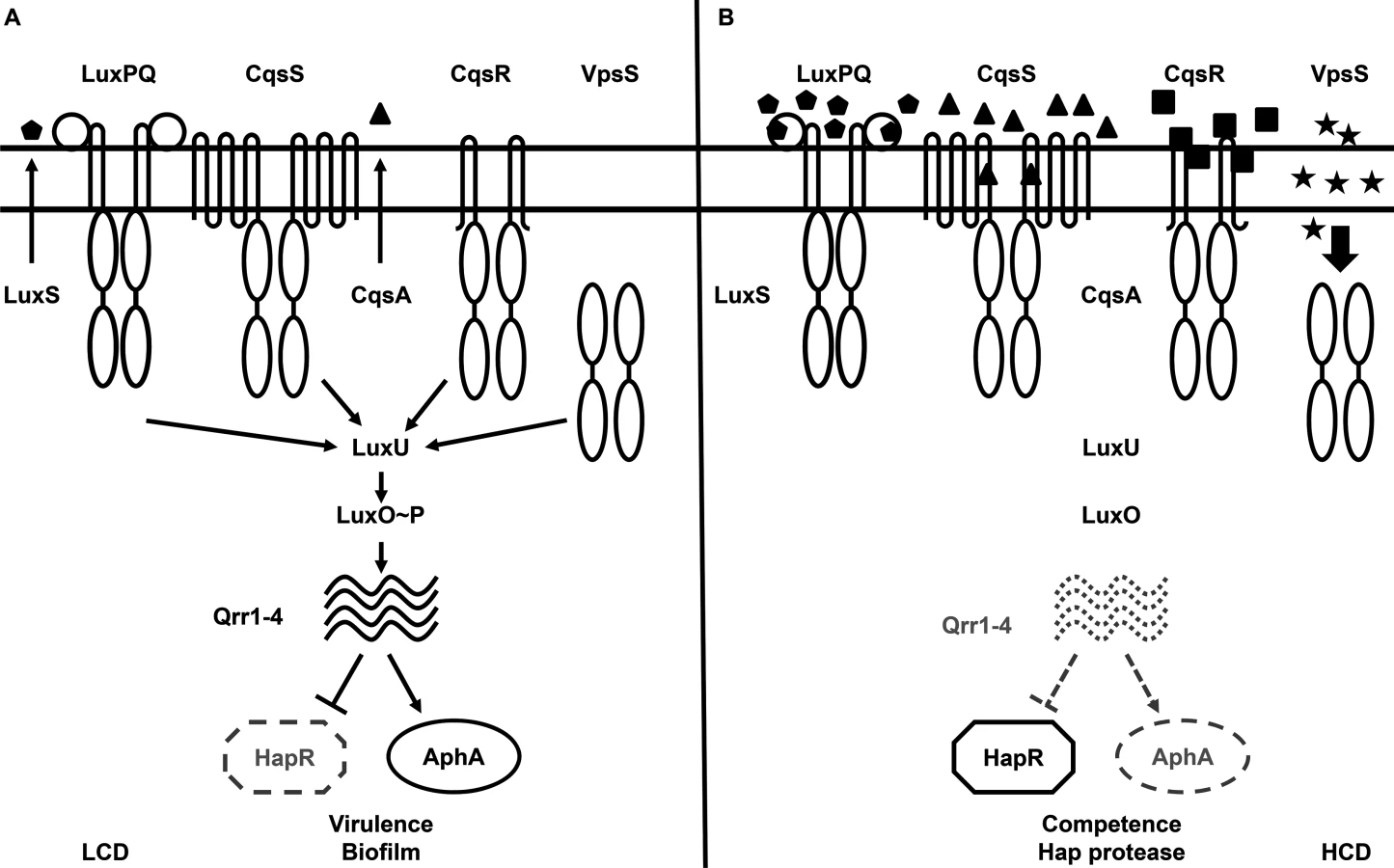
At high cell density (HCD), when autoinducers accumulate to high levels, the kinase activities of CqsS and LuxQ are inhibited by binding of their cognate signals. As a consequence, phosphate flow is reversed, leading to dephosphorylation and deactivation of LuxO. Transcription of qrr1-4 terminates and, hence, HapR, but not AphA, is produced (Fig 1B) [6, 28–30]. Reciprocal production of AphA and HapR at LCD and HCD is central to the switch from individual to group behaviors in Vibrio species [29]. Together these two transcriptional regulators regulate the expression levels of over 100 genes [7, 29].
Although CqsS and LuxQ both contribute to LuxO activation (Fig 1), strikingly, mutants missing both receptors are phenotypically identical to the wild-type and remain virulent [6]. Thus, additional unknown signaling pathways are predicted to activate LuxO [6, 31]. A clue to the identity of a potential LuxO-activation pathway came from a study in which overexpression of a hybrid histidine kinase VpsS leads to a LuxO-dependent up-regulation of the biofilm biosynthetic gene vpsL in V. cholerae, suggesting that VpsS could regulate LuxO activity to control biofilm formation [32]. Moreover, the purified receiver domains of VpsS and another hybrid histidine kinase VC1831 are capable of effectively accepting the phosphoryl group from phosphorylated LuxU in vitro [32]. However, it is unclear if VpsS and VC1831, together with CqsS and LuxQ, function as phosphoryl group donors to activate LuxO and control QS inside V. cholerae cells. Furthermore, it is also unknown if, and to what extent, each of these four histidine kinase receptors is individually contributing to the global control of the QS response in V. cholerae. The role of VpsS and VC1831 in V. cholerae pathogenesis also has not been investigated.
Here we report the connections between VpsS and CqsR and QS in V. cholerae (VC1831 is renamed as CqsR hereafter based on its role as Cholera Quorum Sensing Receptor). VpsS and CqsR function in parallel with CqsS and LuxQ and act upstream of LuxO in the V. cholerae QS signal transduction pathway. Indeed V. cholerae is capable of QS when any single one of these four receptors is present. Furthermore, in addition to CAI-1 and AI-2, additional stimuli whose levels presumably vary depending on cell density, are perceived by VpsS and CqsR to modulate QS. Finally, multiple functionally redundant receptors that control a signal regulator (i.e., LuxO) enable a QS response that is insensitive to perturbations in the cognate sensory cues.
Results
V. cholerae host colonization requires LuxO activation and Qrr sRNAs
Previous studies established that LuxO, the key response regulator in the V. cholerae QS system, is activated by phosphorylation at the conserved Asp61 by CqsS and LuxQ at LCD (Fig 1) [33]. V. cholerae mutants lacking LuxO are unable to express Qrr1-4 sRNAs; as a result, they fail to express AphA and instead produce HapR at all population densities [6, 28]. Therefore, ΔluxO mutants are highly attenuated in colonization of animal hosts [6, 7]. Surprisingly, V. cholerae mutants lacking both CqsS and LuxQ are phenotypically identical to the wild-type and colonize animal hosts effectively. Thus, LuxO appears to be activated by additional mechanisms [6]. Alternatively, these results could be interpreted to mean that unphosphorylated LuxO, but not phosphorylated LuxO, is required for host colonization and there is no additional source of activation. If the former model is correct, a V. cholerae luxOD61A mutant, expressing a form of LuxO that is incapable of being phosphorylated, should be defective in colonizing animal hosts. Indeed, both ΔluxO and luxOD61A mutant cells were out-competed by the wild-type in an infant mouse colonization model, however, there was a 10-fold difference in the CIs observed for these two mutants. In many cases, we did not detect any ΔluxO and luxOD61A mutants inside the animal hosts (Fig 2). Furthermore, V. cholerae cells lacking all 4 Qrr sRNAs, the only known targets of activated LuxO, were defective in host colonization (Fig 2). Together, these in vivo data indicate that phosphorylated LuxO and the downstream Qrr sRNAs are required for V. cholerae host colonization, and further suggest that phosphorylation by CqsS and LuxQ are not the only sources of LuxO activation.

LuxO activation depends on LuxU only
To further explore the pathway for LuxO activation, we focused on the protein that interacts with LuxO in the V. cholerae QS circuit. Only a single histidine phosphotransfer (HPT) protein, LuxU, is known to interact with LuxO and link to LuxO activation ([6], Fig 1). Unexpectedly, V. cholerae mutants lacking LuxU were shown to be active in QS gene regulation and colonize animal hosts effectively [6]. These findings seem to contradict the apparent role of active LuxO in V. cholerae pathogenicity regulation (Fig 2). Alternatively, LuxO could be activated by interacting with other HPT proteins. However, LuxU was found to be required for V. cholerae virulence in two independent genome-scale transposon mutant analyses [34, 35]. To resolve these conflicting results, we revisited the role of LuxU in V. cholerae QS control and pathogenicity regulation. We constructed a new ΔluxU mutant (WN3557) and compared its QS response to that of the ΔluxU mutant (WN3045) previously reported [6]. We used the heterologous Vibrio harveyi luxCDABE luciferase operon to measure QS-dependent gene regulation, because expression of this operon is activated by HapR, whose level is inversely proportional to the amount of activated LuxO inside the cell ([6], Fig 1). Therefore, if LuxU is the major HPT protein that is essential for LuxO activation, mutants lacking LuxU would express a high level of luciferase and be bright. We found that bioluminescence production (shown as specific light production versus cell density) was very different between these two ΔluxU strains. The newly-constructed ΔluxU mutant cells were constitutively bright, indicating that LuxO is inactive and that HapR is constantly produced at all population densities (Fig 3A). In contrast, the original ΔluxU mutant cells were 10 - to 100-fold darker, depending on the cell density at which they were sampled, indicating that less HapR is produced in the original ΔluxU mutant (Fig 3A). To understand these differences, we sequenced the luxOU locus of these ΔluxU strains. Using published V. cholerae genome sequences as a reference, we identified a missense mutation in luxO of the original ΔluxU strain, resulting in a change from glycine to serine at amino acid residue 333 of LuxO. In contrast, no mutation was identified in the luxOU locus of the new ΔluxU strain. To explain the difference in phenotype between the two strains, we hypothesized that LuxOG333S mimics the active form of LuxO such that it does not require LuxU for activation. To test this hypothesis, plasmids expressing luxO+ or luxOG333S were introduced into the new ΔluxU strain, and HapR-dependent bioluminescence from the resulting strains was measured. We found that extra copies of luxO+ did not alter the specific bioluminescence production of the new ΔluxU mutant and the strain remained constitutively bright, similar to the empty plasmid control (Fig 3B). However, when luxOG333S was expressed in the new ΔluxU mutant, the resulting strain was >50-fold darker than the other two strains (Fig 3B). These results indicate that the luxOG333S mutation is dominant to the luxO+ allele and is epistatic to the ΔluxU mutation. Although the activation mechanism is unclear, we suggest that LuxOG333S mimics an active form of LuxO, bypassing the requirement of LuxU in QS signal transduction in the original ΔluxU strain. Consistent with the idea that LuxU is the key HPT protein in QS control, the new ΔluxU mutant cells were out-competed by the wild-type in the infant mouse colonization model, while the original ΔluxU cells were not (Fig 3C).

VpsS and CqsR histidine kinases contribute to LuxO activation
After confirming the importance of LuxU in LuxO activation, we hypothesized that, similar to CqsS and LuxQ, histidine kinases that employ LuxU as an intermediate phosphorelay partner are able to activate LuxO. The isolated receiver domains of two hybrid histidine kinases, VpsS and CqsR (VC1831), are capable of interacting with and effectively removing the phosphoryl group from phosphorylated LuxU in vitro [32]. Thus, we reasoned that full length VpsS and CqsR, when active, could phosphorylate LuxO via LuxU and contribute to its activation in V. cholerae cells. To explore these ideas, we measured HapR-dependent bioluminescence in mutants missing one or more of these histidine kinases. As previously shown [6], wild-type V. cholerae cells produce a U-shaped bioluminescence profile, representing the change in LuxO activity and HapR levels at different cell-densities (Fig 4A). At HCD (OD600 >1), V. cholerae produced a high level of HapR-dependent bioluminescence, indicating that LuxO activity is low in this condition (Fig 4A). When these HCD cells were diluted in fresh medium, specific luciferase activity was high initially since the enzyme had not been turned over from the overnight culture. However, when these diluted cells started to grow, HapR-dependent bioluminescence decreased due to activation of LuxO and repression of HapR production. Light production reached a minimum at OD600 ~0.5. Afterwards, HapR-dependent bioluminescence increased and reached a maximum at OD600 >1 (Fig 4A). Using the same assay, we found that cells missing both CqsS and LuxQ displayed a HapR-dependent bioluminescence profile indistinguishable from that of the wild-type, indicating that LuxO activation is still controlled by a cell density-dependent mechanism in the absence of these two known QS receptors (Fig 4A). Likewise, V. cholerae cells missing both VpsS and CqsR also displayed a HapR-dependent bioluminescence profile similar to that of the wild-type and the ΔcqsS ΔluxQ double QS receptor mutant (Fig 4A).

We constructed four different triple receptor mutants expressing only one of the four possible QS receptors and found that their HapR-dependent bioluminescence profiles were different from each other and from the wild-type (Fig 4B). Although each triple receptor mutant still displayed a U-shaped HapR-dependent bioluminescence profile, switching from low to high light production from LCD to HCD, these mutants all produced more light than the wild-type at LCD (Fig 4B). Mutant cells with only LuxQ showed the largest difference (~100-fold) in relative light production between LCD and HCD, while mutant cells with only CqsR showed the smallest difference (~10-fold). The temporal dynamics of the response in each triple receptor mutant were also distinct. Mutant cells expressing only CqsS switched from low to high light production at the lowest cell density (OD600 ~0.05), while the other three mutant strains switched at around the same cell density (OD600 ~0.5) (Fig 4B). Thus, our results indicate that CqsS, LuxQ, VpsS, and CqsR can each independently activate LuxO, but the influence of each receptor on the overall QS response is not identical.
We then measured the HapR-dependent bioluminescence in a ΔluxQ ΔcqsS ΔvpsS ΔcqsR quadruple receptor mutant and found that the profile was identical to the new ΔluxU mutant (Figs 3A and 4B), indicating that very little, if any, active LuxO is present. To determine if VpsS and CqsR both act upstream to activate LuxO, we introduced the luxOD61E mutation, which renders LuxO constitutively active by mimicking the phosphorylated form of the protein [8, 36], into the quadruple receptor mutant. We predicted that the luxOD61E allele would override the effect of the loss of all four histidine kinases. Indeed, we found that the ΔluxQ ΔcqsS ΔvpsS ΔcqsR luxOD61E strain was constitutively dark, similar to the luxOD61E mutant (S1 Fig). Likewise, LuxO activity could be partially restored when cqsS, luxQ, vpsS, or cqsR was individually overexpressed episomally in the quadruple receptor mutant (S2 Fig). Additionally, we used a qrr4-lux transcriptional fusion to study the contribution of CqsS, LuxQ, VpsS, and CqsR and found that each receptor alone was sufficient to support Qrr4 expression to different degrees at LCD (S3 Fig), while the quadruple receptor, ΔluxO, and new ΔluxU mutants all expressed very little, if any, Qrr4. As expected, the luxOD61E mutation restored Qrr4 expression in the quadruple receptor mutant (S3 Fig).
Previously, CsrA was proposed to enhance LuxO~P activity [31]. VpsS and CqsR could exert their regulatory effects on LuxO by modulating the activity of CsrA. However, a csrA::Tn5 insertion mutation that had been identified before was not sufficient to abolish Qrr4 production in the ΔcqsS ΔluxQ mutant (S4 Fig), arguing against the possibility that VpsS and CqsR signal through CsrA.
A single QS receptor is sufficient to activate LuxO for host colonization
The above studies show that CqsS, LuxQ, VpsS, and CqsR each independently contributes to part of the QS response in V. cholerae growing under laboratory conditions. To determine the minimal requirement of LuxO activation through these histidine kinases that is sufficient for V. cholerae infection of animal hosts, we tested double, triple, and quadruple receptor mutants using an infant mouse colonization model. We found that the two double receptor mutants (ΔluxQ ΔcqsS and ΔvpsS ΔcqsR) and the four triple receptor mutants all colonized the small intestine effectively (Fig 4C). In contrast, the quadruple receptor mutant was highly defective in animal colonization (Fig 4C). While a slight advantage in host colonization (~2 fold) was observed for the luxOD61E mutants in the wild-type genetic background, the luxOD61E mutation was epistatic to the ΔluxQ ΔcqsS ΔvpsS ΔcqsR mutations and restored the colonization defects (>10,000-fold) of the quadruple receptor mutants (Fig 4C). Thus, even though these four receptors contribute to the control of the V. cholerae QS response to different extents under laboratory conditions, any one of the receptors appears to be sufficient to promote LuxO activation enough to support colonization of mice.
Multiple sensory inputs maintains the robustness of V. cholerae QS system
It is curious that V. cholerae integrates four parallel sensory inputs to activate a common response regulator LuxO, even though a single receptor is sufficient for a QS response (Fig 4B and 4C). We hypothesized that by integrating multiple signals, LuxO activation and the downstream QS response is less sensitive to perturbations from any one of the sensory inputs. To test this idea, we first determined that 2μM of synthetic CAI-1 was sufficient to induce a premature QS response in the triple receptor mutant expressing only CqsS (S5 Fig). Then, we measured HapR-dependent bioluminescence in the presence of surplus CAI-1 (20 μM) in the wild-type and different receptor mutants. Consistent with our prediction, we found that extra CAI-1 did not significantly alter the HapR-dependent bioluminescence profiles of the wild-type or any single receptor mutant missing LuxQ or VpsS or CqsR (Fig 5A–5D). We likewise found that extra CAI-1 did not significantly increase light production in strains expressing CqsS and LuxQ (ΔvpsS ΔcqsR) (Fig 5E), but addition of CAI-1 slightly, yet reproducibly, increased light production in strains expressing CqsS and VpsS (ΔluxQ ΔcqsR) (Fig 5F), indicating that inhibition of CqsS kinase activity is compensated for by LuxQ and partially by VpsS (Fig 5E and 5F). In contrast, surplus CAI-1 caused the strains expressing CqsS and CqsR (ΔluxQ ΔvpsS) to produce light constitutively, indicating that CqsR is not sufficient to compensate for the loss of CqsS kinase activity (Fig 5G). Finally, as expected, strains expressing CqsS alone (ΔluxQ ΔcqsR ΔvpsS) constantly produced light in the presence of surplus CAI-1, as no compensating kinase activity is present (Fig 5H). Thus, functionally redundant receptors, particularly LuxQ and VpsS, render V. cholerae cells insensitive to surplus CAI-1. These combined results are consistent with the idea that multiple parallel sensory inputs controlling a single LuxO protein are important for resisting perturbations in signal inputs to maintain the robustness of the V. cholerae QS system.

VpsS and CqsR activities are regulated by extracellular molecules
To achieve QS regulation, the activities of VpsS and CqsR must be controlled by a cell density dependent mechanism. We reasoned that, similar to CqsS and LuxPQ, the autokinase activities of VpsS and CqsR could both be inhibited by binding to specific molecules that accumulate during cell growth (Fig 1). Therefore, we studied the effects of addition of cell-free spent medium harvested from V. cholerae HCD cultures on Qrr sRNA expression in the two triple receptor mutants expressing either VpsS or CqsR with a qrr4-lux reporter. To ensure that any observed regulatory effect from the spent medium was not due to nutrient deprivation after bacterial growth, we replenished any missing ingredients by reconditioning the spent medium (80% v/v) with 20% (v/v) of 5× LB. As expected, when the strains were grown in fresh medium, Qrr4 expression levels were high at LCD and low at HCD (Fig 6A–6D). In contrast, addition of reconditioned spent culture medium decreased LCD Qrr4 production in both strains (Fig 6A and 6B). Qrr4 expression was also repressed by reconditioned spent culture medium harvested from a ΔcqsA ΔluxS double synthase mutant that cannot make CAI-1 and AI-2 (Fig 6C and 6D), indicating that the signals sensed by VpsS and CqsR are different from the two canonical autoinducers. Addition of reconditioned spent culture medium did not alter the growth rates of these two strains (S6 Fig). Moreover, reconditioned spent medium harvested from LCD (OD600 ~ 0.5) V. cholerae did not alter Qrr4 expression in these two strains (S7 Fig). These combined results suggest that additional molecules other than CAI-1 and AI-2 are made and secreted by V. cholerae to regulate VpsS and CqsR kinase activities and ultimately control its QS response.

Discussion
In the current study, we show that the in vitro and in vivo behaviors of the ΔluxU mutant are essentially identical to the ΔluxO mutant, therefore suggesting that all the LuxO-activation inputs, including VpsS and CqsR, must shuttle through LuxU to activate LuxO. Indeed, if VpsS and CqsR do not signal through LuxU to activate LuxO, mutants lacking LuxU would behave like mutants lacking CqsS and LuxQ. However, we demonstrate in multiple assays that this was not the case. Therefore, we propose that LuxO, the key QS regulator, is activated by four independent histidine kinase receptors CqsS, LuxQ, VpsS, and CqsR through HPT protein LuxU to control the QS response in V. cholerae (Fig 1).
Our new model provides additional insights into the V. cholerae QS signal transduction pathway and explains why V. cholerae mutants missing the canonical QS receptors CqsS and LuxQ remain proficient in controlling cell density-dependent genes [6]. The influence on LuxO activation of each of the four histidine kinases is not identical; LuxQ is the strongest and CqsR is the weakest activator of LuxO (Fig 4B). Similarly, Yildiz and coworkers previously showed that overexpression of LuxQ and VpsS, but not CqsR and CqsS, increases vpsL expression and biofilm formation through a LuxO-dependent mechanism [32]. Surprisingly, our results are in contrast to previous studies of autoinducer synthase mutants in which the QS response is affected more by a ΔcqsA mutation than a ΔluxS mutation, arguing that CqsS has a stronger impact than LuxQ on V. cholerae QS [6, 36]. However, it should be noted that our current study was performed under conditions in which autoinducers are produced by V. cholerae cells at their native levels. Thus, the accumulation rate of each cognate signal in the culture and the signal sensitivity of each receptor could influence the contribution of each receptor to QS control.
Unlike the case in laboratory cultures, CqsS, LuxQ, CqsR, or VpsS alone is sufficient to activate LuxO enough for V. cholerae to effectively colonize the mouse small intestine (Fig 4C). That is, the loss of three histidine kinase activities has little effect on V. cholerae colonization of animal hosts, a trait that is strongly dependent on LuxO activation. Perhaps the overall kinase activities of these receptors are substantially stronger in V. cholerae growing inside an animal than in bacteria growing under laboratory conditions. Additionally, the level of the cognate signals for these receptors could be altered in the host environment such that each receptor maintains a longer period of activation. Alternatively, the level of LuxO activation required to repress HapR-dependent bioluminescence and activate biofilm formation under laboratory conditions is higher than the level required for expression of virulence genes in animal hosts, and that could also explain the difference observed between the in vitro and in vivo phenotypes. It is interesting that differential contribution from multiple receptor inputs is observed in other microbial signaling pathways. For instance, the sporulation pathway of Bacillus subtilis is controlled by five histidine kinases, KinA-E [37–40]. These five receptors participate in the phosphorylation of Spo0F, which in turn activates the key response regulator Spo0A. Although all of these kinases can activate Spo0A, only KinA and KinB can activate Spo0A to a level high enough to trigger sporulation, while KinC and KinD kinases are only capable of initiating entry into stationary phase [40, 41]. Thus, this “many-to-one” configuration has evolved independently in multiple bacterial signal transduction pathways to maintain a specific input-output relationship depending on particular environmental parameters [42].
It is not uncommon for bacterial species to possess multiple QS systems for cell-cell communication. These systems can be wired in different configurations to accomplish specific biological goals [43, 44]. We show here that by using four different receptors in parallel to control the overall QS response, the V. cholerae QS circuit is built to resist perturbations in external conditions (Fig 5). This circuit architecture could be especially important to maintain synchronous expression of QS genes in the population, and to prevent premature commitment to HCD gene expression. This set up could also be useful for filtering out signal noise caused by analogous molecules present in the environment. It should be noted that a high level of CAI-1 was tested for the sensitivity of the system and V. cholerae likely will not encounter CAI-1 alone without other autoinducers. However, previous studies showed that molecules with structures drastically different from CAI-1 could inhibit CqsS activity [45], suggesting possibilities for decoy molecules acting alone on a single QS receptor. Such circuitry has been proposed to function as a “coincidence detector” in other QS systems [18, 46]. However, whether the V. cholerae QS circuit is used for coincidence detection requires further investigation. Indeed, we suspect that not all genes in the V. cholerae QS regulon display the same regulatory pattern as the HapR-dependent bioluminescence operon, and we predict that a subset of V. cholerae QS genes could be more sensitive to perturbations. For instance, it has been shown that addition of CAI-1 alone is able to resuscitate viable but non-culturable (VNBC) V. cholerae [47]. Although these VNBC cells are physiologically distinct from cells cultured under laboratory conditions, these results suggest that a single autoinducer input can trigger differential gene expression in certain V. cholerae cell types.
The other advantage of using multiple sensory systems is to allow QS bacteria to decipher distinctive information contained within each specific signal. For instance, V. harveyi detects three autoinducers HAI-1, CAI-1, and AI-2, using LuxN, CqsS, and LuxPQ, respectively, to control its QS response. These circuits are proposed to be used for intra-species, intra-genus, and inter-species communication, respectively [18, 21, 48]. Intriguingly, both VpsS and CqsR are predicted to be capable of detecting small chemical molecules. VpsS is predicted to be cytoplasmic, as it lacks any obvious membrane spanning domain. However, vpsV, a gene upstream of vpsS, could encode the signal-sensing partner [32]. VpsV carries a FIST domain, which could bind small ligands [49]. CqsR, in contrast, is predicted to be membrane-bound and possess a periplasmic CACHE domain, which is often found in receptors that detect amino acids and other molecules [50–52]. Thus, we speculate that the signals detected by VpsS and CqsR are chemical in nature and contain information that is absent from CAI-1 and AI-2. Although VpsS and CqsR are found predominantly in Vibrio species, it remains to be determined if their cognate signals are used for enumeration of species composition or as cell density proxies.
Materials and Methods
Strains, media, and culture conditions
All V. cholerae strains used in this study were derived from C6706str2, a streptomycin-resistant isolate of C6706 (O1 El Tor) [53]. E. coli S17-1 λpir was used as hosts for plasmids. All strains used in this study are described in S1 Table. V. cholerae and E. coli cultures were grown with aeration in Luria-Bertani (LB) broth at 30°C and 37°C, respectively. Unless specified, media was supplemented with streptomycin (Sm, 100 μg/ml), tetracycline (Tet, 5 μg/ml), ampicillin (Amp, 100 μg/ml), kanamycin (Kan, 100 μg/ml), chloramphenicol (Cm, 10 μg/ml) and polymyxin B (Pb, 50 U/ml) when appropriate.
DNA manipulations and mutant construction
All DNA manipulations were performed using standard procedures. High-fidelity PCR was performed using Phusion DNA polymerase (New England Biolabs). Taq DNA polymerase was used for routine screenings. Oligonucleotide sequences used for PCR, site-directed mutagenesis, and sequencing reactions will be provided upon request. Deletions and point mutations were introduced into the V. cholerae genome by allelic exchange using the suicide vector pKAS32 [54]. Mutations carried in vector pKAS32 from E. coli donors were introduced into the V. cholerae genome by conjugation on LB plates. Transconjugants were selected for by plating on Pb/Amp plates. Subsequent recombinants were selected on LB/Sm (5000 μg/ml) plates, followed by single colony isolation on LB/Sm (5000 μg/ml) plates. Mutant strains carrying the desired mutations were screened and confirmed by PCR. All mutant strains were confirmed by sequencing at the Tufts University Core Facility.
Infant mouse colonization model
V. cholerae bacterial cultures were grown aerobically for 16 hr in LB/Sm at 30°C. Mutant strains were then mixed equally with the wild-type ΔlacZ strain and approximately 106 colony forming units (CFU) were fed orally to 3 - to 5-day-old CD-1 mice (Charles River Laboratories). Prior to infection, infant mice were housed with ample food and water for at least 24 hr and monitored in accordance with the regulations of the Department of Laboratory Animal Medicine at Tufts University School of Medicine. Infected infant mice were sacrificed 24 hr post inoculation and their small intestines were harvested and homogenized. V. cholerae colonization in the small intestine was measured by plating serial dilutions of intestinal homogenate on LB/Sm/X-Gal plates and enumerating bacterial colonies the next day. Competitive index (CI) was calculated as the ratio of output to input of the mutant strain relative to the wild-type. A minimum of eight infected animals were used to calculate CI. V. cholerae colonization of the small intestine is presented as a single data point per mouse and data are graphed with the median. If the mutant strains were below the level of detection, it was assumed that there was 1 mutant CFU present at the next lowest dilution of the wild-type sample (indicated by open symbols in the figures).
HapR-dependent bioluminescence assays
V. cholerae strains carrying cosmid pBB1 [6], which harbors the heterologous V. harveyi luxCDABE operon, were first streaked on LB/Tet plates. Individual colonies were then grown aerobically for 16 hr at 30°C in LB/Tet. Cultures were further diluted 1 : 200 in 20 ml of LB/Tet and grown at 30°C with aeration. OD600 (1 ml of culture) and light production (0.1 ml of culture) were measured every 45–60 min for at least 10 hr using a Thermo Scientific Evolution 201 UV-Visible Spectrophotometer and a BioTek Synergy HT Plate Reader, respectively. Light production per cell was calculated from dividing light production by OD600. For the assays that determined the effects of surplus CAI-1, a 100 mM CAI-1 stock dissolved in DMSO was diluted to 20 μM in fresh media, DMSO was used as a negative control. For the assays that determined the effects of LuxO overexpression, IPTG was added to the cultures at 100 μM.
Effect of reconditioned spent culture medium on Qrr4 expression
Spent culture medium was prepared from wild-type V. cholerae or the ΔcqsA ΔluxS double synthase mutant. These two strains were grown in LB at 30°C aerobically to HCD (OD600 >4). Cells were removed by centrifugation and the supernatants were filtered through a 0.2 μm filter. Filtered cell-free spent culture medium (80%, v/v) was reconditioned by adding back 20% (v/v) 5× LB. As a control, fresh medium was prepared by adding 1× LB (80%, v/v) to 20% (v/v) 5× LB. V. cholerae mutants expressing only vpsS or cqsR (vc1831) and carrying pBK1003 (Pqrr4-lux) [55] were inoculated (1 : 1000 dilution) into these two media conditions in triplicate and grown in a 96-well microplate at 30°C with aeration. OD600 and light production were measured every 30 min for at least 10 hr using a BioTek Synergy HT Plate Reader. Light production per cell was calculated from dividing light production by OD600.
Ethics statement
All animal experiments were done in accordance with NIH guidelines, the Animal Welfare Act, and US federal law. The infant mouse colonization experimental protocol B2013-03 was approved by Tufts University School of Medicine's Institutional Animal Care and Use Committee. The mice were housed in a centralized and AAALAC-accredited research animal facility that is fully staffed with trained husbandry, technical and veterinary personnel.
Accession numbers (UniProt)
CqsS Q9KM66
LuxP Q9KLK6
LuxQ Q9KLK7
VpsS Q9KS16
CqsR Q9KR16
LuxO Q9KT84
LuxU Q9KT83
AphA H9L4T0
HapR B2CKP3
LuxS Q9KUG4
CqsA Q9KM65
CsrA Q9KUH3
Supporting Information
Zdroje
1. Williams P, Winzer K, Chan WC, Camara M. Look who's talking: communication and quorum sensing in the bacterial world. Philos Trans R Soc Lond B Biol Sci. 2007;362(1483): 1119–1134. 17360280
2. Novick RP, Geisinger E. Quorum sensing in staphylococci. Annu Rev Genet. 2008;42 : 541–564. doi: 10.1146/annurev.genet.42.110807.091640 18713030
3. Ng WL, Bassler BL. Bacterial Quorum-Sensing Network Architectures. Annu Rev Genet. 2009;43 : 197–222. doi: 10.1146/annurev-genet-102108-134304 19686078
4. Rutherford ST, Bassler BL. Bacterial quorum sensing: its role in virulence and possibilities for its control. Cold Spring Harb Perspect Med. 2012;2(11): a012427. doi: 10.1101/cshperspect.a012427 23125205
5. Schuster M, Sexton DJ, Diggle SP, Greenberg EP. Acyl-homoserine lactone quorum sensing: from evolution to application. Annu Rev Microbiol. 2013;67 : 43–63. doi: 10.1146/annurev-micro-092412-155635 23682605
6. Miller MB, Skorupski K, Lenz DH, Taylor RK, Bassler BL. Parallel quorum sensing systems converge to regulate virulence in Vibrio cholerae. Cell. 2002;110(3): 303–314. 12176318
7. Zhu J, Miller MB, Vance RE, Dziejman M, Bassler BL, Mekalanos JJ. Quorum-sensing regulators control virulence gene expression in Vibrio cholerae. Proc Natl Acad Sci U S A. 2002;99(5): 3129–3134. 11854465
8. Hammer BK, Bassler BL. Quorum sensing controls biofilm formation in Vibrio cholerae. Mol Microbiol. 2003;50(1): 101–104. 14507367
9. Zhu J, Mekalanos JJ. Quorum sensing-dependent biofilms enhance colonization in Vibrio cholerae. Dev Cell. 2003;5(4): 647–656. 14536065
10. Joelsson A, Kan B, Zhu J. Quorum sensing enhances the stress response in Vibrio cholerae. Appl Environ Microbiol. 2007;73(11): 3742–3746. 17434996
11. Tsou AM, Zhu J. Quorum sensing negatively regulates hemolysin transcriptionally and posttranslationally in Vibrio cholerae. Infect Immun. 2010;78(1): 461–467. doi: 10.1128/IAI.00590-09 19858311
12. Zheng J, Shin OS, Cameron DE, Mekalanos JJ. Quorum sensing and a global regulator TsrA control expression of type VI secretion and virulence in Vibrio cholerae. Proc Natl Acad Sci U S A. 2010;107(49): 21128–21133. doi: 10.1073/pnas.1014998107 21084635
13. Antonova ES, Hammer BK. Quorum-sensing autoinducer molecules produced by members of a multispecies biofilm promote horizontal gene transfer to Vibrio cholerae. FEMS Microbiol Lett. 2011;322(1): 68–76. doi: 10.1111/j.1574-6968.2011.02328.x 21658103
14. Suckow G, Seitz P, Blokesch M. Quorum sensing contributes to natural transformation of Vibrio cholerae in a species-specific manner. J Bacteriol. 2011;193(18): 4914–4924. doi: 10.1128/JB.05396-11 21784943
15. Blokesch M. A quorum sensing-mediated switch contributes to natural transformation of Vibrio cholerae. Mob Genet Elements. 2012;2(5): 224–227. 23446800
16. Lo Scrudato M, Blokesch M. The regulatory network of natural competence and transformation of Vibrio cholerae. PLoS Genet. 2012;8(6): e1002778. doi: 10.1371/journal.pgen.1002778 22737089
17. Shao Y, Bassler BL. Quorum regulatory small RNAs repress type VI secretion in Vibrio cholerae. Mol Microbiol. 2014;92(5): 921–930. doi: 10.1111/mmi.12599 24698180
18. Henke JM, Bassler BL. Three parallel quorum-sensing systems regulate gene expression in Vibrio harveyi. J Bacteriol. 2004;186(20): 6902–6914. 15466044
19. Jahan N, Potter JA, Sheikh MA, Botting CH, Shirran SL, Westwood NJ, et al. Insights into the biosynthesis of the Vibrio cholerae major autoinducer CAI-1 from the crystal structure of the PLP-dependent enzyme CqsA. J Mol Biol. 2009;392(3): 763–773. doi: 10.1016/j.jmb.2009.07.042 19631226
20. Kelly RC, Bolitho ME, Higgins DA, Lu W, Ng WL, Jeffrey PD, et al. The Vibrio cholerae quorum-sensing autoinducer CAI-1: analysis of the biosynthetic enzyme CqsA. Nat Chem Biol. 2009;5(12): 891–895. doi: 10.1038/nchembio.237 19838203
21. Ng WL, Perez LJ, Wei Y, Kraml C, Semmelhack MF, Bassler BL. Signal production and detection specificity in Vibrio CqsA/CqsS quorum-sensing systems. Mol Microbiol. 2011;79(6): 1407–1417. doi: 10.1111/j.1365-2958.2011.07548.x 21219472
22. Wei Y, Perez LJ, Ng WL, Semmelhack MF, Bassler BL. Mechanism of Vibrio cholerae Autoinducer-1 Biosynthesis. ACS Chem Biol. 2011;6(4): 356–365. doi: 10.1021/cb1003652 21197957
23. Higgins DA, Pomianek ME, Kraml CM, Taylor RK, Semmelhack MF, Bassler BL. The major Vibrio cholerae autoinducer and its role in virulence factor production. Nature. 2007;450(7171): 883–886. 18004304
24. Surette MG, Miller MB, Bassler BL. Quorum sensing in Escherichia coli, Salmonella typhimurium, and Vibrio harveyi: a new family of genes responsible for autoinducer production. Proc Natl Acad Sci U S A. 1999;96(4): 1639–1644. 9990077
25. Chen X, Schauder S, Potier N, Van Dorsselaer A, Pelczer I, Bassler BL, et al. Structural identification of a bacterial quorum-sensing signal containing boron. Nature. 2002;415(6871): 545–549. 11823863
26. Pereira CS, Thompson JA, Xavier KB. AI-2-mediated signalling in bacteria. FEMS Microbiol Rev. 2013;37(2): 156–181. doi: 10.1111/j.1574-6976.2012.00345.x 22712853
27. Bassler BL, Wright M, Silverman MR. Multiple signalling systems controlling expression of luminescence in Vibrio harveyi: sequence and function of genes encoding a second sensory pathway. Mol Microbiol. 1994;13(2): 273–286. 7984107
28. Lenz DH, Mok KC, Lilley BN, Kulkarni RV, Wingreen NS, Bassler BL. The small RNA chaperone Hfq and multiple small RNAs control quorum sensing in Vibrio harveyi and Vibrio cholerae. Cell. 2004;118(1): 69–82. 15242645
29. Rutherford ST, van Kessel JC, Shao Y, Bassler BL. AphA and LuxR/HapR reciprocally control quorum sensing in vibrios. Genes Dev. 2011;25(4): 397–408. doi: 10.1101/gad.2015011 21325136
30. Shao Y, Bassler BL. Quorum-sensing non-coding small RNAs use unique pairing regions to differentially control mRNA targets. Mol Microbiol. 2012;83(3): 599–611. doi: 10.1111/j.1365-2958.2011.07959.x 22229925
31. Lenz DH, Miller MB, Zhu J, Kulkarni RV, Bassler BL. CsrA and three redundant small RNAs regulate quorum sensing in Vibrio cholerae. Mol Microbiol. 2005;58(4): 1186–1202. 16262799
32. Shikuma NJ, Fong JC, Odell LS, Perchuk BS, Laub MT, Yildiz FH. Overexpression of VpsS, a hybrid sensor kinase, enhances biofilm formation in Vibrio cholerae. J Bacteriol. 2009;191(16): 5147–5158. doi: 10.1128/JB.00401-09 19525342
33. Wei Y, Ng WL, Cong J, Bassler BL. Ligand and antagonist driven regulation of the Vibrio cholerae quorum-sensing receptor CqsS. Mol Microbiol. 2012;83(6): 1095–1108. doi: 10.1111/j.1365-2958.2012.07992.x 22295878
34. Fu Y, Waldor MK, Mekalanos JJ. Tn-Seq analysis of Vibrio cholerae intestinal colonization reveals a role for T6SS-mediated antibacterial activity in the host. Cell Host Microbe. 2013;14(6): 652–663. doi: 10.1016/j.chom.2013.11.001 24331463
35. Kamp HD, Patimalla-Dipali B, Lazinski DW, Wallace-Gadsden F, Camilli A. Gene fitness landscapes of Vibrio cholerae at important stages of its life cycle. PLoS Pathog. 2013;9(12): e1003800. doi: 10.1371/journal.ppat.1003800 24385900
36. Hammer BK, Bassler BL. Regulatory small RNAs circumvent the conventional quorum sensing pathway in pandemic Vibrio cholerae. Proc Natl Acad Sci U S A. 2007;104(27): 11145–11149. 17556542
37. Perego M, Cole SP, Burbulys D, Trach K, Hoch JA. Characterization of the gene for a protein kinase which phosphorylates the sporulation-regulatory proteins Spo0A and Spo0F of Bacillus subtilis. J Bacteriol. 1989;171(11): 6187–6196. 2509430
38. Trach KA, Hoch JA. Multisensory activation of the phosphorelay initiating sporulation in Bacillus subtilis: identification and sequence of the protein kinase of the alternate pathway. Mol Microbiol. 1993;8(1): 69–79. 8497199
39. LeDeaux JR, Grossman AD. Isolation and characterization of kinC, a gene that encodes a sensor kinase homologous to the sporulation sensor kinases KinA and KinB in Bacillus subtilis. J Bacteriol. 1995;177(1): 166–175. 8002614
40. Jiang M, Shao W, Perego M, Hoch JA. Multiple histidine kinases regulate entry into stationary phase and sporulation in Bacillus subtilis. Mol Microbiol. 2000;38(3): 535–542. 11069677
41. LeDeaux JR, Yu N, Grossman AD. Different roles for KinA, KinB, and KinC in the initiation of sporulation in Bacillus subtilis. J Bacteriol. 1995;177(3): 861–863. 7836330
42. Laub MT, Goulian M. Specificity in two-component signal transduction pathways. Annu Rev Genet. 2007;41 : 121–145. 18076326
43. Cornforth DM, Popat R, McNally L, Gurney J, Scott-Phillips TC, Ivens A, et al. Combinatorial quorum sensing allows bacteria to resolve their social and physical environment. Proc Natl Acad Sci U S A. 2014;111(11): 4280–4284. doi: 10.1073/pnas.1319175111 24594597
44. Drees B, Reiger M, Jung K, Bischofs IB. A modular view of the diversity of cell-density-encoding schemes in bacterial quorum-sensing systems. Biophys J. 2014;107(1): 266–277. doi: 10.1016/j.bpj.2014.05.031 24988360
45. Ng WL, Perez L, Cong J, Semmelhack MF, Bassler BL. Broad spectrum pro-quorum-sensing molecules as inhibitors of virulence in vibrios. PLoS Pathog. 2012;8(6): e1002767. doi: 10.1371/journal.ppat.1002767 22761573
46. Mok KC, Wingreen NS, Bassler BL. Vibrio harveyi quorum sensing: a coincidence detector for two autoinducers controls gene expression. Embo J. 2003;22(4): 870–881. 12574123
47. Bari SM, Roky MK, Mohiuddin M, Kamruzzaman M, Mekalanos JJ, Faruque SM. Quorum-sensing autoinducers resuscitate dormant Vibrio cholerae in environmental water samples. Proc Natl Acad Sci U S A. 2013;110(24): 9926–9931. doi: 10.1073/pnas.1307697110 23716683
48. Federle MJ, Bassler BL. Interspecies communication in bacteria. J Clin Invest. 2003;112(9): 1291–1299. 14597753
49. Borziak K, Zhulin IB. FIST: a sensory domain for diverse signal transduction pathways in prokaryotes and ubiquitin signaling in eukaryotes. Bioinformatics. 2007;23(19): 2518–2521. 17855421
50. Anantharaman V, Aravind L. Cache—a signaling domain common to animal Ca(2+)-channel subunits and a class of prokaryotic chemotaxis receptors. Trends Biochem Sci. 2000;25(11): 535–537. 11084361
51. Chen Y, Cao S, Chai Y, Clardy J, Kolter R, Guo JH, et al. A Bacillus subtilis sensor kinase involved in triggering biofilm formation on the roots of tomato plants. Mol Microbiol. 2012;85(3): 418–430. doi: 10.1111/j.1365-2958.2012.08109.x 22716461
52. Shemesh M, Chai Y. A combination of glycerol and manganese promotes biofilm formation in Bacillus subtilis via histidine kinase KinD signaling. J Bacteriol. 2013;195(12): 2747–2754. doi: 10.1128/JB.00028-13 23564171
53. Thelin KH, Taylor RK. Toxin-coregulated pilus, but not mannose-sensitive hemagglutinin, is required for colonization by Vibrio cholerae O1 El Tor biotype and O139 strains. Infect Immun. 1996;64(7): 2853–2856. 8698524
54. Skorupski K, Taylor RK. Positive selection vectors for allelic exchange. Gene. 1996;169(1): 47–52. 8635748
55. Svenningsen SL, Waters CM, Bassler BL. A negative feedback loop involving small RNAs accelerates Vibrio cholerae's transition out of quorum-sensing mode. Genes Dev. 2008;22(2): 226–238. doi: 10.1101/gad.1629908 18198339
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2015 Číslo 4
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Toxin-Induced Necroptosis Is a Major Mechanism of Lung Damage
- Transgenic Fatal Familial Insomnia Mice Indicate Prion Infectivity-Independent Mechanisms of Pathogenesis and Phenotypic Expression of Disease
- Role of Hypoxia Inducible Factor-1α (HIF-1α) in Innate Defense against Uropathogenic Infection
- A Temporal Gate for Viral Enhancers to Co-opt Toll-Like-Receptor Transcriptional Activation Pathways upon Acute Infection
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy