
Global Genome and Transcriptome Analyses of Epidemic Isolate 98-06 Uncover Novel Effectors and Pathogenicity-Related Genes, Revealing Gene Gain and Lose Dynamics in Genome Evolution
Genetic variations in pathogens, such as the causal agent of rice blast Magnaporthe oryzae, often lead to circumvention of disease-resistance cultivars. Previous genome-wide analyses of model organisms suggest that pathogen effectors are also rapidly evolving, especially in regions with high genome plasticity. However, genetic variations among different isolates remain largely unknown in M. oryzae, particularly at the genome and transcriptome levels. In this study, we provided a systematic genomic and interaction transcriptome profile for a dominant rice blast field isolate, resulting in identification of 134 candidate effectors. Two effectors, Iug6 and Iug9, and one pathogenicity-related (PaR) gene product, Iug18, were subjected to functional characterization. We found that Iug6 and Iug9 are located in the biotrophic interfacial complex (BIC) and their overexpression leads to suppression of defense-related gene expression in rice, while Iug18 appears to be a novel PaR protein. Our studies support the hypothesis that isolate-unique genes may serve as a source of genetic variability in the M. oryzae population encountering different environments. Our studies also facilitate further understanding of effectors and genomic variations in pathogenicity of M. oryzae.
Published in the journal:
. PLoS Pathog 11(4): e32767. doi:10.1371/journal.ppat.1004801
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004801
Summary
Genetic variations in pathogens, such as the causal agent of rice blast Magnaporthe oryzae, often lead to circumvention of disease-resistance cultivars. Previous genome-wide analyses of model organisms suggest that pathogen effectors are also rapidly evolving, especially in regions with high genome plasticity. However, genetic variations among different isolates remain largely unknown in M. oryzae, particularly at the genome and transcriptome levels. In this study, we provided a systematic genomic and interaction transcriptome profile for a dominant rice blast field isolate, resulting in identification of 134 candidate effectors. Two effectors, Iug6 and Iug9, and one pathogenicity-related (PaR) gene product, Iug18, were subjected to functional characterization. We found that Iug6 and Iug9 are located in the biotrophic interfacial complex (BIC) and their overexpression leads to suppression of defense-related gene expression in rice, while Iug18 appears to be a novel PaR protein. Our studies support the hypothesis that isolate-unique genes may serve as a source of genetic variability in the M. oryzae population encountering different environments. Our studies also facilitate further understanding of effectors and genomic variations in pathogenicity of M. oryzae.
Introduction
Rice (Oryza sativa) is one of the most important staple food crops for more than half of the global human population [1]. However, the rice production is severely impacted by the blast disease caused by the fungus Magnaporthe oryzae, despite the cultivation of various disease-resistance cultivars [2,3,4]. One of the reasons is that new cultivars often lose their resistance within a few years of introduction. It is thought that genetic variability occurs rapidly within pathogen populations, especially in regions with high genome plasticity, and that the pathogenic traits such as secreted pathogen avirulence (AVR)/effectors evolve rapidly to counteract plant defense [5,6,7,8].
M. oryzae primarily infects rice but can also infects wheat or other small grains [9]. Recent advances in genetic and genomic technology have allowed M. oryzae to be a tractable model for studying the plant-microbe interaction [10]. Jantasuriyarat and colleagues used large-scale expressed sequence tag (EST) sequencing to profile gene expression at the early stage of the M. oryzae-rice interaction and identified four genes to be involved in the interaction out of 13,570 uniESTs [11]. Approximately 100 pathogenesis-related protein genes specific to infection were identified in other studies using EST analysis [10], and four biotrophy-associated secreted (BAS) proteins were also found in M. oryzae using microarray [4]. More recently, several studies have identified the presence of an infection structure, known as the biotrophic interfacial complex (BIC), that is involved in mediating the delivery of pathogen effectors into the rice host cytoplasm [4,12].
Interestingly, studies have also revealed that genome plasticity is closely linked to host-pathogen interaction specificities. Using genome-wide DNA polymorphism existed between M. oryzae field isolate Ina168 and isolate 70–15, one of the progenies from a cross between a weeping lovegrass isolate and the archetypical rice isolate Guy11 [13,14,15], three novel AVR genes were identified [16]. The presence of these AVRs correlates to a 1.68 Mb sequence in Ina168 that is absent from the assembled genome sequence of 70–15 [16]. Studies of two other field isolates, P131 and Y34, also revealed slightly larger genome contents with more genes. It is suggested that the presence of these isolate-specific genes play diverse roles, including conidiation, vegetative growth, or virulence [17].
Genome studies of additional M. oryzae field isolates and identification of novel AVR effectors will help us to further address the mechanism of pathogen and host coevolution. Here, we characterized the genome of the dominant blast isolate 98–06 on several different rice cultivars cultivated in Southeast China (S1 Table). We found that 98–06 contains seven known AVRs, corresponding to its wide field adaptability. AVR PWL1, PWL2, Avr-Pita, Avr1-CO39, and ACE1 are highly polymorphic due to point mutations while AVR Avr-Pia and Avr-Pii are also polymorphic, but with complete deletions. AvrPiz-t and Avr-Pik allele (D) were also found and conserved. In addition, we found that isolate 98–06 contains 1.43 Mb of isolate-specific sequences encoding 238 isolate-specific genes, in comparison to isolate 70–15. In addition, genome-wide expression profiling revealed the presence of a defense network in rice and major expression patterns of pathogenesis-related genes during the M. oryzae-rice interaction. Moreover, we predicted 134 candidate effectors from 1,732 putative secreted proteins and provided evidence to demonstrate that IUG6 and IUG9 are novel effectors.
Results
Sequencing and assembling of the isolate 98–06 genome
The genome of the M. oryzae isolate 98–06 was sequenced using Illumina high-throughput sequencing technology. Four different insert-size libraries (500, 350, 5,000, and 10,000 bp) were generated, which represent 36.8-, 38.2-, 31.9-, and 27.6-fold coverage assemblies spanning 1,560, 1,618, 1,350, and 1,167 Mb, respectively. The sequence coverage that was approximately six-fold greater than that of P131 and Y34 [17], provided a near perfect assembly. The sequence reads were assembled into 1,161 contigs and placed into 284 scaffolds with a combined length of 42.1 Mb using SOAPdenovo [18,19]. The N50 and maximum lengths of scaffolds were 1,530 and 3,511 kb, respectively (Table 1). The sequences have been deposited at GenBank under the submission number of JRBC00000000.

When the scaffolds of isolate 98–06 were aligned with the assembled genome of isolate 70–15, 98.83% of the genome was conserved. However, 98–06 contains 1.4 Mb isolate-specific sequences that are dispersed throughout the genome (Fig 1, S2 Table). Interestingly, blocks of the reverse-alignment sequences and chromosomal breakpoints were found at regions near the telomeres (S1 Fig).

Genome annotation
A total of 14,019 genes including 1,732 secreted protein genes were identified from the annotated isolate 98–06 genome (Table 1). The average length of predicted proteins is ~470 amino acids, and the predicted genes comprise 46% of the assembly genome. In all, the 98–06 genome possesses more genes than isolates 70–15 [15], P131, Y34 [17], FJ, or HN [20]. Further analysis revealed that 238 genes are unique when compared with 70–15 (49 genes if compared with P131 and Y34), and of these 238 genes, 10% encode secreted proteins while 69% have no significant homologs in GenBank (S3 Table). We conducted a Clusters of Orthologous Groups (COG) study and found that some of these specific genes are associated with general biological functions, including secondary metabolism and energy generation processes (S2 Fig). Intriguingly, isolate-specific genes appear to be mainly located at some of the chromosomal ends.
To identify possible gene families, we clustered the predicted proteins of isolates 98–06, 70–15, P131, and Y34 using the OrthoMCL algorithm. A total of 52,177 proteins were grouped into 13,236 clusters, with each cluster containing at least two putative protein orthologs/paralogs (S3 Fig, S4 Table). The mean gene number of each cluster for 98–06, 70–15, P131, and Y34 was 1.11, 1.04, 1.02, and 1.02, respectively. Among these clusters, 12.86% of all 14,019 genes in 98–06 had at least one paralog, while 6.28%, 3.27%, and 3.08% of all genes for 70–15, P131, and Y34 respectively (S4 Table). These results suggested that there are more paralogs in 98–06 than in the other isolates. Meanwhile, there are 455, 310, 165, and 150 gene families with at least two paralogs in 98–06, 70–15, P131, and Y34, respectively. Comparative analysis indicated that 227 gene families are expanded in 98–06 (S4 Table), which is consistent with its larger genome content.
Repeated sequences and transposable elements
Repeated sequences and transposable elements (TEs) often account for a large portion of genomes and therefore are important in genome annotation. Using multiple approaches, we found that 9.3% of the 98–06 genome consists of repetitive sequences. This is consistent with other fungi in which repetitive sequences comprise ≤ 10% proportion of the genomes [21]. However, transposable elements predicted by RepeatMasker [22] are more abundant in isolate 98–06 (2,776) than in isolates 70–15 (2,297), P131 (2,055), or Y34 (2,322) (S5 Table). In a follow-up examination of the larger number of genes (up to 1,732) that encode putative secreted proteins in 98–06, 132 genes containing putative signal peptide sequences were disrupted by TEs (S6 Table).
Identification of effector candidates
Proteins secreted by fungal pathogens during host colonization are generally referred to as effectors [23,24], which are often less than 200 amino acids in length and cysteine-rich. Therefore, we principally focused on small secreted proteins less than 200 amino acids in length, and identified 645 effector candidates out of 1,732 secreted proteins in 98–06 (S7 Table). These candidates, including known AVR genes Avr-Pik (allele D) and AvrPiz-t, have an average cysteine content of 3.11% in comparison to 1.26% of all 1,732 putative secreted proteins. Isolate 98–06 is known to exhibit an incompatible interaction with various rice cultivars harboring 12 known resistance (R-) genes: Pi7, Pi1, Pik, Pik-m, Pi20, Pi9, Pita2, Piz-t, Pik-p, Pish, Piz5, and Pik-h (S1 Table), and the Pi7, Pi1, Pik, Pik-m, Pi-kp, and Pik-h are alleles that could all recognize Avr-Pik (allele D). We focused on the remaining 643 (645–2 = 643) secreted proteins that provide a rich repertoire for the identification of novel effectors.
Interactive transcriptome analyses
Despite that conserved motifs such as the RxLR domain of oomycetes [25] were not found in some M. oryzae effectors, the M. oryzae effectors are often planta-specific and secreted proteins [4]. To study the host-pathogen interaction, we performed RNA sequencing (RNA-Seq) for six stages: mycelium (MY) and conidial infection at 0 h, 8 h, 24 h, 48 h, and 72 h post inoculation (CO-0h to CO-72h). After discarding low-quality raw reads, we obtained 12.8~13.7 million clean reads from each of the six samples, and aligned these reads against the reference genes and genomes (S8 Table). Almost all of the genes in isolate 98–06 and rice (O. sativa) were transcribed (11,075 and 26,452, respectively, S9 Table). The majority of the differentially expressed genes (log2Ratio ≥1 and GFOLD (0.01) >1) in M. oryzae were up-regulated during host-pathogen interaction in comparison to conidial infection at 0 h, suggesting a strong interaction between M. oryzae and rice (S4 Fig).
To confirm the RNA-Seq profiles, qRT-PCR was conducted on nine randomly selected M. oryzae and rice genes that were either induced or repressed. The average match between RNA-Seq and qRT-PCR data of the original sequenced samples was 30 of 41 (73%) (S5 Fig), indicating basic consistency between the two approaches. In addition, the average match between qRT-PCR of another independent sampling and the original sampling, or the RNA-Seq, was 76% and 63%, respectively (S5 Fig). There also appear certain correlations among the sequencing data of samples belonging to different stages, as distinct from the comparative RNA-Seq between wild type and mutant. Moreover, the expression patterns of several previously characterized genes, such as MoACTIN; MoAP1, MoVAM7, and MoBAS1 [4,26,27], in the transcriptome (see S10 Table) were similar to previous studies. These validation tests rectified to certain degree the potential limitation resulting from lack of sample duplication in RNA-Seq analyses.
Since plant hormone responses play an important role in the host defense network during the rice-pathogen interaction [28], we examined the expression of genes involved in salicylic acid (SA), jasmonic acid (JA), ethylene (ET), and mitogen-activated protein kinase (MAPK) signaling cascades. Most of the genes in the SA and ET signaling pathways were up-regulated, meanwhile, JAZ genes that act as transcriptional repressors of JA [29] were induced after 48 hpi. A comparison of aggregate expression levels at 0 hpi versus 8, 24, 48 and 72 hpi in planta is shown in Fig 2. Transcription for the most of the MAPK genes was increased during infection. In addition, several WRKY transcription factors and pattern recognition receptors (PRRs) FLS2 [30] and EFR were also significantly induced at 48 hpi (Fig 2). These findings revealed that multiple host defense signaling pathways were involved in the response to the infection of isolate 98–06.

Finally, to investigate how the M. oryzae genetic program is deployed during infection, we explored 64 known genes relevant to pathogenicity and 10 known effectors of M. oryzae in our interaction transcriptome (S10 Table), and determined their expression patterns by the clustering affinity search technique (CAST) assay through MultiExperiment Viewer 4.6 software package [31]. The distance metric is the default Pearson correlation, and the threshold affinity value is 0.8. Fourteen clusters were generated, and the cluster-a with the most members (23 genes) was illustrated, which provided the major expression pattern of pathogenesis-related genes (Fig 3A, a; S10 Table). The expression pattern-a represents high expression throughout the infection process, with the exception of sharp reduction at 24 hpi. In general, the virulence factors were up-regulated with different waves of expression during infection (Fig 3A). Interestingly, another expression pattern-b for effectors was also distinguished (Fig 3A and 3B; S10 Table).

Expressions of SNARE and effector genes
In transcriptome analysis described above, two genes MoVAM7 and MoSSO1 encoding soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) were also identified. SNARE proteins mediate intracellular transport that is an essential biological process in fungi [32]. Consistent with this, MoVam7 and MoSso1 were found to be important in effector secretion and pathogenesis [27,33]. We gathered 21 SNARE genes and 35 other endocytosis-related (ER) genes through bioinformatics analysis, and found five expression patterns of SNARE genes and eight patterns of ER genes using the CAST method (S6 Fig; S11 and S12 Tables). It is surprising that the key expression patterns of SNARE and ER genes are similar to the pathogenicity pattern-a, suggesting that this typical pathogen–host interaction pattern could be used to explore novel pathogenicity genes.
In a broader definition, effectors are specific types of proteins secreted into the plant by pathogens to interfere with plant defenses, and they most likely play other roles in promoting infection as well [34]. To date, 15 effectors were identified that include nine Avr effectors, six newly identified effectors including four BAS proteins, Slp1, and MC69. In this analysis, AvrPiz-t, BAS1, BAS3, BAS4, and SLP1 genes were clustered into expression pattern-b (Fig 3A), indicating that the expression is highly inducible during the biotrophic invasion. Hierarchical clustering (HCL) analysis also showed two distinct expression patterns that are similar to pattern-a and pattern-b, respectively (Fig 3B).
Since Avr-Pik and AvrPiz-t genes are highly expressed during infection, and studies of two M. oryzae isolate-specific sequences and Verticillium dahliae indicated that lineage-specific genomic regions are enriched in genes encoding new effectors [35], we explored the expression patterns of 645 small candidate effectors in 98–06 through CAST assay. 134 candidate effectors were found to be co-regulated with Avr-Pik and AvrPiz-t (Fig 4; S13 Table). Six known effectors BAS1, BAS2, BAS3, BAS4, SLP1, and PWL1 were among these candidates, providing a compelling argument for the presence of additional effectors among these candidates.

Five candidate effectors suppress the hypersensitive cell death induced by BAX
Many bacterial and fungal pathogen effectors can suppress innate plant immunity, particularly that triggered by pathogen-associated molecular patterns [34,36,37]. For example, M. oryzae AvrPiz-t suppresses the BAX-mediated programmed cell death in tobacco leaves [38]. Among 134 putative secreted proteins identified, most have no predicted functions, including several 98–06 isolate-unique genes (IUG) that were not found in 70–15. We queried the nucleic acid sequences against the genomic sequences of P131 and Y34 and found that IUG6 (Mo_GLEAN_10000617) and IUG9 (Mo_GLEAN_10000765) are specific to 98–06.
To identify Iug6 and Iug9 functions, we used a PVX-based high-throughput transient plant expression system in Nicotiana benthamiana. We also included three randomly selected non-specific proteins Nup1, Nup2, and Nup3 (MGG_07900, MGG_08024, and MGG_04546) for controls. We first removed the signal peptides of IUG6, IUG9, NUP1, NUP2, and NUP3 to enable the genes to be expressed stably in plant cells before cloning into the PVX vector pGR106. Infiltration of N. benthamiana leaves with Agrobacterium tumefaciens cells carrying pGR106:IUG6, pGR106:IUG9, pGR106:NUP1, pGR106:NUP2, pGR106:NUP3, and the negative control pGR106:GFP did not cause any obvious cell-death symptoms (S7 Fig), whereas obvious cell death was observed in N. benthamiana leaves infiltrated with A. tumefaciens cells carrying pGR106:BAX. N. benthamiana leaves infiltrated with A. tumefaciens cells harboring IUG6, IUG9, NUP1, NUP2, and NUP3 genes 24 h prior to infiltration with the pGR106:BAX-harboring cells did not produce symptoms (Fig 5A). The expression of BAX was detected at 48 h after infiltration (Fig 5B), which ruled out the possibility that BAX failed to express. Iug6, Iug9, Nup1, Nup2, and Nup3 all conferred BAX cell-death suppression activity. By analogy, additional effectors may also present in these 134 candidate effectors.
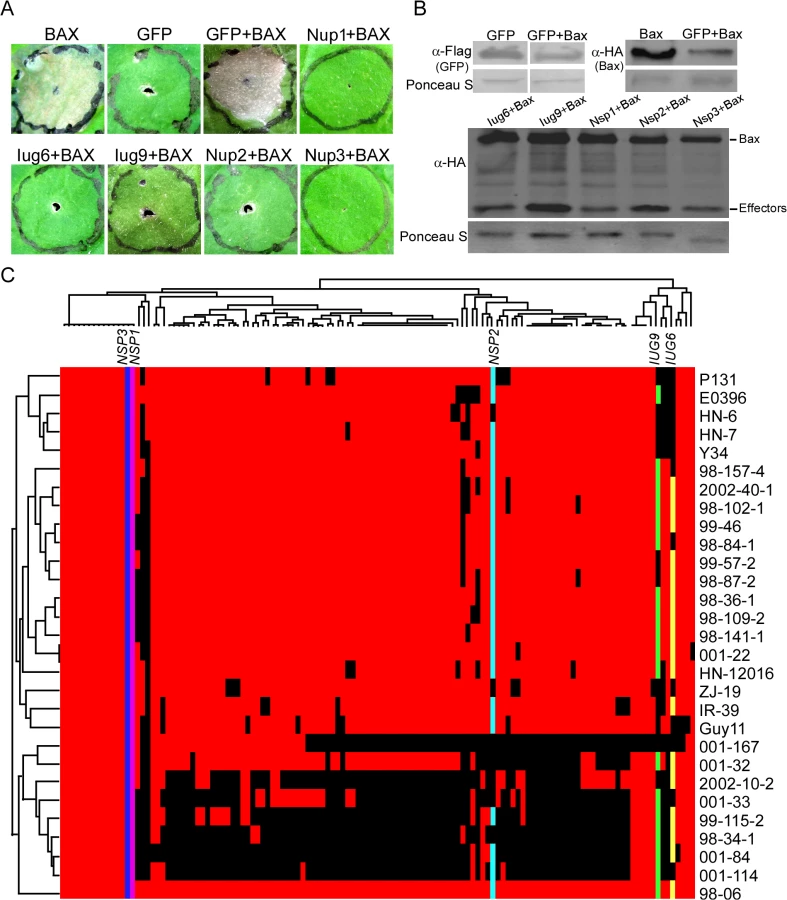
Presence or absence polymorphisms of these 127 candidate effectors (minus six known effectors and one failed to be amplified by PCR) were tested by PCR using 29 M. oryzae field isolates collected from China (Fig 5C). The candidate effectors showed different segregation patterns. Among them, IUG6, IUG9, and NUP2 have varied presence in 29 field stains, whereas NUP1 and NUP3 are detected in all isolates (Fig 5C). A recent report indicates that the presence or absence of putative secreted proteins shows a good correlation with AVR genes in M. oryzae [16]. Since half of the characterized blast AVR effector genes are not present in the isolate 70–15 genome, it may suggest that gene gain or loss events could be a major factor in the evolution of AVR effectors [39].
Functional analysis of isolate-unique genes (IUGs)
Functional characterization of IUGs may reveal novel effectors or other factors significant in the host-pathogen interaction. We thus further characterized the functions of IUG6 and IUG9, along with IUG17, IUG18, IUG34, and IUG37. We found that IUG6 and IUG9 are located near the chromosomal ends (S14 Table), which is consistent with the hypothetic rapid development of novel effectors in plastic regions [40]. IUG17 (Mo_GLEAN_10000632) and IUG18 (Mo_GLEAN_10001404) are predicted to each contain the aminoglycoside phosphotransferase (APH) domain (145–223 amino acids) and the RelA_SpoT domain (96–234 amino acids), while IUG34 (Mo_GLEAN_10000877) and IUG37 (Mo_GLEAN_10002374) are predicted to be associated with pathogenicity-related expression pattern through CAST assays. We also generated respective deletion mutants in 98–06 using the hygromycin-resistance marker gene and complemented the mutants with the respective wild type gene alleles including the endogenous promoter of approximately 1-kb (S8 Fig).
Disruption of IUG6 and IUG9, as well as IUG18, resulted in defects in vegetative hyphal growth and virulence, whereas disruption of IUG17, IUG34, and IUG37 had no obvious effects on colony morphology, conidiation, or virulence (S9 Fig). Iug6 is relatively specific to M. oryzae, since only one homolog was identified in the fungus Gaeumannomyces graminis (GL385397.1, 53% identity, E = 4e-17). Iug9 homologs can be found in Colletotrichum higginsianum (CCF43986.1, 63% identity, E = 7e-29) and C. graminicola (EFQ33033.1, 63% identity, E = 4e-26). In contrast, Iug18 contain the RelA_SpoT domain that is well conserved among other filamentous fungi. Microsynteny analysis revealed that all three genes are located at the genomic inserted regions comparable to 70–15 (S10A Fig). However, the parallel result was unfavorable, probably because the laboratory strain 70–15 was generated from a cross between rice and weeping lovegrass isolates [13,14]. To exclude the possibility that this was an assembly gap leading to gene deletion in the other three isolates, we amplified the genes by PCR from isolates 70–15, Guy11, P131, and Y34 and performed a Southern blotting analysis (S10B Fig). Expression of these three genes was validated by qRT-PCR, which also showed that IUG6 and IUG9 exhibit high expression levels during the pathogen-host interaction (S11 Fig).
The Δiug6, Δiug9, and Δiug18 mutants were examined for phenotypes including conidiogenesis. Conidiation in 10-d-old cultures of the iug6 mutant was reduced dramatically, by ~17-fold, compared with WT (Fig 6B). The Δiug9 and Δiug18 mutants showed approximately 14% and 32% reduction in conidiation on SDC medium (Fig 6B). Microscopic observations showed that Δiug6, Δiug9, and Δiug18 mutants produced significantly fewer conidia than the WT strain (Fig 6A). To determine whether IUG6, IUG9, and IUG18 affect the expression of conidiation-related genes including MoCOM1, MoHOX2, MoCON7, MoCOS1, and MoSTUA [41,42,43,44,45], we measured and found that their expression was decreased (Fig 6C). To determine whether Δiug6, Δiug9, and Δiug18 have defects in pathogenicity, conidial suspensions (5 x 104 spores /ml) were sprayed onto 2-wk-old susceptible rice seedlings (CO-39 & LTH). Only small, necrotic-like dark brown spots were observed in Δiug6-infected rice leaves in comparison to controls (Fig 7A and 7B). When the lesions were excised, surface sterilized with 70% ethanol for 1 min, and incubated with light and humidity on 4% water agar for 2 days, as described in a previous study [46], no fungal growth or conidia occurred. The Δiug9 and Δiug18 mutants also resulted in a reduction in disease symptoms on rice 7 days after inoculation (Fig 8A). The mean lesion density per unit area of the mutants was significantly lower than that of WT (Fig 8B). Disease symptoms of three approximately 6 cm long rice blades from the same parts of plants infected by Δiug9 mutant or WT were also quantified using a ‘lesion-type’ scoring assay [9], which showed that lesion types 4 and 5 (severe, coalescing) were rarely produced by the Δiug9 mutant (Fig 8C). In addition, fungal DNA in rice was significantly lower in infection by Δiug9 and Δiug18 mutants than that by the WT as determined by M. oryzae 28S rDNA quantitation [47] (Fig 8D).



Finally, we performed a detailed phenotypic analysis to investigate the infectious hyphae within the host cells. At 24 hpi in barley epidermal cells and 48 hpi in rice leaf sheaths, both the WT and the Δiug6 mutant strain formed normal appressoria, but the invasive hyphae (IH) of the WT strain freely expanded into host cells. On the other hand, appressoria of Δiug6 failed to develop any penetration structures among 50 examined infection sites in rice and barley, respectively (Fig 7C and 7D). We also analyzed invasive hyphal growth of Δiug9 and Δiug18 mutants at 100 appressorial penetration sites in barley tissues by rating the hyphal growth into four types (type I, no penetration; type II, with penetration peg; type III, with a single invasive hypha; type IV, with extensive hyphal growth) after inoculation with the spore suspensions for 30 hours (Fig 8E). 75% of penetration sites of WT showed III or IV invasive growth type, by contrast 42% of cells were limited in type I invasive hyphal growth in the Δiug9 mutant, and 77% penetration sites of the Δiug18 mutant showed type II or III hyphal growth. A similar result was also observed in rice leaf sheaths, with the WT and complemented strains displaying faster hyphal growth extension to neighboring cells while the Δiug9 and Δiug18 mutants showing lower hyphal growth limited to one cell (Fig 8F).
Iug6 and Iug9 are accumulated in BICs
To further characterize these Iug proteins, we expressed IUG6:GFP, IUG9:GFP, and IUG18:GFP fusion genes under the control of their native promoters in Guy11, respectively. GFP fluorescence was observed in the conidial septum expressing Iug6:GFP and Iug9:GFP, while faint fluorescence was seem in the cytoplasm of conidia expressing Iug18:GFP (S12 Fig).
We next detected whether any GFP signals can be detected in the biotrophic interfacial complex (BIC). To suppress host immunity during biotrophic intracellular growth, fungal effectors of M. oryzae are secreted and accumulated at the BIC or more generally within the Extra-Invasive Hyphal Membrane (EIHM) [4]. When transformants invaded rice sheath cells at ~27 hpi, the fluorescence of Iug6 can be observed in BIC (Fig 9A). Microscopy of the secreted Iug9:GFP protein showed fluorescence that outlines the primary hyphae and BICs at ~27 hpi (Fig 9B), similar to the BIC localization control (Fig 9C), AvrPiz-t:GFP [48]. Both findings suggested that Iug6 and Iug9 could be delivered into the rice cytoplasm and accumulated in BICs to facilitate biotrophic invasion.

To validate the signal peptide prediction of Iug6 and Iug9, we used an assay based on the method previously described by Oh et al. [49]. The constructs with the signal peptides of Iug6 and Iug9 allowed the invertase secretion-deficient yeast strain YTK12 to grow on YPRAA medium, and this invertase activity was confirmed by inclusion of the triphenyl tetrazolium chloride dye (S13 Fig). These results showed that the signal peptides of IUG6 and IUG9 are indeed functional.
We further transformed the IUG6 gene into the virulent isolate Guy11 and used the Δiug9 mutant for pathogenicity comparison tests in four resistant rice cultivars: IRBLz5-CA (Pi-z5), IRBLsh-S (Pi-sh), IRBL20-IR24 (Pi-20), IRBLta2-Re (Pi-ta2), and susceptible cultivar LTH. All of the transformants showed disease symptoms (S14 Fig), excluding the possibility that IUG6 and IUG9 are conventional AVR genes corresponding to these specific resistance genes. However, further studies are underway to address whether or not IUG6 and IUG9 are unknown avirulence genes that are involved in the evolutionary host-pathogen arms race.
SA and ET signaling of rice is suppressed in the presence of Iug6 and Iug9
As Iug6 and Iug9 can suppress the BAX-mediated programmed cell death in tobacco leaves, we investigated whether their expression affects the transcription of the rice defense-related genes. When infected with Guy11 over-expressing IUG6 and IUG9, the pathogenicity of these transformants is not obviously improved, but the expression of PR1a and Cht1 in rice was significantly less than that caused by Guy11 following qRT-PCR analysis (Fig 10B). Similarly, the expression levels of PR1a and Cht1 in rice by infection with isolate 98–06 were also significantly lower than that infected with Guy11 at 24 hpi or 8 hpi, with delayed peak expression. The expression patterns of PR1a and Cht1 in rice when infected with isolates over-expressing IUG6 or IUG9 is similar to that by isolate 98–06, with somewhat lowered peak expression. PR1a and Cht1 are respective SA and ET signaling marker genes. SA is involved in establishing basal defenses, effector-triggered immunity, and systemic acquired resistance in many dicotyledonous species [50,51], as well as in modulating redox balance and protecting rice plants from the oxidative stress caused by M. oryzae [52]. Our above interaction transcriptome analysis also suggested that SA and ET signaling pathways might function positively on rice basal defense against M. oryzae. We hypothesized that Iug6 and Iug9 might target unknown factors in rice, leading to suppression of the SA and ET signaling and promotion of biotrophy.

Discussion
M. oryzae is known for its natural genetic variation in the field, resulting in emergence of new epidemics threatening world food supply. Comparative analyses of multiple new epidemics not only steadily improved the assembly and annotation and the identification of variations in the M. oryzae genome, but also open a door to explore new virulence mechanism of the pathogen for effective blast control [2,9]. In this study, we reported the genome and interactive transcriptome analyses of isolate 98–06 that is known to be one dominant field isolate containing as many as seven AVR genes (Avr-Pik/km/kp, AvrPiz-t, Avr-Piz5, Avr-Pita2, Avr-Pish, Avr-Pi20, and Avr-Pi9). Among them, Avr-Pik/km/kp [16], AvrPiz-t [38], and Avr-Pi9 [53] were characterized. Avr-Pik/km/kp (allele D) and AvrPiz-t were also identified in our study.
Our analysis indicated that isolate 98–06 contains an extra 1.4 Mb of genomic sequences not present in 70–15, while a whole-genome dot-plot alignment of 98–06 and P131 suggested good genome synteny (S15 Fig). To examine whether the extra genomic region is present in other isolates, we searched the genomes of isolates P131 and Y34 that found that 49 genes remain unique to isolate 98–06. COG classification revealed that, while the functions for most of these genes were unknown, those identified exhibit functions relevant to general characteristics, secondary metabolites, and energy metabolism (S2 Fig).
Yoshida et al. [16] observed that presence or absence of effector gene polymorphisms are often associated with unstable genomic regions near the chromosome ends. It is therefore hypothesized that isolate-specific regions located at the chromosomal ends supply new effectors and pathogenicity-related factors to drive genome evolution. The dynamic adaptation of M. oryzae may be primarily achieved by deletion or recovery of AVR genes [54]. Such hypotheses indicate that further genome sequencing of multiple M. oryzae isolates is warranted for characterizing rice pathogenic strains, as key information can be discovered only by exploring beyond the “core” genomes [16]. One good example is that the extensive chromosomal rearrangement in asexual fungus V. dahliae establishes dynamic, lineage-specific regions that provide new effectors as a general mechanism of adaptation [35]. Although sexual reproduction can occur in M. oryzae under laboratory conditions, it has not been observed in nature. Such asexual organisms are often considered to be less flexible than sexual organisms [55,56], so that the expanded genome is likely to represent examples of evolutionary tradeoffs, as the cost of maintaining the extra DNA is counterbalanced by the functional advantages it confers [57]. Since horizontal gene transfer (HGT) enables the acquisition of new genes and functions [57], we hypothesize that some of these isolate-specific genes might also be acquired by HGT to drive the evolutionary process.
Based on the genome data of isolate 98–06, our transcriptome analysis provides a further molecular view of M. oryzae and rice gene expression during infection. The major expression patterns of M. oryzae genes relevant to pathogenicity remain high throughout the infection process, with the exception of a sharp decline at 24 hpi (Fig 3A). The decreased expression pattern might due to low levels of transcript detection at 24 hpi, with only 2,426 genes (M. oryzae) compared with at least 5,161 genes for other infection stages. This is consistent with the previous discovery that expression of known pathogenicity genes was unchanged or down-regulated at 36 hpi [4]. We should point out that our RNA-Seq analyses were carried out without sample duplication that could result in potentially biased interpretation of the transcriptome data. Nevertheless, the result is agreeable with that from validation of several selected genes through qRT-PCR.
Effectors are known key pathogenicity determinants that modulate plant innate immunity and enable disease development in plant-pathogen interactions [58]. Several AVR genes have been previously isolated by map-based cloning, genetic association analysis, or loss-of-function approaches [7,16,59,60,61,62]. Most of these effectors are small secreted proteins lacking homology to known proteins [63,64,65]. In characterizing the isolate 98–06 genome, we found 645 small candidate effectors with an average 3.11% cysteine content from 1,732 putative secreted proteins. Biotrophic invasive-specific expression is one of the best indicators for identifying new blast effectors [65]. Based on the gene expression data, we found that BAS1, BAS3, BAS4, SLP1, and AvrPiz-t effectors all show the similar expression pattern during infection (Fig 3A). Based on the expression of Avr-Pik and AvrPiz-t, 134 candidate effectors were further explored using hierarchical clustering (Fig 4), including six characterized effectors BAS1, BAS2, BAS3, BAS4, SLP1, and PWL1. Among them, two isolate-unique effectors Iug6, Iug9, and three non-isolate unique proteins Nup1, Nup2, Nup3 were selected to show a similar function in blocking plant immunity (Fig 5). Our characterization of three IUG genes indicates that these 134 putative effectors were a rich source for additional effector identification. In our study, a high frequency in suppression of cell death induced by BAX in tobacco leaves was observed that may also be associated with effects of heterologous gene expression or the unfolded protein response (UPR) [66]. Therefore, additional studies are necessary to further characterize these effector candidates. Our functional analysis of IUG6 and IUG9 indeed showed that they are important for full virulence expression of isolate 98–06. In addition, IUG6 and IUG9 characterization revealed the gene absence/presence polymorphism among 29 field stains (Fig 5C), similar to six AVR (Avr-Pita1, Avr-Pik, Avr-Pia, Avr-Pii, PWL2, and ACE1) genes that exhibited a prevalence of presence/absence polymorphisms among 62 M. oryzae isolates [67]. The similarity is consistent with that IUG6 and IUG9 might be AVR effectors.
Functional characterization indicated that Iug6 and Iug9, as well as Iug18, are important for mycelial growth, sporulation, and pathogenicity (Fig 7 and Fig 8). Xue et al. [17] have also observed that several randomly selected isolate-specific genes play important and diverse roles, some affecting virulence while others affecting conidiation and vegetative growth. There are, however, several secreted proteins that are required for pathogenicity, such as MoMpg1, MoEmp1, MoMhp1, MoMsp1, MC69, and Slp1 [62,68,69,70,71]. MC69, which is potentially localized in BIC, is essential for successful appressorial penetration and pathogenicity [68]. Slp1 suppresses chitin-induced host defense responses, thereby facilitating rapid spread of the fungus within the host [71]. Targeted gene disruption of BAS1, BAS2, and BAS3 did not show any effects on pathogenic phenotypes, whereas BAS4 disruption did not provide any viable transformants [4]. Furthermore, over-expressing IUG6 or IUG9 in Guy11 can suppress the expression of PR1a and Cht1 in rice, which are marker genes of SA and ET pathways, respectively. These results are also supportive of the effector role for Iug6 and Iug9. Meanwhile Iug18 appears to be a PaR protein based on the absence of a secretion signal peptide.
Gene deletion and pseudogenization, silencing, amino acid replacements, protein chimerization, and new gene addition are all important factors for altering virulence in filamentous plant pathogens [57]. Our microsyntenic and Southern blotting analyses showed that IUG6, IUG9, and IUG18 are unique to isolate 98–06 compared to 70–15 and Guy11 (S10 Fig). Iug18 is present in several isolates belongs to the Rel-Spo like protein superfamily. Blast search indicates that Iug6 and Iug18 have homologs in G. graminis, while Iug9 has a homolog from C. higginsianum. An emerging question would be how some M. oryzae isolates obtained these extra genes and where these genes come from. An attractive model was proposed to explain how M. oryzae regains deleted avirulence effector genes, in which parasexual exchange of genetic material enabled the recovery of ‘lost’ genes in asexual lineages [54]. A comprehensive population study revealed that the Avr-Pita gene has experienced a number of translocations in M. oryzae and related Pyricularia species, most likely as the consequence of its recovery by lateral transfer (HGT) [54]. Another example of translocation was recently reported, in which the ToxA gene was transferred from Stagonospora nodorum, a wheat pathogen, to another (Pyrenophora tritici-repentis) [72]. All these findings point out the likely source of these IUG genes.
On the other hand, it has been reported that M. oryzae has evolved two distinct secretion systems to deliver apoplastic and cytoplasmic effectors, which outline the entire invasive hyphae (IH) or accumulate in the BIC [33]. Like AvrPiz-t, Iug6 and Iug9 proteins are accumulated at BICs (Fig 9), suggesting that they have high possibility to be translocated into rice cell as effectors. Fungal apoplastic effectors are invariably Cys rich, but cytoplasmic effectors accumulated in BICs are not necessarily Cys rich [73]. Especially, Iug6, a small, secreted protein with four Cys residues accumulating in BICs, is similar to other known Cys-rich blast effectors like Bas2 and AvrPiz-t [4,38]. Effector genes are also often located near the telomeres, which tend to evolve at higher rates than the rest of the genome [10, 64, 65]. IUG6 and IUG9 are located on the subtelomeric regions of chromosome II and chromosome I, respectively (S14 Table), revealing that they could be the result of rapid adaptation to environmental conditions. This discovery further promotes the hypothesis that isolate-specific regions of chromosomal plasticity serve as facilitators of genome evolution in M. oryzae.
Overall, this study provides a systematic genomic and interaction transcriptome analysis of the dominant rice blast field isolate 98–06. Based on bioinformatics and functional analysis, two novel effectors Iug6 and Iug9 and a pathogenicity-related gene IUG18 were characterized. This knowledge prompts the hypothesis that the isolate-unique genes beyond the “core” genome may act as a source for M. oryzae adaptation to the environment. Our analyses will facilitate further study of the roles of effectors and molecular mechanisms of pathogenesis and pathogen-host evolution.
Materials and Methods
Genome sequencing and assembling
The genome of M. oryzae 98–06 isolate was sequenced using a whole-genome shotgun approach. The genomic Illumina paired-end libraries were constructed with insertion size of 350 bp, 500 bp, 5 kb, and 10 kb, respectively, and sequenced at the Beijing Genomic Institute (BGI, China). The short reads of sequencing data (135-fold coverage) were assembled into genome sequence using SOAP denovo (version 1.05, http://soap.genomics.org.cn/soapdenovo.html), and then reads are mapped to contigs for partial assembling and filling gap through paired-end and overlap relationship between the reads [22,74]. The M. oryzae 70–15 genome sequence version 6 was downloaded from the Broad Institute (http://www.broadinstitute.org/annotation/genome/magnaporthe_grisea/MultiDownloads.html). The P131 and Y34 genome sequences were downloaded from the NCBI Genome Database (http://www.ncbi.nlm.nih.gov/genome).
Gene prediction and annotation
CEGMA software was used to predict the core genes of isolate 98–06, which is based on the eukaryotes conservative gene database. We used the core gene set as the training set to predict genes by the Augustus and SNAP softwares [75,76]. In addition, we used the Homolog software to predict genes by using Magnaporthe oryzae isolate 70–15 as the reference sequence [77]. The Glean software was then employed to integrate the above results. Gene functions were predicted by comparison with the NCBI NR protein database and the KEGG [78], COG [79], SwissProt [80], GO [81] databases. Protein domain was predicted using SMART database. Membrane and sub-cellular localization domains were predicted by TMHMM 2.0 [82], SignalP4.0 [83], and NLStradamus [84].
Nucleotide sequences of the predicted isolate 98–06 genes were compared separately with the genomic sequences of P131, Y34, and 70–15 isolates with TBLASTN [85]. Homologous genes with sequence identities of 100%, 80–100%, and 50–80% were defined as identical, similar, and divergent, respectively, while those below 50% were considered non-homologous.
Amino acids of 52,177 proteins from all four isolates were compared with each other using TBLASTN (version 2.2.23) [85], and orthologs/paralogs families were clustered through OrthoMCL (version 1.4) [74].
Transposable elements (TE) analysis and synteny mapping
We used three programs (RepeatMasker, RepeatProteinMasker, and Denovo) to predict transposon sequences [22]. We also used TRF (Tandem Repeat Finder) to identify tandem repeat sequences [86]. To search for genes disrupted by TEs, unique flanking sequences of TEs were used to search against 98–06 genes. Masked genome sequences of the 98–06 isolates were compared with the MUMMER package [87] to construct chromosome sequences for isolate 98–06 based on isolate 70–15 data.
Effector genes prediction
The putative secreted proteins were identified using several prediction algorithms. TargetP 1.1 (http://www.cbs.dtu.dk/services/TargetP/) was used to predict the cleavage sites of the predicted presequences with the ‘‘Perform cleavage site predictions” option. SignalP 3.0 (http://www.cbs.dtu.dk/services/SignalP-3.0/) was used to predict signal peptide cleavage sites. Transmembrane helices were predicted using TMHMM 2.0 (http://www.cbs.dtu.dk/services/TMHMM-2.0/). Proteins that contain signal peptide cleavage sites but not transmembrane helices were selected as putative secreted proteins. Proteins of less than 200 amino acids in length were retained, and the average percentage of cysteine content was calculated.
Interaction transcriptome analysis
Three-week-old rice plants (cv. CO-39) were inoculated with M. oryzae isolates at 1 x 108 spores /ml. The inoculated plants were placed in a sealed plastic box in the dark for 24 h at 25°C, and leaf tissues were collected at 0 h, 8 h, 24 h, 48 h and 72 h after inoculation. Mycelia were grown in shaking culture in complete medium for 36 h at 28°C, 150 rpm and harvested. Total RNA was extracted using the Invitrogen kit as described previously [88]. RNA was extracted from samples that were the mixtures of three independent experiments.
Poly(A) mRNA was isolated from total RNA using oligo(dT) magnetic beads (Invitrogen, Carlsbad, CA, USA). Using RNA as templates, random hexamer primed cDNA synthesis was performed using reverse transcriptase (Invitrogen). Second-strand cDNA was synthesized using RNase H (Invitrogen), DNA polymerase I (New England Biolabs), dNTPs and buffer. DNA was purified using the QIAquick PCR extraction kit and ligated to sequencing adaptors following end repairing and Ploy (A) addition. Finally, the cDNA libraries were loaded onto the flow cell channels of an Illumina HiSeqTM 2000 platform for paired-end 90 bp x 2 sequencing at the Beijing Genomics Institute (BGI), Shenzhen, China [89].
After discarding low-quality raw reads, the clean reads from each library were assembled to M. oryzae and rice genomes separately, and gene sequences were annotated using SOAP2 [90]. Gene expression levels were measured in the RNA-Seq analysis as reads per kilobase of exon model per million mapped reads (RPKM) [91]. Differentially expressed genes and their corresponding P-values were determined using the recent GFOLD algorithm, which could give more stable and biological meaningful gene ranking in comparison with other methods especially for single biological replicate experiments [92]. Fold changes (log2Ratio) were estimated according to the normalized gene expression level in each sample. We used the absolute value of log2Ratio ≥1 and GFOLD (0.01) >1 as the threshold to judge differentially expressed genes.
For KEGG pathway analysis [93], all the differentially expressed genes in the pathways were examined to uncover common expression patterns. For hierarchical clustering, Pearson’s correlation coefficient and Spearman’s rank were used to measure similarities between gene expression profiles and between samples, respectively. The heat map of the clustered genes and samples was generated by complete linkage.
Quantitative RT-PCR assay
For RT-PCR and quantitative real time RT-PCR (qRT-PCR), 5 mg of total RNA was reverse transcribed into first-strand cDNA using the oligo(dT) primer and M-MLV Reverse Transcriptase (Invitrogen). The qRT-PCR reactions were performed following previously established procedures [82]. RNA-Seq expression profiles were validated by quantitative RT-PCR. Primer pairs used in this section are listed in S15 Table.
Culture conditions and plant infection assays
All strains were cultured on complete medium (CM) agar plates at 28°C [69]. The M. oryzae isolate 98–06 was used as the wild type strain for transformation in this study. Protoplasts were prepared and transformed as previously described [94]. Conidiation assay was performed also as previously described [95]. Mycelia were harvested from liquid CM for genomic DNA and RNA extraction. Vegetative growth was measured on CM, minimal medium (MM), straw decoction and corn medium (SDC) and oatmeal medium (OM) plates for 7 days at 28°C [27]. The radial growth was measured after incubation for 7 days and then photographed. All experiments were repeated three times, each with three replicates.
Plant infection and injection assays were performed as previously described by spraying 4 ml of conidial suspensions (5 x 104 spores /ml in 0.2% gelatin) on rice [96]. In order to distinguish resistant levels, spore suspensions were adjusted to 1 x 106 spores/ml for AVR candidate pathogenicity tests. For microscopic observation, rice was inoculated with 100 μl of conidial suspension (5 x 104 spores /ml) on the inner leaf sheath epidermal cells. After 48 h incubation under humid conditions at 28°C, leaf sheaths were collected and observed under a microscope. All experiments were repeated three times.
Gene disruption and complementation
To generate the IUG6 gene replacement vector pCX62, approximately 1 kb upstream and 1 kb downstream fragments were amplified with primer pairs (S15 Table). The resulting PCR products were ligated to the hygromycin resistance cassette released from pCX62, as previously described [97]. Putative mutants were screened by PCR and confirmed by Southern blotting analysis. To complement the Iug6 mutant, a DNA fragment including the putative promoter and the coding sequence was amplified and inserted into pYF11 (bleomycin resistance) by homologous recombination in Saccharomyces cerevisiae. The plasmids were extracted and transformed into E. coli competent cells, and then the plasmids with correct inserts were transferred into protoplasts, as previously described [97]. The same approach was used to generate mutants for isolate-specific genes IUG9, IUG17, IUG18, IUG34, and IUG37. The primer pairs used are listed in S15 Table. To observe secretion in rice cells, the coding sequences of IUG6, IUG9, and AvrPiz-t with their native promoters were fused with GFP in pYF11. To generate over-expression transformants of IUG6 and IUG9 in Guy11, the coding sequences of IUG6 and IUG9 driven by the ribosomal protein P27 promoter were inserted into pYF11, respectively. DNA primers are also listed in S15 Table.
Yeast signal sequence trap system
The yeast signal trap system is based on vector pSUC2, which carries a truncated invertase gene, SUC2, lacking both the initiation Met and the signal peptide [98]. DNA fragments coding for the signal peptides of Iug6 and Iug9 were PCR amplified and introduced into pSUC2 using EcoRI and XhoI restriction sites to create in-frame fusion with the invertase (primers listed in S15 Table). The pSUC2-derived plasmids were then transformed into the invertase negative yeast strain YTK12 through lithium acetate method [98,99]. Following transformation, yeast cells were plated on CMD-W (minus Trp) plates, and positive colonies were transferred to fresh CMD-W plates and incubated at 30°C for two days. For invertase secretion, positive colonies were replica plated on YPRAA plates, supplemented with raffinose instead of sucrose. Growth occurs only when the invertase is secreted. The invertase enzymatic activity was measured by the reduction of TTC to insoluble redcolored triphenylformazan as described previously by Oh et al [49].
Agrobacterium tumefaciens infiltration assays
The IUG and NUP genes without signal peptides were amplified using combinations of primers for the PVX assay. The amplified fragments were cut and ligated into the PVX vector PVX::HA [100,101] before introduction into A. tumefaciens strain GV3101 by electroporation [102]. The PVX::HA transformants were selected using Tetracycline (12.5 μg /ml) and Kanamycin (50 μg /ml). Individual colonies were verified by PCR. For infiltration of PVX::HA and PVX::flag (negative control GFP) into leaves, recombinant strains of A. tumefaciens were grown in LB medium plus 50 μg /ml Kanamycin for 48 h, harvested, and infiltrated as previously described by Yu et al [103]. The experiment was repeated three times with each assay consisting of three plants each with three leaves inoculations.
Microscopy
Green fluorescent protein fluorescence in rice cells was captured using an LSM 710 laser scanning microscope with a 40 x objective lens (Carl Zeiss), with an excitation 480 ± 10 nm and an emission 510 ± 10 nm.
Accession numbers
The genome sequence data of 98–06 was deposited at the NCBI Genome Database (http://www.ncbi.nlm.nih.gov/assembly) under the accession number JRBC00000000. RNA-Seq reads were deposited at the GenBank SRA database under sample number SRS692257 and experiment number SRX689727. The GenBank accession numbers for IUG6, IUG9, and IUG18 are KM522919, KM522920, and KM522921, respectively. The information of other genes with their reference gene ID in 70–15 and gene location in the 98–06 genome is provided in S9 Table.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Jantasuriyarat C, Gowda M, Haller K, Hatfield J, Lu G, et al. (2005) Large-scale identification of expressed sequence tags involved in rice and rice blast fungus interaction. Plant Physiol 138 : 105–115. 15888683
2. Talbot NJ (2003) On the trail of a cereal killer: Exploring the biology of Magnaporthe grisea. Annu Rev Microbiol 57 : 177–202. 14527276
3. Ebbole DJ (2007) Magnaporthe as a model for understanding host-pathogen interactions. Annu Rev Phytopathol 45 : 437–456. 17489691
4. Mosquera G, Giraldo MC, Khang CH, Coughlan S, Valent B (2009) Interaction transcriptome analysis identifies Magnaporthe oryzae BAS1-4 as biotrophy-associated secreted proteins in rice blast disease. Plant Cell 21 : 1273. doi: 10.1105/tpc.107.055228 19357089
5. Levy M, Correavictoria FJ, Zeigler RS, Xu SZ, Hamer JE (1993) Genetic Diversity of the Rice Blast Fungus in a Disease Nursery in Colombia. Phytopathology 83 : 1427–1433.
6. Kumar J, Nelson RJ, Zeigler RS (1999) Population structure and dynamics of Magnaporthe grisea in the Indian Himalayas. Genetics 152 : 971–984. 10388817
7. Orbach MJ, Farrall L, Sweigard JA, Chumley FG, Valent B (2000) A telomeric avirulence gene determines efficacy for the rice blast resistance gene Pi-ta. Plant Cell 12 : 2019–2032. 11090206
8. Gout L, Fudal I, Kuhn ML, Blaise F, Eckert M, et al. (2006) Lost in the middle of nowhere: the AvrLm1 avirulence gene of the Dothideomycete Leptosphaeria maculans. Mol Microbiol 60 : 67–80. 16556221
9. Valent B, Chumley FG (1991) Molecular genetic analysis of the rice blast fungus, Magnaporthe grisea. Annu Rev Phytopathol 29 : 443–467. 18479196
10. Kim S, Park J, Park SY, Mitchell TK, Lee YH (2010) Identification and analysis of in planta expressed genes of Magnaporthe oryzae. BMC Genomics 11 : 104. doi: 10.1186/1471-2164-11-104 20146797
11. Jantasuriyarat C, Gowda M, Haller K, Hatfield J, Lu G, et al. (2005) Large-scale identification of expressed sequence tags involved in rice and rice blast fungus interaction. Plant physiol 138 : 105. 15888683
12. Khang CH, Berruyer R, Giraldo MC, Kankanala P, Park SY, et al. (2010) Translocation of Magnaporthe oryzae effectors into rice cells and their subsequent cell-to-cell movement. Plant Cell 22 : 1388–1403. doi: 10.1105/tpc.109.069666 20435900
13. Leung H, Borromeo ES, Bernardo MA, Notteghem JL (1988) Genetic-analysis of virulence in the rice blast fungus Magnaporthe grisea. Phytopathology 78 : 1227–1233.
14. Chao CCT, Ellingboe AH (1991) Selection for mating competence in Magnaporthe grisea pathogenic to rice. Can J Bot 69 : 2130–2134.
15. Dean RA, Talbot NJ, Ebbole DJ, Farman ML, Mitchell TK, et al. (2005) The genome sequence of the rice blast fungus Magnaporthe grisea. Nature 434 : 980–986. 15846337
16. Yoshida K, Saitoh H, Fujisawa S, Kanzaki H, Matsumura H, et al. (2009) Association genetics reveals three novel avirulence genes from the rice blast fungal pathogen Magnaporthe oryzae. Plant Cell 21 : 1573–1591. doi: 10.1105/tpc.109.066324 19454732
17. Xue M, Yang J, Li Z, Hu S, Yao N, et al. (2012) Comparative analysis of the genomes of two field isolates of the rice blast fungus Magnaporthe oryzae. PLoS Genet 8: e1002869. doi: 10.1371/journal.pgen.1002869 22876203
18. Li R, Li Y, Kristiansen K, Wang J (2008) SOAP: short oligonucleotide alignment program. Bioinformatics 24 : 713–714. doi: 10.1093/bioinformatics/btn025 18227114
19. Li R, Zhu H, Ruan J, Qian W, Fang X, et al. (2010) De novo assembly of human genomes with massively parallel short read sequencing. Genome Res 20 : 265–272. doi: 10.1101/gr.097261.109 20019144
20. Chen C, Lian B, Hu J, Zhai H, Wang X, et al. (2013) Genome comparison of two Magnaporthe oryzae field isolates reveals genome variations and potential virulence effectors. BMC Genomics 14 : 887. doi: 10.1186/1471-2164-14-887 24341723
21. Smith DR, Lee RW (2008) Nucleotide diversity in the mitochondrial and nuclear compartments of Chlamydomonas reinhardtii: investigating the origins of genome architecture. BMC Evol Biol 8 : 156. doi: 10.1186/1471-2148-8-156 18495022
22. Saha S, Bridges S, Magbanua ZV, Peterson DG (2008) Empirical comparison of ab initio repeat finding programs. Nucleic Acids Res 36 : 2284–2294. doi: 10.1093/nar/gkn064 18287116
23. van der Does HC, Rep M (2007) Virulence genes and the evolution of host specificity in plant-pathogenic fungi. Mol Plant Microbe Interact 20 : 1175–1182. 17918619
24. Oliva R, Win J, Raffaele S, Boutemy L, Bozkurt TO, et al. (2010) Recent developments in effector biology of filamentous plant pathogens. Cell Microbiol 12 : 705–715. doi: 10.1111/j.1462-5822.2010.01471.x 20374248
25. Rehmany AP, Gordon A, Rose LE, Allen RL, Armstrong MR, et al. (2005) Differential recognition of highly divergent downy mildew avirulence gene alleles by RPP1 resistance genes from two Arabidopsis lines. Plant Cell 17 : 1839–1850. 15894715
26. Guo M, Chen Y, Du Y, Dong Y, Guo W, et al. (2011) The bZIP transcription factor MoAP1 mediates the oxidative stress response and is critical for pathogenicity of the rice blast fungus Magnaporthe oryzae. PLoS Pathog 7: e1001302. doi: 10.1371/journal.ppat.1001302 21383978
27. Dou X, Wang Q, Qi Z, Song W, Wang W, et al. (2011) MoVam7, a conserved SNARE involved in vacuole assembly, is required for growth, endocytosis, ROS accumulation, and pathogenesis of Magnaporthe oryzae. PLoS One 6: e16439. doi: 10.1371/journal.pone.0016439 21283626
28. De Vleesschauwer D, Gheysen G, Hofte M (2013) Hormone defense networking in rice: tales from a different world. Trends Plant Sci 18 : 555–565. doi: 10.1016/j.tplants.2013.07.002 23910453
29. Yamada S, Kano A, Tamaoki D, Miyamoto A, Shishido H, et al. (2012) Involvement of OsJAZ8 in jasmonate-induced resistance to bacterial blight in rice. Plant Cell Physiol 53 : 2060–2072. doi: 10.1093/pcp/pcs145 23104764
30. Takai R, Isogai A, Takayama S, Che FS (2008) Analysis of flagellin perception mediated by flg22 receptor OsFLS2 in rice. Mol Plant Microbe Interact 21 : 1635–1642. doi: 10.1094/MPMI-21-12-1635 18986259
31. Saeed AI, Sharov V, White J, Li J, Liang W, et al. (2003) TM4: a free, open-source system for microarray data management and analysis. Biotechniques 34 : 374–378. 12613259
32. Wang P, Shen G (2011) The endocytic adaptor proteins of pathogenic fungi: charting new and familiar pathways. Med Mycol 49 : 449–457. doi: 10.3109/13693786.2011.553246 21254965
33. Giraldo MC, Dagdas YF, Gupta YK, Mentlak TA, Yi M, et al. (2013) Two distinct secretion systems facilitate tissue invasion by the rice blast fungus Magnaporthe oryzae. Nat Commun 4 : 1996. doi: 10.1038/ncomms2996 23774898
34. Hogenhout SA, Van der Hoorn RAL, Terauchi R, Kamoun S (2009) Emerging concepts in effector biology of plant-associated organisms. Mol Plant Microbe Interact 22 : 115–122. doi: 10.1094/MPMI-22-2-0115 19132864
35. de Jonge R, Bolton MD, Kombrink A, van den Berg GC, Yadeta KA, et al. (2013) Extensive chromosomal reshuffling drives evolution of virulence in an asexual pathogen. Genome Res 23 : 1271–1282. doi: 10.1101/gr.152660.112 23685541
36. Dou D, Kale SD, Wang X, Chen Y, Wang Q, et al. (2008) Conserved C-terminal motifs required for avirulence and suppression of cell death by Phytophthora sojae effector Avr1b. Plant Cell 20 : 1118–1133. doi: 10.1105/tpc.107.057067 18390593
37. Block A, Li GY, Fu ZQ, Alfano JR (2008) Phytopathogen type III effector weaponry and their plant targets. Curr Opin Plant Biol 11 : 396–403. doi: 10.1016/j.pbi.2008.06.007 18657470
38. Li W, Wang B, Wu J, Lu G, Hu Y, et al. (2009) The Magnaporthe oryzae avirulence gene AvrPiz-t encodes a predicted secreted protein that triggers the immunity in rice mediated by the blast resistance gene Piz-t. Mol Plant Microbe Interact 22 : 411–420. doi: 10.1094/MPMI-22-4-0411 19271956
39. Liu W, Liu J, Ning Y, Ding B, Wang X, et al. (2013) Recent progress in understanding PAMP - and effector-triggered immunity against the rice blast fungus Magnaporthe oryzae. Mol Plant 6 : 605–620. doi: 10.1093/mp/sst015 23340743
40. Raffaele S, Kamoun S (2012) Genome evolution in filamentous plant pathogens: why bigger can be better. Nat Rev Microbiol 10 : 417–430. doi: 10.1038/nrmicro2790 22565130
41. Kim S, Park SY, Kim KS, Rho HS, Chi MH, et al. (2009) Homeobox transcription factors are required for conidiation and appressorium development in the rice blast fungus Magnaporthe oryzae. PLoS Genet 5: e1000757. doi: 10.1371/journal.pgen.1000757 19997500
42. Yang J, Zhao XY, Sun J, Kang ZS, Ding SL, et al. (2010) A novel protein Com1 is required for normal conidium morphology and full virulence in Magnaporthe oryzae. Mol Plant Microbe Interact 23 : 112–123. doi: 10.1094/MPMI-23-1-0112 19958144
43. Shi ZX, Leung H (1995) Genetic analysis of sporulation in Magnaporthe grisea by chemical and insertional mutagenesis. Mol Plant Microbe Interact 8 : 949–959.
44. Zhou Z, Li G, Lin C, He C (2009) Conidiophore stalk-less1 encodes a putative zinc-finger protein involved in the early stage of conidiation and mycelial infection in Magnaporthe oryzae. Mol Plant Microbe Interact 22 : 402–410. doi: 10.1094/MPMI-22-4-0402 19271955
45. Nishimura M, Fukada J, Moriwaki A, Fujikawa T, Ohashi M, et al. (2009) Mstu1, an APSES transcription factor, is required for appressorium-mediated infection in Magnaporthe grisea. Biosci Biotechnol Biochem 73 : 1779–1786. 19661696
46. Chen Y, Zhai S, Zhang H, Zuo R, Wang J, et al. (2014) Shared and distinct functions of two Gti1/Pac2 family proteins in growth, morphogenesis and pathogenicity of Magnaporthe oryzae. Environ Microbiol 16 : 788–801. doi: 10.1111/1462-2920.12204 23895552
47. Jiang CJ, Shimono M, Sugano S, Kojima M, Yazawa K, et al. (2010) Abscisic acid interacts antagonistically with salicylic acid signaling pathway in rice-Magnaporthe grisea interaction. Mol Plant Microbe Interact 23 : 791–798. doi: 10.1094/MPMI-23-6-0791 20459318
48. Park CH, Chen S, Shirsekar G, Zhou B, Khang CH, et al. (2012) The Magnaporthe oryzae effector AvrPiz-t targets the RING E3 ubiquitin ligase APIP6 to suppress pathogen-associated molecular pattern-triggered immunity in rice. Plant Cell 24 : 4748–4762. doi: 10.1105/tpc.112.105429 23204406
49. Oh SK, Young C, Lee M, Oliva R, Bozkurt TO, et al. (2009) In planta expression screens of Phytophthora infestans RXLR effectors reveal diverse phenotypes, including activation of the Solanum bulbocastanum disease resistance protein Rpi-blb2. Plant Cell 21 : 2928–2947. doi: 10.1105/tpc.109.068247 19794118
50. Durrant WE, Dong X (2004) Systemic acquired resistance. Annu Rev Phytopathol 42 : 185–209. 15283665
51. Vlot AC, Dempsey DA, Klessig DF (2009) Salicylic Acid, a multifaceted hormone to combat disease. Annu Rev Phytopathol 47 : 177–206. doi: 10.1146/annurev.phyto.050908.135202 19400653
52. Yang Y, Qi M, Mei C (2004) Endogenous salicylic acid protects rice plants from oxidative damage caused by aging as well as biotic and abiotic stress. Plant J 40 : 909–919. 15584956
53. Wu J, Kou Y, Bao J, Li Y, Tang M, et al. (2015) Comparative genomics identifies the Magnaporthe oryzae avirulence effector AvrPi9 that triggers Pi9-mediated blast resistance in rice. New Phytol.
54. Chuma I, Isobe C, Hotta Y, Ibaragi K, Futamata N, et al. (2011) Multiple translocation of the AVR-Pita effector gene among chromosomes of the rice blast fungus Magnaporthe oryzae and related species. PLoS Pathog 7: e1002147. doi: 10.1371/journal.ppat.1002147 21829350
55. Burt A (2000) Perspective: sex, recombination, and the efficacy of selection—was Weismann right? Evolution 54 : 337–351. 10937212
56. McDonald BA, Linde C (2002) Pathogen population genetics, evolutionary potential, and durable resistance. Annu Rev Phytopathol 40 : 349–379. 12147764
57. Raffaele S, Kamoun S (2012) Genome evolution in filamentous plant pathogens: why bigger can be better. Nat Rev Microbiol 10 : 417–430. doi: 10.1038/nrmicro2790 22565130
58. Kamoun S (2007) Groovy times: filamentous pathogen effectors revealed. Curr Opin Plant Biol 10 : 358–365. 17611143
59. Kang S, Sweigard JA, Valent B (1995) The PWL host specificity gene family in the blast fungus Magnaporthe grisea. Mol Plant Microbe Interact 8 : 939–948. 8664503
60. Sweigard JA, Carroll AM, Kang S, Farrall L, Chumley FG, et al. (1995) Identification, cloning, and characterization of PWL2, a gene for host species specificity in the rice blast fungus. Plant Cell 7 : 1221–1233. 7549480
61. Ahn N, Kim S, Choi W, Im KH, Lee YH (2004) Extracellular matrix protein gene, EMP1, is required for appressorium formation and pathogenicity of the rice blast fungus, Magnaporthe grisea. Molecules and Cells 17 : 166–173. 15055545
62. Kim S, Ahn IP, Rho HS, Lee YH (2005) MHP1, a Magnaporthe grisea hydrophobin gene, is required for fungal development and plant colonization. Mol Microbiol 57 : 1224–1237. 16101997
63. Ellis JG, Rafiqi M, Gan P, Chakrabarti A, Dodds PN (2009) Recent progress in discovery and functional analysis of effector proteins of fungal and oomycete plant pathogens. Curr Opin Plant Biol 12 : 399–405. doi: 10.1016/j.pbi.2009.05.004 19540152
64. Stergiopoulos I, de Wit PJ (2009) Fungal effector proteins. Annu Rev Phytopathol 47 : 233–263. doi: 10.1146/annurev.phyto.112408.132637 19400631
65. Valent B, Khang CH (2010) Recent advances in rice blast effector research. Curr Opin Plant Biol 13 : 434–441. doi: 10.1016/j.pbi.2010.04.012 20627803
66. Bozkurt TO, Schornack S, Banfield MJ, Kamoun S (2012) Oomycetes, effectors, and all that jazz. Curr Opin Plant Biol 15 : 483–492. doi: 10.1016/j.pbi.2012.03.008 22483402
67. Huang J, Si W, Deng Q, Li P, Yang S (2014) Rapid evolution of avirulence genes in rice blast fungus Magnaporthe oryzae. BMC Genet 15 : 45. doi: 10.1186/1471-2156-15-45 24725999
68. Saitoh H, Fujisawa S, Mitsuoka C, Ito A, Hirabuchi A, et al. (2012) Large-scale gene disruption in Magnaporthe oryzae identifies MC69, a secreted protein required for infection by monocot and dicot fungal pathogens. PLoS Pathog 8: e1002711. doi: 10.1371/journal.ppat.1002711 22589729
69. Talbot NJ, Ebbole DJ, Hamer JE (1993) Identification and characterization of MPG1, a gene involved in pathogenicity from the rice blast fungus Magnaporthe grisea. Plant Cell 5 : 1575–1590. 8312740
70. Jeong JS, Mitchell TK, Dean RA (2007) The Magnaporthe grisea snodprot1 homolog, MSP1, is required for virulence. FEMS Microbiol Lett 273 : 157–165. 17590228
71. Mentlak TA, Kombrink A, Shinya T, Ryder LS, Otomo I, et al. (2012) Effector-mediated suppression of chitin-triggered immunity by Magnaporthe oryzae is necessary for rice blast disease. Plant Cell 24 : 322–335. doi: 10.1105/tpc.111.092957 22267486
72. Friesen TL, Stukenbrock EH, Liu Z, Meinhardt S, Ling H, et al. (2006) Emergence of a new disease as a result of interspecific virulence gene transfer. Nat Genet 38 : 953–956. 16832356
73. de Jonge R, van Esse HP, Kombrink A, Shinya T, Desaki Y, et al. (2010) Conserved fungal LysM effector Ecp6 prevents chitin-triggered immunity in plants. Science 329 : 953–955. doi: 10.1126/science.1190859 20724636
74. Li L, Stoeckert CJ Jr., Roos DS (2003) OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res 13 : 2178–2189. 12952885
75. Korf I (2004) Gene finding in novel genomes. BMC Bioinformatics 5 : 59. 15144565
76. Besemer J, Borodovsky M (2005) GeneMark: web software for gene finding in prokaryotes, eukaryotes and viruses. Nucleic Acids Res 33: W451–454. 15980510
77. Birney E, Clamp M, Durbin R (2004) GeneWise and Genomewise. Genome Res 14 : 988–995. 15123596
78. Kanehisa M, Goto S, Kawashima S, Okuno Y, Hattori M (2004) The KEGG resource for deciphering the genome. Nucleic Acids Res 32: D277–280. 14681412
79. Tatusov RL, Fedorova ND, Jackson JD, Jacobs AR, Kiryutin B, et al. (2003) The COG database: an updated version includes eukaryotes. BMC Bioinformatics 4 : 41. 12969510
80. Magrane M, Consortium U (2011) UniProt Knowledgebase: a hub of integrated protein data. Database (Oxford) 2011: bar009.
81. Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, et al. (2000) Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25 : 25–29. 10802651
82. Sonnhammer EL, von Heijne G, Krogh A (1998) A hidden Markov model for predicting transmembrane helices in protein sequences. Proc Int Conf Intell Syst Mol Biol 6 : 175–182. 9783223
83. Petersen TN, Brunak S, von Heijne G, Nielsen H (2011) SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods 8 : 785–786. doi: 10.1038/nmeth.1701 21959131
84. Nguyen Ba AN, Pogoutse A, Provart N, Moses AM (2009) NLStradamus: a simple Hidden Markov Model for nuclear localization signal prediction. BMC Bioinformatics 10 : 202. doi: 10.1186/1471-2105-10-202 19563654
85. Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, et al. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25 : 3389–3402. 9254694
86. Benson G (1999) Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 27 : 573–580. 9862982
87. Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, et al. (2004) Versatile and open software for comparing large genomes. Genome Biol 5: R12. 14759262
88. Guo M, Guo W, Chen Y, Dong S, Zhang X, et al. (2010) The basic leucine zipper transcription factor Moatf1 mediates oxidative stress responses and is necessary for full virulence of the rice blast fungus Magnaporthe oryzae. Mol Plant Microbe Interact 23 : 1053–1068. doi: 10.1094/MPMI-23-8-1053 20615116
89. Zhang C, Wang G, Wang J, Ji Z, Liu Z, et al. (2013) Characterization and comparative analyses of muscle transcriptomes in Dorper and small-tailed Han sheep using RNA-Seq technique. PLoS One 8: e72686. doi: 10.1371/journal.pone.0072686 24023632
90. Li R, Yu C, Li Y, Lam TW, Yiu SM, et al. (2009) SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics 25 : 1966–1967. doi: 10.1093/bioinformatics/btp336 19497933
91. Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B (2008) Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5 : 621–628. doi: 10.1038/nmeth.1226 18516045
92. Feng J, Meyer CA, Wang Q, Liu JS, Shirley Liu X, et al. (2012) GFOLD: a generalized fold change for ranking differentially expressed genes from RNA-seq data. Bioinformatics 28 : 2782–2788. doi: 10.1093/bioinformatics/bts515 22923299
93. Kanehisa M, Araki M, Goto S, Hattori M, Hirakawa M, et al. (2008) KEGG for linking genomes to life and the environment. Nucleic Acids Res 36: D480–484. 18077471
94. Sweigard JA, Chumley FG, Valent B (1992) Disruption of a Magnaporthe grisea cutinase gene. Mol Gen Genet 232 : 183–190. 1557024
95. Zhang H, Zhao Q, Liu K, Zhang Z, Wang Y, et al. (2009) MgCRZ1, a transcription factor of Magnaporthe grisea, controls growth, development and is involved in full virulence. FEMS Microbiol Lett 293 : 160–169. doi: 10.1111/j.1574-6968.2009.01524.x 19260966
96. Zhang H, Xue C, Kong L, Li G, Xu JR (2011) A Pmk1-interacting gene is involved in appressorium differentiation and plant infection in Magnaporthe oryzae. Eukaryot Cell 10 : 1062–1070. doi: 10.1128/EC.00007-11 21642506
97. Chen Y, Zuo R, Zhu Q, Sun Y, Li M, et al. (2014) MoLys2 is necessary for growth, conidiogenesis, lysine biosynthesis, and pathogenicity in Magnaporthe oryzae. Fungal Genet Biol 67 : 51–57. doi: 10.1016/j.fgb.2014.04.001 24731804
98. Jacobs KA, Collins-Racie LA, Colbert M, Duckett M, Golden-Fleet M, et al. (1997) A genetic selection for isolating cDNAs encoding secreted proteins. Gene 198 : 289–296. 9370294
99. Robert H, Schidst I, Andrew R (1995) Studies on the transformation of intact yeast cells by the LiAc/SSDNA/PEG procedure. Yeast 11 : 355–360. 7785336
100. Lu R, Malcuit I, Moffett P, Ruiz MT, Peart J, et al. (2003) High throughput virus-induced gene silencing implicates heat shock protein 90 in plant disease resistance. EMBO J 22 : 5690–5699. 14592968
101. Wang QQ, Han CZ, Ferreira AO, Yu XL, Ye WW, et al. (2011) Transcriptional programming and functional interactions within the Phytophthora sojae RXLR effector repertoire. Plant Cell 23 : 2064–2086. doi: 10.1105/tpc.111.086082 21653195
102. Hellens RP, Edwards EA, Leyland NR, Bean S, Mullineaux PM (2000) pGreen: a versatile and flexible binary Ti vector for Agrobacterium-mediated plant transformation. Plant Mol Biol 42 : 819–832. 10890530
103. Yu X, Tang J, Wang Q, Ye W, Tao K, et al. (2012) The RxLR effector Avh241 from Phytophthora sojae requires plasma membrane localization to induce plant cell death. New Phytol 196 : 247–260. doi: 10.1111/j.1469-8137.2012.04241.x 22816601
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2015 Číslo 4
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Toxin-Induced Necroptosis Is a Major Mechanism of Lung Damage
- Transgenic Fatal Familial Insomnia Mice Indicate Prion Infectivity-Independent Mechanisms of Pathogenesis and Phenotypic Expression of Disease
- Role of Hypoxia Inducible Factor-1α (HIF-1α) in Innate Defense against Uropathogenic Infection
- A Temporal Gate for Viral Enhancers to Co-opt Toll-Like-Receptor Transcriptional Activation Pathways upon Acute Infection
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy