
Recombination in Enteroviruses Is a Biphasic Replicative Process Involving the Generation of Greater-than Genome Length ‘Imprecise’ Intermediates
The rapid evolution of most positive-sense RNA viruses enables them to escape immune surveillance and adapt to new hosts. Genetic variation arises due to their error-prone RNA polymerases and by recombination of viral genomes in co-infected cells. We have developed a novel approach to analyse the poorly understood mechanism of recombination using a poliovirus model system. We characterised the initial viable recombinants and demonstrate the majority are longer than genome length due to an imprecise crossover event that duplicates part of the genome. These viruses are unfit, but rapidly lose the duplicated material and regain full fitness upon serial passage, a process we term resolution. We show this is a replicative recombination process by modifying the fidelity of the viral polymerase, or replication complex coalescence, using methods that have no influence on a previously reported, less efficient, non-replicative recombination mechanism. We conclude that recombination is a biphasic process involving separate generation and resolution events. These new insights into an important evolutionary mechanism have implications for our understanding of virus evolution through partial genome duplication, they suggest ways in which recombination might be modified and provides an approach that may be exploited to analyse recombination in other RNA viruses.
Published in the journal:
. PLoS Pathog 10(6): e32767. doi:10.1371/journal.ppat.1004191
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004191
Summary
The rapid evolution of most positive-sense RNA viruses enables them to escape immune surveillance and adapt to new hosts. Genetic variation arises due to their error-prone RNA polymerases and by recombination of viral genomes in co-infected cells. We have developed a novel approach to analyse the poorly understood mechanism of recombination using a poliovirus model system. We characterised the initial viable recombinants and demonstrate the majority are longer than genome length due to an imprecise crossover event that duplicates part of the genome. These viruses are unfit, but rapidly lose the duplicated material and regain full fitness upon serial passage, a process we term resolution. We show this is a replicative recombination process by modifying the fidelity of the viral polymerase, or replication complex coalescence, using methods that have no influence on a previously reported, less efficient, non-replicative recombination mechanism. We conclude that recombination is a biphasic process involving separate generation and resolution events. These new insights into an important evolutionary mechanism have implications for our understanding of virus evolution through partial genome duplication, they suggest ways in which recombination might be modified and provides an approach that may be exploited to analyse recombination in other RNA viruses.
Introduction
The high levels of genetic variation observed in many RNA viruses has important consequences for viral pathogenesis, host tropism and evolution. Two predominant mechanisms contribute to the generation of genetic diversity of the genome; misincorporation of nucleotides and recombination/reassortment. The absence of proofreading by the majority of viral RNA-dependent RNA-polymerases (RdRp) results in error frequencies of 10−3 to 10−5 per nucleotide polymerized [1], so generating a population of related genomes, the quasispecies [2]. In contrast to the incremental changes (‘drift’) that accumulate from this misincorporation, much more extensive variation can result from exchange or acquisition of large regions of the virus genome through the processes of recombination or, in terms of genetic consequences for the segmented RNA viruses, the analogous process of reassortment.
The enteroviruses are a well characterised genus within the family Picornaviridae. The human enteroviruses (HEV) are particularly well studied as they contain important pathogens responsible for acute flaccid paralysis (including poliomyelitis), myocarditis, encephalitis and a variety of other ailments [3] and have recently been extended to include the three species of human rhinoviruses [4]. The nonsegmented single-stranded positive-sense enterovirus RNA genome is ∼7.5 kb in length, is polyadenylated at the 3′ end and encodes a single polyprotein from an open reading frame flanked by an extensive 5′ and shorter 3′ non-coding region (NCR). The polyprotein is co - and post-translationally proteolytically processed to yield (in order of translation) the viral structural (VP4, 2, 3 and 1; occupying the P1 region, Figure 1) and non-structural proteins (2Apro, 2B, 2C, 3A, VPg, 3Cpro and 3Dpol; comprising the P2 and P3 regions of the genome). The latter control the cellular environment, establish the membrane-bound replication complex, replicate the genome via formation of a negative-stranded intermediate and process the polyprotein.

The HEV are divided into four phylogenetically distinct species (designated A–D; [4]) the members of which were initially differentiated serologically, and are now usually defined by sequence analysis of VP1 [5]–[8]. Extensive sequencing studies have unequivocally demonstrated the time-related accumulation of sequence variation – and consequent antigenic diversification – within the capsid-coding region [9]–[14] and frequent recombination within the regions encoding the non-structural proteins [13], [15]–[33]. Additional recombination events have been characterised between the 5′ NCR and capsid coding region [34], [35]. The HEV genome can therefore be considered as modular, with recombination between the functional entities that drive translation and replication (5′ NCR), that encode the structural proteins (the P1 region) and that encode the non-structural proteins [21], [28]. Recombination within the P1 region has rarely been observed, and only then between very closely related viruses or involving the extreme termini of VP1, presumably due to restrictions imposed on assembly of the icosahedral particle [36]. In contrast, in addition to the extensive documentation of recombination within the non-structural protein coding region, different serotypes exhibit distinct temporal and geographic kinetics in the appearance of the predominant recombinant forms (RF) [23]–[25] although the selection of emergent RFs, whether due to replicative or immunological advantage, has yet to be determined [25].
The majority of characterised HEV recombinants circulating are intraspecies, though interspecies recombination may have occurred in ancestral enteroviruses [37]–[40] and 5′ NCR exchanges constructed in the laboratory can generate viable viruses [41], [42]. The restrictions preventing viable interspecies recombinants between structural and non-structural coding regions are poorly understood. In addition to a requirement to access the same cell type and co-occupy the same replication complex [43], these presumably include the necessity for compatibility in all cis-acting replication functions [44]. For example, recent evidence indicates that particle morphogenesis requires an interaction of the 2C protein and capsid protein VP3 [45], [46].
Despite the importance of recombination as an evolutionary process in enteroviruses, the molecular mechanism involved has received relatively little attention. The favoured replicative mechanism in enteroviruses (and other RNA viruses) involves template-switching of the viral RdRp during negative strand RNA synthesis [47]–[49]. An alternative process, involving the replication-independent joining of RNA molecules has been described by Agol and colleagues [50], [51] which may be mediated by cellular RNA ligases as postulated for analogous studies in pestiviruses and hepatitis C virus [52], [53].
Studies of the viable recombinant progeny from cells co-infected with enteroviruses are confounded by the relatively low frequency with which recombinants are generated and the high levels of parental viruses generally produced from such infections. Together, this necessitates the use of a selection strategy to preferentially isolate recombinants from a mixed population, which likely imposes an additional selection for increased fitness within the recombinant population. To overcome these limitations we have developed an in vitro reverse genetic system that enables the recovery of recombinants alone from dually transfected cells in culture. We have used this to characterise the recombination junctions and subsequently investigate the replication phenotype and stability of the recovered viruses. Our studies suggest that replicative (i.e. polymerase-dependent) recombination is a biphasic process in which an initial chimeric genome is generated that contains a duplication of up to several hundred nucleotides (nt.) of the virus genome. Subsequent replication of these genomes results in a process of resolution in which variants are selected that have undergone an internal deletion to remove the duplicated sequences, resulting in genomes of the expected (i.e. genomic) length. These studies provide important insights into the molecular mechanism of recombination in positive strand RNA viruses and additionally suggest a means by which ‘evolution by duplication’ may occur. The novel experimental strategy we described provides the basis for further studies of the viral and cellular processes that control this important evolutionary process.
Results
A system for generating recombinant enteroviruses
To facilitate the analysis of recombination junctions and early recombination events in enteroviruses we wanted an approach that allowed the production of recombinants in the absence of either replication-competent helper viruses or low frequency revertants of parental genomes bearing genetic lesions such as resistance to guanidine hydrochloride [45], [54]. Since recombination is proposed to occur by a ‘copy-choice’ process involving polymerase template switching during negative strand synthesis [47] we reasoned that one of the parental genomes could be defective in synthesis of positive-strands, a phenotype associated with destabilization of the cis-acting replication element (CRE) located within the 2C coding region of enteroviruses [55]. We have previously demonstrated that a poliovirus genome bearing eight synonymous substitutions within this CRE does not revert, even after extensive blind passage [56], [57]. For the second parental genome we used a replication-competent sub-genomic replicon, in which the capsid-coding region was replaced with a luciferase reporter gene. The rationale was that co-transfection of RNA generated in vitro from these two cDNAs should yield a complete, replication competent genome – bearing both the capsid-coding region and a functional CRE – if a recombination cross-over occurred between the end of the capsid coding region and the CRE (Figure 1a). The reciprocal recombinant – if generated – bearing a luciferase reporter and non-functional CRE would not replicate and neither parental genome was capable of generating progeny virus. To facilitate the recovery of potential recombinants that had not undergone successive rounds of release, re-infection and replication, we used rodent cells – permissive for virus replication but lacking a suitable receptor and so not susceptible to infection – for transfection. This approach facilitated the capture of early recombinant virus progeny whilst minimizing their loss due to continued propagation. The generic recombination system and subsequent isolation strategy is illustrated in Figure 1b. For convenience, and because it refers to the parental genomes used in the assay, we designate this as a CRE-REP recombination assay.
We initially investigated the ability to generate intra-serotypic recombinants using parental genomes derived from poliovirus type 3 as we reasoned that – if sequence identity or protein-protein compatibility influenced the efficiency with which viable recombinants could be recovered – this would impose the minimum constraints on the process. L929 murine fibroblasts were transfected – individually or together – with RNA generated in vitro from pT7Rep3-L and pT7/SL3. Cell-free supernatant was harvested 24–48 hours post-transfection and the presence of infectious virus detected by plaque assay in HeLa cells. Individually, neither parental genome yielded virus capable of forming plaques on the HeLa monolayers. In contrast, undiluted supernatant from co-transfected L929 cells yielded ∼1.1×103 pfu/ml of virus (Figure 1cd), the identity of which was confirmed by PCR analysis and subsequent sequencing. L929 cells yielded no recombinants when the growth media was supplemented with 4 mM guanidine hydrochloride, a known inhibitor of poliovirus negative strand initiation [58].
We sequenced several biologically cloned intra-serotypic recombinants (see below). However, the inevitable sequence identity of the parental viral genomes confounded the identification of recombination junctions in the majority of recovered viruses. Therefore, having demonstrated the recovery of viable progeny after co-transfection of a sub-genomic replicon and CRE mutant we repeated the experiment replacing the poliovirus type 3-derived luciferase-encoding sub-genomic replicon with an equivalent replicon derived from poliovirus type 1 (plasmid pRLucWT; 21% divergent within the potential recombination region shown in Figure 1). In this instance co-transfection of both parental plasmids routinely generated 50–200 pfu/ml (mean of 115 pfu/ml from three independent replicates) from transfected murine L929 cells following transfer of undiluted supernatant to HeLa cell monolayers (Figure 1d). In repeated independent co-transfections of mouse or hamster (L929 or BHK respectively; not shown) monolayers, homologous (PV3/PV3) parental genomes yielded ∼10-fold more progeny than heterologous (PV1/PV3) parental genomes (P<0.05 Mann Whitney U test; Figure 1d). Inter-serotypic poliovirus recombinants were biologically cloned by limit dilution and analysed further.
Sequence analysis of poliovirus recombinants
Fifteen intra-serotype recombinants were analysed after co-transfection of L929 monolayers with RNA generated in vitro from pT7Rep3-L and pT7/SL3. Recovered virus was reverse transcribed using an oligo-dT primer and the region within which a recombination event was targeted to occur was amplified using oligonucleotides PV3-2995F and PV3-5191R (Supplementary Table S1) and subsequently sequenced. The majority (n = 13) of the recovered genomes were indistinguishable from the parental type 3 poliovirus genome. However, to our surprise two genomes included a duplication, of either 27 nt. (PV33393/PV33367; see Materials and Methods for nomenclature used to define recombinants and recombination junctions) or 78 nt. (PV33390/PV33313) within the region encoding the 2A protease (Figure 2a). We designated these types of recombinants as imprecise reflecting the nature of the junction between the donor (assumed to be the 3′ partner encoding the viral polymerase) and recipient parental genomes. Since further analysis of such imprecise recombinants was limited by the sequence identity of the parental viruses we went on to analyse the products from a PV1/PV3 inter-serotypic co-transfection.

A total of 146 viable viruses were biologically cloned by limit dilution of cell-free supernatant after three independent co-transfections of L929 cells with equimolar ratios of RNA generated in vitro from pRLucWT and pT7/SL3. Following reverse transcription, the oligonucleotides PV3-2995F and PV1-5200R (Supplementary Table S1) were used to amplify the intervening region. Initial analysis by gel electrophoresis indicated that a significant proportion of the products were larger that the expected size of 2205 nt. (data not shown). All amplified products were therefore sequenced. Of these, 10 were discarded as the sequence was ambiguous indicating they did not contain a clonal virus population. The remaining 136 viruses all consisted of recombinants in which the 5′ component was derived from the PV3 cDNA pT7/SL3 and the 3′ component was derived from the PV1 sub-genomic replicon, pRLucWT. These were stratified – on the basis of the unique sequence that defined the junction – into 20 distinct groups (illustrated schematically in Figure 2b; Figure S1). Of these, three groups (#4E, #35C and #44B) contained a recombination junction in which no additional sequences were present, located in either the region encoding the C-terminus of 2Apro (#35C) or the 2C protein (#35C and #44B; in the latter group the junction was defined by the mutation engineered into the SL3 parental virus [56]). We designate these as precise junctions i.e. in a sequence alignment of the two parental genomes, the locations of the 3′ nucleotide of the recipient genome and the 5′ nucleotide of the donor are adjacent. The remaining 17 distinct recombination groups contained junctions in which additional sequences of 3 to 321 nt. were present (i.e. imprecise junctions). All imprecise junctions maintained the open reading frame. In eight groups the 5′ nucleotide of the donor was located within the luciferase encoding region of the PV1-derived sub-genomic replicon. These groups, together with groups #32A (in which the PV1 sequence started within the linker between the reporter gene and the 2Apro-coding region; Figure 2b and Figure S1) and #25A (see below), contained an average 254 additional nucleotides and formed a distinct cluster (Cluster 1) in which the imprecise junction spanned the region encoding the VP1/2Apro cleavage site (Figure 2b). A second cluster (Cluster 2), containing six groups with an average 118 additional nucleotides had imprecise recombination junctions spanning the region encoding the 2Apro/2B cleavage site. We included group #9C, which contains a 3 nt. duplication within the region encoding the extreme amino-terminus of the 2B protein, in Cluster 2. In six of the recombination groups it was not possible to unambiguously define the recombination junction due to local sequence identity between the aligned parental genomes at the junction (#11A, #34D, #53A, #67B, #E1 and #4E; Figure S1). Only one group (#25A) contained virus-derived sequences that were not an effective duplication of adjacent sequences at an imprecise junction. This group additionally contained 16 nts. derived from the 5′ NCR (nts. 447–462) of a total of 243 nts. additional sequence present within the crossover region (Figure S1). Since this region of the 5′ NCR was identical in the parental genomes it was not possible to determine which it had been derived from.
Growth characteristics and serial passage of recombinant viruses
Of the 136 heterotypic recombinants sequenced, the majority – both in terms of individual sequences (95/136) or distinct groups (17/20) – contained additional sequences within the recombination region (Figures 2b and S1). Since recombinant polioviruses previously reported do not contain additional sequences we reasoned that these imprecise recombinants might have a growth disadvantage. We therefore investigated the growth characteristics and stability of the virus genome upon serial passage of selected recombinants.
A recombinant from group #51G (PV33874/PV12785, cluster 2; Figure S1) was passaged a dozen times in HeLa cells. The plaque phenotype and single step growth characteristics of the second (P2) and seventh (P7) passage were compared with the parental poliovirus type 1 and type 3 (Figure 3ab). The initial small plaque phenotype (P2) increased by P7 to one close to that of either poliovirus types 1 or 3. In a single step growth analysis the P7 virus was indistinguishable from poliovirus type 1 or type 3, whereas the P2 virus had a lower yield at all time points tested, with a titre at 24 hours ∼1 log10 reduced. This suggested that serial passage of #51G resulted in changes that favoured the selection of viruses with faster replication kinetics and a large plaque phenotype. RNA therefore was purified at P2, P3, P7 and P12, reverse transcribed and the region spanning the recombination junction amplified by PCR using oligonucleotides PV3-2995F and GEN-4615R. In early passages (P2, P3) a single product was amplified of ∼1.7 kb but was largely replaced by P7 (and totally by P12) with a PCR product of a reduced size (Figure 3c). The P7 product was cloned and eight resulting cDNAs selected at random were sequenced.
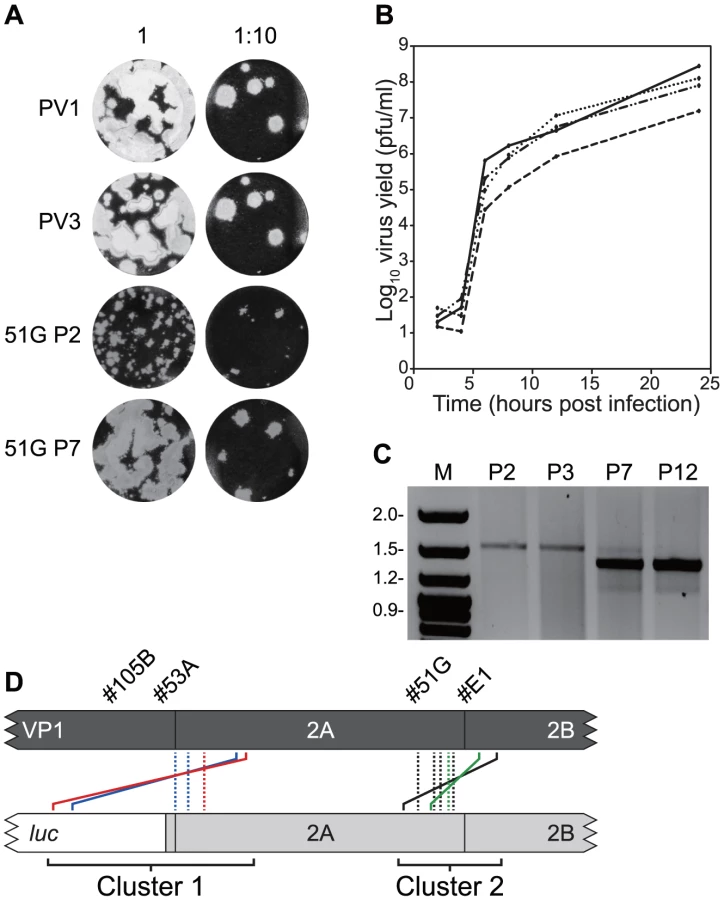
Four distinct junction sequences between poliovirus types 1 and 3 were identified (Figure 3d; #51G-a to –d in Figure S2). In each case the product contained a precise junction in which there were no additional duplicated sequences from either parental genome. The four distinct junctions all mapped within the limits of overlap in the original recombinant PV33874/PV12785 and to the region of the genome encoding the C-terminus of 2Apro (Figure 3d). In each case it was not possible to unambiguously identify the junction due to identity of 2–4 nucleotides at the crossover region.
Three further recombinants, one more from cluster 2 (#E1; PV33849/PV12830) and two from cluster 1 (#53A, PV33485/PV12256 and #105B, PV33471/PV12287) were similarly analysed. In each instance, serial passage (6 to 12 times) in HeLa cells resulted in the internal deletion of sequences present within the region duplicated in the imprecise junction, confirmed by sequence analysis of 8 randomly selected cDNAs (Figure 3d). All eight sequences recovered from recombinant #E1 were identical, as were those from #53A, whereas recombinant #105B yielded two distinct precise junctions (Figure S2). As before, in all cases the exact recombination junction could not be defined due to the conservation of 2–13 nucleotides at the junction (Figure S2). We interpreted these results as indicating that the recombination process in enteroviruses is biphasic – the initial generation of an imprecise recombinant followed by the selection of precise recombinants with enhanced fitness.
Construction and selection of an imprecise recombinant
The process by which initial recombinant genomes were generated – cloning by limit dilution with minimal passage – meant there was a possibility that they were not truly clonal but instead may have contained undetectable levels of precise recombinants. Potentially these subsequently out-competed the imprecise recombinant, rather than being derived from it. To exclude this possibility we constructed a complete cDNA for the #105B recombinant (PV33471/PV12287; see Materials and Methods) and transfected L929 cells with RNA transcribed in vitro. Supernatant media from transfected L929 cells contained ∼8×104 pfu/ml of virus as quantified by plaque assay in HeLa cells. This virus population was subjected to further serial passage and analysed by PCR as before. After four passages a product indistinguishable in size from that of the parental poliovirus genome remained visible together with one or more additional smaller PCR products, intermediate in size between the starting material and the parental poliovirus genome (data not shown). After three further passages the amplified PCR product was cloned and six clones were sequenced. Of these, four (#105B-3; Figure S2) exhibited an identical imprecise junction, PV33381/PV12389, between the PV3 - and PV1 replicon-derived sequences. This differed from that present in the input genome (#105B, PV33471/PV12287; Figure S1) but retained 57 nt. derived from the region encoding the C-terminus of the luciferase reporter gene. The two remaining sequences exhibited different precise junctions (#105B-1, #105B-2; Figure S2). One of these (#105B-1, PV33378/PV12443; Figure S2), exhibited an identical precise recombination junction to that selected previously by serial passage of recombinant viruses cloned by limit dilution from a co-transfection of PV3 and pRLucWT (#105B-a; Figure S2). These results strongly support our contention that the imprecise recombinants isolated from L929 cells using limited passage and serial dilution are the likely progenitors of the precise recombinants subsequently selected by serial passage in HeLa cells. Furthermore, the identification of an imprecise recombinant with a duplicate sequence reduced in length from the input (249 nt. in #105B cf. 57 nt. in #105B-3, see Figure S2) suggests that the generation of precise recombinants may progress via intermediate imprecise recombinants.
Are recombinants generated by a replicative or non-replicative process?
The CRE-REP assay we have developed (Figure 1) was based on the assumption that recombination occurs via a copy-choice mechanism during negative strand synthesis. However, formally the mechanism could have been replication-independent, as has been reported for a number of positive strand RNA viruses including enteroviruses, BVDV and HCV [50], [52], [53]. To clarify this we investigated the consequences of modifying the polymerase error rate, reasoning that a replicative process might be influenced by the characteristics of the polymerase. We therefore investigated the frequency of recombinant generation in the presence of ribavirin, an antiviral which induces a well-characterised increase in the poliovirus polymerase errors rate [59]. We also determined the influence of a G64S substitution in the polymerase, a mutation that confers resistance to ribavirin [60] as a consequence of increased polymerase fidelity [61]. We additionally investigated the influence of the drug nocodazole on recombination as previous studies have shown it inhibits the formation of mixed replication complexes which we presumed are required for replicative recombination [62]. In each instance we also investigated the influence of these various treatments or modifications on the generation of recombinants in a non-replicative assay.
We initially compared the yield of intra-serotype recombinants in the CRE-REP assay we developed (Figure 1) with the yield from truncated – and therefore non-replicating – pT7/SL3Δ and ΔpRLucWT templates (see Materials and Methods for details). Using equimolar amounts of each RNA, and the same total amount of RNA for the CRE-REP or non-replicative transfection of L929 cells, progeny virus was quantified in the supernatant. Under these conditions, the yield of recombinants in the non-replicative assay was only 3.8% of the yield produced in the CRE-REP assay (Figure 4a). Ribavirin concentrations greater than 400 µM eliminated the generation of recombinant progeny following co-transfection of rodent cells with RNA derived from pT7/SL3 and pRLucWT, a result in agreement with previous data on the inhibitory concentration of ribavirin [59]. In contrast, concentrations of ribavirin of 200 µM and 100 µM increased the yield of progeny virus (over that produced in the absence of ribavirin) significantly, by ∼2 fold and ∼3 fold respectively (Figure 4b). However, 100 µM ribavirin did not influence the yield of progeny from a non-replicative assay (Figure 4c) using the truncated pT7/SL3Δ and ΔpRLucWT versions of the cDNA templates. Ribavirin concentrations over the range tested also had no influence on transfection efficiency of L929 cells (Figure S3).

In contrast to the enhancing effect of ribavirin use of donor and recipient genomes bearing a 3Dpol G64S high-fidelity substitution markedly reduced the yield of recombinants in the CRE-REP assay, by ∼20-fold (Figure 4d), despite the mutation having no discernible influence on poliovirus replication ([60]; Figure S4). In a parallel non-replicative recombination assay, using the truncated sub genomic replicon template bearing the same G64S polymerase mutation, there was no significant influence on the yield of progeny recombinants (Figure 4e).
Nocodazole treatment (5 µM) of transiently chilled cells prevents repolymerization of microtubules and the consequent coalescence of replication complexes, yet does not inhibit virus replication or yield ([62], [63]; Figure S5). In the CRE-REP assay, prior treatment of cells with nocodazole reduced the intra-serotypic yield of recombinants by ∼70% (Figure 4f). In contrast, similarly treated L929 cells transfected with RNA from the pT7/SL3Δ and ΔpRLucWT non-replicative versions of the same templates, generated similar levels of recombinant progeny to untreated cells (Figure 4g). Taken together, the influence of the polymerase characteristics and the requirement for replication complex mixing strongly suggests that the generation of recombinants in the CRE-REP assay involves a replicative process.
Analysis of imprecise and precise recombination junctions
Previous studies have implicated local RNA secondary structure, sequence composition or template identity as influencing recombination [64]–[66]. To investigate this further the amount of RNA structure on the positive and negative sense genome was determined by calculating the mean folding energy difference (MFED) between the native sequence and sequence order randomized controls [67] over a sliding 250 nt window spanning the region within which recombination could occur (Figure 5a). MFED values ranged between +5% and −15%, with positive values indicating the presence of sequence order-dependent structure. The average MFED value for sense and antisense strands was 1.58% and −1.56% respectively for the type 1 sub-genomic replicon (the donor genome) and −1.95% and −3.89% respectively for the poliovirus type 3 recipient genome. Neither the precise or imprecise junctions mapped to the regions with maximum or minimum predicted RNA structure (positive and negative values respectively in Figure 5a) between the luciferase coding region and defective CRE. To exclude the possibility that the use of a sliding window during MFED calculations may have obscured limited localized secondary structure, we also determined the MFED value for the 100 nt window spanning each mapped junction in the donor and recipient genomes by comparison of the native sequence to 999 sequence-order randomized controls, using a scrambling algorithm (NDR) that maintained key features, such as dinucleotide composition, of the sequence. Both precise and imprecise junctions exhibited positive MFED (i.e. structured) values in the positive strand of donor sequences (Figure 5b; dark bars), though the MFED values were near an arbitrary 4% cutoff we have previously considered marks the lower limit of reliably predicted RNA structure [68]. In contrast, the negative strand (Figure 5b; pale bars) of both precise and imprecise junctions was largely unstructured, and this was particularly marked in the negative strand of precise recipient sequences.

Sequence identity within the potential recombination region ranged from ∼27% (luciferase and VP1) to almost 98% in the sequence that forms the CRE, and averaged 79.9% within P2-coding region (Figure 6a), distributed as short regions of identity between 2 and 21 nt. in length interspersed with variant nucleotides. There was no correlation between the length of sequence identity and the frequency or distribution of the precise recombination junctions. For example, in the aligned sequences there were 99 distinct conserved dinucleotides (almost all being the first two nucleotides of aligned codons) but only 10 positions with identity of 12 nt. or greater. Of the precise junctions characterised, 50% occurred at positions with identical dinucleotides, with only 10% occurring at regions of identity of more than 12 nt. (Figure 5c, Figure S1). Finally, detailed analysis of the 15 nt. sequences flanking every junction obtained, both precise and imprecise, failed to detect any significant biases whether quantified as individual nucleotides (A,C,G,U), grouped as (A+U) or (C+G) or as purine/pyrimidines (data not shown).

Discussion
The rapid evolution of single-stranded positive-sense RNA viruses can be attributed to their error-prone RNA dependent RNA polymerases (RdRp) and the ability of their modular genomes to undergo recombination. Coupled with their generally short replication cycles and high yields this allows rapid adaptation to altered environmental conditions and the acquisition of novel tissue or host tropisms [69], [70]. In recipients of the live attenuated poliovirus vaccine recombinants are excreted within 7 days [18], [19], [71]. Furthermore, recombination between Sabin vaccine strains and co-circulating – and co-infecting - enteroviruses can generate novel neurovirulent chimeras capable of spreading within a community with low vaccine coverage [27], [72], [73]. Further studies have demonstrated that recombinant forms of particular serotypes of human enteroviruses wax and wane in their relative geographical dominance with characteristic half lives [23]–[25].
Despite recombination being a well defined phenomena in picornaviruses of humans and animals [13], [74], as well as other important positive-strand RNA virus pathogens [44], [75]–[77], the underlying mechanism(s) by which recombinants arise are relatively poorly understood. Studies have suggested that the copy-choice template-switching model originally proposed [47] was additionally influenced by sequence identity between parental genomes, in particular short direct sequence repeats [65], [78] or by the RNA secondary structure of donor or recipient molecules [49], [66], [79], [80].
These analyses have generally assumed that the selected recombinant genome contains a junction that reflects the point at which the polymerase switched from donor to recipient template. To test this we reasoned it would be necessary to isolate recombinants as soon as possible after they had arisen, before additional selection by serial passage and inevitable competition amongst the viral population. To achieve this we investigated the recombination between a poliovirus sub-genomic replicon [81], [82] and a genome defective in positive-strand RNA synthesis due to disruption of the CRE [55], [57], [83], [84]. By transfecting RNA into rodent cells, which lack the receptor for poliovirus, we prevented reinfection and so could analyse early products of recombination. To ensure we were analyzing replication-competent genomes we biologically cloned progeny virus by limit dilution in HeLa cells before genome analysis.
Having determined that neither parental genome generated viable revertants (Figure 1c) we quantified progeny virus generated by intra - (poliovirus type 3) and inter-serotypic (poliovirus type 1 and type 3) recombination. Using standardized conditions intra-serotypic recombinants arose approximately ten-fold more frequently that inter-serotypic recombinants (Figure 1d). Based upon the known recovery (pfu/µg) of type 1 and type 3 poliovirus in L929 cells following RNA transfection (data not shown) it was estimated that intra-serotypic recombinants would represent ∼1-10% of the progeny, broadly in line with figures previously reported [47], [85], [86]. However, as this would be influenced by absolute levels of replication of the unmodified genomes, by the co-transfection efficiency and by the amount of replication post-recombination, this was not further investigated.
The majority of intratypic poliovirus recombinants were indistinguishable from a parental poliovirus type 3 genome. However, intriguingly, two of the sequences contained genome duplications, of 27 or 78 nt. (Figure 2a). This prompted us to analyse an extensive panel of intertypic poliovirus type 1/type 3 recombinants in which the 21% sequence divergence between the selection markers (the luciferase coding region and the mutated CRE) facilitated identification of recombination junctions. Of 136 genomes in which the sequence could be unambiguously identified, 95 (70%) contained additional sequences, forming 17/20 (85%) distinct sequence junctions analysed. We termed these imprecise junctions to distinguish them from genomes bearing no additional sequences (precise junctions). We prefer the use of the terms precise or imprecise – rather than homologous or non-homologous – as they define the characteristics of the recombination junction with regard to the parental virus genome, rather than the mechanism by which recombination occurred.
Recombinant virus progeny were obtained upon co-transfection of rodent (mouse, hamster) and human (HeLa) cell lines, with no strong evidence for cell specificity; we regularly observed a ∼2.5 fold higher yield in hamster cells compared with mouse cells but consider this reflects differences in transfection efficiency and genome replication (data not shown). Both precise and imprecise recombinants were recovered in all the cell types tested. In rodent cells we observed different ratios of precise to imprecise recombinants in repeated parallel co-transfections. We believe this represents the stochastic temporal nature of an individual recombination event and subsequent replication; recombinants arising soon after transfection would be expected to yield genomes that would undergo multiple additional rounds of replication, with the possibility of secondary events (see below) therefore being more likely to yield precise recombinant progeny with enhanced fitness. It should be noted that the CRE-REP assay we describe here involves analysis of progeny 24-48 hours post transfection. During this period, selection for genomes with enhanced replication likely occurs, so the genomes analysed are an indicative, rather than exhaustive, representation of early recombination products. Based upon our demonstration of enhanced replication kinetics after passage (Figure 3ab) we speculate that precise recombinants arise following rapid selection from an initial imprecise recombinant, and that they quickly outcompete the less fit recombinants to prevail in the analysed population. We also note that precise recombinants were usually represented multiple times during the group analysis (Figure S1), suggesting that once they had appeared in the population their enhanced replication meant that they rapidly became the dominant virus present. This interpretation could be addressed by next generation sequencing analysis of the viral RNA population of co-transfected (or infected) cells in future studies.
All recombinants were harvested from transfected cell supernatant and isolated by limit dilution in HeLa cells. This would have imposed a minimal viability criteria on the genome of being encapsidated and capable of being both translated and replicated. By definition, the imprecise recombinants obtained contained partial duplications of parts of the genome. Although the packaging limit of poliovirus is not known, previously constructed dicistronic viruses have contained at least an additional 573 nt. [87] and the genome of foot and mouth disease virus, which possesses a similarly sized particle, is 8.2 kb. It is therefore unlikely that the largest imprecise genome obtained (#67B; 321 nt.; Figure S1) represents any sort of packaging limit, but rather reflects the selection assay used or the mechanism of recombinant generation. Furthermore, since recombinant populations are usually mixtures (generated in vivo in the presence of fully viable parental viruses), both the size and range of the additional sequences may represent genomes which retain sufficient replicative fitness to compete in a mixed infection. How do imprecise recombinants, possibly of lower fitness, compete at all in a natural infection? We think this reflects the stochastic nature of the infection and transmission of enteroviruses. A replication-competent recombinant, even of only limited fitness, shed from the initial dually infected cell would presumably either infect another cell in the same host, or be shed into the environment. Although locally the multiplicity of infection in the original host may be high (and the recombinant poorly, if at all, competitive with parental viruses), transmission to distant sites in the same host or another host may result in a founder effect, in which the recombinant – or subsequent resolved derivatives of it – can proliferate in the absence of parental genomes of potentially greater fitness.
A striking feature of the imprecise recombinants recovered was their clustering in regions encoding the amino and carboxyl termini of 2Apro (Figures 2b, 3d). The consequences of this clustering was that the majority (94/95 individual sequences in 16/17 groups) of imprecise recombinants retained the ability to encode non-chimeric versions of the 2Apro and 2B proteins; the sole exception being #9C in which the additional 3 nt. at the junction occurred within the first few conserved codons of the 2B coding region (Figure S1). The junctions generated in Cluster 1 encoded both 2Apro and 2B from the sub-genomic replicon recombination partner (the polymerase donor) and those in Cluster 2 encoded 2B from the donor and 2Apro from the recipient poliovirus type 3 genome. We propose that this clustering is a consequence of the overriding requirement for functional 2Apro and 2B proteins in the initial recombinant and that this is rarely, if ever, achieved in a single step. Since imprecise recombinants are viable and can serve as the source for subsequent precise recombinants (Figure 3; Figure S2), it seems logical that this mechanism increases the chance by as much as two orders of magnitude (cf. generation of a precise recombinant with an imprecise recombinant containing 3–321 nt. of additional sequence) of generating the latter. It should be noted that the polyprotein encoded by imprecise recombinants would include an additional 2Apro or 3Cpro cleavage site (Figure 2B). Since these provide an authentic context for proteolytic processing we presume this contributes to the viability of such imprecise recombinants. The 16 nt. insert derived from the 5′ NCR present in #25A must have arisen by two relocations of the viral polymerase with regard to the template. Since this sequence is identical in the parental genomes it is not clear which it was derived from, though at least one of the polymerase template reassociations must have been in cis. It is unclear why imprecise recombinants spanning the 2B/2C junction were not recovered in our analysis, whereas precise recombinants in 2C (#4E and #44B; Figure 2b, Figure S1) did occur. One possibility is that the assay used (Figure 1) requires that the hybrid genome is capable of establishing a productive infection in HeLa cells and that imprecise recombinants across 2B/2C are unable to do this due to deficiencies in homodimerisation, membrane permeabilization or formation of the replication complex [88]–[91]. Alternatively, this may indicate that the higher order structure of one or both of these proteins involves a ‘head to tail’ interaction in which the interface is between the amino terminus of one subunit and the carboxyl terminus of the other. Related to this point, it is also interesting to note that there is a reported asymmetry in reciprocal recombinants in the P2 region of poliovirus and related species C coxsackie A viruses which may reflect important protein-protein interactions required for genome replication and particle assembly [45].
The yield of intra-serotype recombinants was ∼10-fold higher than inter-serotype recombinants (Figure 1d) and, although only a limited number were sampled, a greater proportion of the former were precise (87% 13/15 cf. 30% 41/136 of inter-serotype recombinants). Further studies will be needed to determine whether the increased proportion of precise junctions is due to the sequence identity of the parental genomes, reflecting its role in the underlying recombination mechanism. This may also influence the yield of recombinants. However, protein-protein compatibility is also likely to influence both the yield and type of viable recombinants generated. Identical parental genomes inevitably encode proteins that have co-evolved and have presumably achieved optimal compatibility. This may enhance the replication and subsequent resolution (see below) of imprecise intra-serotype recombinants. In contrast, even the limited sequence divergence at the amino acid level between the parental serotypes (4–9% divergence at the amino acid level between poliovirus type 1 and 3 proteins 2Apro, 2B and 2C), might compromise the fitness of inter-serotype recombinants. Future analysis should therefore include looking for adaptive changes potentially some distance from the recombination site.
We propose that the recombination process of enteroviruses is biphasic (Figure 6), involving the generation of an initial imprecise recombinant which – through a process we term resolution – yields precise, genome-length, recombinants. The resolution process may be direct, generating precise recombinants in a single step, or indirect in which imprecise recombinants with shorter genome duplications are generated. For example, the synthesised #105B recombinant cDNA generated both precise and imprecise recombinants upon serial passage (Figure S2). We predict that resolution is an iterative process in which genomes with incrementally increased fitness are selected from the pool of molecules generated during replication. Whether resolution involves intra - or intermolecular polymerase transfer remains to be determined. In practice the resolution event may occur in the same dually infected cell in which the initial recombinant was generated. We think that this is the likely explanation for the generation of precise recombinants in co-transfected L929 murine cells (Figure 2). A biphasic recombination process is likely to have a bearing upon the recombination junctions generated. For example, the processes involved in the generation of imprecise and precise junctions may be influenced – if at all – by different contextual and sequence-dependent attributes. Prompted by the observation that the majority of precise recombinants occur at positions with limited sequence identity between templates (underlined in Figures S1 and S2) we investigated the role of primary sequence and RNA secondary structure. Other than an apparent absence of structure in the negative strand of recipient precise junctions (Figure 6b), which mechanistically seems unlikely to contribute to the polymerase strand-transfer reaction, we found no compelling evidence for a role for either primary sequence or sequence order-dependent RNA structure in influencing the recombination junctions observed. In contrast, Runckel and colleagues have recently reported an elegant next generation sequencing study of precise recombinants of poliovirus partners tagged with synonymous mutations [64] in which sequence composition and RNA structure correlated with, in a predictable and modifiable manner, recombination hotspots. The application of this type of approach to the imprecise junctions that predominate in early recombinant populations is likely to allow the separation of sequence/structural influences on the recombination process per se and the subsequent resolution process. The independent isolation of the same precise recombinant by serial passage of #105B virus recovered from co-transfection and from an engineered cDNA (#105B-a and #105B-1; Figure S2) suggests there may to be sequence-dependent influences on the sites of recombination during the process of resolution. Alternatively, this particular recombinant may have a subtle fitness advantage over others generated, thereby prevailing upon serial passage. Further studies on the relative fitness of precise recombinants, for example by competition studies between known levels of two or more input viruses [92], may help elucidate this.
As a prelude to investigating the influence of the polymerase on the generation of recombinants we compared the relative yield of viable recombinants in the CRE-REP assay and a non-replicative assay [50], [51] primed with the same amounts of truncated variants of the same template RNA (Figure 4a). Under these conditions an intratypic CRE-REP assay generated ∼25 fold more recombinants. We went on to conduct three related studies that indicate that the generation of recombinants in the CRE-REP assay is a replicative process. By increasing (ribavirin) or decreasing (G64S) the error rate of the viral polymerase we respectively enhanced or suppressed the recovery of recombinants (Figure 4bd) in the CRE-REP assay. In contrast, neither ribavirin nor the G64S polymerase mutation influenced the yield of recombinants using a non-replicative assay consisting of truncated RNA templates (Figure 4ce). Further support for the replicative generation of recombinants in the CRE-REP assay was provided by the demonstration that nocodazole reduced the yield of recombinants by ∼70%, but had no effect on either genome replication ([62] and Figure S5) or on the yield of recombinants from the non-replicative assay (Figure 4fg).
The three-fold enhancement of poliovirus recombinant recovery in the presence of ribavirin may have implications for the therapeutic use of this antiviral drug. However, we acknowledge that in hepatitis C patients – in which ribavirin is widely used therapeutically – recombination is considered a relatively rare event [93], [94]. A mechanistic exploration of the enhanced recombination observed in the presence of ribavirin clearly deserves further analysis. In preliminary studies we have observed that 5-fluorouracil also enhances the yield of recombinants in the CRE-REP assay (data not shown). It is likely that extension of these mutagenic inhibitor studies will provide further insight into the precise mechanism of template disengagement and re-engagement by the viral polymerase. Indeed, previous studies have already proposed that mismatches induced by mutagens enhance the dissociation of the template and the polymerase [48], [95]. However, it is interesting to note that in a limited range of imprecise recombinants generated in the presence of ribavirin, nucleotide substitutions were not observed at the recombination junctions sequenced (Woodman and Evans, in preparation).
Since the templates used in the non-replicative assay were identical (other than being either 5′ or 3′ truncated) to those used in the CRE-REP assay, recombination must have had to occur between the same genetic markers – the luciferase reporter gene and the defective CRE (Figure 1) – to generate viable progeny. It is therefore interesting to note that non-replicative recombination generated only ∼4% of the progeny of that produced in the equivalent CRE-REP assay when transfecting equimolar amounts of in vitro synthesized RNA into L929 cells (Figure 6a). This striking difference in yield is further support that the CRE-REP assay is likely mechanistically different from a non-replicative recombination event. Further support for this interpretation includes the lack of inhibition of non-replicative recombination by nocodazole – this implies this process occurs outside the confines of the replication complex, or at least does not require co-occupied replication complexes to occur. Finally, although non-replicative recombination, presumably involving cellular RNA exonucleases and ligases, is well documented in a number of picornaviruses, flaviviruses and alphaviruses [51]–[53], [96] – and may generate the types of junctions we define as imprecise [53] – the relative importance in vivo of replicative and non-replicative mechanisms of recombinant generation remain to be determined.
The CRE-REP assay we describe may also provide an approach to study the restrictions on intra - and interspecific recombination in enteroviruses, the former being far more frequent than the latter. In preliminary studies we have recovered recombinants between species B enteroviruses (echovirus 7; data not shown), suggesting that the mechanism proposed from our studies using poliovirus is likely generic to other enteroviruses, and presumably to other picornaviruses which also exhibit recombination [97]. However, in repeated attempts we have been unable to recover interspecies recombinants between reciprocal transfections of species C and species B enteroviruses (poliovirus type 1 and either echovirus types 6 or 7 respectively; data not shown). Further studies may elucidate whether this is due to direct genomic incompatibility, i.e. any recombinant genomes generated are non-viable, perhaps because they are unable to replicate or undergo proteolytic processing, or due to the absence of protein-protein interactions necessary for encapsidation [46]. Alternatively the rarity of interspecies recombination may reflect a lack of opportunity, perhaps due to occupancy of distinct replication complexes, sub-cellular or cellular compartmentalization.
The suggestion that an imprecise replicative recombination process results in partial duplication of the virus genome also has intriguing implications for our wider understanding of the evolution of positive strand RNA viruses. There are several variously well characterised evolutionary duplication events that have shaped extant picornavirus genomes. These include the capsid proteins (VP1-3) which share a common ‘jelly roll’ structure [98], the three contiguous VPg proteins of foot and mouth disease virus [99] and even the non-contiguous 2A and 3C proteases [100]. In addition, other positive strand RNA viruses exhibit genome duplications [101] such as the multiple VPg proteins of some dicistroviruses [102] or the functionally distinct adjacent leader proteinases of the Closteroviridae [103]. The type of imprecise recombination event we describe, potentially coupled with partial resolution, could account for this ‘evolution by duplication’. Likewise, duplication and subsequent resolution events could explain the permutation of the polymerase palm domain reported in some Alphavirus-like insect tetraviruses [104]. Other than the ancestral evolution of the capsid proteins the majority of these involve duplication of relatively short sequences – this is presumed to reflect intrinsic constraints on genome length [101], but may also reflect the mechanism by which they are generated.
An improved insight into evolution of RNA viruses, including acquisition of extensive regions of the genome by the process of recombination, will improve our understanding of the basic biology of this important group of viruses and help us identify how the process can be controlled, avoided or exploited.
Materials and Methods
Virus and cell culture
HeLa, L929 murine fibroblasts and baby hamster kidney (BSR-T7) cells were grown as monolayers in Dulbecco's Modified Eagle Medium (DMEM) or Glasgow Minimum Essential Medium (GMEM supplemented with G418 antibiotic). Media was supplemented with 100 U/ml penicillin, 100 µg/ml streptomycin, 2 mM L-glutamine and 10% Heat Inactivated (HI)-FBS. All cells were passaged in the presence of trypsin-EDTA. Where stated, ribavirin (Sigma) or guanidine hydrochloride (Sigma) were added to growth media at the concentrations indicated. Nocodazole was used where required by treating pre-chilled cells (10 minutes at 0°C) with a final concentration of 5 µM in DMSO (essentially as described previously; [62]) for two hours prior to transfection. Poliovirus type 1 (Mahoney) and type 3 (Leon) were recovered following transfection of RNA generated in vitro (see below) from full length cDNA. Virus was quantified by plaque assay as described previously [105]. Growth kinetics of viruses were determined following synchronous infection of HeLa cells at a multiplicity of infection (moi) of 10 pfu/cell. Unabsorbed virus was removed by washing with sterile PBS and plates incubated in fresh media at 37°C in an atmosphere containing 5% CO2. Virus in the supernatant was quantified at various time points post infection by plaque assay. Virus competition assays were conducted by co-infection of HeLa cells with an moi of 10 pfu/cell of each virus, removing unabsorbed virus by washing in PBS and harvest of virus from the fresh supernatant at either 24 hours post infection (p.i.) or when cells displayed at least 50% cytopathic effect (cpe). When serially passaging virus, harvested supernatant was diluted 1∶4 with fresh media. Recombinant viruses were biologically cloned by limit (doubling) dilution in 96-well plates seeded with 1×105 HeLa cells/well. Plates were inoculated and incubated at 37°C/5% CO2 for four days after which time virus-containing supernatant was removed (and stored) and the remaining cell monolayer stained with crystal violet. Virus supernatant from the highest dilution causing complete cpe was retained for further analysis.
Plasmids, in vitro transcription and cell transfection
pT7Rep3-L is a poliovirus type 3 (Leon) sub-genomic replicon bearing a luciferase reporter gene inserted in-frame in place of the P1 capsid coding region of the genome. pRLucWT is a pBR-based plasmid containing a cDNA for a poliovirus type 1 sub-genomic replicon in which the capsid coding region is replaced, in frame, with a luciferase reporter gene, as previously described [106]. pT7/SL3 has also been described previously [56] and consists of a full-length poliovirus type 3 (Leon) cDNA bearing 8 synonymous substitutions in the cis-acting replication element (CRE) within the 2C-coding region. A variant of pRLucWT containing a Gly to Ser substitution at residue 64 (hence designated pRLucWTG64S) of the RNA dependent RNA polymerase was generously provided by Craig Cameron and Jamie Arnold. The same mutation was built into the pT7/SL3 cDNA using standard methods. Plasmids pT7/SL3 and pRLucWT were linearised with Sal I and Apa I respectively and transcribed in vitro using T7 RNA Polymerase (Fermentas) following the manufacturer's protocol. Residual DNA template was removed by addition of 2 U DNase Turbo (Ambion) and the RNA transcripts were purified using RNeasy Mini Kit (Qiagen) before spectrophotometric quantification. Unless otherwise stated, 0.5 µg or 1 µg (total) of equimolar amounts of both template RNAs was prepared with Lipofectamine 2000 (Invitrogen; used according to the manufacturers instructions) and transfected into confluent monolayers in 12 or 6 well dishes respectively.
A full length cDNA of the recombinant virus genome designated #105B was constructed using standard molecular biology techniques from the parental pRLucWT and pT7/SL3 cDNAs together with the central region of the genome which was RT-PCR amplified from #105B virus. The compete cDNA sequence was validated before further analysis.
Truncated cDNA templates were generated from pT7Rep3-L, pRLucWT and pT7/SL3 for use in non-replicative recombination assays, essentially as previously described [50]. Briefly, pT7Rep3-L and pRLucWT were digested with Pml I and Pac I and religated, effectively removing the entire IRES and the majority of the luciferase coding region. Similarly, pT7/SL3 was digested with Xho I and Sal I and religated, removing the polymerase coding region. Truncated templates are indicated with a prefix or suffix Δ where appropriate e.g. pT7/SL3Δ. Where required, template cDNAs were modified by inclusion of a G64S substitution in the viral polymerase using standard methods. Non-replicative recombination assays were always conducted in parallel with the CRE-REP assay (see above) and used equimolar ratios of RNA (total 0.5 µg) co-transfected into L929 murine cells in 12 well dishes. Supernatant virus was recovered, 60 hours post-transfection, and quantified by plaque assay on HeLa cell monolayers.
Oligonucleotides, viral RNA isolation and characterisation
Viral RNA was extracted from clarified culture supernatant using a Qiagen RNAeasy Mini kit, reverse transcribed using Superscript II reverse transcriptase (Invitrogen) and an oligo-dT primer at 46°C for 50 minutes (mins.) with the reaction terminated by incubation for 15 mins. at 70°C. PCR amplification of recombination junctions used template cDNA and appropriate oligonucleotides as listed in the results section (all oligonucleotides are listed in Supplementary Table S1) with KOD XL DNA polymerase (Novagen) used according to the manufacturer's protocol. PCR products were sized by agarose gel electrophoresis and sequenced by the University of Warwick Genomics Facility on an ABI PRISM 3130xl Genetic Analyser. Sequence analysis used a combination of Lasergene v.6.0 (DNA*) and Clustal [107] as appropriate.
Nomenclature for recombinant identification
Reference sequences of poliovirus type 3 Leon (Genbank #X00925), type 1 Mahoney (Genbank #V01149) or the type 1 Mahoney-derived luciferase-encoding sub-genomic replicon (pRLucWT). To facilitate definition of junctions defined in clonal groups of recombinant virus genomes a standardized naming scheme was used; the 5′ and 3′ components were numbered with the last or first nucleotide of the relevant parental genome (poliovirus type 3 or the type 1 derived sub-genomic replicon). For reference, the latter consists of a 5′ untranslated region (nucleotides 1–742), the luciferase encoding reporter gene (nt. 743–2410) fused with a short linker to the sequence encoding the last 5 amino acids of VP1. In this cDNA, nonstructural protein coding region starts at nt. 2441. In cases where the recombination junction could not be unambiguously defined – due to sequence identity between aligned parental genomes – the numbering assumes these sequences were derived from the parental genome contributing the 5′ portion of the genome. For example, PV33897/PV12737 (recombination group #14C; Figure 3) consists of nucleotides 1-3897 from PV3 and 2737-polyA from the sub-genomic replicon, pRLucWT.
Bioinformatic analysis
Mean folding energy differences were calculated essentially as described previously [67] using a sequence scrambling algorithm (NDR) that retains compositional features such as the dinucleotide frequencies of the native sequence as implemented in the SSE package [108].
Supporting Information
Zdroje
1. DomingoE, HollandJJ (1997) RNA virus mutations and fitness for survival. Annu Rev Microbiol 51 : 151–178.
2. EigenM (1996) On the nature of virus quasispecies. Trends Microbiol 4 : 216–218.
3. GristNR, BellEJ, AssaadF (1978) Enteroviruses in human disease. Prog Med Virol 24 : 114–157.
4. Knowles NJ, Hovi T, Hyypia T, King AMQ, Lindberg AM, et al. (2012) Picornaviridae. In: King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ, editors. Virus Taxonomy: Classification and Nomenclature of Viruses: Ninth Report of the International Committee on Taxonomy of Viruses. San Diego: Elsevier. pp. 855–880.
5. CaroV, GuillotS, DelpeyrouxF, CrainicR (2001) Molecular strategy for ‘serotyping’ of human enteroviruses. J Gen Virol 82 : 79–91.
6. ObersteMS, MaherK, KilpatrickDR, PallanschMA (1999) Molecular evolution of the human enteroviruses: correlation of serotype with VP1 sequence and application to picornavirus classification. J Virol 73 : 1941–1948.
7. ObersteMS, MaherK, KilpatrickDR, FlemisterMR, BrownBA, et al. (1999) Typing of human enteroviruses by partial sequencing of VP1. J Clin Microbiol 37 : 1288–1293.
8. PalaciosG, CasasI, TenorioA, FreireC (2002) Molecular identification of enterovirus by analyzing a partial VP1 genomic region with different methods. J Clin Microbiol 40 : 182–192.
9. BrownBA, ObersteMS, AlexanderJP, KennettML, PallanschMA (1999) Molecular epidemiology and evolution of enterovirus 71 strains isolated from 1970 to 1998. J Virol 73 : 9969–9975.
10. ChuaBH, McMinnPC, LamSK, ChuaKB (2001) Comparison of the complete nucleotide sequences of echovirus 7 strain UMMC and the prototype (Wallace) strain demonstrates significant genetic drift over time. J Gen Virol 82 : 2629–2639.
11. KünkelU, SchreierE (2000) Genetic variability within the VP1 coding region of echovirus type 30 isolates. Arch Virol 145 : 1455–1464.
12. MartínJ, DunnG, HullR, PatelV, MinorPD (2000) Evolution of the Sabin strain of type 3 poliovirus in an immunodeficient patient during the entire 637-day period of virus excretion. J Virol 74 : 3001–3010.
13. SimmondsP, WelchJ (2006) Frequency and dynamics of recombination within different species of human enteroviruses. J Virol 80 : 483–493.
14. TakedaN, TanimuraM, MiyamuraK (1994) Molecular evolution of the major capsid protein VP1 of enterovirus 70. J Virol 68 : 854–862.
15. AnderssonP, EdmanK, LindbergAM (2002) Molecular analysis of the echovirus 18 prototype: evidence of interserotypic recombination with echovirus 9. Virus Res 85 : 71–83.
16. LindbergAM, AnderssonP, SavolainenC, MuldersMN, HoviT (2003) Evolution of the genome of Human enterovirus B: incongruence between phylogenies of the VP1 and 3CD regions indicates frequent recombination within the species. J Gen Virol 84 : 1223–1235.
17. ChevaliezS, SzendröiA, CaroV, BalanantJ, GuillotS, et al. (2004) Molecular comparison of echovirus 11 strains circulating in Europe during an epidemic of multisystem hemorrhagic disease of infants indicates that evolution generally occurs by recombination. Virology 325 : 56–70.
18. GuillotS, CaroV, CuervoN, KorotkovaE, CombiescuM, et al. (2000) Natural genetic exchanges between vaccine and wild poliovirus strains in humans. J Virol 74 : 8434–8443.
19. CuervoNS, GuillotS, RomanenkovaN, CombiescuM, Aubert-CombiescuA, et al. (2001) Genomic features of intertypic recombinant sabin poliovirus strains excreted by primary vaccinees. J Virol 75 : 5740–5751.
20. LukashevAN, LashkevichVA, KorolevaGA, IlonenJ, HinkkanenAE (2004) Recombination in uveitis-causing enterovirus strains. J Gen Virol 85 : 463–470.
21. LukashevAN, LashkevichVA, IvanovaOE, KorolevaGA, HinkkanenAE, et al. (2005) Recombination in circulating Human enterovirus B: independent evolution of structural and non-structural genome regions. J Gen Virol 86 : 3281–3290.
22. LukashevAN, IvanovaOE, EremeevaTP, GmylLV (2008) Analysis of echovirus 30 isolates from Russia and new independent states revealing frequent recombination and reemergence of ancient lineages. J Clin Microbiol 46 : 665–670.
23. McWilliam LeitchE, BendigJ, CabrerizoM, CardosaJ, HyypiäT, et al. (2009) Transmission networks and population turnover of echovirus 30. J Virol 83 : 2109–2118.
24. McWilliam LeitchEC, CabrerizoM, CardosaJ, HarvalaH, IvanovaOE, et al. (2010) Evolutionary dynamics and temporal/geographical correlates of recombination in the human enterovirus echovirus types 9, 11, and 30. J Virol 84 : 9292–9300.
25. McWilliam LeitchEC, CabrerizoM, CardosaJ, HarvalaH, IvanovaOE, et al. (2012) The association of recombination events in the founding and emergence of subgenogroup evolutionary lineages of human enterovirus 71. J Virol 86 : 2676–2685.
26. NorderH, BjerregaardL, MagniusLO (2002) Open reading frame sequence of an Asian enterovirus 73 strain reveals that the prototype from California is recombinant. J Gen Virol 83 : 1721–1728.
27. KewO, Morris-GlasgowV, LandaverdeM, BurnsC, ShawJ, et al. (2002) Outbreak of poliomyelitis in Hispaniola associated with circulating type 1 vaccine-derived poliovirus. Science 296 : 356–359.
28. ObersteMS, MaherK, PallanschMA (2004) Evidence for frequent recombination within species human enterovirus B based on complete genomic sequences of all thirty-seven serotypes. J Virol 78 : 855–867.
29. ObersteMS, PeñarandaS, PallanschMA (2004) RNA recombination plays a major role in genomic change during circulation of coxsackie B viruses. J Virol 78 : 2948–2955.
30. OprisanG, CombiescuM, GuillotS, CaroV, CombiescuA, et al. (2002) Natural genetic recombination between co-circulating heterotypic enteroviruses. J Gen Virol 83 : 2193–2200.
31. SanttiJ, HyypiäT, KinnunenL, SalminenM (1999) Evidence of recombination among enteroviruses. J Virol 73 : 8741–8749.
32. SanttiJ, VainionpääR, HyypiäT (1999) Molecular detection and typing of human picornaviruses. Virus Res 62 : 177–183.
33. SanttiJ, HarvalaH, KinnunenL, HyypiäT (2000) Molecular epidemiology and evolution of coxsackievirus A9. J Gen Virol 81 : 1361–1372.
34. McIntyreCL, McWilliam LeitchEC, Savolainen-KopraC, HoviT, SimmondsP (2010) Analysis of genetic diversity and sites of recombination in human rhinovirus species. C. J Virol 84 : 10297–10310.
35. KimH, KimK, KimDW, JungHD, Min CheongH, et al. (2013) Identification of Recombinant Human Rhinovirus A and C in Circulating Strains from Upper and Lower Respiratory Infections. PLoS One 8: e68081.
36. MartínJ, SamoilovichE, DunnG, LackenbyA, FeldmanE, et al. (2002) Isolation of an intertypic poliovirus capsid recombinant from a child with vaccine-associated paralytic poliomyelitis. J Virol 76 : 10921–10928.
37. SmuraT, BlomqvistS, PaananenA, VuorinenT, SobotováZ, et al. (2007) Enterovirus surveillance reveals proposed new serotypes and provides new insight into enterovirus 5′-untranslated region evolution. J Gen Virol 88 : 2520–2526.
38. YozwiakNL, Skewes-CoxP, GordonA, SaborioS, KuanG, et al. (2010) Human enterovirus 109: a novel interspecies recombinant enterovirus isolated from a case of acute pediatric respiratory illness in Nicaragua. J Virol 84 : 9047–9058.
39. TapparelC, JunierT, GerlachD, CordeyS, Van BelleS, et al. (2007) New complete genome sequences of human rhinoviruses shed light on their phylogeny and genomic features. BMC Genomics 8 : 224.
40. BolanakiE, KottaridiC, MarkoulatosP, KyriakopoulouZ, MargaritisL, et al. (2007) Partial 3D gene sequences of Coxsackie viruses reveal interspecies exchanges. Virus Genes 35 : 129–140.
41. RohllJ, PercyN, LeyR, EvansD, AlmondJ, et al. (1994) The 5′-untranslated regions of picornavirus RNAs contain independent functional domains essential for RNA replication and translation. J Virol 68 : 4384–4391.
42. GromeierM, AlexanderL, WimmerE (1996) Internal ribosomal entry site substitution eliminates neurovirulence in intergeneric poliovirus recombinants. Proc Natl Acad Sci U S A 93 : 2370–2375.
43. EggerD, BienzK (2002) Recombination of poliovirus RNA proceeds in mixed replication complexes originating from distinct replication start sites. J Virol 76 : 10960–10971.
44. WorobeyM, HolmesEC (1999) Evolutionary aspects of recombination in RNA viruses. J Gen Virol 80 (Pt 10): 2535–2543.
45. JiangP, FaaseJA, ToyodaH, PaulA, WimmerE, et al. (2007) Evidence for emergence of diverse polioviruses from C-cluster coxsackie A viruses and implications for global poliovirus eradication. Proc Natl Acad Sci U S A 104 : 9457–9462.
46. LiuY, WangC, MuellerS, PaulAV, WimmerE, et al. (2010) Direct interaction between two viral proteins, the nonstructural protein 2C and the capsid protein VP3, is required for enterovirus morphogenesis. PLoS Pathog 6: e1001066.
47. KirkegaardK, BaltimoreD (1986) The mechanism of RNA recombination in poliovirus. Cell 47 : 433–443.
48. ArnoldJJ, CameronCE (1999) Poliovirus RNA-dependent RNA polymerase (3Dpol) is sufficient for template switching in vitro. J Biol Chem 274 : 2706–2716.
49. NagyPD, SimonAE (1997) New insights into the mechanisms of RNA recombination. Virology 235 : 1–9.
50. GmylAP, BelousovEV, MaslovaSV, KhitrinaEV, ChetverinAB, et al. (1999) Nonreplicative RNA recombination in poliovirus. J Virol 73 : 8958–8965.
51. GmylAP, KorshenkoSA, BelousovEV, KhitrinaEV, AgolVI (2003) Nonreplicative homologous RNA recombination: promiscuous joining of RNA pieces? RNA 9 : 1221–1231.
52. GalleiA, PankrazA, ThielHJ, BecherP (2004) RNA recombination in vivo in the absence of viral replication. J Virol 78 : 6271–6281.
53. ScheelTK, GalliA, LiYP, MikkelsenLS, GottweinJM, et al. (2013) Productive homologous and non-homologous recombination of hepatitis C virus in cell culture. PLoS Pathog 9: e1003228.
54. EminiEA, LeibowitzJ, DiamondDC, BoninJ, WimmerE (1984) Recombinants of Mahoney and Sabin strain poliovirus type 1: analysis of in vitro phenotypic markers and evidence that resistance to guanidine maps in the nonstructural proteins. Virology 137 : 74–85.
55. GoodfellowIG, PolacekC, AndinoR, EvansDJ (2003) The poliovirus 2C cis-acting replication element-mediated uridylylation of VPg is not required for synthesis of negative-sense genomes. Journal of General Virology 84 : 2359–2363.
56. GoodfellowI, ChaudhryY, RichardsonA, MeredithJ, AlmondJ, et al. (2000) Identification of a cis-acting replication element within the poliovirus coding region. J Virol 74 : 4590–4600.
57. GoodfellowI, KerriganD, EvansD (2003) Structure and function analysis of the poliovirus cis-acting replication element (CRE). RNA 9 : 124–137.
58. BartonDJ, FlaneganJB (1997) Synchronous replication of poliovirus RNA: initiation of negative-strand RNA synthesis requires the guanidine-inhibited activity of protein 2C. J Virol 71 : 8482–8489.
59. CrottyS, MaagD, ArnoldJJ, ZhongW, LauJY, et al. (2000) The broad-spectrum antiviral ribonucleoside ribavirin is an RNA virus mutagen. Nat Med 6 : 1375–1379.
60. PfeifferJK, KirkegaardK (2003) A single mutation in poliovirus RNA-dependent RNA polymerase confers resistance to mutagenic nucleotide analogs via increased fidelity. Proc Natl Acad Sci U S A 100 : 7289–7294.
61. PfeifferJK, KirkegaardK (2005) Increased fidelity reduces poliovirus fitness and virulence under selective pressure in mice. PLoS Pathog 1: e11.
62. EggerD, BienzK (2005) Intracellular location and translocation of silent and active poliovirus replication complexes. J Gen Virol 86 : 707–718.
63. DoedensJ, MaynellLA, KlymkowskyMW, KirkegaardK (1994) Secretory pathway function, but not cytoskeletal integrity, is required in poliovirus infection. Arch Virol Suppl 9 : 159–172.
64. RunckelC, WestessonO, AndinoR, DeRisiJL (2013) Identification and manipulation of the molecular determinants influencing poliovirus recombination. PLoS Pathog 9: e1003164.
65. KingAM (1988) Preferred sites of recombination in poliovirus RNA: an analysis of 40 intertypic cross-over sequences. Nucleic Acids Res 16 : 11705–11723.
66. DedepsidisE, KyriakopoulouZ, PliakaV, MarkoulatosP (2010) Correlation between recombination junctions and RNA secondary structure elements in poliovirus Sabin strains. Virus Genes 41 : 181–191.
67. SimmondsP, TuplinA, EvansD (2004) Detection of genome-scale ordered RNA structure (GORS) in genomes of positive-stranded RNA viruses: Implications for virus evolution and host persistence. RNA 10 : 1337–1351.
68. DavisM, SaganSM, PezackiJP, EvansDJ, SimmondsP (2008) Bioinformatic and Physical Characterizations of Genome-Scale Ordered RNA Structure in Mammalian RNA Viruses. Journal of Virology 82 : 11824–11836.
69. Simon-LoriereE, HolmesEC (2011) Why do RNA viruses recombine? Nat Rev Microbiol 9 : 617–626.
70. HahnCS, LustigS, StraussEG, StraussJH (1988) Western equine encephalitis virus is a recombinant virus. Proc Natl Acad Sci U S A 85 : 5997–6001.
71. CammackN, PhillipsA, DunnG, PatelV, MinorPD (1988) Intertypic genomic rearrangements of poliovirus strains in vaccinees. Virology 167 : 507–514.
72. YangCF, NaguibT, YangSJ, NasrE, JorbaJ, et al. (2003) Circulation of endemic type 2 vaccine-derived poliovirus in Egypt from 1983 to 1993. J Virol 77 : 8366–8377.
73. AduF, IberJ, BukbukD, GumedeN, YangSJ, et al. (2007) Isolation of recombinant type 2 vaccine-derived poliovirus (VDPV) from a Nigerian child. Virus Res 127 : 17–25.
74. HeathL, van der WaltE, VarsaniA, MartinDP (2006) Recombination patterns in aphthoviruses mirror those found in other picornaviruses. J Virol 80 : 11827–11832.
75. De GraziaS, MediciMC, PintoP, MoschidouP, TummoloF, et al. (2012) Genetic heterogeneity and recombination in human type 2 astroviruses. J Clin Microbiol 50 : 3760–3764.
76. TwiddySS, HolmesEC (2003) The extent of homologous recombination in members of the genus Flavivirus. J Gen Virol 84 : 429–440.
77. MooreJ, JironkinA, ChandlerD, BurroughsN, EvansDJ, et al. (2011) Recombinants between Deformed wing virus and Varroa destructor virus-1 may prevail in Varroa destructor-infested honeybee colonies. Journal of General Virology 92 : 156–161.
78. PilipenkoEV, GmylAP, AgolVI (1995) A model for rearrangements in RNA genomes. Nucleic Acids Res 23 : 1870–1875.
79. RomanovaLI, BlinovVM, TolskayaEA, ViktorovaEG, KolesnikovaMS, et al. (1986) The primary structure of crossover regions of intertypic poliovirus recombinants: a model of recombination between RNA genomes. Virology 155 : 202–213.
80. TolskayaEA, RomanovaLI, BlinovVM, ViktorovaEG, SinyakovAN, et al. (1987) Studies on the recombination between RNA genomes of poliovirus: the primary structure and nonrandom distribution of crossover regions in the genomes of intertypic poliovirus recombinants. Virology 161 : 54–61.
81. BarclayW, LiQ, HutchinsonG, MoonD, RichardsonA, et al. (1998) Encapsidation studies of poliovirus subgenomic replicons. J Gen Virol 79 (Pt 7): 1725–1734.
82. PercyN, BarclayWS, SullivanM, AlmondJW (1992) A poliovirus replicon containing the chloramphenicol acetyltransferase gene can be used to study the replication and encapsidation of poliovirus RNA. J Virol 66 : 5040–5046.
83. MorascoBJ, SharmaN, ParillaJ, FlaneganJB (2003) Poliovirus cre(2C)-dependent synthesis of VPgpUpU is required for positive - but not negative-strand RNA synthesis. J Virol 77 : 5136–5144.
84. MurrayKE, BartonDJ (2003) Poliovirus CRE-dependent VPg uridylylation is required for positive-strand RNA synthesis but not for negative-strand RNA synthesis. J Virol 77 : 4739–4750.
85. JarvisTC, KirkegaardK (1992) Poliovirus RNA recombination: mechanistic studies in the absence of selection. EMBO J 11 : 3135–3145.
86. DuggalR, WimmerE (1999) Genetic recombination of poliovirus in vitro and in vivo: temperature-dependent alteration of crossover sites. Virology 258 : 30–41.
87. MollaA, JangSK, PaulAV, ReuerQ, WimmerE (1992) Cardioviral Internal Ribosomal Entry Site Is Functional in a Genetically Engineered Dicistronic Poliovirus. Nature 356 : 255–257.
88. CuconatiA, XiangW, LahserF, PfisterT, WimmerE (1998) A protein linkage map of the P2 nonstructural proteins of poliovirus. J Virol 72 : 1297–1307.
89. BarcoA, CarrascoL (1998) Identification of regions of poliovirus 2BC protein that are involved in cytotoxicity. J Virol 72 : 3560–3570.
90. SuhyDA, GiddingsTH, KirkegaardK (2000) Remodeling the endoplasmic reticulum by poliovirus infection and by individual viral proteins: an autophagy-like origin for virus-induced vesicles. J Virol 74 : 8953–8965.
91. TeterinaNL, LevensonE, RinaudoMS, EggerD, BienzK, et al. (2006) Evidence for functional protein interactions required for poliovirus RNA replication. J Virol 80 : 5327–5337.
92. AtkinsonNJ, WitteveldtJ, EvansDJ, SimmondsP (2014) The influence of CpG and UpA dinucleotide frequencies on RNA virus replication and characterization of the innate cellular pathways underlying virus attenuation and enhanced replication. Nucleic Acids Res 42(7): 4527–4545.
93. YunZ, LaraC, JohanssonB, Lorenzana de RiveraI, SönnerborgA (1996) Discrepancy of hepatitis C virus genotypes as determined by phylogenetic analysis of partial NS5 and core sequences. J Med Virol 49 : 155–160.
94. González-CandelasF, López-LabradorFX, BrachoMA (2011) Recombination in hepatitis C virus. Viruses 3 : 2006–2024.
95. FreistadtMS, VaccaroJA, EberleKE (2007) Biochemical characterization of the fidelity of poliovirus RNA-dependent RNA polymerase. Virol J 4 : 44.
96. RajuR, SubramaniamSV, HajjouM (1995) Genesis of Sindbis virus by in vivo recombination of nonreplicative RNA precursors. J Virol 69 : 7391–7401.
97. JacksonAL, O'NeillH, MareeF, BlignautB, CarrilloC, et al. (2007) Mosaic structure of foot-and-mouth disease virus genomes. J Gen Virol 88 : 487–492.
98. HogleJM, ChowM, FilmanDJ (1985) Three-dimensional structure of poliovirus at 2.9 A resolution. Science 229 : 1358–1365.
99. ForssS, SchallerH (1982) A tandem repeat gene in a picornavirus. Nucleic Acids Res 10 : 6441–6450.
100. Palmenberg A, Neubauer D, Skern T (2010) Genome organisation and encoded proteins. In: Ehrenfeld E, Domingo E, Roos R, editors. The Picornaviruses. Washington DC: ASM Press.
101. Simon-LoriereE, HolmesEC (2013) Gene duplication is infrequent in the recent evolutionary history of RNA viruses. Mol Biol Evol 30 : 1263–1269.
102. NakashimaN, ShibuyaN (2006) Multiple coding sequences for the genome-linked virus protein (VPg) in dicistroviruses. J Invertebr Pathol 92 : 100–104.
103. PengCW, PeremyslovVV, MushegianAR, DawsonWO, DoljaVV (2001) Functional specialization and evolution of leader proteinases in the family Closteroviridae. J Virol 75 : 12153–12160.
104. GorbalenyaAE, PringleFM, ZeddamJL, LukeBT, CameronCE, et al. (2002) The palm subdomain-based active site is internally permuted in viral RNA-dependent RNA polymerases of an ancient lineage. J Mol Biol 324 : 47–62.
105. Minor PD (1985) Growth, Assay and Purification of Picornaviruses. In: Mahy BWJ, editor. Virology: A practical approach. Oxford: IRL Press. pp. 25–41.
106. AndinoR, RieckhofGE, AchacosoPL, BaltimoreD (1993) Poliovirus RNA Synthesis Utilizes an RNP Complex Formed Around the 5′-End of Viral RNA. EMBO Journal 12 : 3587–3598.
107. LarkinMA, BlackshieldsG, BrownNP, ChennaR, McGettiganPA, et al. (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23 : 2947–2948.
108. SimmondsP (2012) SSE: a nucleotide and amino acid sequence analysis platform. BMC Res Notes 5 : 50.
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2014 Číslo 6
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Fungal Nail Infections (Onychomycosis): A Never-Ending Story?
- Profilin Promotes Recruitment of Ly6C CCR2 Inflammatory Monocytes That Can Confer Resistance to Bacterial Infection
- IscR Is Essential for Type III Secretion and Virulence
- Contribution of Specific Residues of the β-Solenoid Fold to HET-s Prion Function, Amyloid Structure and Stability
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy