
A Genome-Wide Tethering Screen Reveals Novel Potential Post-Transcriptional Regulators in
Survival and adaptation of trypanosomatids to new surroundings requires activation of specific gene networks. This is mainly achieved by post-transcriptional mechanisms, and proteins that bind to specific mRNAs, and influence degradation or translation, are known to be important. However, only few such proteins have been characterized to date. The trypanosome genome encodes over 150 proteins with conserved RNA-binding domains, and it is very likely that additional proteins that do not have such domains could also modulate mRNA fate. Here, we report the results of a genome-wide screen to identify mRNA-fate regulators in Trypanosoma brucei. We used a method called “tethering” to artificially attach protein fragments to an mRNA. Our findings confirmed the role of RNA-binding proteins in the regulation of mRNA fate, and also suggested such roles for many other proteins, including some metabolic enzymes. Our results should serve as a useful resource. Moreover, the tethering screen approach could readily be adapted for use in other organisms.
Published in the journal:
. PLoS Pathog 10(6): e32767. doi:10.1371/journal.ppat.1004178
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004178
Summary
Survival and adaptation of trypanosomatids to new surroundings requires activation of specific gene networks. This is mainly achieved by post-transcriptional mechanisms, and proteins that bind to specific mRNAs, and influence degradation or translation, are known to be important. However, only few such proteins have been characterized to date. The trypanosome genome encodes over 150 proteins with conserved RNA-binding domains, and it is very likely that additional proteins that do not have such domains could also modulate mRNA fate. Here, we report the results of a genome-wide screen to identify mRNA-fate regulators in Trypanosoma brucei. We used a method called “tethering” to artificially attach protein fragments to an mRNA. Our findings confirmed the role of RNA-binding proteins in the regulation of mRNA fate, and also suggested such roles for many other proteins, including some metabolic enzymes. Our results should serve as a useful resource. Moreover, the tethering screen approach could readily be adapted for use in other organisms.
Introduction
Kinetoplastid protists are exposed to environmental challenges in the host and vector, necessitating extensive changes in gene expression. In kinetoplastids, most protein-coding genes are transcribed by RNA polymerase II in unidirectional clusters. Individual mRNAs are excised by 5′ trans splicing, with addition of a 39mer spliced leader RNA, and by 3′ polyadenylation [1]. At the level of individual pol II-transcribed open reading frames (ORFs), there is no evidence for control of expression via regulated transcription initiation. Trypanosomes and related parasites are therefore mainly dependent on post-transcriptional mechanisms for the control of gene expression [2], and thus for both survival and pathogenesis.
Control of gene expression at the post-transcriptional level is essential in all organisms, and RNA-binding proteins (RBPs) play critical roles at all stages: RNA processing, transport, stability and translation. The interactions of RBPs with cytosolic mRNAs - often, but not always, with the 3′-untranslated region (3′-UTR) - can affect transcript stability, translational efficiency, or both [3]. In this way, RBPs can modify and regulate every step of RNA metabolism and function. Recent studies in different organisms have revealed that the repertoire of RBPs is far greater than anticipated [4]–[7]. Although members of all classical RBP domain families were abundantly represented in these analyses, several novel and unexpected candidates with no RNA-related ontology or domain homology were seen to interact with RNA. In addition to novel proteins containing low complexity amino acid repeat motifs, metabolic enzymes, and enzymes with potential RNA - and protein-modifying activities were found. However, for most of these novel RBPs, the functional consequences of the mRNA binding remain unknown.
In trypanosomatids, RBPs are key factors in gene expression regulation. Over a hundred RBPs have been predicted to exist in trypanosomes, based on their containing canonical RNA-binding domains: RNA recognition motif (RRM), CCCH zinc fingers, and pumilio or PUF domains. Some of these are already known to be involved in the regulation of differentiation, development, the cell cycle and rRNA processing (recently reviewed in [8]). Most of the characterized RBPs control mRNA abundance by stabilizing the target transcript (for example TbZPF3 [9], TbZC3H11 [10], TbZC3H20 [11], TbPUF9 [12]) or by increasing translation (ALBA domain proteins, [13]). In mammalian cells, several proteins (such as BRF1, TTP, AUF1 and TIA-1) have been shown actively to destabilize mRNAs or to act as translation silencers ([14]. In trypanosomatids, although indirect results suggest that some proteins cause degradation of target transcripts this has never been shown directly (examples are UBP1 [15], PUF6 [16] and PUF5 [17].) There is evidence that one protein, RBP10, can suppress translation when attached to an mRNA [18]. With the single exception of ZC3H11 [19], the mechanisms by which trypanosome proteins determine mRNA fate are not known.
The functions of RBPs in vivo can be studied by “tethering” them to a reporter mRNA. In this technique, the protein under study is attached to the UTR of an mRNA reporter through an artificial RNA-protein interaction [20]. Hence, the functional activity of a protein can be studied independent of its intrinsic ability to bind to RNA. This assay has been applied successfully for numerous proteins in diverse organisms [21] and has proven useful in the analyses of the precise function of essential genes [22], mapping of protein function [10], dissecting functional maps of protein complexes [23] and visualizing tagged mRNAs [24]. The bacteriophage lambda-N protein is often used in the tethered function assay [25]. N-protein regulates bacterial transcriptional anti-termination by binding to a 15-nucleotide RNA hairpin, called boxB, within nascent transcripts [26]. The N-peptide is used in tethering assays because its small size (only 22 amino acids) limits the extent of likely effects on the function of the fused protein, and because of its high affinity interaction with boxB RNA.
In this paper, we use the “tethering” approach in a genome-wide screen for post-transcriptional regulators in Trypanosoma brucei. We have been able to take advantage of the excellent tools for advanced genetics and very high efficiency transfection of trypanosomes [27]. Also, since introns are almost completely absent and coding sequences are closely spaced, all open reading frames could be represented in a library made from genomic DNA. We were able to map functional domains on well-characterized proteins and found evidence for functions of many RBPs. RNA-binding properties were also analysed using a targeted protein array. The results greatly increase our knowledge of the possible regulatory repertoire in trypanosomes and provide functional annotation for many previously uncharacterized genes.
Results
Regulation of selectable markers by tethered proteins
To screen for proteins that increase gene expression, we used a reporter cell line constitutively expressing the blasticidin resistance (BLA) protein. The BLA mRNA contains five copies of the boxB hairpin RNA sequence between the resistance cassette and the actin (ACT) 3′-UTR (BLA-B-ACT). As a secondary reporter, a cassette encoding GFP (GFP-B-ACT) was introduced upstream of the blasticidin reporter (Figure 1A). Cell lines containing the BLA reporter mRNA lacking the boxB element were used as controls. The tandemly arranged reporters were integrated into the trypanosome alpha-beta tubulin repeat, and should be transcribed constitutively by RNA polymerase II reading through from upstream.
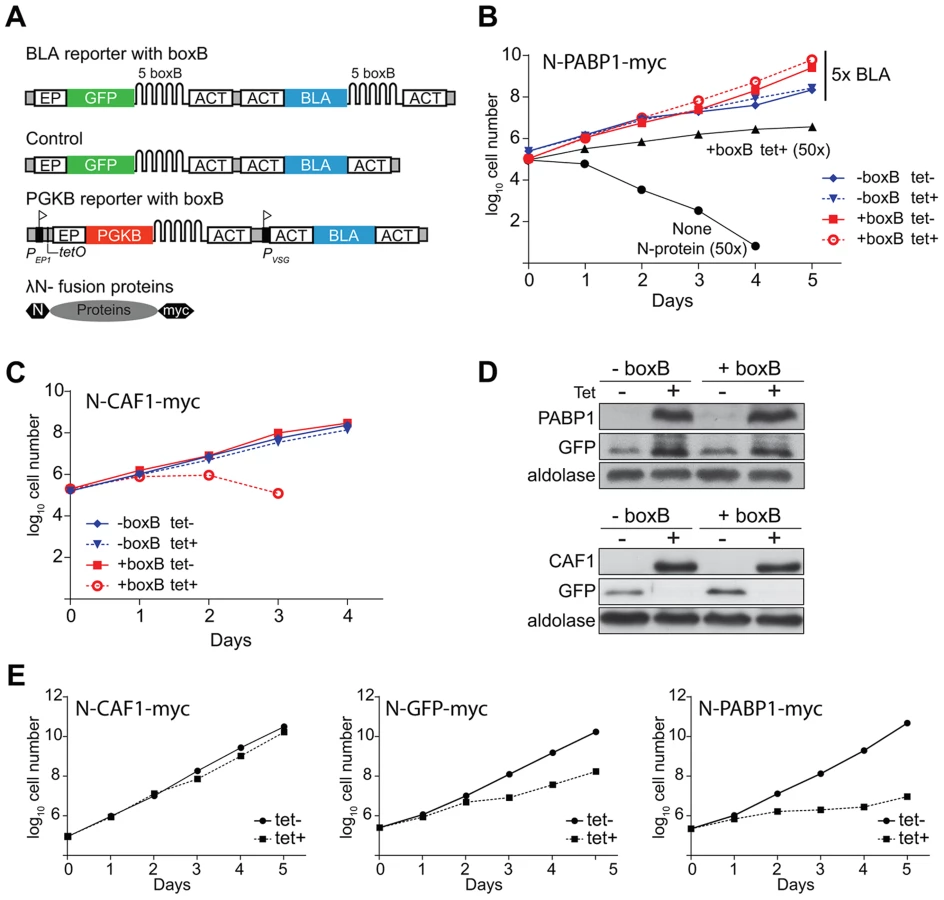
We first introduced a plasmid that inducibly expressed trypanosome poly(A)-binding protein 1 (PABP1) with the lambda-N peptide at the N-terminus and a myc tag at the C-terminus (N-PABP1-myc). Tethering of N-PABP1 to a trypanosome mRNA is known to increase mRNA stability and reporter expression [28]. As we had hoped, co-expression of N-PABP1-myc enabled the cells to grow at 250 µg/ml blasticidin - 50 times the normal concentration - whilst cells expressing only the BLA-B-ACT reporter were rapidly killed (Figure 1B). At 25 µg/ml blasticidin (5-fold normal), cells containing the BLA reporter with the boxB element grew slightly faster than the control (Figure 1B); this was independent of tetracycline addition, probably because a small amount of N-PABP1-myc was expressed in the absence of tetracycline (Figure 1D). As expected, tethering of PABP1-myc also increased GFP expression (Figure 1D). To find out whether we could also detect the activity of proteins that decreased expression, we tethered the deadenylase CAF1, which is known to result in reporter mRNA degradation [29]. Indeed, expression of the N-CAF1-myc decreased cell survival under low (25 µg/ml) blasticidin pressure (Figure 1C). Inhibition of GFP protein expression in both cell lines confirmed specificity (Figure 1D).
We next designed a screen that would positively select for proteins that decreased expression upon tethering. In bloodstream trypanosomes, inducible expression of cytosolic phosphoglycerate kinase B (PGKB) is lethal [30]. We therefore created a cell line with a tetracycline inducible PGKB open reading frame followed by 5 boxB copies at the 3′-UTR (Figure 1A). Upon tetracycline induction, the parasites died (not shown). As expected, co-expression of N-CAF1-myc (“destabilizing”, Figure 1E) expression conferred a selective advantage while N-GFP-myc (“neutral”, Figure 1F) and N-PABP-myc (“stabilizing”, Figure 1G) did not rescue the parasites. We concluded that our two complementary selections could be used to discover proteins affecting mRNA-fate.
Screening for modulators of trypanosome mRNA-fate
To screen for proteins involved in mRNA-fate regulation, a plasmid library was constructed using randomly sheared trypanosome genomic DNA. The plasmid backbone was designed such that transcription of each genomic DNA fragment could be induced with tetracycline, and the resulting proteins would contain the lambda-N peptide at the N-terminus. 3×106 independent clones were obtained with an average insert size (as judged by plasmid-specific PCR) of about 1.2 kbp. Based on the gene density within the coding regions of trypanosome chromosomes we expected approximately one in twelve plasmids to encode an authentic trypanosome protein fragment fused in frame to the N-peptide. This would indicate that our plasmid pool represents a ∼10-fold coverage of the open reading frames.
The inducible library was transfected into bloodstream cells expressing both the BLA-B-ACT and GFP-B-ACT reporters, using site-specific endonuclease-enhanced transfection [31]. In the first experiment (BLA-A), 1.8 million clones were obtained and in a second (BLA-B), 3.3 million clones (Supplementary Figure S1). To determine the sequence coverage of the fusions in the trypanosome populations, the plasmid inserts were amplified by PCR, using primers located within the lambda-N peptide and immediately 3′ to the cloning site. Control experiments with the resistance mRNA lacking the boxB elements were performed once with 3.1 million clones (experiment BLA-C).
We induced N-peptide fusion protein expression for 24 h and then grew cells for four days under different conditions. Clones that were unable to grow after tetracycline induction must express proteins that have intrinsic dominant-negative effects, unrelated to blasticidin resistance (Figure 2, black cloned inserts and trypanosomes). In addition, after 24 h induction we grew the cells for four days in various concentrations of blasticidin. To detect fusion proteins that decrease mRNA stability or translation (Figure 2, blue) we grew the cells in 1x (experiment BLA-A) or 2x (experiment BLA-B) levels of blasticidin. No significant effect on overall population growth was seen (Supplementary Figure 1A, C). To detect fusion proteins that increase mRNA stability or translation (Figure 2, red) we grew the cells in 6x (experiment BLA-A) or 10x or 20x (experiment BLA-B) levels of blasticidin. In this case, growth was impaired but then the cells started to recover, indicating rescue by lambda-N fusions that could enhance BLA expression (Supplementary Figure 1A, C). In parallel, we run a similar experiment with the cells carrying the BLA reporter without boxB elements (Supplementary Figure 1D, experiment BLA-C). The cloned genomic DNA fragments were in each case recovered by plasmid-specific PCR. Examination of small aliquots from the reactions revealed similar-looking smears of many products for all populations except the high blasticidin treated cells (Supplementary Figure 1B, E). All PCR mixtures were subjected to high-throughput sequencing.

To screen for protein fragments that impaired expression, the plasmid library was transfected into bloodstream-form cells expressing the inducible PGKB-B-ACT reporter (Supplementary Figure 2A). Three independent experiments were done, resulting in 0.65 million (Library PGKB-A), 1.2 million (Library PGKB-B) and 4.8 (Library PGKB-C) million independent clones. Expression of both PGKB and the lambda-N fusions was induced with tetracycline and cells were grown in the presence of inducer for 5 days. In the libraries, the induced PGKB only slightly affected growth. Presumably, some of the parasites had lost inducible expression of the lethal protein during the transfection procedures. This is a common problem in trypanosomes that inducibly express toxic RNAs or proteins. Nevertheless, expression of lambda-N fusions did confer a modest growth advantage (Supplementary Figure 2B). Again, the selected plasmid inserts were amplified (Supplementary Figure 2C) and sequenced.
The screen detects known expression activators
Sequencing reads from the blasticidin expression experiment were mapped to the T. brucei reference after trimming of the non-trypanosome boundary sequences. Only reads that were in frame with the lambda-N peptide were considered. Results are tabulated in Supplementary Tables S1, S2, S3, with Supplementary Table S1 showing the reads for all mapped in-frame locations at the nucleotide level. Before selection, reads were fairly uniformly distributed with no strand or coding-region preference. However, under inducing conditions, in-frame coding-region reads on particular genes were strongly enriched. Figure 3A shows an example of aligned reads from the second blasticidin selection experiment, in the ∼12 kb region that surrounds the gene encoding the zinc finger protein ZC3H11. After selection, the accumulation of reads over ZC3H11 was striking. Overall there were about 50-fold more in-frame reads from the ZC3H11 open reading frame after selection than in the unselected population.

We preciously characterized the activity of ZC3H11 using the tethering assay, with a chloramphenicol acetyltransferase (CAT) reporter mRNA. We found that the entire region downstream of residue 185 was required for activity [19]. This portion contains, at residues 196–199, a four-residue motif (HNPY) that is required for interaction with the post-transcriptional regulator MKT1, and beyond that an additional region that interacts with the PABP-interacting protein PBP1 [19]. Consequently, any lambda-N fusion that commenced beyond residue 185 should not be selected, and fusions that commenced earlier, but did not include the C-terminus, should also be excluded. Figure 3B, which shows the individual reads, illustrates this: without selection reads were scattered on forward and reverse strands, but after selection, only fusions that commenced upstream of and including nt 477 (residue 159) showed enrichment. However, detailed analysis showed that a fusion that should have been selected, commencing at residue 167, was not enriched. This particular fusion protein may have non-functional folding pattern, or it may be truncated at the C-terminus. Interestingly, overall, many fusions that were selected by blasticidin were not detected in the population prior to selection.
To further check the results we examined additional genes whose activities had previously been studied in the tethering assay. Both PABP1 and PABP2 yielded multiple clones that increased blasticidin resistance. Averaging the entire open reading frames, PABP1 gave 80-fold more reads after 6-fold or 10-fold blasticidin selection than were present without blasticidin, and PABP2 gave 25-fold read-count enrichment. Pab1p-binding protein, PBP1 - which is also active in the tethering assay [19] - gave over 150-fold enrichment. In each case, it is possible to deduce approximately, from the positions of selected fusions, the portion of the protein that is required for activity in the tethering assay.
Activators include many proteins with RNA-binding domains
We now analyzed the entire dataset from the blasticidin selection experiment (Supplementary Table S1). To do this, we used a list of 6933 non-redundant protein-coding sequences predicted in the assembled T. brucei genome ([32] with modifications) (Supplementary Table S2). We obtained at least one in-frame read for more than 6700 (>96%) of these. The mapped sequence reads represented over 340 thousand independent locations, equivalent to >10 locations and >50 reads for each CDS (Supplementary Table S2). For data analysis we included reads that started from position −36 relative to the ATG, to maximize retention of N-terminal sequences. We also excluded all reads that would include less than 6 amino acids of the protein C-terminus.
To obtain an overview of the data, and a list of candidate activators, we selected proteins from which at least two different fragments showed read-count enrichment of at least 3-fold, and there was also at least 3-fold read-count enrichment when the reads for the whole ORF were considered (see Methods for details). We will henceforth discuss these proteins as activators, although strictly speaking we have only shown activation activity for at least two fragments. If only one or two clones from an ORF contain the entire region required for activity, averaging over the entire ORF will eliminate real effects. Our selection was therefore biased against long proteins, and against any proteins for which regions near the N-terminus are required for activity. A few proteins that were not covered at all in experiment BLA-A were also excluded. For these reasons it is essential to refer to Supplementary Table S1 in individual cases.
Applying the stringent criteria listed above, there were 197 putative up-regulating proteins. We classified them functionally using TritrypDB annotations supplemented by other published information (Supplementary Table S3). Although a huge variety of proteins was classified among the activators (Figure 4A), known activators were included and both RBPs and translation factors were strongly enriched (Figure 4A, Table 1). We also found unexpected enrichment of cytoskeleton-associated proteins. Although a role for the cytoskeleton as translation regulator is possible [33], more probably the enrichment is an artifact of the alignments because the proteins contain sequence repeats (discussed in more detail later).


Proteins containing Pfam domains related to RNA binding (for example, RRM and zinc finger variants) were the most significantly increased group (Figure 4A). This is very encouraging since these proteins might be expected to have activity even when not tethered via lambda-N (Table 1). The 25 active proteins include 15 zinc finger proteins and 10 RRM domain proteins. Several of the RBPs that increased expression in our tethering screen have been shown to increase the abundance or translation of their target mRNAs, but have not previously been analyzed by tethering (Supplementary Figure S3). These include ZC3H20 [11], PTB1/DRBD3 [34], [35], and PTB2/DRBD4 [35]. RBP42, which was also found in the enhancing group, is associated with coding and non-coding regions of abundant mRNAs [36], which would be consistent with a stabilizing function.
Translation initiation factors tethered to the 3′-UTR can increase gene expression
Proteins involved in translation - especially initiation factors - were over-represented in the activating population. The trypanosome genome encodes multiple homologues for the eIF4A (two), eIF4E (four) and eIF4G (five) subunits. Of these, only eIF4E3 and eIF4E4, which are in complex with eIF4G4 and eIF4G3 respectively, are thought to be active in T. brucei translation initiation [37]. Supporting this, we found different fragments of the initiation factors eIF4E3 and 4 to increase reporter expression; but the full set of eIF4Gs appeared to be active (Supplementary Figure S4). Other high-scoring proteins related to translation are listed in Table 1.
Proteins that decrease gene expression: XRNA and the NOT complex
We now examined the fusion protein fragments that were positively selected in the presence of PGKB-B-ACT (for details, see Methods). The strongest suppressors of expression were also negatively selected in the BLA experiments after addition of 5–10 µg/ml (1–2x) blasticidin (Supplementary Tables S4 and S5). We found 127 proteins that gave reproducible repression of gene expression (Supplementary Table S3, and S4 sheets 3 & 4; for definition of “reproducible” see methods). As for the activators, the discussion below refers to whole proteins although we have actually shown activity only for at least two fragments. Reassuringly, three components of the CAF1/NOT deadenylation complex NOT2, NOT11 (C2ORF29) and NOT5 [38] reproducibly suppressed PGKB expression, suggesting these subunits could recruit active NOT/CAF1 complex to associated mRNA. In contrast, neither CAF1 nor UBP1, both of which are active as full-length proteins (this paper, [28], [29]) were selected, and neither were the two CAF/NOT complex proteins CNOT10 or CAF40, presumably because larger fragments or the intact protein are required. The ORF that encodes the 5′-3′ exoribonuclease XRNA [39] is over 4 kb long and three tethered fragments that gave really clear suppression all contained the C-terminal quarter of the protein (Supplementary Figure S5). As shown for both human and Drosophila melanogaster XRN1 [40], we speculate that the C-terminus may recruit other proteins that initiate mRNA degradation.
Repression by RNA-binding proteins and 4E-IP
Multiple potential RBPs were able to suppress PGKB expression. These included RBP10, which was previously shown to inhibits translation if tethered to a reporter [18] and DRBD12 (Tb927.7.5380), which was shown to destabilize ARE-containing targets [41] (Supplementary Figure S5). In total, we found 16 RBPs conferring a clear negative effect on reporter expression, from which 7 were zinc finger proteins, 8 had a RRM motif and one, PUF3, a pumilio domain (Table 2). In mammalian cells, cytosolic pumilio domain proteins are mostly known as translational repressors [42].

One of the most dramatic and reproducible suppressors we found was the eIF4E-interacting protein. The Leishmania homologue of this protein was recently shown to interact with eIF4E1 in a stage-specific fashion [43], and it was suggested that this keeps eIF4E1 inaccessible for translation. Finally, several proteases, peptidases and components of the ubiquitination machinery were found to suppress expression when tethered. We speculate that in these cases the nascent polypeptide was subject to proteolysis.
Identification and validation of novel post-transcriptional regulators
For many of the “hits” in our experiments, there was no previous indication for involvement in post-transcriptional regulation. 68 proteins that decreased expression, and 88 that increased it, have no previously annotated function at all (Figure 4B). There are also some metabolic enzymes in the list. It is tempting to speculate that these have regulatory function in addition to catalytic activity, as suggested for animal cells [44], [45]. Mass spectrometry studies of proteins poly(A)+ RNA in human cells catalogued more than 40 metabolic enzymes [4], [5].
Because of the possibility of false-positives (discussed below), it was important to validate some of our novel results using full-length clones. To do this, trypanosomes that constitutively expressed an mRNA encoding the CAT reporter with 5 boxB elements (CAT-B-ACT) were transfected with inducible lambda-N-myc fusion proteins. Most observed CAT activities were in qualitative agreement with the screening results (Figure 5). Quantitative agreement was not expected, since the measurement methods were so different. Cytidine deaminase was revealed as a false-positive, while ZC3H45 was a false-negative since tethering of full-length ZC3H45 decreased CAT expression. Remarkably, the full-length cap-binding protein eIF4E1 vastly decreased CAT expression, although it was negative in the PGKB experiment and a fragment had increased expression in the BLA screen (Supplementary Figure S4).
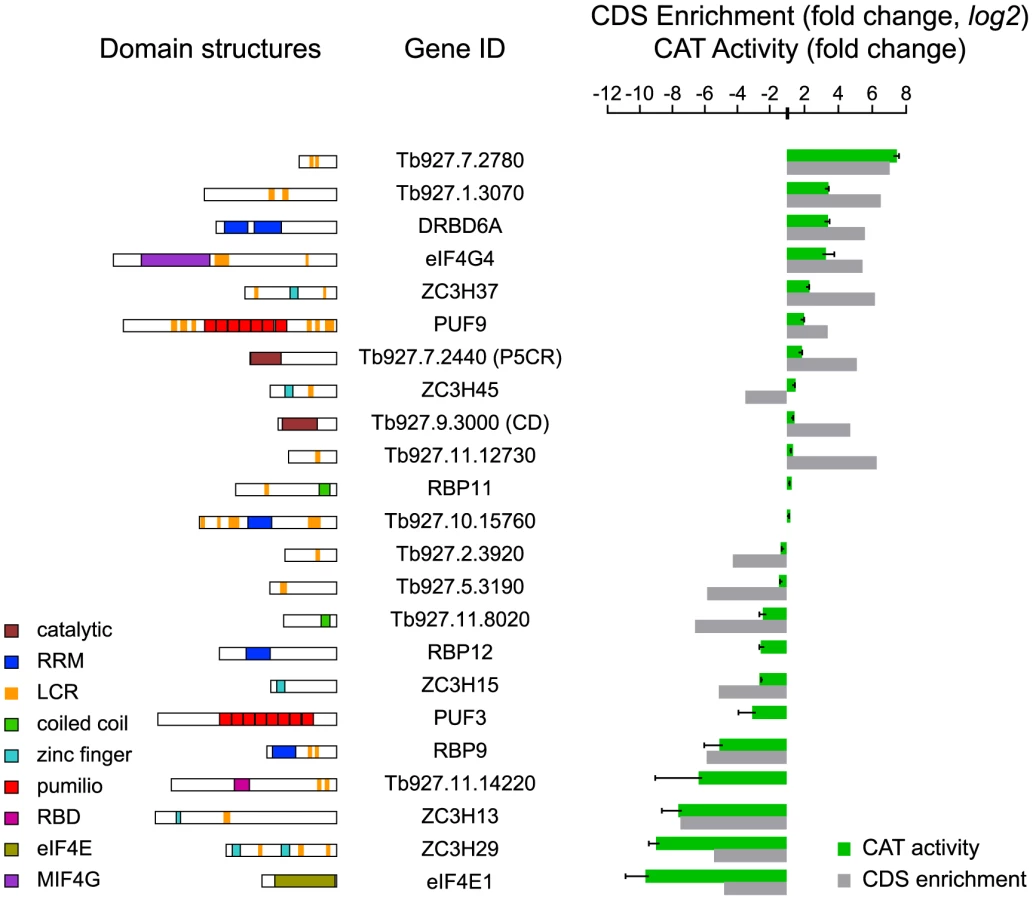
Concentrating on the novel proteins, the Tb927.7.2780 fusion increased reporter CAT activity around 7-fold, as observed for PABP1 [28]. This protein has short poly-glutamine and -histidine tracts (Figure 5A) and was previously found to be essential in a high-throughput RNAi screen [31]. It also co-purifies with two proteins previously shown to increase mRNA stability, ZC3H11 [10] and MKT1 [19]. Tethering of the full-length pyrroline-5-carboxylate reductase (P5CR, not in the list) caused a modest but reproducible 1.6-fold CAT activity increase (Figure 5). The hypothetical proteins Tb927.11.14220 and Tb927.11.8020 decreased reporter expression; interestingly, SCOP analysis detected an RNA-binding domain in Tb927.11.14220.
Some of the novel regulatory proteins can bind RNA
Our screen had identified many proteins that affected mRNA translation or degradation when tethered artificially to an mRNA. In vivo, such proteins might be bound to mRNA directly, or via another protein. Alternatively, they might never be associated with mRNA, in which case the effect we had observed would be an artefact. To assess the RNA-binding properties of potential regulators, we constructed a custom protein array. We chose 384 proteins that are of interest in post-transcriptional regulation, including some translation factors, degradative enzymes, all proteins with identified RNA-binding domains and a subset of proteins that had been identified in the screen but had no known RNA-binding characteristics. For reasons of economy, we also excluded proteins of the mitochondrial inner membrane and matrix, cytoskeleton, vesicular transport pathway, surface proteins, nucleoporins, nuclear proteins, and a few proteins for which the ORFs were too large to be amplified by PCR. Gateway-compatible primers were designed (Supplementary Table S6), the ORFs were amplified, and the lengths of products confirmed by gel electrophoresis. The products were then re-amplified to create DNA templates for protein expression in a prokaryotic in vitro transcription-translation system. The resulting templates were spotted onto glass slides in triplicate, and then a transcription-translation mix was added to create protein arrays (Supplementary Figure S6). In order to minimise disruption of the protein structure our oligonucleotides included the native stop codon. This had the disadvantage that it was not possible to verify production of full-length protein by addition of a C-terminal tag. The templates did, however, encode an N-terminal His tag, production of which was verified (not shown).
The protein arrays were used for studies of RNA interactions by probing them with labelled total or poly(A)+ RNA (in each case two procyclic-form samples and one bloodstream-form sample). The results are summarized in Supplementary Table S7. Negative results cannot be interpreted since we did not test whether the full-length proteins were made and there is no way to know whether RNA-binding domains, if they exist, were correctly folded. A negative result would also be obtained if a protein has very sequence specificity and its target mRNAs have low abundance. For example, the PABPs and pumilio domain proteins were negative. As a positive control, we probed ZC3H11 with its cognate recognition sequence of (UAU) repeats; it showed a clear signal whereas a C→S mutant in the zinc finger failed to bind (not shown).
157 protein spots bound to total RNA in all three replicates, whereas 47 bound to poly(A)+ RNA (of which all but two were also positive on total RNA); 148 spots bound in at least 4 of the 6 experiments. The small RNA binding protein RBP3 gave the strongest signal (Supplementary Table S7); 43 other RNA-binding domain proteins were also positive in at least 5 slides. Proteins that showed some RNA binding included some with confirmed tethering activity (Figure 5) but no known RNA-binding domain: Tb927.10.15760 (5 slides), Tb927.1.3070 (5 slides), Tb927.7.2780 (total RNA only) and pyrroline-5-carboxylate reductase (3 slides). Other rather surprising positives were the two peroxins, PEX13 (3 slides) and PEX14 (5 slides), which are in the glycosomal membrane, and a variety of enzymes. From these results we concluded that many of the proteins that we had identified in our tethering screen - including those with no annotated RNA-binding domains - may indeed be capable of binding RNA. The remainder may influence mRNA or translation via interactions with other RNA-associated proteins.
Discussion
In this study we have shown that genes regulating mRNA turnover and translation can be identified using a functional genomics approach. The screen identified mRNA regulation by canonical RBPs and by proteins that act via multi-protein complexes, and also revealed nearly 100 proteins that can bind RNA but had no previously known RNA-related function. The random shotgun approach has the additional power of allowing delineation of functional domains.
The tethering approach is very powerful but, as in all high-throughput screens, false-positives and -negatives can occur:
-
The assay relies on the tethering of protein fragments to a reporter RNA. Most of the fragments come from proteins that have no way to interact with RNA in vivo, either directly or indirectly. The protein might be in the cytosol, but have no RNA-binding domains and no interactions with any other RNA-binding protein. Alternatively, it may normally be in another compartment. A positive score could arise from a chance interaction of this protein with some other component of the translation or degradation machineries. For example, mitochondrial proteins were found in both positive and negative categories. Proteins with nuclear targeting signals might retain the mRNA in the nucleus, inhibiting translation.
-
The use of fragments can result in abnormal or defective protein folding. This may expose interacting domains that are normally concealed within the full-length, properly folded protein, giving a false-positive result. Alternatively, it may prevent normal function of the protein, giving a false-negative result. The fragments will also give a false-negative result if the entire protein is required for activity. A false-negative result is particularly likely if (i) an open reading frame of more than about 1.5 kb is required; (ii) the domain that is required for activation is very near the N-terminus; or (iii) the protein is very short, so few in-frame fusions are present. For example, full-length MKT1 strongly increases expression in the CAT tethering assay, but both the N - and C-terminal portions are required [19] and the open reading frame of over 3 kb is too long to be included in our library. Consequently, only a single clone showed any selection, in just one experiment; considering the whole open reading frame, no selection for MKT1 was apparent at all. The above disadvantages could be abrogated by using a library of full-length open reading frames: a resource that is already available for budding yeast [46] and humans [47]. N-terminal tagging may however abolish protein activity, for example if a native N-terminus is required for interactions.
-
Scoring of the screen relies on PCR. Genes encoding repetitive proteins could give artifactually high scores since reads map more than once. Some cytoskeletal proteins that scored in our assays might fall into this category. Easily amplified sequences may also be artifactually enriched.
-
A false-negative result will be obtained if the protein acts as part of a complex, but the other components are present in limiting amounts.
Despite these possible problems, the screen was overall highly informative. We envisage that this technique could be adapted to screen libraries for proteins that play a role in post-transcriptional control in other organisms where transfection methods with high efficiency are available. For T. brucei, this forward genetic approach could be easily used to identification of genes and pathways regulating other complex biological phenomena such as quorum sensing, antigenic variation, or drug target identification. Our set of 384 ORFs already provides a Gateway-compatible resource for proteins potentially involved in mRNA metabolism.
The RNA binding of yeast [48] and mammalian [49] proteins has previously been examined using microarrays, but it is not clear which of the identified RNA binders was also found in the datasets obtained by poly(A)+ mRNA precipitation. Protein microarrays have the advantage (unlike poly(A)+ mRNA precipitation) of being unaffected by in vivo protein abundance; however, false negatives may be caused by incorrect protein folding and there is the danger of un-physiological interactions such as electrostatic binding of basic proteins to nucleic acids. Nevertheless, the results do provide preliminary indications which could be followed up by in vivo RNA cross-linking studies. In future, probing with more specific sequences may allow us to determine the RNA-binding specificities of the candidate proteins.
The results described in this paper will be most useful in the context of other available datasets. A genome-wide RNAi screen provided a catalogue of T. brucei genes whose knock-down is detrimental to the parasite under a variety of developmental conditions [31]. This can be now be used, for example, to look for proteins that are essential in only one life-cycle stage, and are implicated in the post-transcriptional control of gene expression (These results are included in Supplementary Table S7). For bloodstream forms, these would include ZC3H5, ZC3H15, and numerous proteins of no known function. An RNAi screen for proteins associated with the AMP/cAMP response identified a number of proteins whose association with either signalling or control of gene expression were not apparent [50]. Of these, Tb927.11.2250 and Tb927.9.4080 are both up-regulators and Tb927.11.2250 binds RNA. Adenylosuccinate synthetase was suggested to affect differentiation via AMP metabolism: we also identified it as a weak expression down-regulator with RNA-binding properties.
The catalogues of mRNA-associated proteins that are already available for mammals [4], [5], [7] and yeast [6] include numerous metabolic enzymes. Application of our methods to Opisthokont systems would clearly facilitate interpretation of these mRNA-associated protein datasets. We have now documented potential regulatory functions for trypanosome cystathione gamma lyase (CTH), deoxyribose-phosphate aldolase, pyrroline-5-carboxylate reductase (P5CR), dihydrofolate reductase-thymidylate synthase (DHFR-TS), adenylosuccinate synthetase and tryparedoxin (TXN); all but the first two also bound RNA in the protein microarray assay. In Hela cells, two other oxidative stress-related enzymes - thioredoxin and peroxiredoxin - have been shown to bind RNA [4]. In animal cells, thymidylate synthase (TS) and dihydrofolate reductase (DHFR) bind to their own mRNAs, causing translational repression [51]. Here we showed that in trypanosomes, the bifunctional trypanosome DHFR-TS protein is also able to bind RNA and, as in animal cells, the functional consequence of its association with mRNA is inhibition of expression.
Results from yeast-two-hybrid interaction screens and affinity purifications will also assist with mechanism prediction. Proteins that showed activity in the screen but cannot themselves bind to RNA, may nevertheless be associated with RNA via interactions with RNA-binding proteins. For example, although the Tb927.9.4080 protein may not bind to RNA, it is known to interact with both DRBD3 [34] and MKT1 [19]. Down regulators might be expected to interact with components of the degradation machinery, or to have inactivating interactions with the translation apparatus. Proteins that enhance gene expression might be expected to be found stably associated with mRNA, and those that enhance translation ought to be at least partially associated with polysomes, so additional datasets of this sort will greatly facilitate interpretation of our data.
Materials and Methods
Plasmid construction and transgenic trypanosomes
The overexpression library was constructed in a derivative plasmid of the tetracycline-regulated pHD678 [52]. It contains a lambda-N peptide sequence cloned as a HindIII-ApaI fragment, and the NEO (neomycin/G418 resistance) gene replaced a HYG (hygromycin resistance). Specific oligonucleotides were ligated into the ApaI and BamHI sites to add a stop codon in all three possible reading frames downstream of the unique XhoI cloning site. Reporter blasticidin plasmids are derivative of the pHD330 [53]. They contain a GFP gene positioned upstream of a blasticidin (BLA) resistance marker and were designed for targeting into the tubulin locus. For the screening, 5 copies of the boxB sequence element were inserted immediately between the reporter sequences and the actin 3′-UTR. A similar construct lacking the boxB element between the BLA and the 3′-UTR was used as control. For the PGKB experiment, the full-length PGKB CDS was cloned into the pHD2300. It confers BLA resistance and was designed for targeting into the RRNA locus. As described, 5 copies of the boxB element were embedded between the PGKB sequence and the actin 3′-UTR. Details of all plasmids and oligonucleotides are provided in Supplementary Table S6, sheet 2. Complete sequences are available from us upon request.
Construction of the DNA expression library
The overexpression library was essentially made as previously described [19]. The plasmid was linearized at the unique XhoI site, filled in with the large fragment of DNA polymerase I in the presence of dTTP, and ligated to the semi-Xho-adapted DNA (size range from 0.7–3 Kbp). The ligation reaction was used to transform Escherichia coli NEB 5-alpha cells by electroporation to generate a library of approximately 3×106 ampicillin-resistant colonies. To assess the quality of the library, E. coli cells were re-transformed with purified library, plated and plasmids purified from individual colonies. After XhoI digestion, inserts were found in 99% of plasmids and the average size was 1.2 Kbp.
T. brucei growth and manipulation
Bloodstream-form T. brucei 2T1 cells were maintained and transfected as described [31] except that Tb-BSF buffer [54] was used for all transfections. The pRPaSce* plasmid was used to derive Sce* cells from 2T1 cells as described [31]. The Sce* strain expresses the tetracycline repressor (TetR), and inducible homing endonuclease (I-SceI) which facilitates site-specific overexpression library integration. Upon transfection, overexpression plasmid library constructs replace the I-SceI gene and cleavage site. Parasite libraries were generated by several rounds of electroporation and for each series, an aliquot of the transfection was diluted to determine the transfection efficiency. The average efficiency was about 5×10−3. To assay for stabilizing proteins, populations expressing the blasticidin resistance mRNA were pre-induced for 24 h in 1 µg/mL tetracycline and then grown with various concentrations of blasticidin (1x = 5 µg/mL) for four days.
DNA sequencing and analysis
The PCR-amplified DNA was fragmented and sequenced using Illumina HiSeq with multiplexing and data analyzed as described previously [10], [55]. We sequenced input trypanosome population twice since the BLA-selected trypanosome libraries were expected to be less complex. SAMtools [56] and custom-made PERL scripts were used to select only coding region or 5′-UTR sequences that were in frame with the lambda-N peptide. The lambda-N sequence was removed, then the remainder was mapped to the T. brucei 927 reference genome (http://tritrypdb.org/tritrypdb) using Bowtie, allowing one base mismatch.
Data analysis
To find proteins that increased blasticidin resistance we chose locations from position −36 relative to the ATG to position −18 relative to the stop codon (Supplementary Table S1). The counts ranged from 166000 to over a million (Supplementary Table S1). Then only positions for a unique gene set ([32], modified) were chosen. For these we counted, for each experiment, the reads per million (RPM). To avoid zero values, 1 was added to each value to give RPM+1 (Supplementary Table S2, sheet 2). After that, for each location, the number of counts for 6-fold increased blasticidin (BLA 6X) in experiment A was divided by the counts for −tet, and separately by the counts for cells with tetracycline but without blasticidin. To reduce the likelihood of identifying PCR artifacts, the lower of these two values was taken to be the relative enrichment for this site. Similar calculations were done for all locations for BLA10X in experiments B and C (Supplementary Table S2, sheet 3). Experiment C was the -boxB control, which should select only for fusions that enhance growth independent of the blasticidin resistance mRNA, and also for PCR artifacts. We therefore removed all locations that gave at least 3x increase in BLA10x in Experiment C. From the remaining list we selected genes for which at least two locations gave a 3x increase in either BLA6x (A) or BLA10x (B) (Supplementary Table S2, sheet 3). The number of such locations per ORF was counted (Supplementary Table S2, sheet 4).
Separately, and using a similar procedure, numbers of total counts for the whole coding regions were extracted (Supplementary Table S2, Sheet 5). For the unique genes, we computed the RPM in each experiment (Supplementary Table S2, Sheet 2). The RPM+1 with 6x or 10x blasticidin, relative to amounts without blasticidin were calculated as before (Supplementary Table S2, Sheet 3). We now deleted any CDS giving at least three-fold enrichment in counts for BLA10x in negative control experiment C. We retained CDSs that gave an overall count enrichment of at least 3-fold in both experiment A (BLA6x) and experiment B (BLA10x). The resulting 197 genes are listed in Supplementary Table S2, sheet S4 and (for readers who want a smaller and simpler file) in Supplementary Table S3, Sheet 1. Table 1 shows only the RBPs and translation factors that reproducibly increased BLA expression.
To find proteins that increased survival after PGKB expression, we took the counts for each location (Supplementary Table S4, sheet 1), computed the RPM and added 1 to every value to avoid zero values (Supplementary Table S4, sheet 2). We then divided the number of counts for +tet by the counts for −tet (Supplementary Table S4, sheet 3). The highest ratio for each location was found. Now, for each unique gene, we counted the number of locations that gave at least 3-fold enrichment, + tetracycline, in at least one experiment (Supplementary Table S4, sheet 3, column D). Separately, numbers of total counts for the whole coding regions were extracted (Supplementary Table S5, Sheet 1). For the unique genes, we computed the RPM in each experiment, added 1 as before, and ratios +tet to −tet were calculated for each experiment (Supplementary Table S5, Sheet 2). These were transferred to Supplementary Table S4, sheet 3 columns E–F. In addition, we looked for the effects of blasticidin selection. Counts per CDS with BLA1x (experiment A) or BLA2x (experiment B) were divided by the values with no blasticidin and the higher value taken. These values were also transferred to Supplementary Table S4, sheet 3, columns H, I and J. We now chose genes that were candidates as down-regulators. First, we selected genes for which at least two locations gave 3-fold enrichment in at least one of the PGK experiments (Supplementary Table S4, sheet 4, column D). From these, we selected genes that gave a 2-fold RPM increase over the whole coding sequence in at least two of the three PGKB experiments. This list of 127 genes is in Supplementary Table S4, sheet 4. The best regulators are those which gave selective advantage in the PGK experiment, and were selected against in the BLA experiment. Supplementary Table S3, sheet 2 has the same information, without the calculations and the raw data.
Protein microarrays
Gene-specific primers were designed and synthesized in 96 well plates (Supplementary Table S6). The corresponding ORFs were then amplified directly from genomic DNA using Q5 high fidelity DNA polymerase (New England Biolabs), with 2 min at 98°C, 35 cycles (98°C for 15 sec, 52°C for 15 sec and 72°C for 2 min) then 10 min at 72°C. Each primer included additional sequence suitable for amplification and directional Gateway cloning. In a second PCR amplification, these extra sequences on the amplification products were used in order to reamplify the genes while adding a T7 promoter and Shine-Dalgarno sequence upstream of the ATG (Supplementary Table S6), with 0.02 µL of the first PCR as template (98°C for 2 min, two cycles of 98°C for 15 sec, 45°C for 15 sec and 72°C for 2 min; then 35 cycles as with the initial PCR). These templates were then used for the production of protein microarrays as previously described [57]. Briefly, 900 pL of DNA template was arrayed in triplicates on Nexterion Epoxy slide E using a GeSIM non-contact nanoplotter. In a subsequent spotting round, 3.6 nL of the S30T7 high yield protein expression system was spotted onto of each template spot. The entire assembly was placed in a deep humidified chamber containing 50 µL of nuclease-free water in each of its wells, and then incubated at 37°C for one hour and a 30°C overnight. Slides were removed from the chambers, cleaned and stored at −20°C until use. Poly(A)+ RNA was isolated from total RNA using Nucleotrap mRNA purification kit as recommended by the manufacturer. Total and poly(A)+ RNA were biotin-labeled using Pierce RNA 3′ End Biotinylation Kit following the manufacturer's instructions.
On-chip protein-RNA interactions
Protein arrays were removed from the freezer and placed directly into a blocking solution containing Hepes-KOH pH 7.9, 10% glycerol, 40 U of RNaseout RNase inhibitor, 1% BSA, 100 µM ZnCl2, 1x Halt Protease and phosphatase cocktail inhibitor, 40 µg/mL heparin, 1 mM DTT 50 µg/mL E. coli tRNA, 50 mM glutamic acid potassium salt, 0.1% Triton X-100 and 8 mM Magnesium acetate. The slides were blocked for 1 hour at room temperature, and then washed in washing buffer (Blocking buffer without BSA). Slides were then incubated with labeled RNA in blocking buffer, containing 0.5 mg/mL E. coli tRNA overnight at 4°C. The slides were removed from the incubation chambers and washed 3x twenty minutes in washing buffer, and 3x five minutes in Nuclease-free water. For detection of total and poly(A)+ RNA, the arrays were probed with cy3-labelled extravidin at a dilution of 1∶100. The slides were air-dried in a ventilated oven at 30°C for one hour and scanned in a Tecan power scanner with 75% laser power and 500% PMT gain. Images were saved as TIFF files and later loaded into Genepix 6.0 for data extraction. The mean background signal for each spot was subtracted from the mean spot intensity, and the average intensity for all triplicates of each sample calculated. As controls, a mutant zinc finger protein (ZC3H11 C→S) which has been shown to have no RNA binding activity [10], as well as the PCR negative control and expression mix alone were used.
Supporting Information
Zdroje
1. PreusserC, JaeN, BindereifA (2012) mRNA splicing in trypanosomes. Int J Med Microbiol 302 : 221–224.
2. ClaytonCE (2002) Life without transcriptional control? From fly to man and back again. EMBO J 21 : 1881–1888.
3. Muller-McNicollM, NeugebauerKM (2013) How cells get the message: dynamic assembly and function of mRNA-protein complexes. Nat Rev Genet 14 : 275–287.
4. CastelloA, FischerB, EichelbaumK, HorosR, BeckmannBM, et al. (2012) Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell 149 : 1393–1406.
5. BaltzAG, MunschauerM, SchwanhausserB, VasileA, MurakawaY, et al. (2012) The mRNA-bound proteome and its global occupancy profile on protein-coding transcripts. Mol Cell 46 : 674–690.
6. MitchellSF, JainS, SheM, ParkerR (2013) Global analysis of yeast mRNPs. Nat Struct Mol Biol 20 : 127–133.
7. KwonSC, YiH, EichelbaumK, FohrS, FischerB, et al. (2013) The RNA-binding protein repertoire of embryonic stem cells. Nat Struct Mol Biol 20 : 1122–1130.
8. KolevNG, UlluE, TschudiC (2014) The emerging role of RNA-binding proteins in the life cycle of Trypanosoma brucei. Cell Microbiol 16 : 482–489.
9. WalradPB, CapewellP, FennK, MatthewsKR (2012) The post-transcriptional trans-acting regulator, TbZFP3, co-ordinates transmission-stage enriched mRNAs in Trypanosoma brucei. Nucleic Acids Res 40 : 2869–2883.
10. DrollD, MiniaI, FaddaA, SinghA, StewartM, et al. (2013) Post-transcriptional regulation of the trypanosome heat shock response by a zinc finger protein. PLoS Pathog 9: e1003286.
11. LingAS, TrotterJR, HendriksEF (2011) A zinc finger protein, TbZC3H20, stabilizes two developmentally regulated mRNAs in trypanosomes. J Biol Chem 286 : 20152–20162.
12. ArcherSK, LuuVD, de QueirozRA, BremsS, ClaytonC (2009) Trypanosoma brucei PUF9 regulates mRNAs for proteins involved in replicative processes over the cell cycle. PLoS Pathog 5: e1000565.
13. ManiJ, GuttingerA, SchimanskiB, HellerM, Acosta-SerranoA, et al. (2011) Alba-domain proteins of Trypanosoma brucei are cytoplasmic RNA-binding proteins that interact with the translation machinery. PLoS One 6: e22463.
14. KishoreS, LuberS, ZavolanM (2010) Deciphering the role of RNA-binding proteins in the post-transcriptional control of gene expression. Brief Funct Genomics 9 : 391–404.
15. D'OrsoI, FraschAC (2002) TcUBP-1, an mRNA destabilizing factor from trypanosomes, homodimerizes and interacts with novel AU-rich element - and Poly(A)-binding proteins forming a ribonucleoprotein complex. J Biol Chem 277 : 50520–50528.
16. DallagiovannaB, CorreaA, ProbstCM, HoletzF, SmircichP, et al. (2008) Functional genomic characterization of mRNAs associated with TcPUF6, a pumilio-like protein from Trypanosoma cruzi. J Biol Chem 283 : 8266–8273.
17. JhaBA, ArcherSK, ClaytonCE (2013) The trypanosome Pumilio domain protein PUF5. PLoS One 8: e77371.
18. WurstM, SeligerB, JhaBA, KleinC, QueirozR, et al. (2012) Expression of the RNA recognition motif protein RBP10 promotes a bloodstream-form transcript pattern in Trypanosoma brucei. Mol Microbiol 83 : 1048–1063.
19. SinghA, MiniaI, DrollD, FaddaA, ClaytonC, et al. (2014) Trypanosome MKT1 and the RNA-binding protein ZC3H11: interactions and potential roles in post-transcriptional regulatory networks. Nucleic Acids Res 42 : 4652–4668.
20. CollerJM, GrayNK, WickensMP (1998) mRNA stabilization by poly(A) binding protein is independent of poly(A) and requires translation. Genes Dev 12 : 3226–3235.
21. CollerJ, WickensM (2007) Tethered function assays: an adaptable approach to study RNA regulatory proteins. Methods Enzymol 429 : 299–321.
22. De GregorioE, PreissT, HentzeMW (1999) Translation driven by an eIF4G core domain in vivo. EMBO J 18 : 4865–4874.
23. Lykke-AndersenJ, ShuMD, SteitzJA (2001) Communication of the position of exon-exon junctions to the mRNA surveillance machinery by the protein RNPS1. Science 293 : 1836–1839.
24. BertrandE, ChartrandP, SchaeferM, ShenoySM, SingerRH, et al. (1998) Localization of ASH1 mRNA particles in living yeast. Mol Cell 2 : 437–445.
25. Baron-BenhamouJ, GehringNH, KulozikAE, HentzeMW (2004) Using the lambdaN peptide to tether proteins to RNAs. Methods Mol Biol 257 : 135–154.
26. ScharpfM, StichtH, SchweimerK, BoehmM, HoffmannS, et al. (2000) Antitermination in bacteriophage lambda. The structure of the N36 peptide-boxB RNA complex. Eur J Biochem 267 : 2397–2408.
27. GloverL, HornD (2009) Site-specific DNA double-strand breaks greatly increase stable transformation efficiency in Trypanosoma brucei. Mol Biochem Parasitol 166 : 194–197.
28. DelhiP, QueirozR, InchausteguiD, CarringtonM, ClaytonC (2011) Is there a classical nonsense-mediated decay pathway in trypanosomes? PLoS One 6: e25112.
29. FarberV, ErbenE, SharmaS, StoecklinG, ClaytonC (2013) Trypanosome CNOT10 is essential for the integrity of the NOT deadenylase complex and for degradation of many mRNAs. Nucleic Acids Res 41 : 1211–1222.
30. BlattnerJ, HelfertS, MichelsP, ClaytonC (1998) Compartmentation of phosphoglycerate kinase in Trypanosoma brucei plays a critical role in parasite energy metabolism. Proc Natl Acad Sci USA 95 : 11596–11600.
31. AlsfordS, TurnerDJ, ObadoSO, Sanchez-FloresA, GloverL, et al. (2011) High-throughput phenotyping using parallel sequencing of RNA interference targets in the African trypanosome. Genome Res 21 : 915–924.
32. SiegelTN, HekstraDR, WangX, DewellS, CrossGA (2010) Genome-wide analysis of mRNA abundance in two life-cycle stages of Trypanosoma brucei and identification of splicing and polyadenylation sites. Nucleic Acids Res 38 : 4946–4957.
33. KimS, CoulombePA (2010) Emerging role for the cytoskeleton as an organizer and regulator of translation. Nat Rev Mol Cell Biol 11 : 75–81.
34. EstevezAM (2008) The RNA-binding protein TbDRBD3 regulates the stability of a specific subset of mRNAs in trypanosomes. Nucl Acids Res 36 : 4573–4586.
35. SternMZ, GuptaSK, Salmon-DivonM, HahamT, BardaO, et al. (2009) Multiple roles for polypyrimidine tract binding (PTB) proteins in trypanosome RNA metabolism. RNA 15 : 648–665.
36. DasA, MoralesR, BandayM, GarciaS, HaoL, et al. (2012) The essential polysome-associated RNA-binding protein RBP42 targets mRNAs involved in Trypanosoma brucei energy metabolism. RNA 18 : 1968–1983.
37. FreireER, DhaliaR, MouraDM, da Costa LimaTD, LimaRP, et al. (2011) The four trypanosomatid eIF4E homologues fall into two separate groups, with distinct features in primary sequence and biological properties. Mol Biochem Parasitol 176 : 25–36.
38. ErbenE, ChakrabortyC, ClaytonC (2014) The CAF1-NOT complex of trypanosomes. Front Genet 4 : 299.
39. LiCH, IrmerH, Gudjonsdottir-PlanckD, FreeseS, SalmH, et al. (2006) Roles of a Trypanosoma brucei 5′-3′ exoribonuclease homolog in mRNA degradation. RNA 12 : 2171–2186.
40. BraunJE, TruffaultV, BolandA, HuntzingerE, ChangCT, et al. (2012) A direct interaction between DCP1 and XRN1 couples mRNA decapping to 5′ exonucleolytic degradation. Nat Struct Mol Biol 19 : 1324–1331.
41. NajafabadiHS, LuZ, MacPhersonC, MehtaV, AdoueV, et al. (2013) Global identification of conserved post-transcriptional regulatory programs in trypanosomatids. Nucleic Acids Res 41 : 8591–8600.
42. QuenaultT, LithgowT, TravenA (2011) PUF proteins: repression, activation and mRNA localization. Trends Cell Biol 21 : 104–112.
43. ZinovievA, LegerM, WagnerG, ShapiraM (2011) A novel 4E-interacting protein in Leishmania is involved in stage-specific translation pathways. Nucleic Acids Res 39 : 8404–8415.
44. HentzeMW, PreissT (2010) The REM phase of gene regulation. Trends Biochem Sci 35 : 423–426.
45. CieslaJ (2006) Metabolic enzymes that bind RNA: yet another level of cellular regulatory network? Acta Biochim Pol 53 : 11–32.
46. HuY, RolfsA, BhullarB, MurthyTV, ZhuC, et al. (2007) Approaching a complete repository of sequence-verified protein-encoding clones for Saccharomyces cerevisiae. Genome Res 17 : 536–543.
47. YangX, BoehmJS, Salehi-AshtianiK, HaoT, ShenY, et al. (2011) A public genome-scale lentiviral expression library of human ORFs. Nat Methods 8 : 659–661.
48. TsvetanovaNG, KlassDM, SalzmanJ, BrownPO (2010) Proteome-wide search reveals unexpected RNA-binding proteins in Saccharomyces cerevisiae. PLoS One 5: e12671.
49. SiprashviliZ, WebsterD, KretzM, JohnstonD, RinnJ, et al. (2012) Identification of proteins binding coding and non-coding human RNAs using protein microarrays. BMC Genomics 13 : 633.
50. MonyBM, MacGregorP, IvensA, RojasF, CowtonA, et al. (2014) Genome-wide dissection of the quorum sensing signalling pathway in Trypanosoma brucei. Nature 505 : 681–685.
51. TaiN, SchmitzJC, LiuJ, LinX, BaillyM, et al. (2004) Translational autoregulation of thymidylate synthase and dihydrofolate reductase. Front Biosci 9 : 2521–2526.
52. BiebingerS, WirtzLE, LorenzP, ClaytonC (1997) Vectors for inducible expression of toxic gene products in bloodstream and procyclic Trypanosoma brucei. Mol Biochem Parasitol 85 : 99–112.
53. WirtzE, HartmannC, ClaytonC (1994) Gene expression mediated by bacteriophage T3 and T7 RNA polymerases in transgenic trypanosomes. Nucleic Acids Res 22 : 3887–3894.
54. Schumann BurkardG, JutziP, RoditiI (2011) Genome-wide RNAi screens in bloodstream form trypanosomes identify drug transporters. Mol Biochem Parasitol 175 : 91–94.
55. FaddaA, FarberV, DrollD, ClaytonC (2013) The roles of 3′-exoribonucleases and the exosome in trypanosome mRNA degradation. RNA 19 : 937–947.
56. LiH, HandsakerB, WysokerA, FennellT, RuanJ, et al. (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics 25 : 2078–2079.
57. AngenendtP, KreutzbergerJ, GloklerJ, HoheiselJD (2006) Generation of high density protein microarrays by cell-free in situ expression of unpurified PCR products. Mol Cell Proteomics 5 : 1658–1666.
58. HendriksEF, RobinsonDR, HinkinsM, MatthewsKR (2001) A novel CCCH protein which modulates differentiation of Trypanosoma brucei to its procyclic form. EMBO J 20 : 6700–6711.
59. PaterouA, WalradP, CraddyP, FennK, MatthewsK (2006) Identification and stage-specific association with the translational apparatus of TbZFP3, a CCCH protein that promotes trypanosome life-cycle development. J Biol Chem 281 : 39002–39013.
60. MittraB, RayDS (2004) Presence of a poly(A) binding protein and two proteins with cell cycle-dependent phosphorylation in Crithidia fasciculata mRNA cycling sequence binding protein II. Eukaryot Cell 3 : 1185–1197.
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2014 Číslo 6
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Fungal Nail Infections (Onychomycosis): A Never-Ending Story?
- Profilin Promotes Recruitment of Ly6C CCR2 Inflammatory Monocytes That Can Confer Resistance to Bacterial Infection
- IscR Is Essential for Type III Secretion and Virulence
- Contribution of Specific Residues of the β-Solenoid Fold to HET-s Prion Function, Amyloid Structure and Stability
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy