
CTL Escape Mediated by Proteasomal Destruction of an HIV-1 Cryptic
Epitope
Cytotoxic CD8+ T cells (CTLs) play a critical role in controlling viral
infections. HIV-infected individuals develop CTL responses against epitopes
derived from viral proteins, but also against cryptic epitopes encoded by viral
alternative reading frames (ARF). We studied here the mechanisms of HIV-1 escape
from CTLs targeting one such cryptic epitope, Q9VF, encoded by an
HIVgag ARF and presented by HLA-B*07. Using PBMCs of
HIV-infected patients, we first cloned and sequenced proviral DNA encoding for
Q9VF. We identified several polymorphisms with a minority of proviruses encoding
at position 5 an aspartic acid (Q9VF/5D) and a majority encoding an asparagine
(Q9VF/5N). We compared the prevalence of each variant in PBMCs of
HLA-B*07+ and HLA-B*07 - patients. Proviruses encoding Q9VF/5D were
significantly less represented in HLA-B*07+ than in HLA-B*07-
patients, suggesting that Q9FV/5D encoding viruses might be under selective
pressure in HLA-B*07+ individuals. We thus analyzed ex
vivo CTL responses directed against Q9VF/5D and Q9VF/5N. Around
16% of HLA-B*07+ patients exhibited CTL responses targeting Q9VF
epitopes. The frequency and the magnitude of CTL responses induced with Q9VF/5D
or Q9VF/5N peptides were almost equal indicating a possible cross-reactivity of
the same CTLs on the two peptides. We then dissected the cellular mechanisms
involved in the presentation of Q9VF variants. As expected, cells infected with
HIV strains encoding for Q9VF/5D were recognized by Q9VF/5D-specific CTLs. In
contrast, Q9VF/5N-encoding strains were neither recognized by Q9VF/5N - nor by
Q9VF/5D-specific CTLs. Using in vitro proteasomal digestions
and MS/MS analysis, we demonstrate that the 5N variation introduces a strong
proteasomal cleavage site within the epitope, leading to a dramatic reduction of
Q9VF epitope production. Our results strongly suggest that HIV-1 escapes CTL
surveillance by introducing mutations leading to HIV ARF-epitope destruction by
proteasomes.
Published in the journal:
. PLoS Pathog 7(5): e32767. doi:10.1371/journal.ppat.1002049
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1002049
Summary
Cytotoxic CD8+ T cells (CTLs) play a critical role in controlling viral
infections. HIV-infected individuals develop CTL responses against epitopes
derived from viral proteins, but also against cryptic epitopes encoded by viral
alternative reading frames (ARF). We studied here the mechanisms of HIV-1 escape
from CTLs targeting one such cryptic epitope, Q9VF, encoded by an
HIVgag ARF and presented by HLA-B*07. Using PBMCs of
HIV-infected patients, we first cloned and sequenced proviral DNA encoding for
Q9VF. We identified several polymorphisms with a minority of proviruses encoding
at position 5 an aspartic acid (Q9VF/5D) and a majority encoding an asparagine
(Q9VF/5N). We compared the prevalence of each variant in PBMCs of
HLA-B*07+ and HLA-B*07 - patients. Proviruses encoding Q9VF/5D were
significantly less represented in HLA-B*07+ than in HLA-B*07-
patients, suggesting that Q9FV/5D encoding viruses might be under selective
pressure in HLA-B*07+ individuals. We thus analyzed ex
vivo CTL responses directed against Q9VF/5D and Q9VF/5N. Around
16% of HLA-B*07+ patients exhibited CTL responses targeting Q9VF
epitopes. The frequency and the magnitude of CTL responses induced with Q9VF/5D
or Q9VF/5N peptides were almost equal indicating a possible cross-reactivity of
the same CTLs on the two peptides. We then dissected the cellular mechanisms
involved in the presentation of Q9VF variants. As expected, cells infected with
HIV strains encoding for Q9VF/5D were recognized by Q9VF/5D-specific CTLs. In
contrast, Q9VF/5N-encoding strains were neither recognized by Q9VF/5N - nor by
Q9VF/5D-specific CTLs. Using in vitro proteasomal digestions
and MS/MS analysis, we demonstrate that the 5N variation introduces a strong
proteasomal cleavage site within the epitope, leading to a dramatic reduction of
Q9VF epitope production. Our results strongly suggest that HIV-1 escapes CTL
surveillance by introducing mutations leading to HIV ARF-epitope destruction by
proteasomes.
Introduction
Multiple lines of evidence suggest that CD8+ cytotoxic T lymphocytes (CTLs) play a critical role in controlling HIV-1 replication. During acute infection, expansion of HIV-specific CD8+ T cells (HS-CTL), before appearance of neutralizing antibodies, is associated with decreased viremia [1] and most likely determines the viral set point during chronic infection [2], [3]. Resistance to disease progression correlates with the detection of Gag-specific CTLs and with the presence of particular HLA alleles, such as HLA-B*57 and –B*27 [4], [5]. HIV rapidly mutates to evade virus-specific CD8+ T lymphocyte responses, underlying the selection pressure exerted by CTLs [2], [6], [7]–[11]. In large part due to its error prone reverse transcriptase activity, HIV possesses a unique capacity to mutate and evade CTL responses. During acute and chronic HIV infection, CTL escape mutations have been well documented [9], [12], [13]. In most cases, these mutations are intra-epitopic and affect HLA binding and/or alter TCR interactions leading to loss of CTL activation or more subtle effects [14]. However, interference with antigen processing may also lead to a reduced generation of precursor peptides and consequently peptide/MHC-I complex formation and T cell activation. This could occur at any stage of the processing pathway. Mutations in epitope-flanking regions might affect proteasomal processing or N-terminal trimming leading to escape from CTL recognition [15]–[20].
CTLs recognize peptides originating from proteasomal processing of viral proteins or truncated misfolded viral polypeptides, also called DRiPS (for defective ribosomal products) [21]–[23]. These viral polypeptides are classically derived from the fifteen HIV-1 viral proteins encoded by the nine primary open reading frames [24]. However CTLs also target peptides translated from alternative reading frames or ARFs (also called cryptic epitopes). ARF-derived peptides (ARFPs) result from a differential usage of the three-letter codon alphabet during protein synthesis. How this change of reading frame occurs remains elusive but various mechanisms have been proposed. Ribosomes can initiate translation at an internal initiation codon (Met or Cys), change reading frame by shifting, or translate alternatively spliced mRNA. Nonetheless, ARF polypeptides are processed in cells and thus constitute an important source of cryptic epitopes for MHC-I presentation [25]. CTL responses directed against these cryptic epitopes have been detected in autoimmune disease [26], in tumors [27], [28] but also in several infectious diseases, including influenza virus [29], murine AIDS [30], SIV [31] and importantly HIV infections [32]–[35].
We previously described six ARFPs presented by HLA-B*0702 overlapping the alternative reading frames of HIV-1 gag, pol or env genes [32]. CTL responses specific for these ARF-derived peptides were detected in the blood of HIV+ patients. In addition, HIV-infected cells were recognized by CTLs specific for the gag-overlapping ARF epitope (so called Q9VF/5D epitope). Importantly, we showed that the introduction of a stop codon within gag-ARF abrogated Q9VF/5D epitope generation and Q9VF/5D–specific CTL activation [32]. Recent studies further highlighted the in vivo relevance of ARFP-specific CTL responses [33], [34], [36]. In two independent cohorts studies, Bansal et al. and Berger et al. investigated the association between specific HLA alleles and HIV sequence polymorphisms within ARFs. This “HLA class I footprint approach” allowed the prediction of numerous ARFPs within the HIV-1 genome, both from sense and antisense transcripts. On a restricted number of ARFPs, they also demonstrated that these cryptic epitopes induced CTL responses during natural infection that might contribute to viral control in vivo [33], [34].
In the present work, we bring to light a novel mechanism of CTL escape altering the processing and presentation of the Q9VF epitope encoded by the gag-overlapping ARF. In PBMCs of HLA-B*07+ and HLA-B*07 - HIV-infected individuals, we first compared the prevalence of QPRSNTHVF (Q9VF/5N) and QPRSDTHVF (Q9VF/5D) variants of the gag-ARFP. To this end, we PCR amplified and sequenced twenty HIV proviral genomes per individuals. We noticed that the proportion of proviruses encoding Q9VF/5D was significantly lower in HLA-B*07+ than in HLA-B*07 - patients, suggesting that Q9FV/5D encoding viruses might be under selective pressure in HLA-B*07+ individuals. In HLA-B*07+ and HLA-B*07 - patients, we analyzed ex vivo CTL responses directed against Q9VF/5D and Q9VF/5N and we dissected the immunogenicity of Q9VF variants. We observed that cells infected with HIV-1 strains encoding Q9VF/5N were neither recognized by Q9VF/5N - nor Q9VF/5D-specific CTLs. We demonstrate that this single amino acid (AA) variation is responsible for the lack of CD8+ T cell recognition. We show that HIV can escape CTL surveillance by introducing mutations leading to epitope destruction by proteasomes.
Results
Analysis of Q9VF gag proviral sequences and Q9VF-specific CTL responses in HLA-B*07+ patients
Q9VF was originally predicted from the sequence of the consensus HIVHxB2 (HIVLAI) isolate [32]. HIVLAI bears an asparagine (N) to aspartic acid (D) substitution at position 5 (Q9VF/5D) representing less than 5% of HIV-1 clade B strains retrieved from Genbank. We decided to extend these observations by sequencing HIV proviral sequences isolated from 10 HLA-B*07+ and 10 HLA-B*07 - patients. HLA-typing, virological and clinical characteristics of these patients are presented in Table 1. Both groups were age-matched and did not present any significant differences in terms of CD4 counts, viral loads or treatments (not shown). From the PBMCs of each patient, we cloned and sequenced at least 20 HIV-proviral sequences encompassing the gag-ARF DNA region (Figure 1A and Supplementary Figure S1). The isolated HIV sequences encoded either Q9VF/5N (present in 16 out of 20 patients, representing 62% of all isolates), Q9VF/5N variants (exhibiting within the epitope an additional AA difference from the consensus sequence, 9 out of 20 patients, 14% of all isolates) or Q9VF/5D (7 out of 20 patients, 15% of all isolates) and Q9VF/5D variants (2 out of 20 patients, 1% of all isolates) (Table 2). Between Q9VF/5N and Q9VF/5N-variants, Q9VF/5N was the major variant representing 80% of proviral sequences in this group. Q9VF/5D was the major sequence representing 94% of proviral sequences among Q9VF/5D and Q9VF/5D-variants. Note that these mutations did not impact the translation of classical gag ORF (Supplementary Figure S1 and not shown). In contrast, HIV proviruses harboring a STOP codon prior to Q9VF (8% of all isolates) that most likely abolishes Q9VF translation were also identified (Figure 1A). HIV proviral sequences encoding Q9VF/5N and Q9VF/5N-variants were predominant in both HLA-B*07+ and HLA-B*07 - patients. Q9VF/5D or Q9VF/5D-variant HIV proviral sequences could be retrieved in two out of the ten HLA-B*07+ patients and in six out of the ten HLA-B*07 - donors. Taking into consideration the diversity of HIV sequences per donor with regard to their HLA-B7 status, we observe a significant lower proportion of Q9VF/5D+ HIV strains in HLA-B*07+ than in HLA-B*07 - donors (p<0.04, mean value 3% vs 29% of proviral sequences in HLA-B*07+ and HLA-B*07 - donors, respectively, Figure 2B). Altogether, these results suggested that Q9VF/5D-encoding HIV strains might be under negative selective pressure in HLA-B*07+ donors. We thus analyzed CTL responses directed against Q9VF/5D and Q9VF/5N epitopes in PBMCs of patients including the 10 HLA-B*07+ patients used for the analysis of HIV proviral sequences.




PBMCs from 31 HLA-B*07+ patients were loaded with various peptides and submitted to IFNγ-ELISpot (Figure 1C and not shown). Incubations with peptides corresponding to well-characterized HLA-B*0702-restricted immunodominant epitopes from HIV-1 Gag classical ORF (SPRTLNAWV, TPQDLNTML, YPLASLRSLF) induced a significant IFNγ-release, demonstrating that in the course of natural infection the donors mounted CTL responses to HIV-1 antigens. Five out of the 31 HLA-B*07+ donors showed a low but significant activation with Q9VF/5D and Q9VF/5N peptides (Figure 1C). Note that donors reacted to both peptides or reacted to none and that the frequencies of CTL responding to Q9VF/5D and Q9VF/5N peptides were in the same order of magnitude (from 150 to 300 CTL per million of PBMCs), suggesting that the reactivity to one or the other peptide might be due to cross reactivity. We previously demonstrated that CTL lines raised against Q9VF/5N were indeed cross-reactive on Q9VF/5D and vice versa ([32] and Supplementary Figure S2).
Viruses encoding Q9VF/5D were not isolated from PBMCs of the five Q9VF responders (Figure 1), with the exception of patients P1 that harbored proviruses encoding a Q9VF/5D variant (QPRGDTHVF, representing 16% of sequences in this donor). These data prompt us to study the immunogenicity of the Q9VF/5N and Q9VF/5D epitope variants.
Q9VF/5D to 5N substitution abrogates CTL recognition of HIV-infected cells
We asked whether the Q9VF/5N epitope was processed and presented to HS-CTLs by HIV-infected cells. HLA-B*0702+ cells were infected with HIVLAI and HIVNL-AD8 strains encoding Q9VF/5D or Q9VF/5N respectively. Five days post-infection (pi), 50 and 47% of the cells were productively infected by HIVLAI and HIVNL-AD8 respectively (as monitored by intracellular Gag-p24 FACS-staining (not shown)). Infected cells were then co-cultured with HIV-specific CTL lines and T cell activation measured using IFNγ-ELISpot assays (Figure 2). HLA-transgenic mice offer a rapid and convenient model to identify human T cell epitopes [24] and to generate CTL lines specific for peptides of unknown immunogenicity in humans, such as Q9VF/5N. For this reason, Q9VF/5D - and Q9VF/5N-specific CTL lines were generated by peptide immunization of HLA-B*0702+ transgenic mice and in vitro restimulations [32], [37]. As expected, Q9VF/5D - and Q9VF/5N - specific CTLs secreted high levels of IFNγ in response to Q9VF/5D and Q9VF/5N peptide loaded cells respectively (Figure 2A). Note that Q9VF/5D - and Q9VF/5N-specific CTL lines displayed similar capacity to recognize peptide-loaded cells (Supplementary Figure S2), suggesting that the Q9VF/5N variant affects neither MHC nor TCR binding of the peptide. As we previously reported [32], HIVLAI-infected cells induced a robust activation of Q9VF/5D-specific CTLs. Due to their capacity to cross-react on Q9VF/5D peptide (Supplementary Figure S2 and [32]), Q9VF/5N-specific CTLs were also stimulated by HIVLAI-infected cells, thus demonstrating that these CTL lines are fully competent in recognizing HIV-infected cells. In contrast, Q9VF/5D - and Q9VF/5N-specific CTLs were not activated upon co-culture with HIVNL-AD8-infected cells (Figure 2A). This is not due to the incapacity of HIVNL-AD8-infected cells to activate HS-CTLs since CTL clones specific for an HLA-B*0702-restricted HIV-1 Nef epitope (F10LR), raised as a control in these experiments, were activated upon co-culture with HIVLAI - and HIVNL-AD8-infected cells.
To extend these observations to other HIV-1 isolates, HLA-B*0702+ cells were also infected with HIVMN that encodes for Q9VF/5N and used as target cells to activate Q9VF/5D - and Q9VF/5N-specific CTLs (Supplementary Figure S3). HIVNL-AD8 - and HIVMN-infected cells did not induce Q9VF/5D - nor Q9VF/5N-specific CTL activation. Overall, these results suggested that HIV-infected cells did not present the Q9VF/5N peptide.
Epitope flanking regions have a direct impact on antigen processing and presentation [38]. Thereafter, to exclude the possibility that HIV sequence variations outside the Q9VF/5N peptide might be responsible for the lack of presentation, we introduced in HIVLAI a D to N mutation within the Q9VF epitope (so called HIVLAI-5D>5N). This mutation did not affect the primary open reading frame of Gag (Supplementary Figure S1) and did not alter viral replication in T cell lines or primary CD4+ T cells (Figure 2B). However, cells infected with HIVLAI-5D>5N could not activate Q9VF/5D - nor Q9VF/5N-specific CTLs (Figure 2C). Thereafter, this single amino acid substitution was sufficient to abrogate CTL recognition, thus indicating that this asparagine alters Q9VF MHC-I presentation. We then sought to dissect the mechanism responsible for the lack of Q9VF/5N MHC-I presentation.
Q9VF/5N binds TAP pumps and HLA-B*0702 molecules
The capacity of antigenic peptides to bind to a given HLA allele is determined by the so-called anchor residues [39]. Mutating an anchor residue abrogates peptide HLA-binding and subsequent T cell activation, a strategy often used by viruses to escape viral-specific T cell responses. The anchor residues of HLA-B*0702 reside at position 2 and 9 of the peptide-ligands. Thereafter, the D to N substitution at position 5 was not predicted to influence Q9VF peptide binding to HLA-B*0702 [40]. However, besides anchor residues, auxiliary residues might affect peptide binding, we thus compared the capacity of Q9VF/5D and Q9VF/5N peptides to bind HLA-B*0702. To this end, T2-HLA-B*0702 cells were loaded O/N with Q9VF/5D or Q9VF/5N peptides and binding to HLA-B*0702 molecules at the cell surface monitored by FACS (Figure 3A, left panel). Q9VF/5D and Q9VF/5N peptides exhibited similar capacities to bind HLA-B*0702 with a relative affinity (RA, based on the reference peptide) of 2.6 and 1.5 respectively (Figure 3A, left panel). To further characterize the impact of the 5D to 5N substitution on peptide-MHC interactions, we compared the capacity of the peptides to stabilize HLA-B*0702 molecules at the cell surface of T2-HLA-B*0702 (Figure 3A, right panel). To this end, T2-HLA-B*0702 were cultured O/N at 26°C to allow surface expression of peptide-receptive MHC molecules, loaded with a high concentration of peptides, shifted to 37°C and the stability of HLA-B*0702-peptide complexes monitored by FACS at various time points. An exponential regression of HLA-B*0702 mean fluorescence intensity (MFI) vs. time reveals that the stability (t1/2) of HLA-B*0702 pulsed with an irrelevant peptide (S9L) is 22 min while binding of Q9VF/5D and Q9VF/5N peptides prolongs the t1/2 to 211 and 641 min respectively (Figure 3A, right panel). Thereafter, Q9VF/5D and Q9VF/5N peptides are very good HLA-B*0702-binders and 5D to 5N substitution tends to prolong surface expression of HLA-B*0702.

Precursor peptides are transported by the TAP pumps (transporter associated with antigen processing) from the cytosol into the endoplasmic reticulum (ER), and then loaded on nascent MHC-I molecules [41]. N-terminally extended peptide precursors are also transported and further trimmed in the ER by the endoplasmic reticulum aminopeptidase ERAAP and bound to MHC-I molecules [42], [43]. We asked whether the absence of Q9VF/5N peptide presentation by HLA-B*0702 within infected cells might be the result of inefficient ER-translocation of the Q9VF/5N epitope and/or Q9VF/5N-peptide precursors by TAP. Hence, we used a TAP-binding assay [44] to evaluate the affinities of Q9VF/5D and Q9VF/5N and their precursors with TAP. Q9VF/5D and Q9VF/5N exhibited a poor affinity for TAP (Figure 3B), most likely due to the presence of a proline at position 2 that negatively impacts on TAP-mediated peptide transport [44]. In contrast, their N-terminally extended peptide precursors EGF-Q9VF/5D and EGF-Q9VF/5N showed at least a two-log increased efficiency to compete for TAP with an equal 1/IC50 of 0.15. Whatever the precursor, Q9VF/5D and Q9VF/5N containing peptides did not show differences in their capacity to bind human TAP molecules.
Overall, these data demonstrated that the D to N substitution within Q9VF does not impact on TAP transport and HLA binding. In contrast, the 5N substitution might prolong epitope presentation on the cell surface.
Q9VF/5D epitope generation is dependent on proteasomal cleavages
The proteasomes, that are the major catalytic enzymes involved in antigen processing, generate the carboxyl termini of most MHC-bound peptides [38], [45]. We thus asked whether the generation of Q9VF/5D was dependent on proteasomal processing. To this end, HLA-B*0702+ cells were infected with HIVLAI. Five days pi, infected cells were incubated with a potent and selective proteasome inhibitor, epoxomicin [46], treated with a citrate-phosphate buffer to remove residual MHC-peptide complexes, washed and cultured with Q9VF/5D-specific CTLs as previously described. Epoxomicin treatment abolished the capacity of HIVLAI-infected cells to activate Q9VF/5D-specific CTLs, as measured in IFNγ-ELISpot (Figure 3C, left panel). Note that epoxomycin inhibition affected neither MHC-density (as monitored by FACS, not shown) nor the capacity of treated cells to present exogenous peptide (at 0.1 µg/ml) (Figure 3C, right panel). Thereafter, these results demonstrated that the generation of Q9VF epitope depends on proteasomal processing.
5N introduces an aberrant proteasomal cleavage site within Q9VF epitope
Proteasomes might also destroy CTL epitopes by generating aberrant cleavages within the epitope [47] or in epitope-flanking regions [19], [48]. We thus asked whether aberrant proteasomal cleavages might be responsible for the lack of Q9VF/5N presentation.
The proteasome is a large multicatalytic protease composed of standard and inducible subunits that replace the standard subunits upon exposure to IFNγ and form the so-called “immunoproteasomes” (IP). IP is found in most cell types after IFNγ-exposure, but is constitutive in APCs and is induced in HIV-infected T cells [49]. Standard (SP) and IP proteasomes display discrete differences in their capacity to cleave a given peptide substrate [50]. We submitted the full-length polypeptides from the gag-overlapping ARF to IP processing. 27mer peptides encompassing Q9VF/5D or Q9VF/5N peptides were synthesized and incubated with IP purified from T2.27 cells [51]. After 1 h incubation, the digestions were analyzed by mass spectrometry (RP-HPLC SI) and peptide fragments identified by MS/MS (Figure 4A). IP digestion of Q9VF/5D encompassing peptide showed the presence of major proteasomal cleavage sites after amino acids F10, F19, I22 and R24 representing around 80% of total cleavages. The cleavage at position F19 generated the C-terminal cut of the N-extended precursors of Q9VF (M1-F19). After 1 h incubation, when comparing the IP digestion profiles of Q9VF/5D and Q9VF/5N encompassing peptides, we noticed the presence of a new cleavage site within the Q9VF/5N epitope. This cut at position N15 was the most prevalent among Q9VF/5N representing up to 28% of total IP cleavages. These results demonstrated that the D to N substitution introduces a major cleavage site within the Q9VF/5N epitope. Nonetheless the C-terminal cut necessary for the generation of Nt-extended Q9VF/5N precursors was also detected following 1 h of proteasomal digestion.
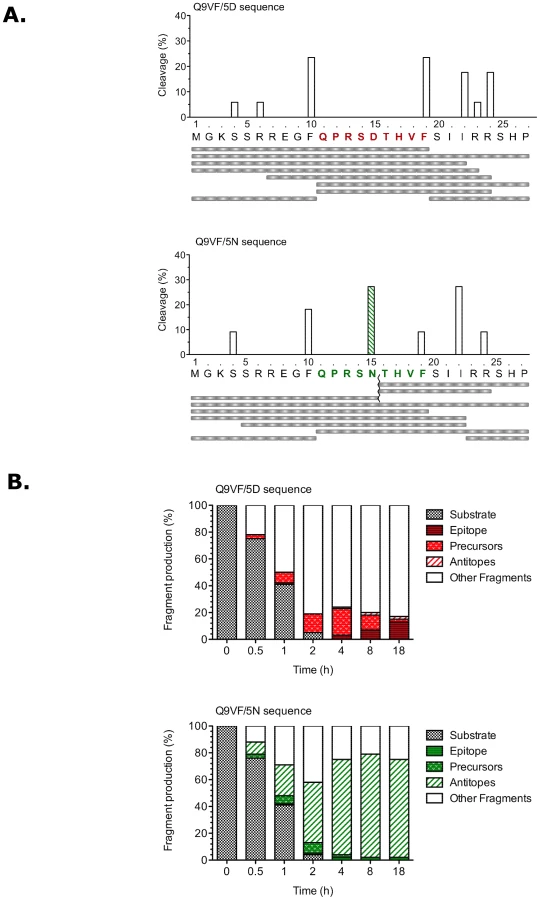
Thereafter, we sought to evaluate the amount of cleavage products generated during Q9VF/5D and Q9VF/5N digestions. To this end, we performed kinetics of IP digestion where aliquots were regularly collected and submitted to mass spectrometry analysis as before (Figure 4B). To compare the amounts of cleavage products, we used the MS fragment intensity as a surrogate marker for quantity since these two parameters correlate significantly [15]. The variations among the different fragments generated are presented as the relative intensity of peptides that exhibit a Q9VF C-terminal cut (epitope or precursors) or peptides issued from cleavages within the Q9VF epitope (referred to as the antitopes) (Figure 4B). Kinetics of digestion of peptides encompassing either Q9VF/5D or Q9VF/5N were identical: 24%, 59% and 96% of both substrates was degraded after 30 min, 1 h and 2 h respectively. At latter time points, both 27mers were undetectable. In the course of Q9VF/5D substrate digestion, the precursor (M1-F19) was readily produced starting from 30 min with a peak at 4 h digestion (representing 20% of digested products). The epitope was detected starting from 1 h digestion and accumulated reaching 13% of all peptide fragments at time 18 h. At latter time points, Q9VF/5D epitopes and precursors represented up to 14% of all peptide fragments detected. An antitope corresponding to a cleavage at position S14 was also generated but represented less than 2% of detected fragments at each time point. In contrast, during Q9VF/5N substrate digestion, the antitopes corresponding to the cleavage at position N15 were already produced after 30 min of digestion and reached around 77% of all peptides from 4 to 18 h, further demonstrating that N15 is a major cleavage site within Q9VF/5N. Interestingly, during Q9VF/5N digestion, the epitope was barely detected even at latter time points (less than 2% of digested products). The precursor M1-F19 accumulated from 30 min to 2 h (8% of digested products) but was undetectable after 4 h, suggesting that the cleavage at position N15 destroyed this peptide. Overall, the amounts of Q9VF/5N epitope and precursors produced were markedly reduced as compared to Q9VF/5D digestion.
Taken together, these results demonstrate that the Q9VF/5D epitope is efficiently produced by proteasomes and accumulates with time. In contrast, the D to N substitution introduces a major cleavage site within the epitope leading to the destruction of the Q9VF/5N epitope and thus the absence of MHC-I binding and presentation.
Discussion
The three-letter codon alphabet allows protein synthesis in six possible overlapping reading frames. A vast number of ARFs have the potential to encode proteins or epitopic peptides (ARFPs). Using an “HLA class I footprint” approach, Bansal et al and Berger et al recently predicted the existence of numerous ARFPs within HIV-1 genome [33], [34]. We have previously shown that ARFP-specific CTLs are induced during natural infection [32]. These CTL responses might contribute to viral control driving HIV evolution at the population level. ARFPs can mutate during the first year of infection, suggesting a possible selection of escapes variants [33], [34]. Such a scenario has been highlighted in the macaque model of SIV infection [31]. Mamu-B*17+ macaques generate strong CTL responses against SIV ARF-encoded epitopes leading to ARF mutation affecting epitope binding to Mamu-B*17 molecules and subsequent SIV replication rebound [31]. In the present study, we characterized a novel mechanism of ARFP-specific CTL escape resulting from HIV epitope destruction by the proteasomes. We suggest that ARFP-specific CTLs exert a selection pressure leading to negative selection of targeted HIV strains. Overall, our work shows that CTL escape mutations are not limited to epitopes encoded by classical ORF, highlighting the role of ARFP-specific CTLs in the control of HIV infection.
We previously identified a panel of epitopes encoded by ARFs within HIV-1 gag, pol and env genes [32]. The gag-overlapping ARF encoding for the Q9VF epitope presented by HLA-B*0702 drew our attention due to its polymorphism. In a cross-sectional cohort study, we report that proviruses encoding the Q9VF/5D epitope (and 5D variants) are rare and significantly under-represented in PBMCs of HLA-B*07+ patients, thus suggesting Q9VF/5D-specific CTLs might exert a negative selection pressure on HIV strains encoding Q9VF/5D variants. In HIV-1 gag ARF, the virus might escape CTL immune pressure by introducing a 5D to 5N substitution or Stop codons but prior the epitope. We thus analyzed CTL responses directed against Q9VF/5D and Q9VF/5N epitopes in PBMCs of patients. Q9VF/5D and Q9VF/5N peptides induced CTL responses in 16% of HLA-B*07+ individuals tested. Donors reacted to both peptides or reacted to none. The frequencies of CTLs responding to Q9VF/5D and Q9VF/5N peptides were about the same magnitude, suggesting that the reactivity to one or the other peptide might be due to cross reactivity. The frequency and magnitude of Q9VF/5D responses in HLA-B*07+ patients were rather low as compared to immunodominant HLA-B*07-restricted responses (Figure 1 and [24]). This might be due to the fact that the patients included in the study were under retroviral therapy that might affect the expression of ARF during residual HIV-1 translation (Table 1). Alternatively in our assays, we are most likely monitoring memory responses to Q9VF/5D that are usually of low magnitude. This possibility is supported by the observation from Bansal et al that ARFP encoding sequences mutate during the first year of infection [33]. Overall, the low representation of Q9VF/5D encoding HIV proviral sequences in PBMCs of HLA-B*07+ individuals and the low frequency and magnitude of CTL responses to Q9VF/5D strongly supported our initial hypothesis that 5N substitution is an escape mutation.
We dissected the immunogenicity of the Q9VF/5N epitope. We showed that cells infected with HIV-1 strains encoding Q9VF/5N (HIVNL-AD8 and HIVMN) were not recognized by Q9VF/5N-specific CTLs. In contrast, Q9VF/5N - and Q9VF/5D-specific CTLs were activated by HIV-1 strains encoding Q9VF/5D (HIVLAI). We demonstrated that the single AA substitution from 5D to 5N in HIVLAI sequence is sufficient and required to abrogate CTL recognition of HIV-infected cells. Thereafter, the acquisition of this 5N mutation by HIV might help the virus to interfere with Q9VF epitope expression or processing and presentation.
Viruses can interfere with antigen expression to escape CTL lysis [23]. Various mechanisms have been proposed for the biosynthesis of ARF-derived polypeptides. Ribosomes can scan through conventional initiation codons [29], initiate translation at an internal initiation non-AUG-codons (Leu or Cys) [34], [52], change reading frame by shifting [53], or translate alternatively spliced mRNA (for review see [25]). We previously described the presence of a conserved slippery motif (UUUAAAU) upstream of gag-ARF start codon that may facilitate ribosomal slippage and thus Q9VF synthesis [32]. Interestingly, a structured region (hairpin) in HIV-1 RNA has been identified downstream of this slippery motif [53]. This highly structured RNA region might cause ribosomal pausing during gag translation thus facilitating ribosomal slippery and Q9VF expression. The D to N substitution within the Q9VF epitope is translated from a codon that is located in the flexible loop of the RNA hairpin structure [53]. Although it remains to be formally proven, this D to N substitution most likely does not impact the RNA structure and hence Q9VF expression.
Viruses also manipulate antigen processing and presentation to escape CTL responses. Interference with antigen presentation could arise at any stage in the pathway, including processing by proteasomes, binding of epitope-precursors to TAP, destruction of these precursors by peptidases in the ER or cytosol and peptide binding to the MHC-I molecule. HIV-specific CTL responses have been shown repeatedly to select for intra-epitope mutations that affect HLA-binding or TcR recognition. In addition, HIV escape mutations outside the epitope (extra-epitope mutations) can interfere with antigen processing by proteasomes [17]–[19], [47], [54], [55] or by the ER aminopeptidase ERAAP [16]. To our knowledge, intra-epitope mutations affecting antigen processing have not been described thus far. Several studies proposed that intra-epitope variation might affect processing but did not provide a mechanism [34], [20]. The only evidence that intra-epitope mutations might affect proteasomal processing of viral antigens comes from mouse models [47], [56].
We provide several lines of evidence strongly suggesting that the D to N substitution within the Q9VF epitope impacts neither TcR recognition nor MHC binding: i) Q9VF/5N - and Q9VF/5D-specific CTLs can be generate upon peptide immunization of HLA-B*07-transgenic mice and cross-react to the alternate peptide ([32] and Supplementary Figure S2); and ii) Q9VF/5N and Q9VF/5D peptides bind HLA-B*0702 (Figure 3A). In addition, we show that Q9VF/5N and Q9VF/5D peptide and their precursors (elongated on the N-termini) efficiently bind TAP, thus demonstrating that the D to N substitution does not affect peptide translocation into the ER. As previously observed with peptides bearing a proline at position 2 [44], the optimal Q9VF/5N - and Q9VF/5D epitopes had a reduced capacity to bind TAP as compared to their Nt-extended precursors (Figure 3B), suggesting that in the ER peptide-trimming is required for proper HLA-B*0702 binding. The ER aminopeptidase ERAAP provides peptides for many MHC-I molecules but has been also implicated in the destruction of CTL epitopes [16]. However, ERAAP cannot process X-P motifs in peptide sequences [42]. Thereafter, though it cannot be formally excluded, a role of ERAAP in the destruction of Q9VF/5N is very unlikely. Overall, these data support the concept that the intra-epitope D to N substitution interferes with proteasomal processing. Using in vitro proteasomal digestions, we demonstrate that the D to N substitution introduces a major cleavage site within the Q9VF epitope (at position N15). Note that at 1 h-digestion time point we identify mainly primary cleavage products since less than 50% of the peptide substrates (the 27mer) have been digested (Figure 4A). To further highlight the potential impact of this N15 cleavage site in the generation of the Q9VF epitope, we performed kinetics of peptide digestion using IP. We observed that amounts of Q9VF/5N epitope and precursors produced were markedly reduced as compared to Q9VF/5D. These results strongly suggest that proteasome cleavages at position N15 destroy the Q9VF/5N epitope and precursors resulting in the lack of MHC-I presentation and CTL activation. In conclusion, a single amino acid variation within HIV epitope can result in epitope destruction and absence of HIV-specific CTL activation.
Mutation in HIV-1 genome can be silent or can differentially impact the fitness of the virus. Due to the redundancy of the codon alphabet, the 5D to 5N substitution in Q9VF does not impact the primary gag-ORF and thus viral replication (Figure 2B). Nevertheless, considering the multitude of existing ARFs, some mutations within ARF encoding sequences most likely affect viral fitness and these ARF sequences might be unavoidably conserved throughout HIV-1 isolates. Thereafter, the great diversity of ARF epitopes produced during HIV infection offers a vast panel of therapeutic targets to stimulate CTL responses. It is interesting to note that ARF-specific CD8+ T cells can performed multiple functions [33], [34] and control viral replication in vitro, characteristics that correlate with slow disease progression [57]. In addition, CTLs targeting ARF-derived epitopes can be induced upon vaccination [58] and tumor infiltrating CTLs specific for ARFPs have been also identified in various cancers, including melanoma and breast cancers [25]. Such responses against crytptic epitopes represent a great potential for future immunotherapeutic strategies.
Materials and Methods
Study population
HIV-1-infected peripheral blood mononuclear cells (PBMCs) were obtained from HCV (Hepatitis C virus) negative French ALT-ANRS-CO15 cohort patients [59]. The 31 HLA-B*07+ and 10 HLA-B*07 - individuals were identified using the anti-HLA-B*07 antibody ME.1. HLA status was further confirmed by genotyping using PCR [60] or using the Luminex xMAP technology [61]. HLA-typing, virological and clinical characteristics of the ten HLA-B*07+ and ten HLA-B*07 - patients included in the study are presented in Table 1.
Ethics statement
Patient samples were collected according to French Ethical rules. Written informed consent and approval by institutional review Board at the Pitié-Salpêtrière Hospital were obtained.
Animals were bred at the Pasteur Institute. The Office of Laboratory Animal Care at Pasteur Institute reviewed and approved protocols for compliance with the French and European regulations on Animal Welfare and with Public Health Service recommendations (Directive 2010/63/EU).
Human CTL assays
PBMCs were isolated by ficoll-centrifugation, pulsed with Q9VF peptides (1 µM, 1 h at 37°C), and submitted to IFNγ-ELISpot assays as previously described [46]. The HLA-B*0702-restricted peptides used were: HCV-derived epitope G9AT (GPRLGVRAT), CMV-derived epitope T10AM (pp65 417TPRVTGGGAM426) used as negative and positive control respectively and a pool of known Gag HIV-1-derived epitopes (p24 16SPRTLNAWV24, p24 48TPQDLNTML56, p2p7p1p6 121YPLASLRSLF130) as control for HIV reactivity [24]. Responses were considered positive when IFNγ production was superior to 50 spots/106 PBMCs and at least threefold higher than background (measured with the HCV peptide).
Mouse CTL recognition of infected T1 cells
Mouse CTL lines were derived from splenocytes of peptide immunized HLA-B*07mα3 transgenic mice. In brief, these mice express HLA-B*0702 heavy chain with a murine α3 domain and their H-2Kb and H-2Db class Ia genes have been inactivated [37]. Cytolytic activity of splenocyte cultures was first assessed in a51Cr release assay [32]. Peptide specific CTL lines were stimulated in vitro (5 µg/mL of peptide) and cultured in RPMI 1640 medium supplemented with 10% FCS, 0.5 µM 2-β-mercaptoethanol (Sigma), 100 IU/mL penicillin and 100 µg/mL streptomycin (Gibco-BRL). Ten days later, 2×103, 400 and 80 CTLs in triplicates were stimulated by 105 HIV-1-infected T1-B7 cells and IFNγ release was detected by ELISpot assay. Cross-reactivity of Q9VF/5D - and Q9VF/5N-specific CTLs was tested in IFNγ-ELISpot and Cr51-release assays [32] using T1-B7 peptide-loaded cells. Mouse CTL lines specific for the HLA-B*0702-restricted HIV-1 Nef-derived epitope F10LR (Nef 68FPVTPQVPLR77; [22]) were used as controls. When stated, HIV-infected T1-B7 cells were treated with epoxomicin (6 h, 1 µg/ml, Calbiochem). To remove residual MHC-peptide complexes, epoxomycin-exposed cells were treated with a citrate-phosphate buffer (pH 3.3) containing 1% BSA and washed twice, prior co-culture with CTLs for an additional 8 h.
Virus and infections
HIVLAI 5D>5N was generated by a single amino acid mutation in HIVLAI provirus. The GAT codon (D) of Gag-ARF (AA in position 15) was replaced by an AAT codon (N) without affecting the primary Gag AA coding sequence, using the following primer (5′-GGC TTT CAG CCC AGA AGT AAT ACC CAT GTT TTC AGC) and Quickchange XL Site-directed Mutagenesis Kit (Stratagene). HIVLAI, HIVLAI-5D>5N, HIVNL-AD8 and HIVMN were produced by transfection of 293T cells using routine procedures [62]. T1 cells (174xCEM, CCR5+ LTR-GFP+) stably transfected with the HLA-B v T1-B7 cells, [53]) were infected and used as antigen-presenting cells. 5×106 T1-B7 cells were infected with 500 ng of p24 for 3 h in culture medium containing 10 mM Hepes and 4 µg/ml DEAE-dextran. 2 to 5 days p.i., infected T1-B7 cells were used as antigen-presenting cells in IFNγ-ELISpot assay. For the infection kinetics, T1-B7 cells were infected with the indicated viruses according to the same procedure using 1, 10 or 100 ng/ml of p24. Primary CD4+ T cells were isolated from the blood of healthy donors using ficoll centrifugation and magnetic beads (Miltenyi) and activated using PHA (1 µg/ml, PAA) and rhIL-2 (50 IU/ml, Chiron) [62]. Seven days post activation, CD4+ PHA blasts were infected with various doses of HIV (from 1 to 100 ng/ml of p24). HIV infection was monitored by FACS (Becton Dickinson) using intracellular HIV p24 staining (KC57 Ab, Beckman Coulter) or p24-Elisa (PerkinElmer).
Sequencing of the Gag-ARF encoding region from clonal HIV-1 populations
Total DNA was extracted from PBMCs of HLA-B*07+ and HLA-B*07 - HIV+ patients using QIAampblood DNA minikit (Qiagen). To analyze the diversity of HIV-1 proviruses in the PBMCs of patients, a 267-bp fragment encompassing the Gag-ARF coding sequence was amplified by nested PCRs as followed: 5 min of initial denaturation at 94°C, 1 min at 94°C, 1 min at 57°C, and 1 min at 72°C for 30 cycles, followed by 7 min at 72°C. The outer primer pair used was (5′ - ATC AAG CTT GCA CAG CAA GCA GCA GCT GAC) and (5′ - CAG GAA CTA CTA GTA CCC TTC AGG AAT TCG G), and the inner primer pair was (5′ - TAC CCT ATA GTG CAG AAC ATC CAG GG) and (5′ - GAT AGA GTG CAT CCA GTG CAT GCA). Samples were treated separately and negative controls were systematically included. Purified PCR products were cloned using a TOPO-TA cloning kit (Invitrogen). Twenty clones per patient were isolated and gag-ARF inserts from each clonal DNA plasmid were amplified by PCR using M13 primers and sequenced (Applied Biosystem).
HLA-B*07.02-peptide binding and stabilization assays
The capacity of the peptides to bind HLA-B*0702 was determined using a classical HLA stabilization assays with the TAP-deficient cell line T2 HLA-B*0702+ [37]. Briefly, cells were incubated overnight with 100, 10, 1 and 0.1 µM of peptide in serum-free medium at room temperature. Cells were then stained with the anti-HLA-B*07 ME.1 antibody and HLA-B*07 surface expression analyzed by FACS (Becton Dickinson). The concentration needed to reach 50% of the maximal fluorescence (as defined with the R10TV peptide (CMV pp65 265RPHERNGFTV274) was calculated (IC50). The relative affinity (RA) is the IC50 ratio of the tested and R10TV reference peptide (the lower the relative affinity, the stronger the binding). The HLA-A*02-restricted peptide S9L (HIV-1 p17 77SLYNTVATL85) was used as negative control. To monitor the capacity of the peptides to stabilize HLA-B*0702, T2-HLA-B*0702 were cultured O/N at 26°C and pulsed the last 2 h with peptide (100 µM) in presence of β2-microglubilin (Sigma, 1 µg/ml) and brefeldin-A (BFA, Sigma, 10 µg/ml). Cells were then shifted to 37°C for 1 h, washed to remove unbound peptides and incubated at 37°C in presence of BFA (0.5 µg/ml). Samples were removed to 0°C at the indicated time points. Cells were then stained at 4°C using the ME.1 antibody and analyzed by FACS. Data (HLA-B*0702 expression) are expressed as MFI vs. time. The capacity of each peptide to stabilize HLA-B*07 (t1/2)is deduced from an exponential regression (one phase decay) using Prism software. A constrain corresponding to the MFI value obtained for the irrelevant peptide (S9L) at the latest time point was applied to the plateaus. T10AM (pp65 417TPRVTGGGAM426) and T9ML (p24 48TPQDLNTML56) peptides were used as positive controls.
TAP-binding assay
The capacity of the peptides to bind TAP was measured in a competitive binding assay as described previously [44]. Briefly, microsomes were purified from Sf9 insect cells expressing human TAP1–TAP2 complexes, pulsed with the iodinated reporter peptide R9L (RRYNASTEL) at 300 nM, and loaded with a dilution of competitor test peptides (0.1 to 1,000 fold molar excess relative to radioactive reporter peptide). TAP affinities were determined as the concentrations required to inhibit 50% of reporter peptide binding (IC50). Results are expressed as 1/IC50 ratios and are mean values from three independent experiments. The highest the 1/IC50 ratio, the highest the affinity.
In vitro proteasome digestions
Immunoproteasomes were isolated from T2.27mp cells (that stably express all three immunosubunits) as previously described [51]. Purified proteasomes were analyzed by SDS-PAGE. The yield was calculated at 90–95%. The 27mer peptides encompassing Q9VF/5D or Q9VF/5N were synthesized using standard Fmoc method on an Applied Biosystems 433A automated synthesizer. The peptides were purified by HPLC and analyzed by mass spectrometry. Three nmol of peptides were digested in vitro using 1 µg of proteasomes (for 0.5, 1, 2, 4, 8 and 18 h) in 100 µl of buffer containing 20 mM Hepes/KOH, pH 7.8, 2 mM magnesium acetate and 2 mM dithiothreitol. Reactions were stopped by the addition of trifluoroacetic acid to a final concentration of 0.3%. The digestions were analyzed, by mass spectrometry (RP-HPLC ESI) and the products were identified by MS/MS.
Statistical analysis
A standard two-tailed nonparametric Mann-Whitney U-test (with P<0.05 considered significant) was used to perform statistical comparison of HIV-1 proviral sequences frequencies using statistical analysis Prism software (GraphPad).
Supporting Information
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. WangYELiBCarlsonJMStreeckHGladdenAD
2009
Protective HLA class I alleles that restrict acute-phase
CD8+ T-cell responses are associated with viral escape mutations
located in highly conserved regions of human immunodeficiency virus type
1.
J Virol
83
1845
1855
2. GoonetillekeNLiuMKSalazar-GonzalezJFFerrariGGiorgiE
2009
The first T cell response to transmitted/founder virus
contributes to the control of acute viremia in HIV-1
infection.
J Exp Med
206
1253
1272
3. KoupRASafritJTCaoYAndrewsCALeodGM
1994
Temporal association of cellular immune responses with the
initial control of viremia in primary human immunodeficiency virus type 1
syndrome.
J Virol
68
4650
4655
4. KiepielaPLeslieAJHoneyborneIRamduthDThobakgaleC
2004
Dominant influence of HLA-B in mediating the potential
co-evolution of HIV and HLA.
Nature
432
769
775
5. KiepielaPNgumbelaKThobakgaleCRamduthDHoneyborneI
2007
CD8+ T-cell responses to different HIV proteins have
discordant associations with viral load.
Nat Med
13
46
53
6. BrummeZLJohnMCarlsonJMBrummeCJChanD
2009
HLA-associated immune escape pathways in HIV-1 subtype B Gag, Pol
and Nef proteins.
PLoS One
4
e6687
7. DudaALee-TurnerLFoxJRobinsonNDustanS
2009
HLA-associated clinical progression correlates with epitope
reversion rates in early human immunodeficiency virus
infection.
J Virol
83
1228
1239
8. GoulderPJBranderCTangYTremblayCColbertRA
2001
Evolution and transmission of stable CTL escape mutations in HIV
infection.
Nature
412
334
338
9. KawashimaYPfafferottKFraterJMatthewsPPayneR
2009
Adaptation of HIV-1 to human leukocyte antigen class
I.
Nature
458
641
645
10. KoizumiHHashimotoMFujiwaraMMurakoshiHChikataT
2010
Different in vivo effects of HIV-1 immunodominant
epitope-specific CTLs on selection of escape mutant viruses.
J Virol
84
5508
19
11. PhilipsRERowland-JonesSNixonDFGotchFMEdwardsJP
1991
Human immunodeficiency virus genetic variation that can escape
cytotoxic T cell recognition.
Nature
354
453
459
12. BorrowPLewickiHWeiXHorwitzMSPefferN
1997
Antiviral pressure exerted by HIV-1-specific cytotoxic T
lymphocytes (CTLs) during primary infection demonstrated by rapid selection
of CTL escape virus.
Nat Med
3
205
211
13. GoulderPJPhillipsREColbertRAMcAdamSOggG
1997
Late escape from an immunodominant cytotoxic T-lymphocyte
response associated with progression to AIDS.
Nat Med
3
212
217
14. GoulderPJWatkinsDI
2004
HIV and SIV CTL escape: implications for vaccine
design.
Nat Rev Immunol
4
630
640
15. TenzerSWeeEBurgevinAStewart-JonesGFriisL
2009
Antigen processing influences HIV-specific cytotoxic T lymphocyte
immunodominance.
Nat Immunol
10
636
646
16. DraenertRLe GallSPfafferottKJLeslieAJChettyP
2004
Immune selection for altered antigen processing leads to
cytotoxic T lymphocyte escape in chronic HIV-1 infection.
J Exp Med
199
905
915
17. Le GallSStamegnaPWalkerBD
2007
Portable flanking sequences modulate CTL epitope
processing.
J Clin Invest
117
3563
3575
18. MilicicAPriceDAZimbwaPBoothBLBrownHL
2005
CD8+ T cell epitope-flanking mutations disrupt proteasomal
processing of HIV-1 Nef.
J Immunol
175
4618
4626
19. ZimbwaPMilicicAFraterJScribaTJWillisA
2007
Precise identification of a human immunodeficiency virus type 1
antigen processing mutant.
J Virol
81
2031
2038
20. YokomakuYMiuraHTomiyamaHKawana-TachikawaATakiguchiM
2004
Impaired processing and presentation of cytotoxic-T-lymphocyte
(CTL) epitopes are major escape mechanisms from CTL immune pressure in human
immunodeficiency virus type 1 infection.
J Virol
78
1324
1332
21. SchubertUAntonLCGibbsJNorburyCCYewdellJW
2000
Rapid degradation of a large fraction of newly synthesized
proteins by proteasomes.
Nature
404
770
774
22. CasartelliNGuivel-BenhassineFBouziatRBrandlerSSchwartzO
2010
The antiviral factor APOBEC3G improves CTL recognition of
cultured HIV-infected T cells.
J Exp Med
207
39
49
23. CardinaudSStarckSRChandraPShastriN
2010
The synthesis of truncated polypeptides for immune surveillance
and viral evasion.
PLoS One
5
e8692
24. CardinaudSBouziatRRohrlichPSTourdotSWeissL
2009
Design of a HIV-1-derived HLA-B07.02-restricted polyepitope
construct.
Aids
23
1945
1954
25. HoOGreenWR
2006
Alternative translational products and cryptic T cell epitopes:
expecting the unexpected.
J Immunol
177
8283
8289
26. SaulquinXScotetETrautmannLPeyratMAHalaryF
2002
+1 Frameshifting as a novel mechanism to generate a cryptic
cytotoxic T lymphocyte epitope derived from human interleukin
10.
J Exp Med
195
353
358
27. WangRFParkhurstMRKawakamiYRobbinsPFRosenbergSA
1996
Utilization of an alternative open reading frame of a normal gene
in generating a novel human cancer antigen.
J Exp Med
183
1131
1140
28. GodetYMoreau-AubryAGuillouxYVignardVKhammariA
2008
MELOE-1 is a new antigen overexpressed in melanomas and involved
in adoptive T cell transfer efficiency.
J Exp Med
205
2673
2682
29. BullockTNEisenlohrLC
1996
Ribosomal scanning past the primary initiation codon as a
mechanism for expression of CTL epitopes encoded in alternative reading
frames.
J Exp Med
184
1319
1329
30. MayrandSMSchwarzDAGreenWR
1998
An alternative translational reading frame encodes an
immunodominant retroviral CTL determinant expressed by an
immunodeficiency-causing retrovirus.
J Immunol
160
39
50
31. ManessNJValentineLEMayGEReedJPiaskowskiSM
2007
AIDS virus specific CD8+ T lymphocytes against an
immunodominant cryptic epitope select for viral escape.
J Exp Med
204
2505
2512
32. CardinaudSMorisAFevrierMRohrlichPSWeissL
2004
Identification of cryptic MHC I-restricted epitopes encoded by
HIV-1 alternative reading frames.
J Exp Med
199
1053
1063
33. BansalACarlsonJYanJAkinsikuOTSchaeferM
2010
CD8 T cell response and evolutionary pressure to HIV-1 cryptic
epitopes derived from antisense transcription.
J Exp Med 207 : 51-59,
S51-53
34. BergerCTCarlsonJMBrummeCJHartmanKLBrummeZL
2010
Viral adaptation to immune selection pressure by HLA class
I-restricted CTL responses targeting epitopes in HIV frameshift
sequences.
J Exp Med 207 : 61-75,
S61-12
35. SchweighardtBWrinTMeiklejohnDASpottsGPetropoulosCJ
2010
Immune escape mutations detected within HIV-1 epitopes associated
with viral control during treatment interruption.
J Acquir Immune Defic Syndr
53
36
46
36. GarrisonKEChampiatSYorkVAAgrawalATKallasEG
2009
Transcriptional errors in human immunodeficiency virus type 1
generate targets for T-cell responses.
Clin Vaccine Immunol
16
1369
1371
37. RohrlichPSCardinaudSFiratHLamariMBriandP
2003
HLA-B*0702 transgenic, H-2K(b)D(b) double-knockout mice:
phenotypical and functional characterization in response to influenza
virus.
Int Immunol
15
765
772
38. KloetzelPM
2001
Antigen processing by the proteasome.
Nat Rev Mol Cell Biol
2
179
187
39. FalkKRotzschkeOStevanovicSJungGRammenseeHG
1991
Allele-specific motifs revealed by sequencing of self-peptides
eluted from MHC molecules.
Nature
351
290
296
40. RammenseeHBachmannJEmmerichNPBachorOAStevanovicS
1999
SYFPEITHI: database for MHC ligands and peptide
motifs.
Immunogenetics
50
213
219
41. LauvauGKakimiKNiedermannGOstankovitchMYotndaP
1999
Human transporters associated with antigen processing (TAPs)
select epitope precursor peptides for processing in the endoplasmic
reticulum and presentation to T cells.
J Exp Med
190
1227
1240
42. SerwoldTGawSShastriN
2001
ER aminopeptidases generate a unique pool of peptides for MHC
class I molecules.
Nat Immunol
2
644
651
43. SaricTBeningaJGraefCIAkopianTNRockKL
2001
Major histocompatibility complex class I-presented antigenic
peptides are degraded in cytosolic extracts primarily by thimet
oligopeptidase.
J Biol Chem
276
36474
36481
44. van EndertPMRiganelliDGrecoGFleischhauerKSidneyJ
1995
The peptide-binding motif for the human transporter associated
with antigen processing.
J Exp Med
182
1883
1895
45. CascioPHiltonCKisselevAFRockKLGoldbergAL
2001
26S proteasomes and immunoproteasomes produce mainly N-extended
versions of an antigenic peptide.
Embo J
20
2357
2366
46. MorisANobileCBuseyneFPorrotFAbastadoJP
2004
DC-SIGN promotes exogenous MHC-I-restricted HIV-1 antigen
presentation.
Blood
103
2648
2654
47. OssendorpFEggersMNeisigARuppertTGroettrupM
1996
A single residue exchange within a viral CTL epitope alters
proteasome-mediated degradation resulting in lack of antigen
presentation.
Immunity
5
115
124
48. ChapiroJClaverolSPietteFMaWStroobantV
2006
Destructive cleavage of antigenic peptides either by the
immunoproteasome or by the standard proteasome results in differential
antigen presentation.
J Immunol
176
1053
1061
49. GavioliRGalleraniEFortiniCFabrisMBottoniA
2004
HIV-1 tat protein modulates the generation of cytotoxic T cell
epitopes by modifying proteasome composition and enzymatic
activity.
J Immunol
173
3838
3843
50. ToesRENussbaumAKDegermannSSchirleMEmmerichNP
2001
Discrete cleavage motifs of constitutive and immunoproteasomes
revealed by quantitative analysis of cleavage products.
J Exp Med
194
1
12
51. KuckelkornUFerreiraEADrungILiewerUKloetzelPM
2002
The effect of the interferon-gamma-inducible processing machinery
on the generation of a naturally tumor-associated human cytotoxic T
lymphocyte epitope within a wild-type and mutant p53 sequence
context.
Eur J Immunol
32
1368
1375
52. SchwabSRLiKCKangCShastriN
2003
Constitutive display of cryptic translation products by MHC class
I molecules.
Science
301
1367
1371
53. WattsJMDangKKGorelickRJLeonardCWBessJWJr
2009
Architecture and secondary structure of an entire HIV-1 RNA
genome.
Nature
460
711
716
54. AllenTMAltfeldMGeerSCKalifeETMooreC
2005
Selective escape from CD8+ T-cell responses represents a
major driving force of human immunodeficiency virus type 1 (HIV-1) sequence
diversity and reveals constraints on HIV-1 evolution.
J Virol
79
13239
13249
55. ParcejDTampeR
2010
ABC proteins in antigen translocation and viral
inhibition.
Nat Chem Biol
6
572
580
56. BaslerMYouhnovskiNVan Den BroekMPrzybylskiMGroettrupM
2004
Immunoproteasomes down-regulate presentation of a subdominant T
cell epitope from lymphocytic choriomeningitis virus.
J Immunol
173
3925
3934
57. AlmeidaJRPriceDAPapagnoLArkoubZASauceD
2007
Superior control of HIV-1 replication by CD8+ T cells is
reflected by their avidity, polyfunctionality, and clonal
turnover.
J Exp Med
204
2473
2485
58. ManessNJWilsonNAReedJSPiaskowskiSMSachaJB
2010
Robust, vaccine-induced CD8(+) T lymphocyte response against
an out-of-frame epitope.
J Immunol
184
67
72
59. MartinezVCostagliolaDBonduelleON'goNSchnurigerA
2005
Combination of HIV-1-specific CD4 Th1 cell responses and IgG2
antibodies is the best predictor for persistence of long-term
nonprogression.
J Infect Dis
191
2053
2063
60. BunceMO'NeillCMBarnardoMCKrausaPBrowningMJ
1995
Phototyping: comprehensive DNA typing for HLA-A, B, C, DRB1,
DRB3, DRB4, DRB5 & DQB1 by PCR with 144 primer mixes utilizing
sequence-specific primers (PCR-SSP).
Tissue Antigens
46
355
367
61. DunbarSA
2006
Applications of Luminex xMAP technology for rapid,
high-throughput multiplexed nucleic acid detection.
Clin Chim Acta
363
71
82
62. MorisAPajotABlanchetFGuivel-BenhassineFSalcedoM
2006
Dendritic cells and HIV-specific CD4+ T cells: HIV antigen
presentation, T-cell activation, and viral transfer.
Blood
108
1643
1651
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2011 Číslo 5
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Crystal Structure and Functional Analysis of the SARS-Coronavirus RNA Cap 2′-O-Methyltransferase nsp10/nsp16 Complex
- Lymphoadenopathy during Lyme Borreliosis Is Caused by Spirochete Migration-Induced Specific B Cell Activation
- The OXI1 Kinase Pathway Mediates -Induced Growth Promotion in Arabidopsis
- : An Emerging Cause of Sexually Transmitted Disease in Women
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy