
A Wide Extent of Inter-Strain Diversity in Virulent and Vaccine Strains of Alphaherpesviruses
Alphaherpesviruses are widespread in the human population, and include herpes simplex virus 1 (HSV-1) and 2, and varicella zoster virus (VZV). These viral pathogens cause epithelial lesions, and then infect the nervous system to cause lifelong latency, reactivation, and spread. A related veterinary herpesvirus, pseudorabies (PRV), causes similar disease in livestock that result in significant economic losses. Vaccines developed for VZV and PRV serve as useful models for the development of an HSV-1 vaccine. We present full genome sequence comparisons of the PRV vaccine strain Bartha, and two virulent PRV isolates, Kaplan and Becker. These genome sequences were determined by high-throughput sequencing and assembly, and present new insights into the attenuation of a mammalian alphaherpesvirus vaccine strain. We find many previously unknown coding differences between PRV Bartha and the virulent strains, including changes to the fusion proteins gH and gB, and over forty other viral proteins. Inter-strain variation in PRV protein sequences is much closer to levels previously observed for HSV-1 than for the highly stable VZV proteome. Almost 20% of the PRV genome contains tandem short sequence repeats (SSRs), a class of nucleic acids motifs whose length-variation has been associated with changes in DNA binding site efficiency, transcriptional regulation, and protein interactions. We find SSRs throughout the herpesvirus family, and provide the first global characterization of SSRs in viruses, both within and between strains. We find SSR length variation between different isolates of PRV and HSV-1, which may provide a new mechanism for phenotypic variation between strains. Finally, we detected a small number of polymorphic bases within each plaque-purified PRV strain, and we characterize the effect of passage and plaque-purification on these polymorphisms. These data add to growing evidence that even plaque-purified stocks of stable DNA viruses exhibit limited sequence heterogeneity, which likely seeds future strain evolution.
Published in the journal:
. PLoS Pathog 7(10): e32767. doi:10.1371/journal.ppat.1002282
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1002282
Summary
Alphaherpesviruses are widespread in the human population, and include herpes simplex virus 1 (HSV-1) and 2, and varicella zoster virus (VZV). These viral pathogens cause epithelial lesions, and then infect the nervous system to cause lifelong latency, reactivation, and spread. A related veterinary herpesvirus, pseudorabies (PRV), causes similar disease in livestock that result in significant economic losses. Vaccines developed for VZV and PRV serve as useful models for the development of an HSV-1 vaccine. We present full genome sequence comparisons of the PRV vaccine strain Bartha, and two virulent PRV isolates, Kaplan and Becker. These genome sequences were determined by high-throughput sequencing and assembly, and present new insights into the attenuation of a mammalian alphaherpesvirus vaccine strain. We find many previously unknown coding differences between PRV Bartha and the virulent strains, including changes to the fusion proteins gH and gB, and over forty other viral proteins. Inter-strain variation in PRV protein sequences is much closer to levels previously observed for HSV-1 than for the highly stable VZV proteome. Almost 20% of the PRV genome contains tandem short sequence repeats (SSRs), a class of nucleic acids motifs whose length-variation has been associated with changes in DNA binding site efficiency, transcriptional regulation, and protein interactions. We find SSRs throughout the herpesvirus family, and provide the first global characterization of SSRs in viruses, both within and between strains. We find SSR length variation between different isolates of PRV and HSV-1, which may provide a new mechanism for phenotypic variation between strains. Finally, we detected a small number of polymorphic bases within each plaque-purified PRV strain, and we characterize the effect of passage and plaque-purification on these polymorphisms. These data add to growing evidence that even plaque-purified stocks of stable DNA viruses exhibit limited sequence heterogeneity, which likely seeds future strain evolution.
Introduction
Alphaherpesviruses are widespread in the human population, with herpes simplex virus 1 (HSV1) and 2 causing oral and genital lesions, respectively, while varicella zoster virus (VZV) causes chicken pox and shingles [1]–[3]. In the agricultural industry, a related veterinary alphaherpesvirus, pseudorabies virus (PRV), causes similar disease in swine and significant economic cost due to weight loss in infected adults and reproductive losses during pregnancy and suckling [4], [5]. As occurs with HSV and VZV, PRV infection has higher morbidity and mortality rates for neonates, with decreasing severity of disease as the age at onset of infection increases [2], [4], [6]. PRV and VZV primarily infect via the respiratory mucosa, while HSV-1 primarily infects at the oral mucosa. VZV infection includes a viremic phase that yields widespread vesicular lesions, while PRV and HSV are usually non-viremic and spread predominantly by mucosal infection and neuronal innervation. These alphaherpesviruses are widespread in the population because of their tendency to infect neurons: they establish lifelong latency in the host peripheral nervous system. These latent neuronal infections may occasionally reactivate and spread back the mucosal surfaces where the infection initiated. After further replication, the viruses can spread to new hosts.
Among alphaherpesviruses, vaccines are available for VZV and PRV, but not HSV [7], [8]. Despite considerable effort and recent progress, no broadly effective vaccine candidates have yet emerged for HSV infection [9]–[11]. The co-morbidities of HSV-1 and HSV-2 with human immunodeficiency virus (HIV), which include increased acquisition of HIV due to the inflammation and lesions caused by HSV infection, have added impetus to the search for a vaccine [10]–[13]. PRV serves as a useful model for HSV pathogenesis and vaccine development, because of their similar infectious cycle and ability to infect a variety of animal models [4], [5], [8], [14]–[17]. In contrast, VZV has a more restricted tropism for human cells that complicates its study in animal models [18]–[20]. The agricultural importance of PRV and relative ease of vaccine testing has led to the development of several PRV vaccine strains, whose genetic characteristics have been determined by mapping isolated genomic fragments and sequencing of select regions [8], [21]–[23]. Of note, the vaccine strain Bartha has a well-characterized deletion of several viral proteins that attenuates its virulence and also limits its spread in neurons, which led to its subsequent development as a tool for trans-neuronal tracing [21], [24]–[27]. Like several other early vaccine strains, PRV Bartha was attenuated by extensive passage in the laboratory, thus making the full discovery of its genome-wide mutations a priority [22], [23], [28], [29]. Because the only available PRV genome sequence to date is a mosaic of six strains [30], it has been difficult to discern whether mutations detected in PRV Bartha and other vaccine strains are unique or represent ordinary sequence diversity, i.e. are found in other wild-type genomes [31]–[35]. We therefore applied our recent success in using Illumina high-throughput sequencing (HTS) to obtain HSV-1 strain genomes to determining the sequence diversity in the PRV vaccine strain Bartha.
In addition to sequence polymorphisms, insertions, and deletions, another major class of variation between nucleic acid sequences lies in copy number variation, either of coding sequences or of repeated structural elements. Herpesvirus genomes have long been known to contain several sites with tandem short sequence repeats (SSRs) or reiterations [36]–[40]. Variation in these elements has been described both within and between herpesvirus strains, but their functions were largely unexplored [22], [35], [41]–[43]. SSRs can be transcription factor binding sites, chromatin insulators, protein folding motifs, or other regulatory elements [44], [45]. Recent studies have shown that SSR expansion and contraction, most likely through recombination or polymerase slippage, can generate phenotypic variation [46]–[49]. A range of human diseases result from SSR expansion or contraction, including the transcriptional silencing of the gene FMR1 via an upstream SSR, which causes Fragile X syndrome, and the poly-glutamine tract expansion in huntingtin protein, which causes Huntington's disease [50]–[53]. Limited explorations of repetitive elements in viral genomes suggest that SSRs in viral genomes likewise play functional roles [54]–[57]. To explore SSR prevalence and function in herpesviruses, we initiated a global SSR assessment and comparison across viral species, as was recently done for a variety of fungal and bacterial pathogens [49], [58]. These data highlight the contribution of SSRs to overall sequence diversity in viruses, and through the presence of these elements in both coding and non-coding regions, suggest that viral SSRs may likewise have the potential to affect gene expression and protein functions.
We sequenced three widely-studied PRV isolates by HTS: the attenuated vaccine strain Bartha and the virulent strains Kaplan and Becker. This analysis reveals genome-wide sequence diversity between strains, both in the PRV proteome and also in many SSRs. Our comparison of protein coding sequences revealed that 46 of 67 PRV proteins have changes in the vaccine strain Bartha which are not found in the virulent Kaplan or Becker strains. We mapped homologous SSRs in all three strains and provide a comprehensive overview of inter-strain variation in SSR length. We compared the proportion of SSRs in PRV to those found in HSV-1, VZV, the human betaherpesvirus cytomegalovirus (HCMV) and gammaherpesviruses Epstein-Barr virus (EBV) and Kaposi's sarcoma-associated herpesvirus (KSHV), and Mimivirus. We find that SSRs are likely to be a common property of these large DNA viruses. Finally, we examined the limited number of polymorphic bases detected in these plaque-purified virus stocks, and tested the rate of polymorphism occurrence in purified and non-purified virus populations. These data on sequence variation in PRV strains expand our understanding of viral genome diversity and how attenuated strains lead to successful anti-viral vaccines.
Results/Discussion
Sequencing and assembly of multiple PRV strain genomes
We used Illumina deep sequencing and bioinformatic analyses to assemble millions of sequence reads into three completed genomes of PRV Kaplan, Becker, and Bartha. To produce genetically homogeneous stocks for sequencing, we purified a single plaque from each virus stock, plated it out, selected a progeny plaque, and repeated the process. These plaque-purified stocks were then used to produce viral nucleocapsid DNA for Illumina genomic DNA libraries. Over 15 million Illumina sequence reads were combined for each strain (details of HTS sequence reads for each strain are listed in Table S1 in Text S1). High quality viral sequence data were used for a 3-phase de novo assembly process (see Methods for details): 1) the automated generation of large blocks of continuous sequence, or contigs, from Illumina sequence data (usually 0.1–30 kilobase pairs (kb) in length), 2) the automated generation of super-contigs (1–60 kb) using a long-read assembler, and 3) the manual curation of gaps, joins, and annotations. Assembly quality was checked by BLAST-based alignment of each new genome versus the prior mosaic reference. PCR-validation confirmed regions of the assembly with greatest divergence from the mosaic strain, and guided genome correction in selected regions of the assembly (Figure S1 and Table S2 in Text S1). The resulting genomes resembled the original mosaic genome in overall size and gene content (Figure 1A). The PRV genome is organized into a unique long (UL) region and a unique short (US) region, with large inverted and terminal repeats (IR, TR) flanking the US region. Overall, DNA sequences are largely conserved between PRV Kaplan, Becker, and Bartha, with the greatest foci of divergence occurring in IR/TR and noncoding regions (Figure 1B). Phylogenetic comparison of the three full-length genomes revealed a closer relationship between PRV strains Kaplan and Bartha than PRV Becker (Figure 1D).

To ascertain the quality and depth of coverage of these new genomes, sequence reads were aligned back to the assembled genomes. Median coverage was very high: 3,704 sequence reads per base for PRV Kaplan, 4,145 reads/base for Becker, and 4,137 reads/base for Bartha (see also Table S1 in Text S1). This coverage was reduced in genome regions with extremely high or low G/C content, as has been observed for both eukaryotic and bacterial genomes (Figure S2A,B in Text S1) [59], [60]. In addition to analyzing coverage depth, the resulting genomes were used to predict restriction digest patterns, which were compared to actual restriction fragment length polymorphism (RFLP) patterns (Figure 2). Digest patterns match the predicted fragment sizes, with the exception of two classically variable fragments (BamHI 10 and 12; Figure 2) that have been observed to differ even between repeated passages of the same strain [22], [41], [42].

Genetic differences and pathogenicity in the vaccine strain PRV Bartha
PRV Bartha displays the most divergent phenotype of the PRV strains sequenced here, with severe attenuation of virulence in vivo conferring its suitability for use as a vaccine strain. We compared all protein coding regions of PRV Bartha and the two wild-type strains PRV Kaplan and Becker, to search for novel sequence differences corresponding to potential effects on pathogenicity and attenuation of the vaccine strain (Tables 1–3). Prior studies mapped a deletion in the Bartha US region that removes all of gE (US8) and US9 and creates an fusion of gI (US7) and US2, as well as subtle variations in gC (UL44), gM (UL10), and UL21 [21], [28], [31], [61]–[65]. Our de novo assembled Bartha genome confirms the boundary of the US region deletion (position 120,927 on the Bartha genome) as originally mapped by Maxam-Gilbert sequencing [66]; this region spans 3,482 bases on the reference PRV Kaplan genome (positions 120,363–123,845; see also Figure 1B). Adding to these previously reported findings, we identified a total of 46 proteins with coding differences that are unique to PRV Bartha and not found in either wild-type strain (Table 1 and Figure 3). Several of these amino acid (AA) changes are conservative, such as a minor Ala13Val change in Bartha's VP18.8 (UL13), or represent expansions or contractions associated with AA repeats (e.g. VP1/2/UL36, ICP4/IE180, AN/UL12). Many mutations affect loosely mapped functional protein domains, for instance two differences in the 300 AA chemokine-binding domain of Bartha's gG [67]. Further studies will be necessary to define any functional effects in these regions.




Several unique Bartha mutations are located within functional domains of proteins not previously considered to affect Bartha's virulence and spread phenotypes, including gH (UL22), gB (UL27), and gN (UL40.5). The core fusion process of most alphaherpesviruses consists of receptor binding via gD (US6), followed by fusion mediated by gB (UL27) and the gH-gL (UL1) heterodimer. PRV gH has recently been crystallized, as have the homologous gH proteins of HSV-2 and Epstein-Barr virus (EBV) [68]-[70]. PRV Bartha has a Pro438Ser change in gH. In the recent crystal structure of PRV gH, this proline was highlighted as a key residue, because it mediates a bend at the end of an alpha helix in the gH core (domain III), which is necessary to allow one of four disulfide bonds in the protein [70]. This proline and the neighboring disulfide-bonded cysteine are absolutely conserved across all known herpesvirus sequences, including the evolutionarily distant beta - and gamma -herpesviruses [70]. In Western blot analysis of infected cell lysates (Figure 4), PRV Bartha produces two bands of gH protein that are comparable to those of the PRV Kaplan and Becker strains. There is no obvious difference in gH produced by these PRV strains.

We also detected three changes to the key fusion protein gB (UL27) coding sequence in PRV Bartha, which affect several residues immediately adjacent to gB's furin cleavage site (Ser506Ala, Pro507Ala, and Pro509Gln). Furin cleavage of gB has been shown to affect cell-cell spread of PRV and in vivo virulence of VZV [71], [72]. Transfer of just 11 AAs surrounding this furin cleavage site, corresponding to residues 497–507 of the PRV Kaplan gB sequence (PAAARRARRSP), are sufficient to confer protease-cleavage when inserted into PRV gC [73]. As noted previously [31], gB is still cleaved in PRV Bartha-infected cells in vitro (Figure S3 in Text S1), but it is unknown whether these changes in gB affect cleavage efficiency or other aspects of gB function in specialized cell types such as neurons.
Finally, PRV Bartha has a Leu7Pro alteration in the signal sequence of gN (UL49.5) that may affect glycoprotein processing and/or packaging [62], [74]. A previously detected Leu14Pro difference in Bartha's gC also affects the signal sequence, leading to inefficient maturation of gC, and reduced incorporation of gC into virions [62]. PRV gN is normally packaged into virions and affects the rate of virion penetration into cells [74], [75]. If this signal sequence mutation affects gN maturation or virion inclusion in a parallel way to that of the gC signal sequence mutation, it may well contribute to the delayed penetration kinetics and cell-to-cell spread phenotype of the attenuated PRV Bartha vaccine strain.
Amino acid variation between strains of PRV, HSV-1, and VZV
The genomes of alphaherpesviruses have long been thought to be quite stable with limited sequence variation among strains [76], [77]. This idea was well supported when the genome-wide comparison of 18 VZV strains revealed inter-strain coding variation of 1% or less [78], [79]. The four HSV-1 genome sequences available show modestly increased inter-strain protein-coding variation [80]-[83]. Surprisingly, we find that protein coding variation between PRV strains is higher than that observed for either HSV-1 or VZV (average of 1.6% for PRV, vs. 1.3% for HSV-1 or 0.2% for VZV; Figure 5 and Table S6) [78], [81]. When the coding sequences for each protein of these three new PRV genomes are compared, the inter-strain variation in AA sequence (number of AA residues varying between strains, normalized for protein length) reaches as high as 13%. Starting on the low end of variation, we found eight invariant proteins across these PRV strains (Figure 3), including the viral DNA polymerase UL30, the minor capsid proteins VP19c (UL38) and VP23 (UL18), the nuclear egress components UL20, UL31, and UL37, and the functionally uncharacterized proteins UL24 and UL56 (ORF-1). In contrast, ICP22 (US1) displays 13% inter-strain variation; this protein has transactivating and regulatory functions in related alphaherpesviruses [84], [85], but has only been studied at the level of transcript expression in PRV [86], [87]. In a similar comparison of AA sequence differences between 3 strains of HSV-1, the inter-strain variation peaked at 7% (for ICP34.5 (RL1) and US11; Table S6) [81]. VZV strains show even less variation in protein coding sequences, with a maximum of 1.2% AA variation (in ORF-1) between strains, and just two additional proteins with variation greater than 0.5% [78]. One of these two VZV proteins is ORF 62/71, which is homologous to PRV IE180 and HSV-1 ICP4; this protein is among the most variable across all known strains of these alphaherpesviruses. IE180 is the sole gene expressed with immediate-early kinetics in PRV, and is a key transactivator of viral gene expression [88]–[90]. In contrast, the nuclear egress proteins UL20 and UL31 thus far shows no inter-strain variation in all known genomes of PRV and HSV-1, while UL31 shows zero coding variation in VZV as well.
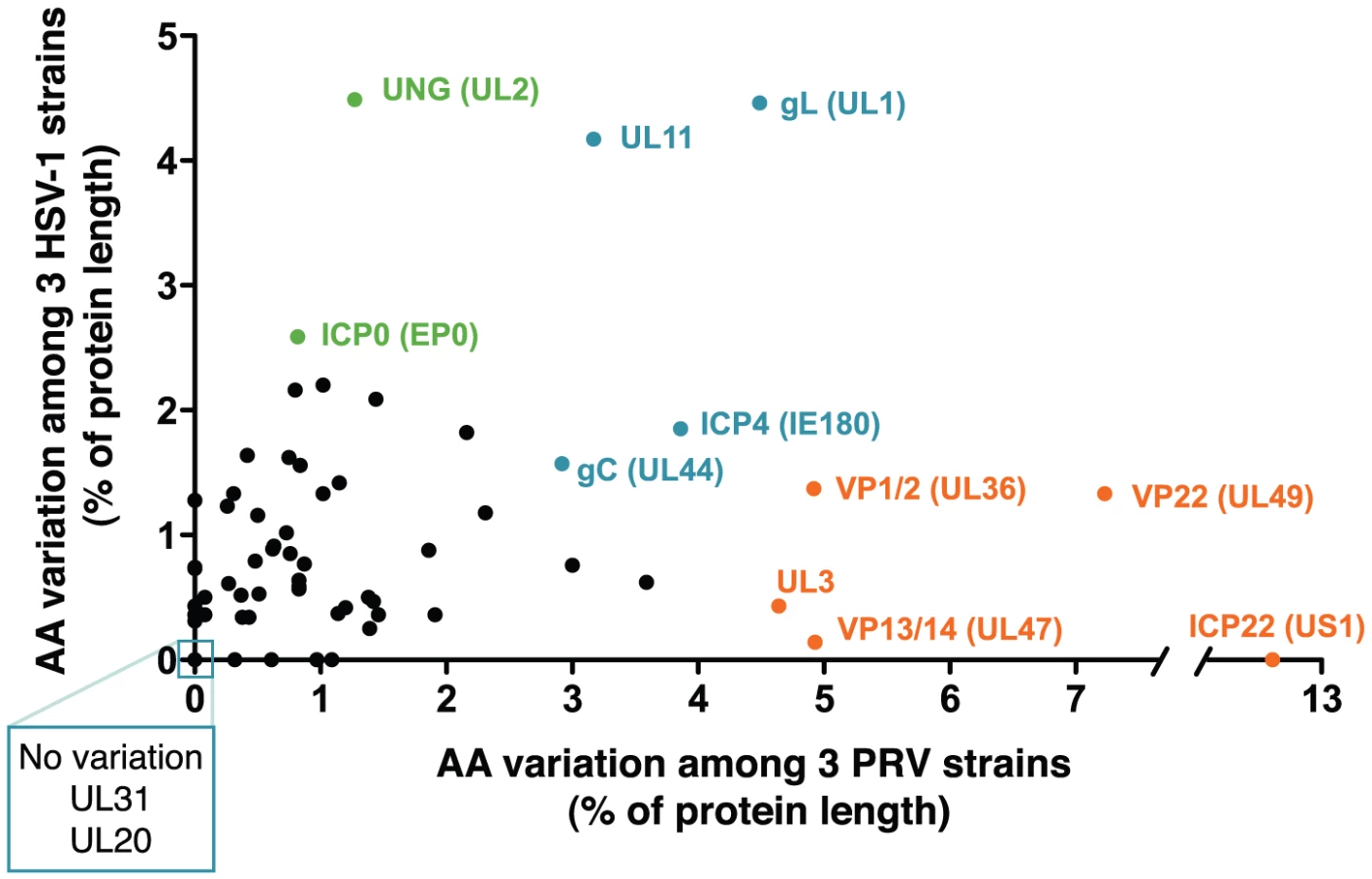
A comparison of the inter-strain variation in homologous proteins of PRV and HSV-1 (Figure 5 and Table S6) highlights several proteins that appear to vary more substantially in one virus than the other. Although ICP22 is the most variable protein in PRV, it is completely invariant among HSV-1 strains 17, F, and H129, as well among the previously described 18 strains of VZV [78], [79], [81]. Likewise, the viral egress protein VP13/14 (UL47) is among the most variant in PRV, but it is well-conserved in HSV-1, while the opposite is true for HSV-1 proteins uracil-DNA glycosylase UNG (UL2) and the ubiquitin E3 ligase ICP0 (EP0) (Figure 5, orange vs. green highlighting). Several proteins, which do not have homologs between HSV-1 and PRV, are also highly variable; these include PRV's viral egress protein UL3.5, which has the third-highest variability of PRV proteins after ICP22 and the tegument protein VP22 (UL49), and the two most variable HSV-specific proteins, which are the neurovirulence-associated protein ICP34.5 (RL1) and the PKR-antagonist US11.
Short sequence repeats (SSRs) are prevalent in the PRV genome
SSRs are widespread in eukaryotic genomes, and mediate functional effects by serving as DNA-binding domains in promoters, protein folding motifs in coding sequences, and sites of inter-molecular recombination [44]–[47]. Since AA repeats generated several examples of inter-strain coding diversity above (Tables 1–3), we investigated the prevalence of SSRs in the PRV genome. SSRs are generally grouped into three main categories: homopolymers, which include a short run of the same base; microsatellites, where the repeating unit is less than 10 bases; and minisatellites, which have a repeating unit of 10–500 bases [44], [45]. The initial description of the PRV genome mapped 26 minisatellite SSRs using a DNA identity scoring matrix [30]. Using software designed to identify all size classes of SSRs and include both perfect and imperfect repeats (see Methods for details), we detected a significantly larger number of repeats, a total of 953 distributed across the PRV Kaplan genome (Table 4 and Table S7; minimum homopolymer length 6). SSRs in PRV occur in both coding and non-coding regions, promoters and open intergenic space, with similar proportions in all three PRV strains (Table 5 and Figure 6A). SSRs of all size classes are distributed throughout the genome, with a slightly higher accumulation of all types in the IR-US-TR region (Figure 1C and Figure S4 in Text S1). The majority of all SSRs in PRV (62%) contain triplet-based repeats (e.g. the repeat unit is a 3-mer, 9-mer, 21-mer, etc.). Likewise, 69% of homopolymers have a triplet-based length. Half of all SSRs are in coding sequences (474/953), and these are largely triplet-based (72%). Triplet-based repeats, as well as insertions or deletions (indels) and partial repeat units of non-triplet-SSRs, help preserve the coding content in the SSR-laden PRV genome because variation in these repeats (addition or removal of repeat units) does not change the reading frame of the downstream sequence.
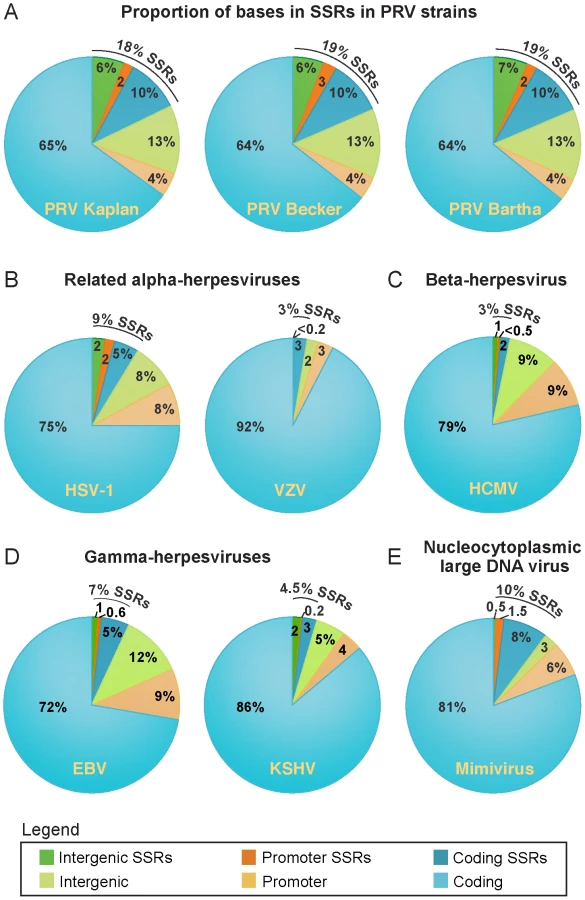


All coding sequences, except the small UL11 gene, contain SSRs (Figure 1C). However it is interesting to note that nineteen genes are free of homopolymers, a size class where expansion or contraction of the SSR is likely to disrupt the reading frame (Table S7). Likewise another 20 genes have regions of at least 1 kb that are homopolymer-free. For instance, the large tegument protein VP1/2 (UL36; 9.2 kb in length) has no homopolymers in its initial 5.5 kb (Figure 1A,C), which contains several domains affecting capsid transport, replication, and neuroinvasion [91]–[95]. In contrast, VP1/2's homopolymer-rich C-terminal region has been previously shown to be dispensable for viral replication [96]. Of the 25 core genes found across multiple families of Herpesviridae that are essential for growth in cell culture [76], 18 have no homopolymers or regions >1 kb that are homopolymer-free. As additional sequences become available for phylogenetic comparison, it may be possible to determine whether this is a chance occurrence or the result of purifying selection.
Since SSRs have not been comprehensively examined in other DNA virus families, we extended these analyses to include the genomes of a wide variety of human herpesviruses, including HSV-1, VZV, HCMV, EBV, and KSHV (Table 4 and Figure 6B–D). To ascertain if these results hold for non-nuclear, non-mammalian viruses, we selected as an outgroup for comparison the nucleocytoplasmic large DNA virus Mimivirus, which infects pathogenic amoebae (Figure 6E) [97]. PRV has the highest overall SSR burden, with short repeats encompassing 18% of the genome, which is roughly double the proportion found in HSV-1, EBV, and Mimivirus, and 5–6 times that of VZV, HCMV or KSHV. In all of these viruses, more than half the SSRs fall into coding regions (Figure 6), creating potential effects on protein structure if these SSRs vary in length between strains. SSRs also occupy a noticeable fraction of the intergenic and promoter regions in PRV and other genomes (Figure 6). For those genomes with a biased nucleotide content, the bias is exaggerated in SSRs (Table 4). PRV's overall genome is 74% G/C, but this level is 79% when all SSR sequences are pooled together. This is similar in HSV-1 (68% G/C overall; 84% in SSRs) and EBV (59% G/C overall; 77% in SSRs), and mirrored in reverse in the A/T-rich genome of Mimivirus (72% A/T overall; 80% in SSRs). PRV thus provides a rich set of SSRs for analysis of a phenomenon that extends to many other viruses.
Previous work in yeast, humans, and other organisms has demonstrated that variation in SSR length, either between individuals or during evolutionary adaptation, can result in phenotypic effects [47]–[50]. Although the overall proportions of SSRs are similar in the PRV Kaplan, Becker, and Bartha genomes (Figure 6A), a comparison across PRV strains revealed that homologous SSRs vary in length between strains (Table 5). Previously, variation in a selection of microsatellites (≤6 bases in length) has been shown for HSV-1, HCMV, and HIV [98]–[100], but the genome-wide complement of all SSR types has not been analyzed. The comparison of homologous SSRs reveals that not all SSRs can be recognized in all three strains (e.g. SSRKa151, SSRKa2093, and SSRKa62103 in Table 5). However the majority of those that do occur in all strains vary in the number of repeating units (of 861 SSRs found in all three strains, 539 vary in number of repeating units). If these SSRs contain transcription factor binding sites or occur in protein coding regions, then these inter-strain differences in SSR copy number may influence gene expression or protein folding domains, and thus lead to phenotypic differences between strains.
Inter-strain variation in SSRs containing CTCF DNA-binding sites
One of the best characterized biological roles for SSRs in herpesviruses are the CCCTC-binding factor (CTCF) binding sites that flank latency-associated transcripts in the genomes of HSV-1 and the gammaherpesviruses EBV and KSHV [101]–[108]. In each of these cases, CTCF binds to motifs within SSRs found near loci that are transcriptionally active during latency; this interaction is proposed to have chromatin insulating and/or silencing effects that maintain a repressed state in flanking genes. CTCF-binding sites occur in several additional conserved locations throughout alphaherpesvirus genomes, as shown by Amelio et al. in a comparison that included HSV-1, VZV, and PRV [104]. Because many PRV SSRs showed inter-strain variation in copy number or length, we investigated CTCF-binding sites in PRV Kaplan, Becker, and Bartha. Of the 17 CTCF binding sites mapped by Amelio et al, 12 were mapped as falling into SSRs in our inter-strain comparison (Table 5; CTCF-binding sites in the repeat-unit consensus are underlined and in bold). All of these vary in repeat-unit length between strains (e.g. Table 5: SSRKa31884, SSRKa115550). Although several have diverged enough to be listed as separate SSRs, their overall location and CTCF-binding ability are preserved (e.g. Table 5: SSRKa115377 and SSRBe115911; see Table S7 for orthologous SSRBa115943). The greatest inter-strain variation in SSR length occurs at SSRKa15795, between UL46 and gB (UL27), where PRV Becker has three times as many repeating-units as either PRV Kaplan or Bartha. This SSR contains both CTCF-binding sites and a non-canonical Egr1/2 binding site, both of which have repressive effects on expression of nearby genes in HSV-1 [57], [104], [109]–[111]. Initial studies show that gB levels in PRV Becker-infected lysates do not appear significantly lower than those in PRV Kaplan or Bartha (Figure S3 in Text S1). Further work will be required to determine if the flanking SSR length affects gB expression and function.
In the only previous publication comparing full-length genomes of HSV-1 (strains 17, H129, and F), the length of fourteen major SSRs throughout the genome were not determined and were instead set to match the reference genome length [81]. These fourteen SSRs, classically termed reiterations in the HSV literature [37], [38], [82], [83], correspond to the fourteen CTCCC-domain-containing SSRs defined by Amelio et al. [104]. To discern if inter-strain variation such as that observed in the PRV genomes is found in HSV-1 as well, we PCR amplified and sequenced two of these SSRs from the HSV-1 strains F and H129. Both SSRs displayed inter-strain variation in copy number, with the reference strain 17 (GenBank Accession NC_001806) having more SSR units at both sites than either the clinical isolate H129 or the laboratory strain F (IRS reiteration 3 [CTRS3 in Amelio et al.]: 6.5 copies in strain 17, 4.7 copies in H129, 1.7 copies in F; US reiteration 1 [CTUS1 in Amelio et al.]: 10 copies in strain 17, 2 copies in H129, 2 copies in F). These data suggest that inter-strain variation in SSR length may affect CTCF-binding efficiency in HSV-1 and could contribute to inter-strain differences in related phenotypes.
Estimation of selected SSRs by Coverage Adjusted Perfect Repeat Expansion (CAPRE)
Annotation of SSRs in the draft PRV genome assemblies had revealed several discrete areas in each genome where peaks of very high coverage coincided exactly with perfect SSRs: for example a peak of over 100,000-fold coverage around an SSR at position 15,600 in the PRV Becker genome (Figure S1 in Text S1 and Table 5). This very high coverage (>2 standard deviations above the median) occurred at three SSR sites in PRV Kaplan, three SSRs in PRV Becker, and four SSRs in PRV Bartha. (Figure S1 and Table S3 in Text S1, also noted in Table 5). De novo assembly methods cannot distinguish whether repeated sequence reads originate from perfect, extended copies of an SSR unit, or from additional coverage depth of a single unit, and the software therefore creates a final assembly with the minimal number of repeating units supported by the data [112]. In fact, the high coverage peak in PRV Becker coincides with the largest SSR array of perfect repeats in the original mosaic PRV genome, which had 39 copies of a 15-mer at this site [30], suggesting that this peak might result from de novo-assembly compression of the homologous SSR in PRV Becker. The short unit size of this SSR (15-mer) meant that its copy number could only be estimated by RFLP and Southern blotting, and the likely amount of perfect repeating units could lead to laddering and polymerase slippage errors in PCR analysis. We therefore devised an approach to computationally estimate the length of these perfect tandem repeats that demonstrate potential compaction during assembly, in order to facilitate future HTS-genome assemblies and preserve coverage-based information on inter-strain variation in SSR length.
Coverage-Adjusted Perfect Repeat Expansion (CAPRE) is based on methods used for copy number variant estimation in HTS data [113], [114], which is used in larger genomes to detect duplications of chromosome regions or individual genes. As in copy number estimations, CAPRE takes into account the observed coverage depth and estimates the length of intergenic SSRs based on the expected sequence depth for its G/C nucleotide content (Figure S2A in Text S1). In order to estimate SSR length conservatively, CAPRE predicts SSR length based on the median coverage expected for a given G/C content, and can also be used to predict potential upper - and lower-range estimates based on the upper and lower quartile ranges of this coverage (Figure S2A in Text S1). Because it is imprecise, we applied this method sparingly, and used it only at intergenic sites where coverage depth exceeded two standard deviations from the median and coincided with a perfect SSR. We used CAPRE to expand the lengths of three SSRs in PRV Becker, three in Kaplan, and four in Bartha (Figure S1 and Table S3 in Text S1). This did not affect the overall count of SSRs in Table 4, but did affect the length of several SSRs included in Table 5 (e.g. SSRKa15795; these are marked). We incorporated these CAPRE-expanded SSRs into the overall assembly of each genome before final annotation and comparisons. The CAPRE method provided a means to estimate the length of these repeats and yielded a more even distribution of sequence read coverage at these sites in the final genome (Figure S1 in Text S1).
To test whether the CAPRE script provides a reasonable estimation of SSR length, we compared the CAPRE-expanded SSRs to alternative sources of data on actual SSR length. First, we compared the three CAPRE-expanded SSRs of PRV Kaplan (Table S3 in Text S1) to their counterparts in the original PRV mosaic genomes. Each of these SSRs falls into areas of the mosaic genome that were originally derived from the Kaplan strain, facilitating comparison of our estimated lengths to SSR lengths that were determined in strain Kaplan by traditional Sanger sequencing. For SSRKa107138, the CAPRE-estimated length nearly matches that of the Sanger-sequenced Kaplan isolate (12.5 copies here vs. 10.5 copies in the mosaic), while for the other two it provides a conservative under-estimate (SSRKa2093 is 8.3 copies here but was 17.3 in the mosaic; SSRKa17595 is 13.7 copies here, but was 39 copies in the mosaic).
Next, we used RFLP and Southern blot analysis to estimate the length of the most divergent SSR between strains (Table 5, SSRKa15795); this SSR is also the only one expanded by CAPRE for all three strains (Table S3 in Text S1; SSRKa15795, SSRBe15739, SSRBa15751). We hybridized a probe to this SSR against SalI-digested DNA from PRV Kaplan, Becker, and Bartha (Figure 7). The size of the SalI fragment reflects a much larger size in PRV Becker than in Kaplan and Bartha, and further reveals that this SSR varies in length even within the purified PRV-Becker stock. A prior Southern blot analysis by Simon et al. showed that this same SSR varied in length between strains and within plaque isolates of a given PRV strain [115]. As occurs here with strain Becker, those authors found that the strain Phylaxia had a wide and blurry band of probe hybridization, while other PRV strains (Kaplan and Dessau) had tight bands [115], suggesting strain-specific differences in SSR length stability. To investigate the stability of this SSR, we serially passaged the plaque-purified PRV Becker stock ten times in culture (potentially 20–30 cycles of replication at low multiplicity of infection (MOI); see Methods for details). RFLP analysis of this stock, termed Becker p10, differed from the parental PRV Becker only in the classically variable BamHI fragments 10 and 12 (Figure 2B and 7A), which have been shown to vary with repeated passages [22], [35], [41], [42]. However the band distribution of SSRBe15739 shifted slightly in the Becker p10 stock (Figure 7). The upper length estimate for SSRBe15739 (Table S3 in Text S1) falls into the band distribution observed in Figure 7B, and the predicted ratios across strains (Table 5) likewise mirror the observed differences. Thus the CAPRE script met our goal of conservative length estimation, and allowed correct prediction of the extreme inter-strain size differential of the homologous SSR that falls between UL46 and gB (UL27).

PCR validations reveal homopolymers as mutational hotspots
We also used PCR sequencing to refine and validate selected areas in the assembly (Tables S2 and S4, and Figure S1 in Text S1). The majority of these PCR products confirmed divergence in the newly sequenced strains from the previous mosaic reference genome, while the remainder corrected SSR-based issues in the assembly, e.g. for Becker UL3.5 and VP1/2 (UL36), and Bartha VP1/2 (Tables 1–3 and Table S2 in Text S1). To assess sequence stability in PRV genomes over time, we PCR-amplified and sequenced the same regions of parental stocks of these plaque-purified isolates. We found no base pair differences between 8.8 kb of the parental and progeny genomes, in ten spatially distributed PCR comparisons (Table S2 in Text S1).
We and others have previously demonstrated that direct Sanger sequencing of PCR products, vs. cloning and subsequent sequencing, provides useful and sensitive detection of minority variants in a population [78], [81]. In a prior sequencing study, we detected variation at a C6 homopolymer in an HSV-1 stock; plaques picked from this stock reproduced either homogeneous C6 or C5 variants [81]. Although we were not searching for minority variants, all of the above PCR sequences were visually screened for any evidence of such variation. We detected two such sites, one each in PRV Becker and Bartha, in different homopolymers upstream of ICP22 (US1). ICP22 has a high concentration of homopolymers in its upstream region (Figure 1A,C). At a C10 site upstream of ICP22, the majority of the PRV Becker PCR products reflected a homopolymer length of ten, while a minority of the products had a length of nine (Figure S2C in Text S1); these may represent the contributions of viral nucleocapsid DNA population used as a template. Likewise, at a different C10 homopolymer upstream of ICP22, PCR sequencing of PRV Bartha revealed homopolymer variants of nine, ten, and eleven (data not shown). Although these variants could reflect polymerase slippage during PCR or Sanger-sequencing of the PCR products, both PCR products contain nearby C8 homopolymers that show no minority products. The homopolymer variants described here, along with accumulating evidence from other alphaherpesviruses, suggests that homopolymers are mutational hotspots in PRV as well [78], [81], [116]–[119].
Sequence polymorphisms in plaque-purified and passaged strains
There is limited evidence for sequence polymorphisms in large DNA virus genomes; these include several studies that noted SSR-based variation in clonal stocks of herpesviruses [35], [78], [120], several recent studies of variation in HCMV DNA from both clinical and lab-passaged strains [121]–[124], and the recent observation of a small number of polymorphic bases scattered throughout the large DNA genome of Mimivirus [125]. We therefore used single-nucleotide polymorphism (SNP) detection software to check for any variation in base calls when HTS data from each strain were aligned back to the finished genome (see Methods for details). A small number of bases (0.004–0.03% of each genome) were indeed called as polymorphic in each plaque-purified isolate (22 in PRV Kaplan, 37 in Becker, 6 in Bartha). Unlike HTS genomes with low coverage depth, HTS data for these viral genome sequences provides deep coverage and a strong likelihood that these base variations are not sequence errors. An examination of the percent of reads contributing to each polymorphic base calls revealed that in most cases, the alternative base was present in a minority of the sequence reads, from 1–20% (Figure 8A).

PRV Becker was the only strain with several polymorphic bases approaching 50–50 variation in the primary versus the alternative base (Figure 8A). We therefore investigated the stability of these polymorphic bases in the serially passaged Becker p10 strain. Nucleocapsid DNA from the Becker p10 stock was sequenced and aligned to the PRV Becker genome for SNP analysis (see Table S1 in Text S1 for details of HTS data generated). We found no increase in the overall number of polymorphic base calls after serial passage (Becker: 37, Becker p10 : 30), and only a slight shift in the frequency of observation of the secondary base call (Figure 8B). Many polymorphic sites in the Becker p10 stock (28 of 30) were in the same position as in the parental, purified Becker stock but had shifted in allele frequency. An additional 9 polymorphic sites either were lost or gained during the passaging that produced the Becker p10 stock. The four most polymorphic sites in the original PRV Becker stock were still called as polymorphic in Becker p10, but had shifted in allele frequency (Figure 8D). Interestingly, only one SNP in any of these strains affected a coding sequence, and this one (P2172A) occurred in the proline-alanine rich region of Kaplan VP1/2 (UL36) that is dispensable for viral replication in vitro [95], [96]. The SNPs in these plaque-purified and limited-passage strains were almost exclusively located in non-coding regions.
Since serial passaging of a plaque-purified population had little effect on these polymorphisms, we examined variation in one of the non-purified viral stocks that gave rise to these plaque-purified isolates. Here we sequenced the oldest viral stock available in the lab, which is the parent of the plaque-purified PRV Kaplan used for these studies [62], [126]. RFLP profiles of this PRV Kaplan stock, termed Kaplan n.p. (not purified), matched that of the plaque-purified PRV Kaplan isolate (Figure 2B). HTS data for Kaplan n.p. was aligned to the PRV Kaplan genome and used for SNP calling (see Table S1 in Text S1 for details of HTS data generated). This stock possessed 547 polymorphic sites relative to the plaque-purified genome (0.39% of the genome; Figure 8C and Figure S5 in Text S1). As found for SNPs in the plaque-purified strains, most alternative base calls resulted from variants present at 1–20% (Figure 7C). Strikingly, the majority of these SNPs occur in coding regions, and are well-distributed across the PRV genome (Figure S5 in Text S1). Because these data cannot distinguish how many polymorphisms are present in any one viral genome of the Kaplan n.p. stock, versus distributed across the entire viral population in that stock, we cannot determine the extent of selection that occurred during plaque-purification. Future sequencing technologies that can examine single genomes will be required to address this. Together with the results above, we suggest that subtle variations such as these SNPs and homopolymer length variants provide the genetic diversity to help these strains adapt to future evolutionary pressures.
Defining a new reference genome for PRV
The genome currently used as a reference for PRV is a mosaic of six strains [30] We therefore propose that the PRV Kaplan genome presented here (GenBank Accession JF797218) serve as a new reference genome for PRV. Strain Kaplan contributed 86% of the sequence in the mosaic reference genome, while the remainder included sequences from strains Becker, Rice, Indiana-Funkhauser, NIA-3, and TNL. Accordingly, we compared our complete PRV Kaplan genome to that of the original mosaic reference genome. Not surprisingly, the majority of protein coding differences between Kaplan and the mosaic genome (81%; 141 of 173 amino acid (AA) differences) occur in twelve of the thirteen proteins that were originally sequenced from non-Kaplan strains: gB (UL27), ICP18.5 (UL28), ICP8 (UL29), UL43, gC (UL44), TK (UL23), ICP0 (EP0), gG (US4), gI (US7), gE (US8), US9, US2 (see Table S5 in Text S1 for specific AA differences).
Several of these sequence differences significantly affect the resulting protein because of frameshifts in the strains used for the mosaic genome. The largest frame-shift changes 46 AAs in the extracellular domain of gG (US4), which has been mapped as a chemokine-binding region [67]. The gG sequence in the mosaic genome was derived from PRV strain Rice. Alignment of the three new PRV strain genomes, along with two geographically distinct gG sequences deposited in GenBank (Ea, China: AY319929, NIA-3, Ireland: EU518619), revealed that the PRV Rice strain included in the original mosaic genome is the only one to possess this frame-shift sequence and cannot be representative of most PRV strains. Similarly, all three new genomes share a common sequence of ICP8 (UL29; only 1 AA difference in PRV Becker; Table 3), which is a single-stranded DNA binding protein that functions in both replication and recombination of the viral genome [127], [128]. This new ICP8 sequence differs from the TNL strain sequence of ICP8 found in the mosaic PRV reference at a total of 20 residues (Table S5 in Text S1), including a compensated frame-shift that affects a stretch of 8 amino acids immediately flanking the zinc finger domain [129].
Conclusions
Herpesvirus genomes: a microcosm of HTS eukaryotic genome assembly
Herpesviruses are among the largest DNA virus genomes and cause significant human disease, making the characterization of their sequence diversity a priority. While viral discovery screens using HTS often produce sufficient data to assemble entire RNA virus genomes [130]–[136], the ten-fold larger size of herpesvirus genomes means that only directed sequencing projects have thus far produced data on new strains [81], [120], [137], [138]. Herpesvirus genomes represent a microcosm of the features found in eukaryotic and bacterial genomes: abundant SSRs, histone modifications, splice sites, and microRNAs, among others, with frequent recombination at the large inverted repeats. An improved understanding of how these elements vary in these viral genomes may shed light on related sequence features in larger genomes, where sequencing of repeated generations or multiple related isolates may be prohibitive in cost or computational time. For instance, while the G/C coverage bias seen in these herpesvirus genomes has been previously observed in higher organisms [59], [60], there has not yet been sufficient depth of coverage and variety of G/C-rich sequence structures to correlate specific sequences with specific coverage-depth consequences. The G/C-bias of PRV and HSV-1 genomes along with their deep sequence coverage (>2,000-fold on average) provide data for future exploration of these issues, which will then provide insight relevant to all future sequencing endeavors.
Multiple glycoprotein mutations in the PRV Bartha vacfcine strain
Herpesvirus virions are coated in glycoproteins, which play a major role in viral spread from cell to cell and host to host, and are thus crucial to pathogenesis and vaccination strategies in vivo [8], [9], [139]–[141]. PRV has 11 glycoproteins, with functions including fusion (gH, gL, and gB), cellular attachment (gC, gD), rate of virion penetration (gN, gM), triggers of host immunity (gG, gI), viral transport in axons (gE), and virion egress (gK). The genome of PRV Bartha reveals mutations in genes that encode the majority of this suite of glycoproteins. Previously known changes in PRV Bartha that affect glycoproteins included the US-region deletion that removes gE and gI, a signal sequence mutation of gC, and a residue change affecting the N-glycosylation site of gM; all of these have also been shown to affect PRV Bartha's spread in culture, and the role of gE and gI have been confirmed to affect the attenuation of PRV Bartha's virulence in vivo [21], [28], [31], [61]–[65], [142]. To this list, we now add several mutations in the coding sequences of gN, gB, gH, gG, and gD, which are unique to PRV Bartha and are not seen in the virulent PRV Kaplan or Becker strains. Future work can now explore the relevance of these sequence differences to the attenuation of PRV Bartha's virulence in vivo, and their potential use in aiding the development of an HSV-1 vaccine strain.
SSRs and homopolymers fuel inter - and intra-strain diversity
Prior to this study, few PRV SSRs had ever been analyzed for potential inter-strain variation [86], [115], [143]–[145]. For decades, researchers have known that certain regions of the PRV and HSV-1 genomes are variable by RFLP analysis of repeatedly passaged virus stocks [22], [35], [42], [43], but little work has been done to elucidate the basis of this variation. The most variable sections of the PRV genome by RFLP analysis are located within BamHI fragments 10 and 12 (Figure 2), which represent the IR and TR copies of ICP22 (US1) and its upstream region. ICP22 has the highest inter-strain variability of any PRV protein (Figure 5). This region includes both areas of homopolymer length variation found in the plaque-purified strains (see above), has a large complement of SSRs of all size classes (Figure 1C), contains several SNPs in its flanking untranslated regions in every strain (Figure S5 in Text S1), and was highly refractory to PCR analysis (data not shown). Taken together, this region shows uniquely high variability that extends well beyond the prior RFLP observations. The ICP22 (US1) protein of PRV has been virtually unstudied at the protein level, so that further work is required to understand its role and the significance of its variability between strains [86], [87]. Our analysis thus reveals a likely target for the historical variability of restriction-digest fragments of this region of the PRV genome, and suggests that similar features could be associated with the classically-variable fragments of the HSV-1 genome as well.
Although larger SSRs are more noticeable to the eye, homopolymers of six or more consecutive bases are the most abundant class of SSRs in PRV and all viral genomes thus far examined. These numbers would only increase if we included homopolymers of five or fewer. Homopolymers have been previously suggested as mutational hotspots for HSV, but only in the context of two genes where they have been well-studied. First, resistance to the drug acyclovir and related nucleoside analogs is often mediated by changes in homopolymers of the TK (UL23) gene, an observation documented in several alphaherpesvirus species [117]–[119], [146]–[148]. Second, variation in the human antibody response to HSV occurs because of homopolymer mutations in the gG (US4) gene [149], [150].
We now suggest that homopolymers across the genome are mutational hotspots for evolutionary diversity in all alphaherpesvirus strains, and potentially in other virus families as well. Examples from the literature support this, with a wide array of examples mentioned in passing as part of other studies: the C4→C6 (wild-type→mutant) shift in HSV-1 strain 17 that caused early struggles in recognizing ICP34.5 (RL1) as a valid gene [151], a C7→C6 deletion in the vhs (UL41) gene of the HSV-2 HG52 strain [152], a T7→T6 mutation in UL5 of an attenuated Marek's disease virus genome [153], a spontaneous G7→G8 insertion in gE (US8) in an engineered strain of PRV [154], among others [78], [81], [99], [100], [155]–[158]. These examples, in conjunction with the clinical examples in TK and gG above, and our own data presented here, demonstrate the homopolymer mutations can occur throughout the herpesvirus genome. The aforementioned studies of TK and gG sequences in clinical samples demonstrate that homopolymer mutations occur readily during human infection. Together these data suggest that this highly abundant class of SSRs could provide a major source of adaptive variation for viral strain divergence. Beyond these viruses, homopolymer variation has been previously found in organisms from yeast to worms to humans [45], [159]–[162]. A significant proportion of cancer-associated mitochondrial DNA mutations occur at homopolymers [163]–[165]. As described earlier, changes in SSR length have been demonstrated to affect gene expression, protein interactions, and chromatin binding, among other functions [45], [47]–[50]. Future study of homopolymeric and SSR-based variation in herpesviruses may help to reveal the evolutionary fitness contributions of these mutational hotspots.
Methods
Virus stocks and passaging
PRV Bartha is a highly passaged vaccine strain, derived from the original Aujeszky strain which was isolated in Hungary [29]. PRV Becker is a virulent field isolate from dog, originally isolated at Iowa State University (USA), with subsequent laboratory passage [166]. PRV Kaplan is a virulent strain with extensive laboratory passage, likely derived from the Aujeszky strain [126], [167]. All viral stocks were grown and titered on monolayers of PK-15 pig kidney cells (ATCC cell line CCL-33). Stocks of each virus were triple-plaque-purified, expanded, and used to infect cells for a nucleocapsid DNA preparation. Viral nucleocapsid DNA was prepared by previously published methods [81], [168], [169].
A passaged PRV Becker strain (Becker p10) was produced by infecting a monolayer of cells with the plaque-purified stock at a multiplicity of infection (MOI) of 0.01. At full cytopathic effect (CPE), a small aliquot of this virus was used to directly infect a fresh monolayer of cells, and this procedure was repeated a total of ten times. The resulting stock was used to prepare nucleocapsid DNA for sequencing and RFLP analysis.
Illumina library preparation and sequencing
DNA sequencing was carried out according to manufacturer protocols and reagents, using an Illumina Genome Analyzer II with SCS 2.3 software at the Princeton University's Lewis-Sigler Institute Microarray Facility. Five micrograms of nucleocapsid DNA was sequenced for each strain, using either one (PRV Kaplan, Becker p10) or two (Becker, Bartha, Kaplan n.p.) flowcell lanes. All sequencing runs were 75 cycles in length, except for one Becker and one Bartha lane of 51 cycles. The total number of sequence reads generated for each strain are listed in Table S1 (in Text S1). All Illumina sequence data has been deposited at the NCBI Short Read Archive under Accession ID SRA035246.1.
Initial data processing and quality control
Initial data processing included several steps: 1) Illumina output converted to a standard file format, 2) library adaptor contaminants removed, 3) host genome sequences removed, 4) mononucleotide reads removed, 5) duplicate runs combined, and 6) quality and length trimming applied. All data and scripts described here are available at a genome-browser (http://viro-genome.princeton.edu) and data analysis website (http://genomics-pubs.princeton.edu/prv) hosted by Princeton University's Lewis Sigler Institute.
First, a script from the FASTX-toolkit developed by the Hannon lab (http://hannonlab.cshl.edu/fastx_toolkit/) was used to remove adaptor sequences resulting from the Illumina library preparation. Next, because these PRV viruses were grown in pig kidney cells, we used the Bowtie software package [170] to compare the sequence data against the Sus scrofa pig genome (NCBI build 1.1) and remove any sequences perfectly matching the host genome. The percent of contaminating host DNA is listed for each strain in Table S1 (in Text S1). Finally, we filtered out any reads that were entirely mononucleotides, which we previously found can confound genome assembly [81]. Finally, where relevant, we concatenated sequence data from two sequencing runs.
Two scripts were then used to remove poor-quality base calls from the end of the Illumina short-sequence reads. First, we used an adapted version of the quality-trimming script (TQSfastq.py) from the SSAKE de novo assembly software package [171]. We modified the parameters for quality threshold (T) and consecutive bases (C) above threshold, producing trimmed datasets for each strain with the default settings of T10, C20 or a more stringent quality control trimming of T20, C25. We then used the more stringently-filtered dataset as the input to a universal length trimmer from the FASTX toolkit, which truncated all sequences in the data file at a specified length, in this case either 41 or 51 bp. This generated four quality-filtered and trimmed datasets for each strain.
De novo assembly
The SSAKE de novo assembler [171] was used to join the short single-end Illumina reads into longer blocks of continuous sequence, or contigs. Each of the four FASTQ files generated above was assembled by SSAKE under two independent conditions. First the default settings of SSAKE were used. Then the trim option was applied to each of the four input files during assembly, to trim two bases from the end of each contig once all possible other joins had been exhausted. This produced a total of eight SSAKE assemblies for each viral strain. These eight alternative sets of SSAKE contigs were combined and used as inputs to a long-read assembler, based on an approach used successfully for HTS assembly of HCMV genomes [137].
The Staden DNA sequence analysis package was used for further genome assembly of the long sequence contigs generated by SSAKE [172], [173]. The Pregap function was used to process and rename all contigs, which were then assembled using the standard “independent assembly” function of Gap4, with default settings. Contigs were sorted into descending size order and outputted as a normal consensus. This generated a multi-line FASTA formatted file that we inputted to NCBI's blast2seq program [174], for comparison to the PRV mosaic reference genome (Accession number NC_006151) [30]. This program produced pairwise alignments of each contig against the reference genome, allowing us to order the contigs along the genome and to flag potential bad joins generated by the assemblers. Contigs with suspicious joins were visually inspected in the Gap4 Contig Editor. These joins often occurred at extended runs of Gs or Cs, where disparate regions of the genome were joined solely as a result of overlapping mono-nucleotide stretches. The final assembly was created in gap4 by manually joining the minimum possible number of contigs. Final genome assemblies were further improved by PCR validation and repeat expansion, and verified by RFLP analysis (see below). All genome sequences are deposited with annotations (described below) in the NCBI Nucleotide (GenBank) collection: PRV Bartha: JF797217, PRV Kaplan: JF797218, PRV Becker: JF797219.
Annotation of genes and coding sequences
Annotation of the new PRV genome sequences was created by BLAST homology-based transfer of annotations from the prior mosaic reference genome (NC_006151) to PRV Kaplan, using previously described scripts [81], [174]. Annotations of PRV Kaplan were then similarly transferred to PRV Becker and Bartha. Scripts for automated annotation transfer are available for download at http://genomics-pubs.princeton.edu/prv. Annotation transfer can fail when several base pairs of divergence or indels occur at the gene boundaries; these instances were addressed by manually varying the BLAST parameters to improve alignment and/or visually inspecting a pairwise alignment of the new strain against the reference. Entrez Gene IDs for all PRV, HSV-1 and VZV genes are listed in text format in Text S1, as well as hyperlinked in Table S6.
Sequence alignment
The completed PRV genomes were aligned using the mVista genomics analysis tool with global LAGAN alignment [175], [176]. The VISTA Browser was used to visualize genome-wide conservation based on this alignment. The VZV genome (NC_001348) was used as an outgroup for tree generation in MacVector v11.1.2 (MacVector, Inc.) by the neighbor-joining method. One thousand rounds of bootstrap analysis provided confidence values for the branch points. Similar trees were obtained using alternative methods, such as clustering by the unweighted pair-group method with arithmetic mean (UPGMA) or following the precedent of single-gene comparison of the variable gC (UL44) nucleotide sequence [33], [34], [177].
RFLP and Southern Blot analysis
Digestion of nucleocapsid DNA was performed to verify predicted fragment sizes corresponding to the newly assembled genomes. RFLP reactions utilized 4 µg nucleocapsid DNA per reaction, while Southern Blot digests used 1 µg nucleocapsid DNA. Reactions included viral nucleocapsid DNA, BamHI or SalI High Fidelity restriction enzymes (New England BioLabs), and supplied buffers and reagents as directed by the manufacturer; these were incubated at 37°C overnight. The addition of 5 µg/ml of ethidium bromide to an 0.8% agarose gel and to the 1X TAE running buffer allowed for enhanced UV visualization of fragments. Gel electrophoresis of the digested samples ran at 30 volts for approximately 48 hours at 4°C.
Southern blotting used the NEB Phototope-Star detection kit for nucleic acids (New England BioLabs) according to manufacturer's instructions. Briefly, the SalI RFLP gel was transferred to a nylon membrane and UV crosslinked. After blocking, the boiled probe was hybridized to the membrane overnight at 68°C, and detected by sequential application of streptavidin, biotinylated alkaline phosphatase, and finally the chemiluminescent reagent CDP-Star (New England BioLabs). The biotinylated probe was synthesized and HPLC-purified (Integrated DNA Technologies/IDT) to match SSRKa15795 and the homologous SSRs in other strains. The probe consisted of three tandem copies of the SSR unit (a 15 mer), using the reverse-strand sequence of the SSR to allow for the incorporation of a biotinylated thymidine (T*, one per oligonucleotide): 5′-TCTCCCCTCCGTCCCTCTCCCCT*CCGTCCCTCTCCCCTCCGTCCC-3′.
PCR validation of selected regions
Primers were designed for the amplification of several genes from nucleocapsid genomic DNA of all three PRV strains and their parental lysate DNA. Primer pairs are listed in Table S4 (in Text S1). To allow for easier PCR access, template DNA was boiled for 5 minutes and immediately cooled on ice. Initial PCRs were executed in 50 µl volumes using 1 µl of template. The reaction setup contained 1X Advantage 2 DNA polymerase (Clontech), 1X buffer as supplied by the manufacturer, 2% dimethyl sulfoxide, 1.2 M betaine (Sigma), each primer at a concentration of 0.5 µM, and each deoxynucleoside triphosphate at a concentration of 250 µM. Initial PCR conditions using an Eppendorf thermocycler are as follows: Initial denaturation at 95°C for 3 minutes, followed by 25 cycles of denaturation at 95°C for 30 seconds, primer annealing at 50°C for 30 seconds, and primer extension at 68°C for 2 minutes, with a final extension step at 68°C for 10 minutes. For more difficult gene amplifications an alternate reaction setup was used: 0.6 U Takara Ex Taq polymerase (Takara); 1X buffer as supplied by the manufacturer; 5% dimethyl sulfoxide; each primer at a concentration of 1 µM; each deoxynucleoside triphosphate, with equal amounts of dGTP and 7-deaza-2′-dGTP (Sigma Aldrich), at a concentration of 200 µM; and 1 µl of template DNA for a total reaction volume of 25 µl. Alternate PCR conditions were also used: Initial denaturation at 95°C for 5 minutes, followed by 40 cycles of denaturation at 95°C for 1 minute, gradient primer annealing temperatures from 55–75°C for 1 minute, and primer extension at 72°C for 2 minutes, with a final extension step at 72°C for 7 minutes.
For PCR validations of PRV Becker and Bartha parental DNA, we used lysates from the oldest available laboratory stocks of each virus. HTS data had already revealed that the oldest available stock of PRV Kaplan in the lab contained several hundred polymorphic base calls (described in Results and Figure 7C), so we instead compared results from PCR amplification of a stock of gH-null PRV Kaplan provided by Mettenleiter and colleagues [178]. By selecting these stocks, all of which were historically separated from the sequenced strains by multiple passages, we aimed to maximize the opportunity to detect sequence divergence relative to the new genomes.
Western blot analysis
Cell lysates from PK15 cells were collected at 12 and 24 hours post infection into ice cold PBS and centrifuged for 3 minutes to pellet the cells and allow aspiration of the supernatant. The cells were lysed with RIPA light buffer (50 mM Tris/HCl (pH 8.0), 150 mM NaCl, 5 mM EDTA, 1% NP-40, 0.1% SDS, 0.1% Triton X-100). Insoluble cell debris was pelleted by centrifugation at 4°C, and the supernatant was collected for protein measurement. 50 µg of protein from the RIPA supernatant was brought up to a common volume using Laemli buffer (100 mM Tris/HCl (pH 6.8), 4% SDS, 200 mM DTT, 0.2% bromophenol blue, 20% glycerol) for each sample. These were boiled for 5 minutes at 95°C, electrophoresed through a 10% SDS-PAGE gel, and transferred to a nitrocellulose membrane (Whatman PROTRAN) using a Bio-Rad semi-dry transfer cell. The membranes were blocked using 5% non-fat milk and PBS-T. Primary and secondary antibodies were diluted in 1% non-fat milk in PBS-T. Proteins were visualized using rabbit polyclonal antibodies for gH (UL22) (1 : 2000) and VP1/2 (UL36) (1 : 10,000); mouse monoclonal antibodies for gB (UL27) (1 : 1000), VP5 (UL19) (1 : 1000) and β-actin (1 : 1000); goat horseradish peroxidase-conjugated secondary antibodies; and SuperSignal chemiluminescence reagents (Thermo Scientific) as indicated by the manufacturer's instructions. Band intensities were measured using the ImageJ (NIH) Gel Analyzer module.
Coverage depth and polymorphic base detection
For quality control assessment of the finished genome assemblies, we used the Bowtie [170] and Samtools [179] software packages to assess the depth of sequence coverage and check for variant base calls. First, Bowtie (option –best) was used to align the Illumina sequence reads used for assembly against the finished genomes. Then three Samtools commands (view, sort, and pileup, with default options) were used to format the Bowtie alignment output and measure the depth of sequence read coverage (a pileup file) at each base of the finished genome sequence. The Integrated Genome Browser (IGB, [180]) was used to visualize each pileup graphically (a wiggle or wig plot; Figure S1 in Text S1). Finally, the Samtools varFilter command (default options, depth 40,000) was used to detect any variant base calls in the alignment of sequence reads back to the finished genomes. Assessment of polymorphic bases in the passaged (Becker p10) and non-purified (Kaplan n.p.) genomes was done by aligning sequence data for these stocks against the finished genome from the matching plaque-purified stock (i.e. Beckerp10 was aligned to the finished PRV Becker genome, and Kaplan n.p. to the PRV Kaplan genome).
Additional filtering was used to remove potential erroneous SNP calls [181]. These filters were based on a manual examination of all SNPs in strains Kaplan and Bartha. First, SNP locations were screened and flagged if they met any of the following criteria: adjacent to homopolymers of length ≥6, directional strand bias >85%, or overall coverage depth <100. All flagged SNPs were manually examined using the Integrative Genomics Viewer (IGV) to display sequence reads aligned to the genome sequence [182]. SNPs with likely homopolymer-based alignment error, unidirectional sequence read support, or signs of site-specific error were discarded [181]. Both filtered and unfiltered lists of DNA polymorphisms are available for download at http://genomics-pubs.princeton.edu/prv. Frequency distributions of polymorphic base calls were plotted using Prism v5.0 (GraphPad Software, Inc.).
Estimation of G/C coverage bias
To measure G/C coverage bias, we followed the method of Frazer and colleagues [59] (Figure S2A in Text S1). Briefly, each genome was divided into sequential 10-mers. The coverage depth of each 10-mer was determined by taking the average coverage depth of the bases in the 10-mer. These were placed into bins according to G/C content, i.e. the number of G or C bases in the 10-mer. We recorded the number of 10-mers and the median coverage depth in each bin.
Coverage Adjusted Perfect Repeat Expansion (CAPRE)
We used the coincidence of very high sequence coverage at perfect repeats in each PRV genome to estimate the actual length of these SSRs. The CAPRE script was applied only to selected regions meeting these criteria: an intergenic region, with coverage more than two standard deviations from the median, and centered on a perfect SSR with repeating units exceeding the median length of the filtered Illumina sequence reads. For each intergenic region meeting these criteria, an SSR unit that most closely matched the median Illumina read length was defined, and its genome position boundaries noted. The CAPRE script first determined the G/C content of the inputted SSR unit and used the G/C coverage bins above to obtain the expected median coverage depth for this SSR unit. The script then took the defined SSR unit boundaries and measured their observed sequence coverage. The script then estimated how many copies of the defined SSR unit would be needed to achieve the expected coverage depth, and inserted the appropriate number of SSR units into the genome sequence. The position of subsequent CAPRE regions was iteratively adjusted to account for expansion of the preceding region. To produce upper and lower estimates of SSR length, we ran the CAPRE script again and estimated the SSR length according to the upper and lower quartiles of observed sequence coverage (Figure S2 in Text S1) for each G/C content, instead of the median.
Short Sequence Repeats (SSRs) comparison between strains
The location of SSRs throughout the PRV genome was mapped using MsatFinder and Tandem Repeat Finder (TRF) [183], [184]. MsatFinder detects perfect tandem repeats from homopolymers (1 repeating base) to hexamers (6 bases long). We searched for homopolymers of at least 6 bases long, and the following minimum number of repeating units for larger microsatellites: 5 units for di-, 4 units for tri-, and 3 units for quadri - to hexa-mers. TRF finds larger repeating units, and was designed to detect imperfect repeats that include minor base variations and indels. We ran TRF v4.04 with the following parameters: match 2, mismatch 5, delta 5, PM 80, PI 10, minScore 40, and maxPeriod 500. TRF output was pruned to remove overlapping repeats, preserving the SSR with higher alignment score. We utilized only TRF output with an alignment score of at least 40. This value is commonly used for other genome analyses, and we validated this cutoff for PRV by analyzing the number of repeats that would occur by chance in a shuffled version of the PRV Kaplan genome. Analysis of this shuffled genome detected 73 TRF SSRs in the randomized genome, vs. 637 in the PRV Kaplan genome. Thus approximately 1 out of every 10 TRF repeats might occur by chance, due to nucleotide composition.
Mapping and comparison of homologous SSRs on related PRV genomes were done by previously described methods [49], [125]. Briefly, we first aligned the complete PRV genomes using the mVista genomics analysis tool (LAGAN alignment option) [175], [176]. Sections of this alignment containing SSRs, as mapped in the PRV Kaplan genome, were screened for comparable SSRs in the orthologous regions of the Becker and Bartha genomes. This process was repeated using the lists of SSRs found in the PRV Becker and Bartha genomes. Screening of SSRs using all three genomes as a starting point allowed detection of SSRs that do not occur in all three strains, whose length or purity of repeating units is below threshold in PRV Kaplan but detectable in Becker or Bartha, or whose sequence is divergent enough to be scored as a separate SSR. Table S7 contains a full list of SSRs found in these three genomes. The identifier for each SSR denotes the genome from which its mapping was derived, as well as its starting position in that genome (e.g. SSRKa151).
Supporting Information
Zdroje
1. RoizmanBPellettPE 2001 The family Herpesviridae: A brief introduction. KnipeDMHowleyPM Fields Virology 4 ed Philadelphia Lippincott Williams & Wilkins 2381 2397
2. SteinerIKennedyPGPachnerAR 2007 The neurotropic herpes viruses: herpes simplex and varicella-zoster. Lancet Neurol 6 1015 1028
3. ArvinAM 1996 Varicella-zoster virus. Clin Microbiol Rev 9 361 381
4. PomeranzLEReynoldsAEHengartnerCJ 2005 Molecular biology of pseudorabies virus: impact on neurovirology and veterinary medicine. Microbiol Mol Biol Rev 69 462 500
5. MettenleiterTCKeilGMFuchsW 2008 Molecular Biology of Animal Herpesviruses. MettenleiterTCSobrinoF Animal viruses: molecular biology xii Norfolk, UK Caister Academic Press 531
6. MullerWJJonesCAKoelleDM 2010 Immunobiology of herpes simplex virus and cytomegalovirus infections of the fetus and newborn. Curr Immunol Rev 6 38 55
7. ArvinAMGershonAA 1996 Live attenuated varicella vaccine. Annu Rev Microbiol 50 59 100
8. MettenleiterTC 1996 Immunobiology of pseudorabies (Aujeszky's disease). Vet Immunol Immunopathol 54 221 229
9. KoelleDMCoreyL 2003 Recent progress in herpes simplex virus immunobiology and vaccine research. Clin Microbiol Rev 16 96 113
10. KoelleDMCoreyL 2008 Herpes simplex: insights on pathogenesis and possible vaccines. Annu Rev Med 59 381 395
11. NikolicDSPiguetV 2009 Vaccines and microbicides preventing HIV-1, HSV-2, and HPV mucosal transmission. J Invest Dermatol 130 352 361
12. StrickLBWaldACelumC 2006 Management of herpes simplex virus type 2 infection in HIV type 1-infected persons. Clin Infect Dis 43 347 356
13. RamaswamyMGerettiAM 2007 Interactions and management issues in HSV and HIV coinfection. Expert Rev Anti Infect Ther 5 231 243
14. WatsonRJEnquistLW 1985 Genetically engineered herpes simplex virus vaccines. Prog Med Virol 31 84 108
15. WeisJHEnquistLWSalstromJSWatsonRJ 1983 An immunologically active chimaeric protein containing herpes simplex virus type 1 glycoprotein D. Nature 302 72 74
16. BrittleEEReynoldsAEEnquistLW 2004 Two modes of pseudorabies virus neuroinvasion and lethality in mice. J Virol 78 12951 12963
17. SimmonsANashAA 1984 Zosteriform spread of herpes simplex virus as a model of recrudescence and its use to investigate the role of immune cells in prevention of recurrent disease. J Virol 52 816 821
18. ArvinAM 2006 Investigations of the pathogenesis of Varicella zoster virus infection in the SCIDhu mouse model. Herpes 13 75 80
19. WhiteTMGildenDHMahalingamR 2001 An animal model of varicella virus infection. Brain Pathol 11 475 479
20. MyersMGConnellyBL 1992 Animal models of varicella. J Infect Dis 166 Suppl 1 S48 50
21. LomnicziBWatanabeSBen-PoratTKaplanAS 1987 Genome location and identification of functions defective in the Bartha vaccine strain of pseudorabies virus. J Virol 61 796 801
22. ToddDMcFerranJB 1985 Restriction endonuclease analysis of Aujeszky's disease (pseudorabies) virus DNA: comparison of Northern Ireland isolates and isolates from other countries. Arch Virol 86 167 176
23. MettenleiterTCLukacsNRzihaHJ 1985 Pseudorabies virus avirulent strains fail to express a major glycoprotein. J Virol 56 307 311
24. Aston-JonesGCardJP 2000 Use of pseudorabies virus to delineate multisynaptic circuits in brain: opportunities and limitations. J Neurosci Methods 103 51 61
25. EnquistLW 2002 Exploiting circuit-specific spread of pseudorabies virus in the central nervous system: insights to pathogenesis and circuit tracers. J Infect Dis 186 Suppl 2 S209 214
26. EkstrandMIEnquistLWPomeranzLE 2008 The alpha-herpesviruses: molecular pathfinders in nervous system circuits. Trends Mol Med 14 134 140
27. GranstedtAESzparaMLKuhnBWangSSEnquistLW 2009 Fluorescence-based monitoring of in vivo neural activity using a circuit-tracing pseudorabies virus. PLoS One 4 e6923
28. LomnicziBBlankenshipMLBen-PoratT 1984 Deletions in the genomes of pseudorabies virus vaccine strains and existence of four isomers of the genomes. J Virol 49 970 979
29. BarthaA 1961 Experimental reduction of virulence of Aujeszky's disease virus. Magy Allatorv Lapja 16 42 45
30. KluppBGHengartnerCJMettenleiterTCEnquistLW 2004 Complete, annotated sequence of the pseudorabies virus genome. J Virol 78 424 440
31. LymanMGDemminGLBanfieldBW 2003 The attenuated pseudorabies virus strain Bartha fails to package the tegument proteins Us3 and VP22. J Virol 77 1403 1414
32. ChristensenLSMedveczkyIStrandbygaardBSPejsakZ 1992 Characterization of field isolates of suid herpesvirus 1 (Aujeszky's disease virus) as derivatives of attenuated vaccine strains. Arch Virol 124 225 234
33. MullerTKluppBGFreulingCHoffmannBMojciczM 2010 Characterization of pseudorabies virus of wild boar origin from Europe. Epidemiol Infect 138 1590 1600
34. HahnECFadl-AllaBLichtensteigerCA 2010 Variation of Aujeszky's disease viruses in wild swine in USA. Vet Microbiol 143 45 51
35. GielkensALVan OirschotJTBernsAJ 1985 Genome differences among field isolates and vaccine strains of pseudorabies virus. J Gen Virol 66 Pt 1 69 82
36. WatsonRJUmeneKEnquistLW 1981 Reiterated sequences within the intron of an immediate-early gene of herpes simplex virus type 1. Nucleic Acids Res 9 4189 4199
37. UmeneKWatsonRJEnquistLW 1984 Tandem repeated DNA in an intergenic region of herpes simplex virus type 1 (Patton). Gene 30 33 39
38. MocarskiESRoizmanB 1981 Site-specific inversion sequence of the herpes simplex virus genome: domain and structural features. Proc Natl Acad Sci U S A 78 7047 7051
39. WagnerMJSummersWC 1978 Structure of the joint region and the termini of the DNA of herpes simplex virus type 1. J Virol 27 374 387
40. GivenDYeeDGriemKKieffE 1979 DNA of Epstein-Barr virus. V. Direct repeats of the ends of Epstein-Barr virus DNA. J Virol 30 852 862
41. Ben-PoratTKaplanAS 1985 Molecular Biology of Pseudorabies Virus. RoizmanB The Herpesviruses New York Plenum Press 105 173
42. WathenMWPirtleEC 1984 Stability of the pseudorabies virus genome after in vivo serial passage. J Gen Virol 65 Pt 8 1401 1404
43. DavisonAJWilkieNM 1981 Nucleotide sequences of the joint between the L and S segments of herpes simplex virus types 1 and 2. J Gen Virol 55 315 331
44. RichardGFKerrestADujonB 2008 Comparative genomics and molecular dynamics of DNA repeats in eukaryotes. Microbiol Mol Biol Rev 72 686 727
45. GemayelRVincesMDLegendreMVerstrepenKJ 2010 Variable tandem repeats accelerate evolution of coding and regulatory sequences. Annu Rev Genet 44 445 477
46. BuschiazzoEGemmellNJ 2006 The rise, fall and renaissance of microsatellites in eukaryotic genomes. Bioessays 28 1040 1050
47. SchmidtALAndersonLM 2006 Repetitive DNA elements as mediators of genomic change in response to environmental cues. Biol Rev Camb Philos Soc 81 531 543
48. VerstrepenKJJansenALewitterFFinkGR 2005 Intragenic tandem repeats generate functional variability. Nat Genet 37 986 990
49. VincesMDLegendreMCaldaraMHagiharaMVerstrepenKJ 2009 Unstable tandem repeats in promoters confer transcriptional evolvability. Science 324 1213 1216
50. BrouwerJRWillemsenROostraBA 2009 Microsatellite repeat instability and neurological disease. Bioessays 31 71 83
51. KrobitschSKazantsevAG 2010 Huntington's disease: From molecular basis to therapeutic advances. Int J Biochem Cell Biol 43 20 24
52. OrrHTZoghbiHY 2007 Trinucleotide repeat disorders. Annu Rev Neurosci 30 575 621
53. WillemsenRLevengaJOostraB 2011 CGG repeat in the FMR1 gene: size matters. Clin Genet 80 214 225
54. PfisterLALetvinNLKoralnikIJ 2001 JC virus regulatory region tandem repeats in plasma and central nervous system isolates correlate with poor clinical outcome in patients with progressive multifocal leukoencephalopathy. J Virol 75 5672 5676
55. KauferBBJarosinskiKWOsterriederN 2011 Herpesvirus telomeric repeats facilitate genomic integration into host telomeres and mobilization of viral DNA during reactivation. J Exp Med 208 605 615
56. BatesPADeLucaNA 1998 The polyserine tract of herpes simplex virus ICP4 is required for normal viral gene expression and growth in murine trigeminal ganglia. J Virol 72 7115 7124
57. BedadalaGRPinnojiRCHsiaSC 2007 Early growth response gene 1 (Egr-1) regulates HSV-1 ICP4 and ICP22 gene expression. Cell Res 17 546 555
58. LegendreMPochetNPakTVerstrepenKJ 2007 Sequence-based estimation of minisatellite and microsatellite repeat variability. Genome Res 17 1787 1796
59. HarismendyONgPCStrausbergRLWangXStockwellTB 2009 Evaluation of next generation sequencing platforms for population targeted sequencing studies. Genome Biol 10 R32
60. DohmJCLottazCBorodinaTHimmelbauerH 2008 Substantial biases in ultra-short read data sets from high-throughput DNA sequencing. Nucleic Acids Res 36 e105
61. MettenleiterTCZsakLKaplanASBen-PoratTLomnicziB 1987 Role of a structural glycoprotein of pseudorabies in virus virulence. J Virol 61 4030 4032
62. RobbinsAKRyanJPWhealyMEEnquistLW 1989 The gene encoding the gIII envelope protein of pseudorabies virus vaccine strain Bartha contains a mutation affecting protein localization. J Virol 63 250 258
63. KluppBGLomnicziBVisserNFuchsWMettenleiterTC 1995 Mutations affecting the UL21 gene contribute to avirulence of pseudorabies virus vaccine strain Bartha. Virology 212 466 473
64. DijkstraJMMettenleiterTCKluppBG 1997 Intracellular processing of pseudorabies virus glycoprotein M (gM): gM of strain Bartha lacks N-glycosylation. Virology 237 113 122
65. CuranovicDLymanMGBou-AbboudCCardJPEnquistLW 2009 Repair of the UL21 locus in pseudorabies virus Bartha enhances the kinetics of retrograde, transneuronal infection in vitro and in vivo. J Virol 83 1173 1183
66. PetrovskisEATimminsJGGiermanTMPostLE 1986 Deletions in vaccine strains of pseudorabies virus and their effect on synthesis of glycoprotein gp63. J Virol 60 1166 1169
67. Viejo-BorbollaAMunozATabaresEAlcamiA 2010 Glycoprotein G from pseudorabies virus binds to chemokines with high affinity and inhibits their function. J Gen Virol 91 23 31
68. MatsuuraHKirschnerANLongneckerRJardetzkyTS 2010 Crystal structure of the Epstein-Barr virus (EBV) glycoprotein H/glycoprotein L (gH/gL) complex. Proc Natl Acad Sci U S A 107 22641 22646
69. ChowdaryTKCairnsTMAtanasiuDCohenGHEisenbergRJ 2010 Crystal structure of the conserved herpesvirus fusion regulator complex gH-gL. Nat Struct Mol Biol 17 882 888
70. BackovicMDuBoisRMCockburnJJSharffAJVaneyMC 2010 Structure of a core fragment of glycoprotein H from pseudorabies virus in complex with antibody. Proc Natl Acad Sci U S A 107 22635 22640
71. OliverSLSommerMZerboniLRajamaniJGroseC 2009 Mutagenesis of varicella-zoster virus glycoprotein B: putative fusion loop residues are essential for viral replication, and the furin cleavage motif contributes to pathogenesis in skin tissue in vivo. J Virol 83 7495 7506
72. OkazakiK 2007 Proteolytic cleavage of glycoprotein B is dispensable for in vitro replication, but required for syncytium formation of pseudorabies virus. J Gen Virol 88 1859 1865
73. WhealyMERobbinsAKEnquistLW 1990 The export pathway of the pseudorabies virus gB homolog gII involves oligomer formation in the endoplasmic reticulum and protease processing in the Golgi apparatus. J Virol 64 1946 1955
74. JonsAGranzowHKuchlingRMettenleiterTC 1996 The UL49.5 gene of pseudorabies virus codes for an O-glycosylated structural protein of the viral envelope. J Virol 70 1237 1241
75. JonsADijkstraJMMettenleiterTC 1998 Glycoproteins M and N of pseudorabies virus form a disulfide-linked complex. J Virol 72 550 557
76. McGeochDJRixonFJDavisonAJ 2006 Topics in herpesvirus genomics and evolution. Virus Res 117 90 104
77. MinsonACDavisonAJDesrosiersRCFleckensteinBMc-GeochDJ 2000 Herpesviridae. van RegenmortelMHVClaudeMFBishopDHLCarstensEBEstesMK Virus taxonomy New York Academic Press 203 255
78. TylerSDPetersGAGroseCSeveriniAGrayMJ 2007 Genomic cartography of varicella-zoster virus: a complete genome-based analysis of strain variability with implications for attenuation and phenotypic differences. Virology 359 447 458
79. PetersGATylerSDGroseCSeveriniAGrayMJ 2006 A full-genome phylogenetic analysis of varicella-zoster virus reveals a novel origin of replication-based genotyping scheme and evidence of recombination between major circulating clades. J Virol 80 9850 9860
80. UshijimaYLuoCGoshimaFYamauchiYKimuraH 2007 Determination and analysis of the DNA sequence of highly attenuated herpes simplex virus type 1 mutant HF10, a potential oncolytic virus. Microbes Infect 9 142 149
81. SzparaMLParsonsLEnquistLW 2010 Sequence variability in clinical and laboratory isolates of herpes simplex virus 1 reveals new mutations. J Virol 84 5303 5313
82. McGeochDJDalrympleMADavisonAJDolanAFrameMC 1988 The complete DNA sequence of the long unique region in the genome of herpes simplex virus type 1. J Gen Virol 69 Pt 7 1531 1574
83. McGeochDJDolanADonaldSBrauerDH 1986 Complete DNA sequence of the short repeat region in the genome of herpes simplex virus type 1. Nucleic Acids Res 14 1727 1745
84. OgleWORoizmanB 1999 Functional anatomy of herpes simplex virus 1 overlapping genes encoding infected-cell protein 22 and US1.5 protein. J Virol 73 4305 4315
85. DerbignyWAKimSKJangHKO'CallaghanDJ 2002 EHV-1 EICP22 protein sequences that mediate its physical interaction with the immediate-early protein are not sufficient to enhance the trans-activation activity of the IE protein. Virus Res 84 1 15
86. ZhangGLeaderDP 1990 The structure of the pseudorabies virus genome at the end of the inverted repeat sequences proximal to the junction with the short unique region. J Gen Virol 71 Pt 10 2433 2441
87. FuchsWEhrlichCKluppBGMettenleiterTC 2000 Characterization of the replication origin (Ori(S)) and adjoining parts of the inverted repeat sequences of the pseudorabies virus genome. J Gen Virol 81 1539 1543
88. WuCLWilcoxKW 1991 The conserved DNA-binding domains encoded by the herpes simplex virus type 1 ICP4, pseudorabies virus IE180, and varicella-zoster virus ORF62 genes recognize similar sites in the corresponding promoters. J Virol 65 1149 1159
89. VlcekCKozmikZPacesVSchirmSSchwyzerM 1990 Pseudorabies virus immediate-early gene overlaps with an oppositely oriented open reading frame: characterization of their promoter and enhancer regions. Virology 179 365 377
90. TaharaguchiSInoueHOnoEKidaHYamadaS 1994 Mapping of transcriptional regulatory domains of pseudorabies virus immediate-early protein. Arch Virol 137 289 302
91. LeeJISollarsPJBaverSBPickardGELeelawongM 2009 A Herpesvirus Encoded Deubiquitinase Is a Novel Neuroinvasive Determinant. PLoS Pathog 5 e1000387
92. LuxtonGWHaverlockSCollerKEAntinoneSEPinceticA 2005 Targeting of herpesvirus capsid transport in axons is coupled to association with specific sets of tegument proteins. Proc Natl Acad Sci U S A 102 5832 5837
93. MohlBSBottcherSGranzowHFuchsWKluppBG 2010 Random transposon-mediated mutagenesis of the essential large tegument protein pUL36 of pseudorabies virus. J Virol 84 8153 8162
94. BottcherSMareschCGranzowHKluppBGTeifkeJP 2008 Mutagenesis of the active-site cysteine in the ubiquitin-specific protease contained in large tegument protein pUL36 of pseudorabies virus impairs viral replication in vitro and neuroinvasion in vivo. J Virol 82 6009 6016
95. BottcherSGranzowHMareschCMohlBKluppBG 2007 Identification of functional domains within the essential large tegument protein pUL36 of pseudorabies virus. J Virol 81 13403 13411
96. BottcherSKluppBGGranzowHFuchsWMichaelK 2006 Identification of a 709-amino-acid internal nonessential region within the essential conserved tegument protein (p)UL36 of pseudorabies virus. J Virol 80 9910 9915
97. La ScolaBAudicSRobertCJungangLde LamballerieX 2003 A giant virus in amoebae. Science 299 2033
98. ChenMTanZJiangJLiMChenH 2009 Similar distribution of simple sequence repeats in diverse completed Human Immunodeficiency Virus Type 1 genomes. FEBS Lett 583 2959 2963
99. WalkerAPetheramSJBallardLMurphJRDemmlerGJ 2001 Characterization of human cytomegalovirus strains by analysis of short tandem repeat polymorphisms. J Clin Microbiol 39 2219 2226
100. DebackCBoutolleauDDepienneCLuytCEBonnafousP 2009 Utilization of microsatellite polymorphism for differentiating herpes simplex virus type 1 strains. J Clin Microbiol 47 533 540
101. TemperaIWiedmerADheekolluJLiebermanPM 2010 CTCF prevents the epigenetic drift of EBV latency promoter Qp. PLoS Pathog 6 e1001048
102. ChauCMZhangXYMcMahonSBLiebermanPM 2006 Regulation of Epstein-Barr virus latency type by the chromatin boundary factor CTCF. J Virol 80 5723 5732
103. BloomDCGiordaniNVKwiatkowskiDL 2010 Epigenetic regulation of latent HSV-1 gene expression. Biochim Biophys Acta 1799 246 256
104. AmelioALMcAnanyPKBloomDC 2006 A chromatin insulator-like element in the herpes simplex virus type 1 latency-associated transcript region binds CCCTC-binding factor and displays enhancer-blocking and silencing activities. J Virol 80 2358 2368
105. ChenQLinLSmithSHuangJBergerSL 2007 CTCF-dependent chromatin boundary element between the latency-associated transcript and ICP0 promoters in the herpes simplex virus type 1 genome. J Virol 81 5192 5201
106. KangHLiebermanPM 2009 Cell cycle control of Kaposi's sarcoma-associated herpesvirus latency transcription by CTCF-cohesin interactions. J Virol 83 6199 6210
107. StedmanWKangHLinSKissilJLBartolomeiMS 2008 Cohesins localize with CTCF at the KSHV latency control region and at cellular c-myc and H19/Igf2 insulators. EMBO J 27 654 666
108. TemperaILiebermanPM 2009 Chromatin organization of gammaherpesvirus latent genomes. Biochim Biophys Acta 1799 236 245
109. BadisGBergerMFPhilippakisAATalukderSGehrkeAR 2009 Diversity and complexity in DNA recognition by transcription factors. Science 324 1720 1723
110. Portales-CasamarEThongjueaSKwonATArenillasDZhaoX 2009 JASPAR 2010: the greatly expanded open-access database of transcription factor binding profiles. Nucleic Acids Res 38 D105 110
111. ElementoOSlonimNTavazoieS 2007 A universal framework for regulatory element discovery across all genomes and data types. Mol Cell 28 337 350
112. SchatzMCDelcherALSalzbergSL 2010 Assembly of large genomes using second-generation sequencing. Genome Res 20 1165 1173
113. YoonSXuanZMakarovVYeKSebatJ 2009 Sensitive and accurate detection of copy number variants using read depth of coverage. Genome Res 19 1586 1592
114. MedvedevPStanciuMBrudnoM 2009 Computational methods for discovering structural variation with next-generation sequencing. Nat Methods 6 S13 20
115. SimonAMettenleiterTCRzihaHJ 1989 Pseudorabies virus displays variable numbers of a repeat unit adjacent to the 3′ end of the glycoprotein gII gene. J Gen Virol 70 Pt 5 1239 1246
116. NugentJBirch-MachinISmithKCMumfordJASwannZ 2006 Analysis of equid herpesvirus 1 strain variation reveals a point mutation of the DNA polymerase strongly associated with neuropathogenic versus nonneuropathogenic disease outbreaks. J Virol 80 4047 4060
117. ChiboDDruceJSasadeuszJBirchC 2004 Molecular analysis of clinical isolates of acyclovir resistant herpes simplex virus. Antiviral Res 61 83 91
118. SasadeuszJJTufaroFSafrinSSchubertKHubinetteMM 1997 Homopolymer mutational hot spots mediate herpes simplex virus resistance to acyclovir. J Virol 71 3872 3878
119. WangKMahalingamGHooverSEMontEKHollandSM 2007 Diverse herpes simplex virus type 1 thymidine kinase mutants in individual human neurons and Ganglia. J Virol 81 6817 6826
120. SpatzSJRueCA 2008 Sequence determination of a mildly virulent strain (CU-2) of Gallid herpesvirus type 2 using 454 pyrosequencing. Virus Genes 36 479 489
121. BradleyAJLurainNSGhazalPTrivediUCunninghamC 2009 High-throughput sequence analysis of variants of human cytomegalovirus strains Towne and AD169. J Gen Virol 90 2375 2380
122. DarganDJDouglasECunninghamCJamiesonFStantonRJ 2010 Sequential mutations associated with adaptation of human cytomegalovirus to growth in cell culture. J Gen Virol 91 1535 1546
123. GorzerIGuellyCTrajanoskiSPuchhammer-StocklE 2010 Deep sequencing reveals highly complex dynamics of human cytomegalovirus genotypes in transplant patients over time. J Virol 84 7195 7203
124. RenzetteNBhattacharjeeBJensenJDGibsonLKowalikTF 2011 Extensive genome-wide variability of human cytomegalovirus in congenitally infected infants. PLoS Pathog 7 e1001344
125. LegendreMSantiniSRicoAAbergelCClaverieJM 2011 Breaking the 1000-gene barrier for Mimivirus using ultra-deep genome and transcriptome sequencing. Virol J 8 99
126. KaplanASVatterAE 1959 A comparison of herpes simplex and pseudorabies viruses. Virology 7 394 407
127. FalkenbergMEliasPLehmanIR 1998 The herpes simplex virus type 1 helicase-primase. Analysis of helicase activity. J Biol Chem 273 32154 32157
128. GaoMKnipeDM 1991 Potential role for herpes simplex virus ICP8 DNA replication protein in stimulation of late gene expression. J Virol 65 2666 2675
129. MapelliMPanjikarSTuckerPA 2005 The crystal structure of the herpes simplex virus 1 ssDNA-binding protein suggests the structural basis for flexible, cooperative single-stranded DNA binding. J Biol Chem 280 2990 2997
130. BekalSDomierLLNiblackTLLambertKN 2011 Discovery and initial analysis of novel viral genomes in the soybean cyst nematode. J Gen Virol 92 1870 1879
131. ChevalJSauvageVFrangeulLDacheuxLGuigonG 2011 Evaluation of High Throughput Sequencing for identifying known and unknown viruses in biological samples. J Clin Microbiol 49 3268 3275
132. GreningerALRunckelCChiuCYHaggertyTParsonnetJ 2009 The complete genome of klassevirus - a novel picornavirus in pediatric stool. Virol J 6 82
133. KreuzeJFPerezAUntiverosMQuispeDFuentesS 2009 Complete viral genome sequence and discovery of novel viruses by deep sequencing of small RNAs: a generic method for diagnosis, discovery and sequencing of viruses. Virology 388 1 7
134. PrestiRMZhaoGBeattyWLMihindukulasuriyaKAda RosaAP 2009 Quaranfil, Johnston Atoll, and Lake Chad viruses are novel members of the family Orthomyxoviridae. J Virol 83 11599 11606
135. LohJZhaoGPrestiRMHoltzLRFinkbeinerSR 2009 Detection of novel sequences related to african Swine Fever virus in human serum and sewage. J Virol 83 13019 13025
136. Al RwahnihMDaubertSGolinoDRowhaniA 2009 Deep sequencing analysis of RNAs from a grapevine showing Syrah decline symptoms reveals a multiple virus infection that includes a novel virus. Virology 387 395 401
137. CunninghamCGathererDHilfrichBBaluchovaKDarganDJ 2010 Sequences of complete human cytomegalovirus genomes from infected cell cultures and clinical specimens. J Gen Virol 91 605 615
138. LeeSWMarkhamPFMarkhamJFPetermannINoormohammadiAH 2011 First complete genome sequence of infectious laryngotracheitis virus. BMC Genomics 12 197
139. AwasthiSLubinskiJMEisenbergRJCohenGHFriedmanHM 2008 An HSV-1 gD mutant virus as an entry-impaired live virus vaccine. Vaccine 26 1195 1203
140. AwasthiSLubinskiJMFriedmanHM 2009 Immunization with HSV-1 glycoprotein C prevents immune evasion from complement and enhances the efficacy of an HSV-1 glycoprotein D subunit vaccine. Vaccine 27 6845 6853
141. KoelleDMMagaretAMcClurkanCLRemingtonMLWarrenT 2008 Phase I dose-escalation study of a monovalent heat shock protein 70-herpes simplex virus type 2 (HSV-2) peptide-based vaccine designed to prime or boost CD8 T-cell responses in HSV-naive and HSV-2-infected subjects. Clin Vaccine Immunol 15 773 782
142. KluppBGKernHMettenleiterTC 1992 The virulence-determining genomic BamHI fragment 4 of pseudorabies virus contains genes corresponding to the UL15 (partial), UL18, UL19, UL20, and UL21 genes of herpes simplex virus and a putative origin of replication. Virology 191 900 908
143. CheungAMaesR 1993 Nucleotide sequence variations surrounding the standard recombination junction of pseudorabies viruses. Nucleic Acids Res 21 5522 5523
144. FuchsWBauerBMettenleiterTCRzihaHJ 1994 A novel intergenic site for integration and expression of foreign genes in the genome of pseudorabies virus. J Virol Methods 46 95 105
145. DeMarchiJMLuZQRallGKupershmidtSBen-PoratT 1990 Structural organization of the termini of the L and S components of the genome of pseudorabies virus. J Virol 64 4968 4977
146. MittalSKFieldHJ 1989 Analysis of the bovine herpesvirus type 1 thymidine kinase (TK) gene from wild-type virus and TK-deficient mutants. J Gen Virol 70 Pt 4 901 918
147. GreyFSowaMCollinsPFentonRJHarrisW 2003 Characterization of a neurovirulent aciclovir-resistant variant of herpes simplex virus. J Gen Virol 84 1403 1410
148. SauerbreiADeinhardtSZellRWutzlerP 2010 Phenotypic and genotypic characterization of acyclovir-resistant clinical isolates of herpes simplex virus. Antiviral Res 86 246 252
149. LiljeqvistJASvennerholmBBergstromT 1999 Herpes simplex virus type 2 glycoprotein G-negative clinical isolates are generated by single frameshift mutations. J Virol 73 9796 9802
150. RekabdarETunbackPLiljeqvistJALindhMBergstromT 2002 Dichotomy of glycoprotein g gene in herpes simplex virus type 1 isolates. J Clin Microbiol 40 3245 3251
151. DolanAMcKieEMacLeanARMcGeochDJ 1992 Status of the ICP34.5 gene in herpes simplex virus type 1 strain 17. J Gen Virol 73 Pt 4 971 973
152. EverettRDFenwickML 1990 Comparative DNA sequence analysis of the host shutoff genes of different strains of herpes simplex virus: type 2 strain HG52 encodes a truncated UL41 product. J Gen Virol 71 Pt 6 1387 1390
153. SpatzSJ 2010 Accumulation of attenuating mutations in varying proportions within a high passage very virulent plus strain of Gallid herpesvirus type 2. Virus Res 149 135 142
154. TirabassiRSEnquistLW 1999 Mutation of the YXXL endocytosis motif in the cytoplasmic tail of pseudorabies virus gE. J Virol 73 2717 2728
155. TylerSSeveriniABlackDWalkerMEberleR 2011 Structure and sequence of the saimiriine herpesvirus 1 genome. Virology 410 181 191
156. HwangCBChenHJ 1995 An altered spectrum of herpes simplex virus mutations mediated by an antimutator DNA polymerase. Gene 152 191 193
157. LaBoissiereSTrudelMSimardC 1992 Characterization and transcript mapping of a bovine herpesvirus type 1 gene encoding a polypeptide homologous to the herpes simplex virus type 1 major tegument proteins VP13/14. J Gen Virol 73 Pt 11 2941 2947
158. NorbergPBergstromTRekabdarELindhMLiljeqvistJA 2004 Phylogenetic analysis of clinical herpes simplex virus type 1 isolates identified three genetic groups and recombinant viruses. J Virol 78 10755 10764
159. LynchMSungWMorrisKCoffeyNLandryCR 2008 A genome-wide view of the spectrum of spontaneous mutations in yeast. Proc Natl Acad Sci U S A 105 9272 9277
160. DenverDRMorrisKKewalramaniAHarrisKEChowA 2004 Abundance, distribution, and mutation rates of homopolymeric nucleotide runs in the genome of Caenorhabditis elegans. J Mol Evol 58 584 595
161. LevySSuttonGNgPCFeukLHalpernAL 2007 The diploid genome sequence of an individual human. PLoS Biol 5 e254
162. BentleyDRBalasubramanianSSwerdlowHPSmithGPMiltonJ 2008 Accurate whole human genome sequencing using reversible terminator chemistry. Nature 456 53 59
163. LeeHCYinPHLinJCWuCCChenCY 2005 Mitochondrial genome instability and mtDNA depletion in human cancers. Ann N Y Acad Sci 1042 109 122
164. BianchiNOBianchiMSRichardSM 2001 Mitochondrial genome instability in human cancers. Mutat Res 488 9 23
165. BallEVStensonPDAbeysingheSSKrawczakMCooperDN 2005 Microdeletions and microinsertions causing human genetic disease: common mechanisms of mutagenesis and the role of local DNA sequence complexity. Hum Mutat 26 205 213
166. PlattKBMareCJHinzPN 1979 Differentiation of vaccine strains and field isolates of pseudorabies (Aujeszky's disease) virus: thermal sensitivity and rabbit virulence markers. Arch Virol 60 13 23
167. HaffRF 1964 Inhibition of the Multiplication of Pseudorabies Virus by Cyclohexamide. Virology 22 430 431
168. SmithGAEnquistLW 1999 Construction and transposon mutagenesis in Escherichia coli of a full-length infectious clone of pseudorabies virus, an alphaherpesvirus. J Virol 73 6405 6414
169. EnquistLWMaddenMJSchiop-StanleyPVande WoudeGF 1979 Cloning of herpes simplex type 1 DNA fragments in a bacteriophage lambda vector. Science 203 541 544
170. LangmeadBTrapnellCPopMSalzbergSL 2009 Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10 R25
171. WarrenRLSuttonGGJonesSJHoltRA 2007 Assembling millions of short DNA sequences using SSAKE. Bioinformatics 23 500 501
172. StadenRJudgeDPBonfieldJK 2001 Sequence assembly and finishing methods. Methods Biochem Anal 43 303 322
173. StadenR 1996 The Staden sequence analysis package. Mol Biotechnol 5 233 241
174. AltschulSFGishWMillerWMyersEWLipmanDJ 1990 Basic local alignment search tool. J Mol Biol 215 403 410
175. FrazerKAPachterLPoliakovARubinEMDubchakI 2004 VISTA: computational tools for comparative genomics. Nucleic Acids Res 32 W273 279
176. BrudnoMDoCBCooperGMKimMFDavydovE 2003 LAGAN and Multi-LAGAN: efficient tools for large-scale multiple alignment of genomic DNA. Genome Res 13 721 731
177. FonsecaAAJrCamargosMFde OliveiraAMCiacci-ZanellaJRPatricioMA 2009 Molecular epidemiology of Brazilian pseudorabies viral isolates. Vet Microbiol 141 238 245
178. BabicNKluppBGMakoscheyBKargerAFlamandA 1996 Glycoprotein gH of pseudorabies virus is essential for penetration and propagation in cell culture and in the nervous system of mice. J Gen Virol 77 Pt 9 2277 2285
179. LiHHandsakerBWysokerAFennellTRuanJ 2009 The Sequence Alignment/Map format and SAMtools. Bioinformatics 25 2078 2079
180. NicolJWHeltGABlanchardSGJrRajaALoraineAE 2009 The Integrated Genome Browser: free software for distribution and exploration of genome-scale datasets. Bioinformatics 25 2730 2731
181. NakamuraKOshimaTMorimotoTIkedaSYoshikawaH 2011 Sequence-specific error profile of Illumina sequencers. Nucleic Acids Res 39 e90
182. RobinsonJTThorvaldsdottirHWincklerWGuttmanMLanderES 2011 Integrative genomics viewer. Nat Biotechnol 29 24 26
183. ThurstonMIFieldD 2005 MsatFinder: detection and characterization of microsatellites. Oxford, United Kingdom CEH Oxford
184. BensonG 1999 Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 27 573 580
185. BaoYFederhenSLeipeDPhamVResenchukS 2004 National center for biotechnology information viral genomes project. J Virol 78 7291 7298
186. DavisonAJScottJE 1986 The complete DNA sequence of varicella-zoster virus. J Gen Virol 67 Pt 9 1759 1816
187. DolanACunninghamCHectorRDHassan-WalkerAFLeeL 2004 Genetic content of wild-type human cytomegalovirus. J Gen Virol 85 1301 1312
188. BaerRBankierATBigginMDDeiningerPLFarrellPJ 1984 DNA sequence and expression of the B95-8 Epstein-Barr virus genome. Nature 310 207 211
189. HatfullGBankierATBarrellBGFarrellPJ 1988 Sequence analysis of Raji Epstein-Barr virus DNA. Virology 164 334 340
190. ParkerBDBankierASatchwellSBarrellBFarrellPJ 1990 Sequence and transcription of Raji Epstein-Barr virus DNA spanning the B95-8 deletion region. Virology 179 339 346
191. JeangKTHaywardSD 1983 Organization of the Epstein-Barr virus DNA molecule. III. Location of the P3HR-1 deletion junction and characterization of the NotI repeat units that form part of the template for an abundant 12-O-tetradecanoylphorbol-13-acetate-induced mRNA transcript. J Virol 48 135 148
192. de JesusOSmithPRSpenderLCElgueta KarsteglCNillerHH 2003 Updated Epstein-Barr virus (EBV) DNA sequence and analysis of a promoter for the BART (CST, BARF0) RNAs of EBV. J Gen Virol 84 1443 1450
193. RezaeeSACunninghamCDavisonAJBlackbournDJ 2006 Kaposi's sarcoma-associated herpesvirus immune modulation: an overview. J Gen Virol 87 1781 1804
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2011 Číslo 10
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Severe Acute Respiratory Syndrome Coronavirus Envelope Protein Regulates Cell Stress Response and Apoptosis
- The SARS-Coronavirus-Host Interactome: Identification of Cyclophilins as Target for Pan-Coronavirus Inhibitors
- Biochemical and Structural Insights into the Mechanisms of SARS Coronavirus RNA Ribose 2′-O-Methylation by nsp16/nsp10 Protein Complex
- Evolutionarily Divergent, Unstable Filamentous Actin Is Essential for Gliding Motility in Apicomplexan Parasites
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy