
Immunotoxin Complementation of HAART to Deplete Persisting HIV-Infected Cell Reservoirs
article has not abstract
Published in the journal:
. PLoS Pathog 6(6): e32767. doi:10.1371/journal.ppat.1000803
Category:
Opinion
doi:
https://doi.org/10.1371/journal.ppat.1000803
Summary
article has not abstract
Achievements and Limitations of Antiretroviral Therapy
The development of combinations of drugs that potently suppress HIV replication, collectively given the acronym HAART (highly active antiretroviral therapy), has transformed the lives of people with HIV infection, particularly in high-income countries [1]. Though modern HAART regimens can drive HIV plasma viral loads below the detection limits of standard clinical assays (≥50 copies HIV RNA/ml), long-term treatment fails to eradicate infectious virus as revealed by the persistence of HIV proviral DNA and infectious HIV in peripheral blood and lymphoid tissue, as well as by low level viremia (1–50 RNA copies/ml) in the majority of treated people as detected by ultrasensitive single copy assays [2]–[4]. Moreover, reservoirs of latently infected resting memory CD4+ T lymphocytes are established early after infection and persist throughout treatment with exceedingly slow decay rates; these latent reservoirs are unlikely to be eliminated by HAART alone, and thus have the potential to re-ignite the infection if activated after therapy is halted. A further complication is the existence of multiple sanctuaries of infection in cell types from various lineages (monocyte-macrophages, dendritic cells, hemapoietic stem cells, etc.) detected in distinct anatomical compartments (blood, peripheral lymph nodes, gut mucosa, central nervous system, genital tract, etc.). These findings raise a number of critical inter-related questions: Does the extreme stability of the latently infected cell reservoirs reflect simply the long intrinsic half-life of memory CD4+ T lymphocytes, and/or are the reservoirs continuously reseeded by low level ongoing replication? To what extent does residual viremia reflect incomplete suppression of replication versus virus output from stable (perhaps renewable) infected cell reservoirs? What is the source(s) and significance of intermittent viremia blips, and from where does HIV rebound upon cessation of HAART? Will deliberate activation of resting CD4+ T lymphocytes under continued HAART provide a clinical benefit by depleting latently infected cell reservoirs? While these issues remain controversial, a major practical consequence is irrefutable: cessation of HAART results in rapid virus rebound, in many cases to pre-treatment levels. As a result, treatment must be long-term, presumably for life.
A Renewed Focus on HIV Eradication
The profound viral suppression achievable with modern-day HAART regimens coupled with the limitations and concerns of prolonged treatment (cumulative side effects, adherence difficulties, emergence of drug resistance, high costs) have revitalized serious consideration of the prospect for eradicating HIV from the body, or at least of achieving a “functional cure” whereby therapy can be stopped without viral rebound [3]–[9]. The latently infected CD4+ T cell reservoirs have generally been viewed as the major obstacle to eradication; hence there has been considerable focus on therapeutic strategies to drive the proviral genome out of latency, including cytokines (e.g., IL-2), histone deacetylase inhibitors (e.g., valproic acid, SAHA), nontumorogenic phorbol esters (e.g., prostratin), anti–T cell antibodies (e.g., OKT3), and kinase agonists. It is typically argued that augmenting HAART with deliberate activation should result in the eventual death of all productively infected T cells by a combination of natural mechanisms including viral cytopathic effects, the inherently short life span of activated T cells, and various immune effector mechanisms. Yet to date, trials testing of such approaches have shown no clinical benefit, with at best a reduction in the frequency of latently infected T cells in a subset of patients [2]–[9]. Thus, clinical trials based strictly on flushing out quiescent HIV to purge the infected cell reservoirs have proven disappointing. Further complicating the issue are recent studies suggesting that in most patients, the residual viremia is invariant and genetically distinct from proviruses in resting and activated CD4+ T cells; this has led to a hypothesis whereby most of the residual viremia arises from a an unknown cell type, perhaps a stem cell of the monocyte-macrophage lineage, with the capacity for proliferation and continuous release of virus [4].
Rationale for Targeted Cytotoxic Treatment as a Complement to HAART
Whatever the source(s) and underlying mechanism(s) for the persisting HIV, a major point emphasized herein is that all drugs in the current HAART arsenal share one major feature: their efficacy results from blocking specific steps of the HIV replication cycle, thus preventing new rounds of infection of naïve cells. What they fail to do, at least directly, is to kill cells that are already infected. The theme to be developed here is straightforward: Why not complement the HAART-induced suppression of HIV replication with a treatment that directly kills infected cells? A direct means of achieving this is based on display of the HIV envelope glycoprotein (Env) on the external surface of productively infected cells, where it can be recognized by a specific binding molecule such as an antibody or a soluble fragment of the CD4 receptor. The Env-targeting moiety can be linked to various types of cytotoxic agents, yielding novel molecules that selectively kill HIV-infected cells. This “magic bullet” concept has been prominent in the cancer field, with consideration given to domains of protein toxins, low MW cytotoxic molecules, and radionuclides as alternative cytotoxic payloads [10], [11]. The first successes came a decade ago, with the US Food and Drug Administration's approval of ONTAK (IL-2 linked to the catalytic domain of diphtheria toxin) for cutaneous T cell lymphoma [12], and Mylotarg (a humanized anti-CD33 monoclonal antibody linked to calicheamicin) for relapsing acute myeloid leukemia in elderly patients [13]. The concept may have a particular advantage for infectious diseases [14], since the targeted molecule is encoded by the pathogen, thereby minimizing side effects encountered in anti-cancer applications associated with killing of normal cells expressing low levels of the targeted human antigen. Of course, selective killing requires that the target antigen of the infecting pathogen be expressed on the surface of the infected cell, raising obvious complexities for applications such as HIV infection that are characterized by the presence of latently infected cells.
Immunotoxin Approaches against HIV in the Pre-HAART Era
Not long after the recognition of the retroviral nature of the AIDS etiologic agent [15], several groups developed cytotoxic agents targeted to HIV Env using antibodies or soluble CD4 linked biochemically or genetically to effector domains of bacterial or plant protein toxins [16]–[19]. Alternative strategies have been considered whereby toxins are targeted to cellular proteins endogenously expressed on T cells, such as the IL-2 receptor on activated CD4+ T lymphocytes [20]–[22] or CD45RO on memory T cells [23].
Over the past two decades, our research groups have collaborated to develop HIV Env-targeted toxins based on Pseudomonas aeruginosa exotoxin A (PE). Discrete structural domains within the linear sequence of PE are associated with specific functions [24]. This domain organization has been exploited for cancer therapy by engineering recombinant immunotoxins in which the native N-terminal cell binding domain is replaced by an antibody fragment (typically a single chain SCFv or a disulfide-linked variable region construct) directed against an antigen overexpressed on the specific malignant cell type of interest [25]. To apply this strategy to HIV (Figure 1), we first designed CD4(178)-PE40 (hereafter referred to as CD4-PE) in which the targeting moiety is the first two domains of CD4, which binds directly to the gp120 subunit of Env [16]. Specific cytotoxicity against Env-expressing cells was demonstrated in two types of in vitro systems: a) direct killing assays, in which Env-expressing cells (either stable transfectants or constitutively HIV-infected cell lines) were potently killed in dose-dependent fashion, whereas the corresponding parental cells lacking Env were unaffected [16], [26]–[28], and b) spreading infection inhibition assays, in which infectious HIV-1 is added to permissive target cells, and virus production is measured (p24 or reverse transcriptase) [27], [29]–[32]. CD4-PE inhibited at concentrations where minimal effects were observed with sCD4 (alone or linked to a PE moiety containing an inactivating mutation), thereby demonstrating that the observed activities were due to selective killing of infected cells rather than merely to virus neutralization by the sCD4 moiety. Spreading infection of primary isolates was inhibited [32]–[34] in primary cell types especially relevant to in vivo infection, i.e., peripheral blood mononuclear cells and monocyte-derived macrophages [31], [32], [34]. The latter are particularly noteworthy in view of their extremely low levels of surface Env, as well as the postulated role of macrophages in HIV persistence during HAART given their relatively slow decay kinetics and refractoriness to HIV-mediated cytopathic effects [35], [36].

Based on these promising in vitro findings, CD4-PE was tested in Phase 1 clinical trials in the pre-HAART era [37], [38]. No antiviral or immune-enhancing effects were observed at the maximum tolerated dose of 10–15 µg/kg, which was well below the 40 µg/kg ×3 doses typically given for PE-based cancer immunotoxins. The major dose-limiting toxicity was reversible hepatocellular injury. These disappointing results with CD4-PE greatly diminished enthusiasm for immunotoxins against HIV, and no additional clinical trials have been conducted since.
Immunotoxin Approaches against HIV: Why Now?
The failed clinical trials with CD4-PE were conducted in the pre-HAART era and thus essentially represented monotherapy (although some individuals also received nucleoside reverse transcriptase (RT) inhibitors that failed to suppress viral loads [37]). The development of HAART prompted us to suggest reconsideration of the Env-targeted toxin concept [39]. In the present report, we propose that experimental and technical advances in the ensuing decade have made this argument even more compelling in several critical ways: a) the persistence of HIV in the face of highly suppressive HAART reveals the need for approaches to augment the depletion of infected cell reservoirs, b) experiments in vitro and in animal models reveal a crucial point: the immunotoxins have limited efficacy in blocking spreading HIV infection when used alone; however, they show dramatic synergistic activities when used in combination with HIV replication inhibitors (discussed below), c) new methods are available to assess various efficacy parameters upon complementing HAART with immunotoxins, d) clinical trials with PE-based immunotoxins against certain leukemias have shown impressive results [40], [41], and e) we have developed an improved immunotoxin with greatly enhanced potency and minimal hepatoxicity potential.
We designed a second PE-based immunotoxin, 3B3(Fv)-PE38 (hereafter referred to as 3B3-PE) [27]. The targeting moiety is the 3B3 SCFv, an affinity-maturated variant of Fab b12 directed against the highly conserved CD4 binding site on gp120; compared to b12, 3B3 displays improved binding affinity and greater breadth of reactivity against Envs from HIV-1 primary isolates [42]. We compared the potencies of the two PE-based immunotoxins in several in vitro systems. In direct cell killing assays against Env-expressing cell lines, 3B3-PE displayed significantly enhanced potency (IC50 0.03–0.04 nM) compared to CD4-PE (IC50 0.6–1.5 nM); neither agent was cytotoxic against the corresponding Env-negative cell lines [28]. Both immunotoxins inhibited spreading infection of all the HIV-1 primary isolates tested (clade B), again with 3B3-PE showing greater potency than CD4-PE [43]. Like the original immunotoxin, 3B3-PE inhibited HIV-1 spreading infection in monocyte-derived macrophages. Most importantly, an extremely high intravenous dose of 3B3-PE (250 µg/kg ×3) caused no hepatotoxicity in rhesus macaques, in contrast with the elevation of serum hepatic enzymes induced by CD4-PE at the same dosage [43]. We had previously speculated [39] that the dose-limiting hepatotoxicity observed in the CD4-PE Phase 1 trials might have been due to the CD4 moiety of the chimeric toxin binding to free gp120 released from virions and infected cells, leading to nonspecific liver uptake perhaps via the asialoglycoprotein receptor on hepatocytes recognizing oligosaccharide chains on gp120. Subsequent studies argue against this hypothesis. First, the distinct macaque hepatotoxicity profiles noted above (none for 3B3-PE; significant for CD4-PE) have also been seen in simian human immunodeficiency virus (SHIV)-infected animals that expressed very high viral loads (W. Wagner, M. G. Lewis, E. A. Berger, and I. Pastan, unpublished data); if hepatotoxicity reflected binding to released gp120, both chimeric toxins should have caused similar effects. Second, animal studies with other PE-based immunotoxins have revealed that hepatoxicity is associated with a high isoelectric point of the Fv [44]; the highly basic nature of the CD4 moiety (isoelectric point 8.86) likely underlies the hepatotoxicity of CD4-PE. We conclude that 3B3-PE is a much more promising agent than CD4-PE, with a significantly improved therapeutic window due to its enhanced specific cytotoxic potency and greatly reduced likelihood for dose-limiting hepatotoxicity.
Potent Synergy between Immunotoxins and Inhibitors of HIV Replication
Our cell culture studies demonstrated significant activity of CD4-PE and 3B3-PE against spreading HIV-1 infection. However, we observed limitations that we now view as critical for our current thinking about how immunotoxins should be considered for clinical use. First, while both CD4-PE and 3B3-PE inhibited spreading infection, the effects were relatively inefficient compared to activities in direct killing assays; at best the viral peak was reduced and delayed and the killing of target cells was slowed, but the effects were never complete [30], [43]. Moreover, the IC50 values in spreading infection assays were significantly weaker than in direct killing assays against target cells uniformly expressing Env. These findings can be readily understood in terms of the mode of action of these agents; they cannot kill a newly infected cell until surface Env is expressed, by which time the virus infection has already begun to spread. Therefore it can be predicted that the presence of replication inhibitor would render immunotoxin action more similar to what is observed in direct killing assays, i.e., greater potency and complete activity. Indeed, early cell culture studies showed marked synergy between RT inhibitors and CD4-PE, with the former agents dramatically reducing the IC50 values of the immunotoxin (and vice versa); combination treatment completely eradicated HIV-1 from the culture [30]. Subsequent experiments in the SCID-hu (thy,liv) mouse model gave parallel results [45], as shown in Figure 2. Combination of replication inhibitors (zidovudine plus lamivudine plus ritonavir) alone greatly suppressed HIV levels in the human tissue implant after a 30-day treatment period, but the loads rebounded as measured at 30 days after cessation of treatment; CD4-PE or 3B3-PE alone only minimally suppressed viral loads at the end of the treatment period, and at 30 days post-treatment cessation. The results were strikingly different with the combination of HAART plus either CD4-PE or 3B3-PE: viral loads were strongly suppressed not only at the end of the 30-day treatment period, but also at 30 days after treatment cessation. These results highlight the particular value of combining HAART drugs, which potently block HIV replication, with Env-targeted toxins, which kill cells that are already infected.
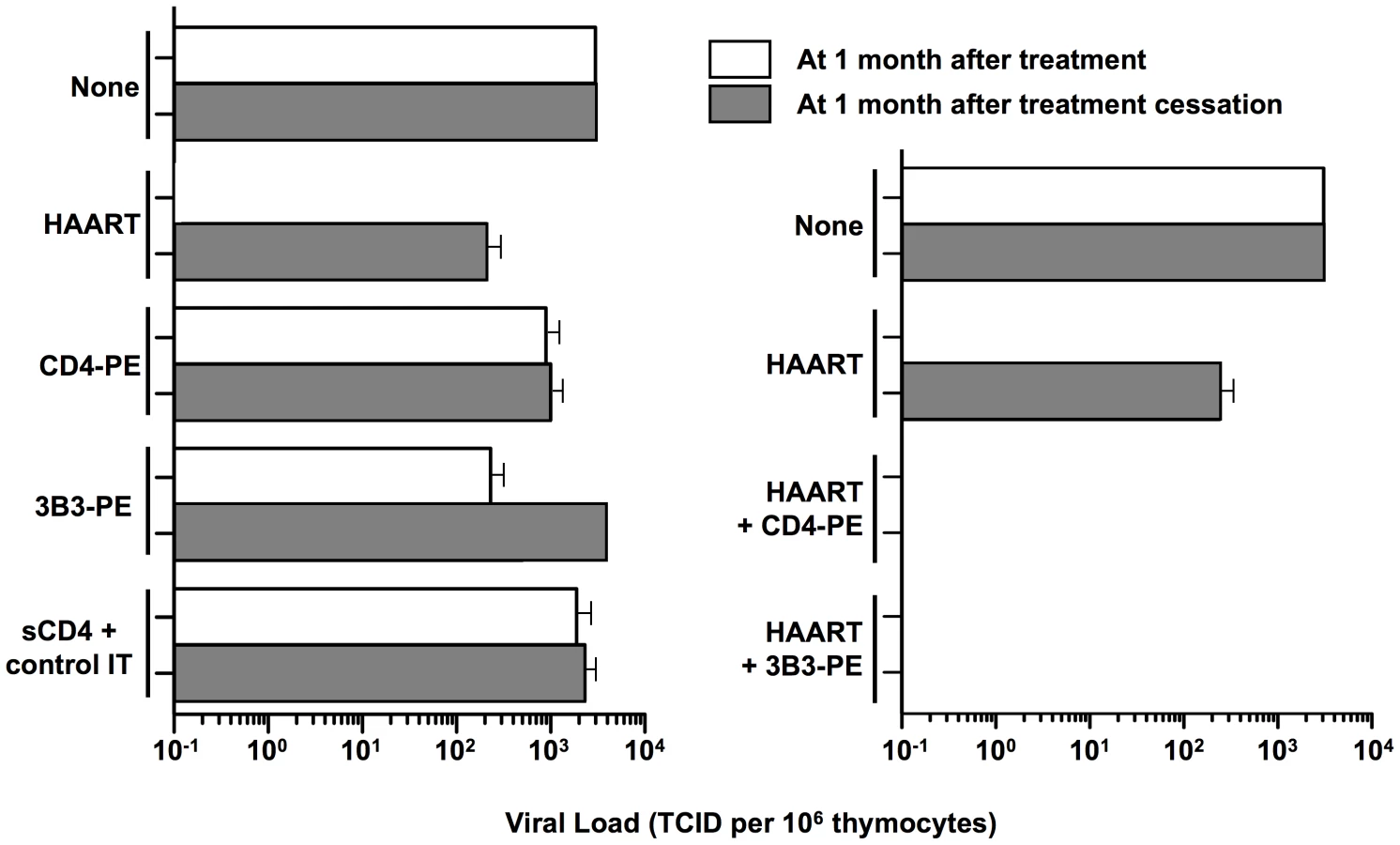
The Way Forward
Based on the considerations outlined above, we believe the time has come for clinical trials of an Env-targeted cytotoxin as a means to deplete infected cell reservoirs persisting in the face of suppressive HAART. This proposal differs fundamentally from HAART “intensification”, whereby a new HIV replication inhibitor is added to an already suppressive antiretroviral regimen. Instead, it represents “complementation” of one class of agents that blocks viral replication (HAART drugs) with a second class that kills those cells already infected (immunotoxin).
“Proof-of-concept” preclinical studies can be conducted in nonhuman primate models of HIV therapy and persistence [46]–[50]. Of particular interest is an Env-SHIV model in which CD4+ T lymphocytes are rapidly depleted, but high viral loads are generated from infected tissue macrophages that appear to be refractory to the viral cytopathic effects and resistant to the antiviral activity of the nucleoside RT inhibitor PMPA [47]. Would immunotoxin treatment deplete this pool of virus-producing cells? Perhaps more relevant to conditions under which immunotoxins might be used in humans are the recently developed chimeric SIVs harboring the HIV-1 reverse transcriptase (RT-SHIVs) [46], [48], [49]. This model permits testing of the same nucleoside and nonnucleoside RT inhibitors currently used in modern HAART regimens. However, the SIV Envs in such SHIVs are not reactive with the 3B3 antibody, thus precluding analysis of 3B3-PE; the alternative CD4-PE could be examined, since it is active against SIV Envs [27] and would cause negligible hepatotoxicity in macaques at the doses that would be employed (40 µg/kg, ×3, unpublished data). While such studies might prove interesting, it is presently unclear whether any of the macaque models will faithfully replicate the mechanisms of HIV persistence in humans, thus arguing against delaying immunotoxin clinical trials until macaque efficacy studies are performed.
3B3-PE is the agent of choice for Phase 1 clinical trials of immunotoxin complementation of HAART, due to its likely improved therapeutic window compared to CD4-PE. The most straightforward approach would involve a one-two punch, i.e., administration of 3B3-PE to patients whose plasma viremia has been well suppressed by HAART. Would persisting HIV be lowered significantly, perhaps to undetectable levels (unlike what has been reported recently for residual viremia with HAART intensification using additional replication inhibitors [51], although contradictory findings with the integrase inhibitor raltegravir have been presented recently [52], [53])? The potential impact of immunotoxin complementation is particularly intriguing in view of the hypothesis noted above that residual viremia may derive predominantly from an unknown cell type capable of proliferation and continuous virus release [54], [55]; immunotoxin treatment might be uniquely suited to target such virus-producing cells. However, the fact that immunotoxin activity is restricted to cells expressing surface Env is almost certain to compromise its ultimate efficacy in the context of persisting reservoirs of latently infected CD4+ T cells. Thus, durable benefits might require a three-tiered attack, i.e., combining HAART plus immunotoxin with a treatment to deliberately trigger HIV expression from latent proviral genomes. In vitro and ex vivo experiments have documented the ability of CD4-PE [26] and 3B3-PE [56] to selectively kill latently infected CD4+ T cells after induction of virus expression; similarly, activating agents have been shown to increase the immunotoxin susceptibility of cultured macrophages [57]. Given the limited understanding and conflicting viewpoints regarding the mechanisms underlying HIV persistence and rebound, we believe it is essential to remain open-minded regarding the testing of two-tiered versus three-tiered approaches. In fact, augmenting HAART with candidate HIV-inducing regimens and immunotoxins, separately and in combination, might provide mutually informative insights. For example, it is possible that an inducing regimen deemed minimally effective in previous clinical trials might have had benefits that went unnoticed in the absence of targeted killing of the newly activated infected cells; conversely, the duration of immunotoxin-mediated benefits might be negligible without prior purging of latently infected cells.
3B3-PE treatment will presumably be limited to short periods (probably three intravenous doses weekly for 1–2 weeks, based on cancer protocols), since the immunotoxin will likely elicit activity-blocking antibodies against the highly immunogenic PE moiety (as was observed in the CD4-PE Phase 1 trial [37]). Should promising results be obtained and additional treatment be desired, the potential exists for switching to Env-targeted cytotoxins based on alternate bacterial or plant protein toxin moieties as noted above, or newly described agents employing low molecular weight drugs [58] or radionuclides [59] as the cytotoxic payloads. Efforts to delete B cell epitopes from PE38 [60] may also prove useful in this context. Regarding enhancements and variations of the targeting moiety, structure-based design has been used to improve the potency of 3B3-PE [61]. However, it must be noted that not all HIV-1 primary isolates are susceptible to neutralization by the 3B3 antibody [42]; thus, it will be important to assess for each potential clinical trial participant whether their virus is recognized by 3B3 (using virus and/or cells obtained from CD8-depleted activated peripheral blood mononuclear cells). The possibility must also be considered for using alternate immunotoxins containing different targeting moieties, such as those described against gp41 [62], [63] or new immunotoxins based on well-studied [64] or newly described [65] broadly neutralizing monoclonal antibodies.
How might immunotoxin efficacy be assessed? Beyond analysis of peripheral blood (proviral DNA and infectious virus in CD4+ T cells; residual viremia detected by single copy assay), the profound involvement of gut-associated lymphoid tissue during both acute HIV infection and viral persistence under HAART [66] highlights the critical importance of examining this compartment. Indeed, HIV DNA levels have been shown to be highly elevated in gut biopsy tissue compared to those in peripheral blood from patients on HAART [67].
Ultimately, immunotoxins will be of value in HIV treatment only if they can enable patients to stop HAART for prolonged periods, sufficient to provide a meaningful quality of life benefit. This is a stringent demand in view of the ongoing improvements in the efficacy and acceptability of antiretroviral drugs, and the risks associated with intermittent cessation of HAART [68]. The ultimate question underlying the potential value of immunotoxins is: must every last infected cell in the body be eliminated in order to achieve a meaningful therapeutic benefit, or is it possible that the infected cell load can be reduced below a threshold such that natural immune effector mechanisms, perhaps enhanced by therapeutic vaccines, can keep the infection in check without the need for ongoing drug treatment? Only a focused effort on clinical evaluation, with all its associated complexities, will provide an answer.
Zdroje
1. BroderS
2010 The development of antiretroviral therapy and its impact on the HIV-1/AIDS pandemic. Antivir Res 85 1 18
2. ContrerasX
LenasiT
PeterlinBM
2006 HIV latency: present knowledge and future directions. Future Virol 1 733 745
3. GeeraertL
KrausG
PomerantzRJ
2008 Hide-and-seek: the challenge of viral persistence in HIV-1 infection. Annu Rev Med 59 487 501
4. ShenL
SilicianoRF
2008 Viral reservoirs, residual viremia, and the potential of highly active antiretroviral therapy to eradicate HIV infection. J Allergy Clin Immunol 122 22 28
5. StevensonM
2008 Can HIV be cured? Sci Am 299 78 83
6. FauciA
2008 The future of AIDS research. XVII International AIDS Conference; 3–8 August 2008; Mexico City, Mexico. Avaliable: http://www.kaisernetwork.org/health_cast/hcast_index.cfm?display=detail&hc=2912. Accessed 12 May 2010.
7. MarsdenMD
ZackJA
2009 Eradication of HIV: current challenges and new directions. J Antimicrob Chemother 63 7 10
8. RichmanDD
MargolisDM
DelaneyM
GreeneWC
HazudaD
2009 The challenge of finding a cure for HIV infection. Science 323 1304 1307
9. MurphyRL
AutranB
KatlamaC
BruckerG
DebreP
2009 A step ahead on the HIV collaboratory. Science 324 1264 1265
10. StrebhardtK
UllrichA
2008 Paul Ehrlich's magic bullet concept: 100 years of progress. Nat Rev Cancer 8 473 480
11. FuchsH
BachranC
2009 Targeted tumor therapies at a glance. Curr Drug Targ 10 89 93
12. DuvicM
TalpurR
2008 Optimizing denileukin diftitox (Ontak (R)) therapy. Future Oncol 4 457 469
13. StasiR
EvangelistaML
BuccisanoF
VendittiA
AmadoriS
2008 Gemtuzumab ozogamicin in the treatment of acute myeloid leukemia. Cancer Treat Rev 34 49 60
14. DadachovaE
CasadevallA
2008 Host and microbial cells as targets for armed antibodies in the treatment of infectious diseases. Curr Opin Investig Drugs 9 184 188
15. GalloRC
MontagnierL
2003 Retrospective: the discovery of HIV as the cause of AIDS. N Eng J Med 349 2283 2285
16. ChaudharyVK
MizukamiT
FuerstTR
FitzGeraldDJ
MossB
1988 Selective killing of HIV-infected cells by recombinant human CD4-Pseudomonas exotoxin hybrid protein. Nature 335 369 372
17. TillMA
GhetieV
GregoryT
PatzerEJ
PorterJP
1988 HIV-infected cells are killed by rCD4-ricin A chain. Science 242 1166 1168
18. PincusSH
WehrlyK
ChesebroB
1989 Treatment of HIV tissue culture infection with monoclonal antibody-ricin A chain conjugates. J Immunol 142 3070 3075
19. AulloP
AlcamiJ
PopoffMR
KlatzmannDR
MurphyJR
1992 A recombinant diphtheria toxin related human CD4 fusion protein specifically kills HIV infected cells which express gp120 but selects fusion toxin resistant cells which carry HIV. EMBO J 11 575 583
20. FinbergRW
WahlSM
AllenJB
SomanG
StromTB
1991 Selective elimination of HIV-1-infected cells with an interleukin-2 receptor-specific cytotoxin. Science 252 1703 1705
21. ZhangL
WatersC
NicholsJ
CrumpackerC
1992 Inhibition of HIV-1 RNA Production by the Diphtheria Toxin - Related IL-2 Fusion Proteins DAB(486)IL-2 and DAB(389)IL-2. J Acquir Immune Defic Syndr 5 1181 1187
22. RamiloO
BellKD
UhrJW
VitettaES
1993 Role of CD25+ and CD25 - T-cells in acute HIV-infection invitro. J Immunol 150 5202 5208
23. McCoigC
Van DykeG
ChouCS
PickerLJ
RamiloO
1999 An anti-CD45RO immunotoxin eliminates T cells latently infected with HIV-1 in vitro. Proc Natl Acad Sci U S A 96 11482 11485
24. WolfP
Elsasser-BeileU
2009 Pseudomonas exotoxin A: From virulence factor to anti-cancer agent. Int J Med Microbiol 299 161 176
25. PastanI
HassanR
FitzGeraldDJ
KreitmanRJ
2006 Immunotoxin therapy of cancer. Nat Rev Cancer 6 559 565
26. BergerEA
ChaudharyVK
ClouseKA
JaraquemadaD
NicholasJA
1990 Recombinant CD4-Pseudomonas exotoxin hybrid protein displays HIV-specific cytotoxicity without affecting MHC class II-dependent functions. AIDS Res Hum Retroviruses 6 795 804
27. AshornP
MossB
BergerEA
1992 Activity of CD4-Pseudomonas exotoxin against cells expressing diverse forms of the HIV and SIV envelope glycoproteins. J Acquir Immune Defic Syndr 5 70 77
28. BeraTK
KennedyPE
BergerEA
BarbasCFI
PastanI
1998 Specific killing of HIV-infected lymphocytes by a recombinant immunotoxin directed against the HIV-1 envelope glycoprotein. Mol Med 4 384 391
29. BergerEA
ClouseKA
ChaudharyVK
ChakrabartiS
FitzGeraldDJ
1989 CD4-Pseudomonas exotoxin hybrid protein blocks the spread of human immunodeficiency virus infection in vitro and is active against cells expressing the envelope glycoproteins from diverse primate immunodeficiency retroviruses. Proc Natl Acad Sci U S A 86 9539 9543
30. AshornP
MossB
WeinsteinJN
ChaudharyVK
FitzGeraldDJ
1990 Elimination of infectious human immunodeficiency virus from human T-cell cultures by synergistic action of CD4-Pseudomonas exotoxin and reverse transcriptase inhibitors. Proc Natl Acad Sci U S A 87 8889 8893
31. AshornP
EnglundG
MartinMA
MossB
BergerEA
1991 Anti-HIV activity of CD4-Pseudomonas exotoxin on infected primary human lymphocytes and monocyte/macrophages. J Infect Dis 163 703 709
32. KennedyPE
MossB
BergerEA
1993 Primary HIV-1 isolates refractory to neutralization by soluble CD4 are potently inhibited by CD4-Pseudomonas exotoxin. Virology 192 375 379
33. VerhoefJ
GekkerG
EriceA
PetersonPK
BalfourHH
1992 Quantitative assay for testing susceptibility of HIV isolates to zidovudine and sCD4 (178)-PE40. Eur J Clin Microbiol Infect Dis 11 715 721
34. WintersMA
MeriganTC
1993 Continuous Presence of CD4-PE40 Is Required for Antiviral Activity Against Single-Passage HIV Isolates and Infected Peripheral Blood Mononuclear Cells. AIDS Res Hum Retroviruses 9 1091 1096
35. GorryPR
ChurchillM
CroweSM
CunninghamAL
GabuzdaD
2005 Pathogenesis of macrophage tropic HIV-1. Curr HIV Res 3 53 60
36. ColemanCM
WuL
2009 HIV interactions with monocytes and dendritic cells: viral latency and reservoirs. Retrovirology 6 51
37. DaveyRT
BoenningCM
HerpinBR
BattsDH
MetcalfJA
1994 Use of recombinant soluble CD4 Pseudomonas exotoxin, a novel immunotoxin, for treatment of persons infected with human immunodeficiency virus. J Infect Dis 170 1180 1188
38. RamachandranRV
KatzensteinDA
WoodR
BattsDH
MeriganTC
1994 Failure of short-term CD4-PE40 infusions to reduce virus load in human immunodeficiency virus-infected persons. J Infect Dis 170 1009 1013
39. BergerEA
MossB
PastanI
1998 Reconsidering targeted toxins to eliminate HIV infection: You gotta have HAART. Proc Natl Acad Sci U S A 95 11511 11513
40. KreitmanRJ
WilsonWH
BergeronK
RaggioM
Stetler-StevensonM
2001 Efficacy of the anti-CD22 recombinant immunotoxin BL-22 in chemotherapy-resistant hairy-cell leukemia. N Engl J Med 345 241 247
41. KreitmanRJ
Stetler-StevensonM
MarguliesI
NoelP
FitzGeraldDJP
2009 Phase II trial of recombinant immunotoxin RFB4(dsFv)-PE38 (BL22) in patients with hairy cell leukemia. J Clin Oncol 27 2983 2990
42. BarbasCF
HuD
DunlopN
SawyerL
CababaD
1994 In vitro evolution of a neutralizing human antibody to human immunodeficiency virus type 1 to enhance affinity and broaden strain cross-reactivity. Proc Natl Acad Sci U S A 91 3809 3813
43. KennedyPE
BeraTK
WangQC
GalloM
WagnerW
2006 Anti-HIV-1 immunotoxin 3B3(Fv)-PE38: enhanced potency against clinical isolates in human PBMCs and macrophages, and negligible hepatotoxicity in macaques. J Leukoc Biol 80 1175 1182
44. OndaM
NagataS
TsutsumiY
VincentJJ
WangQC
2001 Lowering the isoelectric point of the Fv portion of recombinant immunotoxins leads to decreased nonspecific animal toxicity without affecting antitumor activity. Cancer Res 61 5070 5077
45. GoldsteinH
Pettoello-MantovaniM
BeraTK
PastanIH
BergerEA
2000 Chimeric toxins targeted to the human immunodeficiency virus type 1 envelope glycoprotein augment the in vivo activity of combination antiretroviral therapy in thy/liv-scid-hu mice. J Infect Dis 181 921 926
46. UberlaK
StahlhennigC
BottigerD
MatzrensingK
KaupFJ
1995 Animal model for the therapy of acquired-immunodeficiency syndrom with reverse transcriptase inhibitors. Proc Natl Acad Sci U S A 92 8210 8214
47. IgarashiT
BrownCR
EndoY
Buckler-WhiteA
PlishkaR
2001 Macrophage are the principal reservoir and sustain high virus loads in rhesus macaques after the depletion of CD4(+) t cells by a highly pathogenic simian immunodeficiency virus/hiv type 1 chimera (shiv): implications for hiv-1 infections of humans. Proc Natl Acad Sci U S A 98 658 663
48. AmbroseZ
PalmerS
BoltzVF
KearneyM
LarsenK
2007 Suppression of viremia and evolution of human immunodeficiency virus type 1 drug resistance in a macaque model for antiretroviral therapy. J Virol 81 12145 12155
49. Van RompayKKA
JohnsonJA
BlackwoodEJ
SinghRP
LipscombJ
2007 Sequential emergence and clinical implications of viral mutants with K70E and K65R mutation in reverse transcriptase during prolonged tenofovir monotherapy in rhesus macaques with chronic RT-SHIV infection. Retrovirology 4 25
50. DinosoJB
RabiSA
BlanksonJN
GamaL
MankowskiJL
2009 A simian immunodeficiency virus-infected macaque model to study viral reservoirs that persist during highly active antiretroviral therapy. J Virol 83 9247 9257
51. DinosoJB
KimSY
WiegandAM
PalmerSE
GangeSJ
2009 Treatment intensification does not reduce residual HIV-1 viremia in patients on highly active antiretroviral therapy. Proc Natl Acad Sci U S A 106 9403 9408
52. JonesJ
McMahonD
WiegandA
KearneyM
PalmerS
2009 No decrease in residual viremia during raltegravir intensification in patients on standard ART. 16th Conference on Retroviruses and Opportunistic Infections; 8–11 February 2009; Montreal, Canada. CROI 2009. [Abstract 423b]
53. BuzonM
LlibreJ
DomingoP
ParedesR
PalmerS
2009 Transient increase in episomal viral cDNA following raltegravir intensification of a stable HAART Regimen. 16th Conference on Retroviruses and Opportunistic Infections; 8-11 February 2009; Montreal, Canada. CROI 2009. [Abstract 423a]
54. BaileyJR
SedaghatAR
KiefferT
BrennanT
LeePK
2006 Residual human immunodeficiency virus type 1 viremia in some patients on antiretroviral therapy is dominated by a small number of invariant clones rarely found in circulating CD4(+) T cells. J Virol 80 6441 6457
55. BrennanTP
WoodsJO
SedaghatAR
SilicianoJD
SilicianoRF
2009 Analysis of human immunodeficiency virus type 1 viremia and provirus in resting CD4(+) T cells reveals a novel source of residual viremia in patients on antiretroviral therapy. J Virol 83 8470 8481
56. BrooksDG
HamerDH
ArlenPA
GaoLY
BristolG
2003 Molecular characterization, reactivation, and depletion of latent HIV. Immunity 19 413 423
57. MarsdenMD
XuJ
HamerD
ZackJA
2008 Activating stimuli enhance immunotoxin-mediated killing of HIV-infected macrophages. AIDS Res Hum Retroviruses 24 1399 1404
58. JohanssonS
GoldenbergDM
GriffithsGL
WahrenB
HinkulaJ
2006 Elimination of HIV-1 infection by treatment with a doxorubicin-conjugated anti-envelope antibody. AIDS 20 1911 1915
59. DadachovaE
PatelMC
ToussiS
ApostolidisC
MorgensternA
2006 Targeted killing of virally infected cells by radiolabeled antibodies to viral proteins. PLoS Med 3 e427 doi:10.1371/journal.pmed.0030427
60. OndaM
BeersR
XiangL
NagataS
WangQC
2008 An immunotoxin with greatly reduced immunogenicity by identification and removal of B cell epitopes. Proc Natl Acad Sci U S A 105 11311 11316
61. McHughL
HuS
LeeBK
SantoraK
KennedyPE
2002 Increased affinity and stability of an anti-HIV-1 envelope immunotoxin by structure-based mutagenesis. J Biol Chem 277 34383 34390
62. PincusSH
FangH
WilkinsonRA
MarcotteTK
RobinsonJE
2003 In vivo efficacy of anti-glycoprotein 41, but not anti - glycoprotein 120, immunotoxins in a mouse model of hiv infection. J Immunol 170 2236 2241
63. RootMJ
HamerDH
2003 Targeting therapeutics to an exposed and conserved binding element of the HIV-1 fusion protein. Proc Natl Acad Sci U S A 100 5016 5021
64. MontefioriD
SattentauQ
FloresJ
EsparzaJ
MascolaJ
2007 Antibody-based HIV-1 vaccines: recent developments and future directions. PLoS Med 4 e348 doi:10.1371/journal.pmed.0040348
65. WalkerLM
PhogatSK
Chan-HuiPY
WagnerD
PhungP
2009 Broad and potent neutralizing antibodies from an African donor reveal a new HIV-1 vaccine target. Science 326 285 289
66. BrenchleyJM
DouekDC
2008 HIV infection and the gastrointestinal immune system. Mucosal Immunol 1 23 30
67. ChunTW
NickleDC
JustementJS
MeyersJH
RobyG
2008 Persistence of HIV in gut-associated lymphoid tissue despite long-term antiretroviral therapy. J Infect Dis 197 714 720
68. LundgrenJD
BabikerA
El-SadrW
EmeryS
GrundB
2008 Inferior clinical outcome of the CD4(+) cell count guided antiretroviral treatment interruption strategy in the SMART study: Role of CD4(+) cell counts and HIV RNA levels during follow-up. J Infect Dis 197 1145 1155
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2010 Číslo 6
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Requirement of NOX2 and Reactive Oxygen Species for Efficient RIG-I-Mediated Antiviral Response through Regulation of MAVS Expression
- Formation of Complexes at Plasmodesmata for Potyvirus Intercellular Movement Is Mediated by the Viral Protein P3N-PIPO
- Insight into the Mechanisms of Adenovirus Capsid Disassembly from Studies of Defensin Neutralization
- Two Novel Point Mutations in Clinical Reduce Linezolid Susceptibility and Switch on the Stringent Response to Promote Persistent Infection
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy