
Human Cytomegalovirus UL29/28 Protein Interacts with Components of the NuRD Complex Which Promote Accumulation of Immediate-Early RNA
Histone deacetylation plays a pivotal role in regulating human cytomegalovirus gene expression. In this report, we have identified candidate HDAC1-interacting proteins in the context of infection by using a method for rapid immunoisolation of an epitope-tagged protein coupled with mass spectrometry. Putative interactors included multiple human cytomegalovirus-coded proteins. In particular, the interaction of pUL38 and pUL29/28 with HDAC1 was confirmed by reciprocal immunoprecipitations. HDAC1 is present in numerous protein complexes, including the HDAC1-containing nucleosome remodeling and deacetylase protein complex, NuRD. pUL38 and pUL29/28 associated with the MTA2 component of NuRD, and shRNA-mediated knockdown of the RBBP4 and CHD4 constituents of NuRD inhibited HCMV immediate-early RNA and viral DNA accumulation; together this argues that multiple components of the NuRD complex are needed for efficient HCMV replication. Consistent with a positive acting role for the NuRD elements during viral replication, the growth of pUL29/28 - or pUL38-deficient viruses could not be rescued by treating infected cells with the deacetylase inhibitor, trichostatin A. Transient expression of pUL29/28 enhanced activity of the HCMV major immediate-early promoter in a reporter assay, regardless of pUL38 expression. Importantly, induction of the major immediate-early reporter activity by pUL29/28 required functional NuRD components, consistent with the inhibition of immediate-early RNA accumulation within infected cells after knockdown of RBBP4 and CHD4. We propose that pUL29/28 modifies the NuRD complex to stimulate the accumulation of immediate-early RNAs.
Published in the journal:
. PLoS Pathog 6(6): e32767. doi:10.1371/journal.ppat.1000965
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1000965
Summary
Histone deacetylation plays a pivotal role in regulating human cytomegalovirus gene expression. In this report, we have identified candidate HDAC1-interacting proteins in the context of infection by using a method for rapid immunoisolation of an epitope-tagged protein coupled with mass spectrometry. Putative interactors included multiple human cytomegalovirus-coded proteins. In particular, the interaction of pUL38 and pUL29/28 with HDAC1 was confirmed by reciprocal immunoprecipitations. HDAC1 is present in numerous protein complexes, including the HDAC1-containing nucleosome remodeling and deacetylase protein complex, NuRD. pUL38 and pUL29/28 associated with the MTA2 component of NuRD, and shRNA-mediated knockdown of the RBBP4 and CHD4 constituents of NuRD inhibited HCMV immediate-early RNA and viral DNA accumulation; together this argues that multiple components of the NuRD complex are needed for efficient HCMV replication. Consistent with a positive acting role for the NuRD elements during viral replication, the growth of pUL29/28 - or pUL38-deficient viruses could not be rescued by treating infected cells with the deacetylase inhibitor, trichostatin A. Transient expression of pUL29/28 enhanced activity of the HCMV major immediate-early promoter in a reporter assay, regardless of pUL38 expression. Importantly, induction of the major immediate-early reporter activity by pUL29/28 required functional NuRD components, consistent with the inhibition of immediate-early RNA accumulation within infected cells after knockdown of RBBP4 and CHD4. We propose that pUL29/28 modifies the NuRD complex to stimulate the accumulation of immediate-early RNAs.
Introduction
Human cytomegalovirus (HCMV) is a ubiquitous β-herpesvirus that causes life threatening disease in immunocompromised adults, specifically individuals undergoing solid organ or hematopoietic cell transplant and individuals with Acquired Immunodeficiency Syndrome (AIDS) [1]. In addition, congenital HCMV infections cause life-long disabilities in a significant number of children. In recent years, chronic infection has also been linked to cardiovascular disease (reviewed in [2]) and correlated with a decrease in life expectancy [3]; and the virus has been found in several types of human tumors and it expresses gene products with oncogenic potential (for a review see [4]).
The lytic HCMV replication cycle proceeds through a highly coordinated series of events. At the very start of infection, cellular defenses are inhibited and viral immediate-early gene expression is facilitated by proteins and RNAs that are delivered to cells as constituents of virions [5]–[7]. As soon as the viral genome reaches the nucleus, it expresses immediate-early gene products [8], [9], which also help to establish a permissive environment for replication and activate downstream elements of the viral gene expression cascade [1]. Early genes are expressed next, encoding proteins responsible for viral DNA replication as well as products regulating cellular responses to infection; and, finally, late genes encode for proteins needed to assemble infectious viral particles [1].
Upon entry, the HCMV genome rapidly becomes associated with cellular histones [8], which then undergo dynamic changes in their modification state [9]. During the immediate-early phase of the replication cycle, high levels of histone acetylation are detected by 3 h postinfection (hpi) at immediate-early promoters, including the major immediate-early promoter (MIEP). A slight reduction in MIEP histone acetylation occurs at 12 hpi. The change is mediated by the virus-coded IE2 protein binding to the so-called cis-repressive sequence within the promoter and histone deacetylase 1 (HDAC1) activity [9], [10]. Following the onset of viral DNA replication, a general increase in histone occupancy on the genome occurs [8] with high levels of histone acetylation at early and late promoters, coincident with their enhanced activity [9]. In addition to dynamic histone modifications during the lytic replication cycle, increased histone acetylation at the MIEP correlates with reactivation from latency [11].
HDACs have been indentified within multi-protein complexes. HDAC1 is found in the nucleosome remodeling and deacetylase (NuRD) [12], coREST[13], and Sin3 [14] complexes. NuRD is a large complex containing HDAC1 or 2 and chromatin remodeling ATPases, CHD4 and CHD3 [12]. In addition to these proteins, the core NuRD complex includes RBBP4, RBBP7, MBD3, P66A and P66B, and several members of the metastasis-associated (MTA) protein family. The histone demethylase, LSD1, has also been found within the NuRD complex [15]. The recruitment of the core NuRD complex to specific promoters occurs through interactions with transcription factors [12]. NuRD has been proposed to remodel chromatin through histone deacetylation by HDAC1 and 2, as well as by mediating nucleosome mobility via a CHD3 - and CHD4-modulated sliding mechanism [16]. In addition to modifications of histones, the complex deacetylates non-histone proteins, including p53 [17] and HIF-1α [18], which play critical roles in regulating gene expression [12]. Disruption of the NuRD complex has a profound impact on cellular differentiation and proliferation and is associated with metastatic cancer [12].
Regulation of HDACs plays important roles in viral replication and pathogenesis. In HCMV, the virus-coded IE2 protein interacts with HDAC1 [10] and 2 [19] and auto-regulates viral gene expression [10]. Inhibition of HDACs during CMV infection hyperactivates immediate-early gene expression [20]–[24], overcomes the repressive effects of hDaxx at the MIEP [25] and substantially rescues the growth of an IE1-deficient virus [26]. HCMV-coded IE1 protein binds HDAC3 and promotes viral replication by antagonizing histone deacetylation [27]. Proteins from several other herpesviruses bind to and modulate HDACs, including herpes simplex type 1 virus proteins ICP8 [28] and ICP0 [29], [30] and Epstein-Barr virus protein EBNA3C [31], [32]. Beyond herpesviruses, modulation of deacetylases occurs during human papillomavirus (HPV) infection. HPV E7 protein binds [33], [34] and modulates HDAC1, resulting in the activation of E2F-responsive genes [35].
We recently reported that constituents of the NuRD complex can be captured from infected cell extracts with a tagged HCMV UL38-coded protein at 24 hpi [36], identifying a possible interaction between the viral protein and the NuRD complex. To better understand the relationship between histone modification and HCMV gene transcription, we have now performed a global search for putative HDAC1-interacting proteins in the context of infection. We validated several of the interactions, including the formation of complexes between components of the NuRD complex with the HCMV-coded proteins, pUL38 and pUL29/28. pUL38 is an early protein that controls cellular stress responses; it blocks apoptosis [37] and influences both the unfolded protein response [38] and mTOR signaling [36]. pUL29/28 is necessary for efficient viral replication [39], [40] and a pUL29/28-deficient virus fails to express normal levels of immediate-early gene products [41]. Our studies show that the interaction between pUL38 and HDAC requires pUL29/28, and that blocking expression of NuRD components inhibits replication of a wild-type virus. Finally, we demonstrate that full activation of the HCMV MIEP requires pUL29/28 but not pUL38. We propose that HCMV co-opts NuRD function at least in part to enhance expression of its immediate-early genes.
Materials and Methods
Cell culture, viral strains and plasmids
All cells were propagated in Dulbecco's modified Eagle medium supplemented with 10% fetal bovine serum and penicillin-streptomycin (Invitrogen) in 5% CO2 at 37°C. Primary human foreskin fibroblast (HF; passages 6 to 15) [42] and human MRC-5 embryonic lung fibroblast cells (American Type Culture Collection) were used for HCMV infections, except for the initial identification of HDAC1-interacting proteins, which utilized life-extended human foreskin fibroblasts [43]. Luciferase experiments were completed using the human osteosarcoma cell line, U-2 OS (American Type Culture Collection). A matched pair of pUL38-expressing and control U-2 OS cells were constructed using retrovirus stocks made by transfecting pLXSN (Clontech) or pLXSN-UL38 into Phoenix Ampho cells [44] using FuGENE 6 transfection reagent (Roche). The control U-2 OS cells received an empty retrovirus vector, while U-2 OS pUL38 cells contained a retrovirus expressing HCMV pUL38. Transduced U-2 OS cells were selected for presence of the retrovirus by using Geneticin (Invitrogen). Human embryonic kidney 293T cells (American Type Culture Collection) were used for generating shRNA-expressing lentiviruses as described in RNA interference (RNAi) analysis.
Wild-type HCMV, BADwt, was obtained from an infectious bacterial artificial chromosome clone of the AD169 strain, termed pAD/Cre [45]. The BADinUL29F and BADinUL38Tap viruses as well as the pUL28-, pUL29 - and pUL38-deficient mutants, BADsubUL28, BADsubUL29 and BADsubUL38, have been previously described [36], [37], [39], [41]. Viruses were propagated on fibroblasts with the exception of BADsubUL38 which was propagated on pUL38-expressing fibroblasts [37]. Virus preparations were partially purified and concentrated by centrifugation through a sorbitol cushion (20% D-sorbitol, 50 mM Tris-HCl [pH 7.2], 1 mM MgCl2). Viral titers were determined by using an immunofluorescence assay to quantify pUL123 (IE1)-expressing cells [37]. Briefly, serial dilutions of virus samples were plated on fibroblasts and cells were fixed and permeabilized in methanol at −20°C for 15 min at 36 hpi. pUL123-positive cells were quantified by using an antibody to pUL123 and a secondary antibody conjugated to Alexa 488 (Invitrogen). For several experiments, cell-free virus was collected from supernatants and infectivity was determined. To determine DNA levels and particle to PFU ratios in virus stocks, quantitative PCR (qPCR) was performed as previously described [37]. The use of the histone deacetylase (HDAC) inhibitor, trichostatin A (TSA) (Cell Signaling Technologies) in HCMV replication studies has been described by Nevels et al. [26]. Briefly, fibroblasts were grown to confluency and pretreated for 24 h in the presence or absence of the drug. Treated cells were infected and then cultured in the continuous presence of 300 nM TSA for virus rescue studies or 500 nM TSA for IE1 RNA expression studies [26].
The identification of HDAC1-interacting proteins used fibroblasts (HF-HD1gfp cells) stably expressing an HDAC1-enhanced green fluorescent protein (gfp) fusion protein, in which the gfp was fused at the C-terminal end of the cell protein. These cells were generated by transduction using a retrovirus produced from pLXSN-HDAC1gfp and a pure population of GFP-positive cells was isolated by using a FACSVantage flow cytometer (BD Biosciences). pLXSN-HDAC1gfp was constructed by PCR amplification using Expand High Fidelity PCR System (Roche) of the HDAC1 gene (primers: 5′-ATAGAATTCAAGCTTGCCACCATGGCGCAGACGCAGGGC-3′ and 5′-CCTTGCTCACGGCCAACTTGACCTCCTC-3′) from an expression plasmid kindly provided by E. Seto (University of South Florida). PCR amplification of gfp used an enhanced GFP expression plasmid (Clontech) (primers: 5′-CTCCTCCAGTTCAACCGGGTGAGCAAGGGCGAGGAGCTG-3′ and 5′-TTAAGATCTTACTTGTACAGCTCGTCCA-3′) (Integrated DNA Technologies). The PCR products were combined, amplified using the terminal primers to each gene, and introduced into pLXSN using the restriction enzymes, EcoRI and BamHI. The pUL38 expression vector, pLXSN-UL38, was produced by PCR amplification of the UL38 gene using HCMV genomic DNA (primers: 5′-ATAGAATTCGCCACCATGACTACGACCACGCATAG-3′ and 5′-ATACTCGAGCTAGACCACGACCACCATCTG-3′) and introduced into pLXSN. The myc-tagged pUL29/28 expression vector, pLXSN-UL29/28myc, was constructed by PCR amplification UL29/28 from cDNA produced from RNA isolated at 6 hpi [41] (primers: 5′-ATAGTTAACGCCACCATGGAACAAAAGCTAATATCAGAAGAAGACCTATCCGGCCGTCGCAAG-3′ and 5′-ATAGGATCCTCACGACGCCCCCGTGC-3′). Additional plasmids used in these studies have been described elsewhere, including pCGN-UL29/28 and pCGN-UL29 [41], MIEP-luciferase [41], NFκB-luciferase (Clontech) and ISRE-luciferase (Stratagene).
Isolation of HDAC1 complexes
HF-HD1gfp cells were grown to ∼70% confluency, infected with BADwt at a multiplicity of 3 PFU/cell, harvested at 24 hpi and frozen in liquid nitrogen as cell pellets as described [36], [46]. Cell lysis was carried out by cryogenic grinding (Retch MM301 Mixer Mill, Retch Inc) at 30 Hz frequency using 10 cycles of 3 min each [47]. The resulting cell powder (0.42 g) was suspended in 5 ml optimized lysis buffer (20 mM K-HEPES pH 7.4, 0.1 M potassium acetate, 2 mM MgCl2, 0.1% (v/v) Tween-20, 1 µM ZnCl2, 1 µM CaCl2, 0.5% Triton-X100, 250 mM NaCl, 1/100 protease inhibitor cocktail (Sigma)), homogenized, and insoluble material was removed by centrifugation. The supernatant was used for affinity purification by incubation with 6 mg magnetic beads pre-conjugated with anti-GFP antibodies for 1 h at 4°C, as described [47]. Isolated proteins were eluted with 700 µl 0.5 N NH4OH, 0.5 mM EDTA solution, resolved by 1-D gel electrophoresis on a 4–12% NuPAGE Novex Bis-Tris gel (Invitrogen), and stained with Coomassie Blue (Pierce).
As a control (Figure S1), an equivalent isolation was performed using extracts of HF cells expressing free GFP that were prepared at 24 hpi at a multiplicity of 3 PFU/cell with BADwt.
Mass spectrometry analyses
Gel lanes were cut into 25 sections, diced, protein was digested with trypsin (Promega), and the resulting peptides were concentrated on reverse phase resin (POROS 20 R2, Applied Biosystems) and eluted onto a MALDI (matrix assisted laser desorption ionization) target as described [47]. Mass spectrometric analyses were carried out by MALDI MS and MS/MS analyses (single-stage and tandem mass spectrometric analyses, respectively) using a MALDI Qq-TOF (QqTOF Centaur, Sciex) [48] and a MALDI ion trap (LCQDECAXPPLUS, Thermo Finnigan) [49], as described [47]. Interacting protein candidates were identified by incorporating the mass spectrometric data in a search against the National Center for Biotechnology Information nonredundant protein database (version 06/10/16) for Homo sapiens (152010 sequences) and Viruses (346953 sequences) species using the XProteo (http://www.xproteo.com) algorithm, as described [50]. The limited number of candidate proteins allowed for manual verification of the results, and only those proteins confirmed by MS/MS analyses for at least 2 peptides were considered present (Fig. 1, Table S1). The number of detected peptides, protein sequence coverage, database search score and sequences of confirmed peptides are provided for the isolated protein as supplementary material (Table S1).

Analysis of protein expression and interactions
Immunoprecipitation experiments were performed as previously described [36]. Briefly, frozen cell pellets were resuspended in 1 ml RIPA buffer (Tris-HCl, 50 mM, pH 7.4; 1% NP-40; 0.25% Na-deoxycholate; 150 mM NaCl; 1 mM EDTA), sonicated and incubated on ice for 30 min. Insoluble material was pelleted by centrifugation and lysates were precleared for 30 min with Protein A/G Sepharose (Santa Cruz) at 4°C. Primary antibody was added for 1 h at 4°C followed by Protein A/G Sepharose for 1 h at 4°C. Following washes, Sepharose-protein complexes were resuspended in sample buffer, boiled at 100°C for 5 min, and samples were resolved on 10% SDS-containing polyacrylamide gels. After transferring proteins to membrane, specific proteins were identified by Western blot performed using indicated antibodies in 1× TBST with 1% BSA as previously described [36]. Secondary antibody conjugated to HRP (Jackson ImmunoResearch) was used at 1∶5000 dilution in 1× TBST plus 1% BSA, and antibody complexes were visualized using ECL Reagent (GE Healthcare).
Chromatin immunoprecipitation (ChIP) analysis was performed as previously described [9]. In brief, at 48 hpi nuclear proteins were cross-linked to DNA with 1% formaldehyde in culture medium for 10 min, after which the reaction was quenched with glycine (0.125 M). Cells were washed and resuspended in buffer containing 10 mM Tris-HCl [pH 8.0], 100 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 0.1% sodium deoxycholate, 0.5% N-lauroylsarcosine, and protease inhibitors [Roche]; and then cells were disrupted and DNA was fragmented by sonication. Aliquots of lysate containing 3 µg of viral DNA were diluted 1∶10 in 0.01% sodium dodecyl sulfate [SDS], 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris-HCl [pH 8.0], 167 mM NaCl, and protease inhibitors and incubated with antibody overnight at 4°C. The immunocomplexes were captured with protein G-agarose, eluted from the beads at room temperature with buffer containing 1% SDS and 100 mM NaHCO3, crosslinks were reversed by incubation at 65°C for 5 h, proteins were digested with proteinase K (40 µg/ml) for 2 h at 55°C, and DNA recovered by using QiaQuick purification columns (Qiagen) and assayed by quantitative PCR (qPCR) with a Power SYBR green PCR kit (Applied Biosystems) and a primer pair specific for the HCMV MIEP (5′-AACAGCGTGGATGGCGTCTCC-3′ and 5′-GGCACCAAAATCAACGGGACTTT-3′). The efficiency of amplification for the primers was determined by generating a standard curve with 10-fold serial dilutions of a known concentration of viral DNA. The slope values of the standard curve for the primer pair amplicons ranged from −3.5 to −3.2, indicating 90 to 100% efficiency. Dissociation curves were performed to ensure that the primers generated a single amplification product, a template control was included, and each sample was run in duplicate.
For immunofluorescent analysis, fibroblasts were grown to confluence on glass coverslips, infected with HCMV at a multiplicity of 0.01 PFU/cell and fixed for 15 min in 2% paraformaldehyde at 24 hpi. Slides were treated for 15 min in 0.1% Triton X-100, washed with PBST (PBS containing 0.2% Tween 20), and incubated for 30 min in PBS-blocking buffer (PBS containing 2% bovine serum albumin and 0.2%Tween 20). Proteins were identified by incubating slides with the indicated primary antibody in PBS blocking buffer for 1 h at room temperature. After further washing with PBST, slides were incubated for 30 min using a secondary conjugated to Alexa 488 or 546 (Invitrogen). Cells were washed with PBST, mounted and viewed using a Zeiss LSM510 confocal microscope (Carl Zeiss). Quantitation of protein colocalization was performed using the Velocity software package (Improvision).
Antibodies used in these studies were: mouse anti-HDAC1 IgG1 (Millipore), rabbit anti-HDAC1 polyclonal (Millipore), rabbit anti-MTA2 polyclonal (Santa Cruz), mouse anti-FLAG M2 IgG1 (Sigma), mouse anti-Myc IgG1(Millipore), rabbit anti-mSin3A polyclonal (Santa Cruz), rabbit anti-TSC2 polyclonal (Santa Cruz), mouse anti-HA IgG1 (Sigma), mouse anti-tubulin IgG1 (Sigma), rabbit anti-protein A (Sigma), mouse anti-IE1 (clone 1B12) [51] mouse anti-pUL38 (clone 8D6) [37] and mouse anti-pUS24 [52].
Quantitative PCR analysis
RNA levels in infected cells were determined by quantitative real-time reverse transcription-PCR (qRT-PCR) as previously described [37], [41]. Briefly, total RNA was isolated by using TRIzol reagent (Invitrogen), and contaminating DNA was removed from 1.0 µg of total RNA by using DNA-free reagent (Ambion). cDNAs were synthesized from this RNA using Superscript III reverse transcriptase and random hexamers according to the manufacturer's instructions (Invitrogen). DNA was isolated from infected monolayer cultures by harvesting cells and resuspending cell pellets in lysis buffer (400 mM NaCl, 10 mM Tris [pH 8.0], 10 mM EDTA, 0.1 mg/ml proteinase K, 0.2% sodium dodecyl sulfate [SDS]) at 37°C for 12 h. DNA was extracted with phenol-chloroform, extracted again with chloroform, precipitated with ethanol and resuspended in 50 µl water. Real-time PCR analysis was completed using 1 µl of either DNA or cDNA with SYBR green PCR master mix (Applied Biosystems) and primers specific to the sequence of interest. Primers used within these studies have been previously published [41] with the exception of cellular RBBP4 (set 1 : 5′-ATAGCCAGTGTGCAGCTCCCTAAT-3′ and 5′-AACGGGCCCTGTTTACTTCTCCTT-3′; set 2 : 5′-AGGAAGGCTATGGGCTTTCTTGGA-3′ and 5′-TGCCGTATGCCCTGTAAAGATGGT-3′) and CHD4 (set 1 : 5′-AGTGCTGCAACCATCCATACCTCT-3′ and 5′-ATGCCCACCCTCCTTAAGGTTCTT-3′; set 2 : 5′-TCGTAAGGGCCTGCGGAATGATAA-3′ and 5′-TTCTCTGATTTGCCTCGCAGGTCT-3′), and firefly luciferase gene (5′-ATTTATCGGAGTTGCAGTTGCGCC-3′ and 5′-GCTGCGAAATGCCCATACTGTTGA-3′). PCR quantitation was performed using the 7900HT Fast Real-Time PCR System (Applied Biosystems). Quantities for unknown samples were defined relative to a standard curve consisting of 10-fold serial dilutions of a single sample and completed for each primer pair. The results were normalized to either β-actin DNA or GAPDH cDNA levels within each sample.
RNA interference (RNAi) analysis
Disruption of cellular CHD4 and RBBP4 expression utilized short hairpin RNAs (shRNA) stably expressed in primary human fibroblasts or U-2 OS cells. The shRNA clones were expressed from the lentivirus vector, pLKO.1-Puro (Sigma Mission Clones), containing an shRNA sequence against either CHD4 (5′-CCGGGCTGACACAGTTATTATCTATCTCGAGATAGATAATAACTGTGTCAGCTTTTT-3′) or RBBP4 (5′-CCGGCCCTTGTATCATCGCAACAAACTCGAGTTTGTTGCGATGATACAAGGGTTTTTG-3′). The control shRNA consisted of a scrambled sequence (5′-TCAGTCGCGTTTGCGACTGGTTCAAGAGACCAGTCGCAAACGCGACTGTTTTTGGAAAC-3′) and was a kind gift from P. Traktman (Medical College of Wisconsin). Recombinant lentiviruses were produced by co-transfecting 293T cells with lentivirus expression plasmid and packaging plasmids [53], [54] using FuGENE 6 transfection reagent. Infectious lentiviruses were harvested at 48 and 72 hpi, cleared of 293T cells and frozen at −80°C. The infection of fibroblasts and U-2 OS cells with shRNA lentivirus was carried out by adding lentivirus to the cell culture medium and incubating with target cells for 24 h. shRNA-expressing cells were selected by using puromycin at 1 µg/ml (Invitrogen). The inhibition of CHD4 and RBBP4 expression was quantified by qRT-PCR and two separate sets of primers to each gene as listed above.
Luciferase assays
Control or pUL38-expressing U-2 OS cells were seeded onto 12-well plates and transfected using FuGENE 6 according to the manufacturer's instructions. Cells were transfected with 50 ng of pGL3-MIEP reporter plasmid and 10, 100, or 500 ng of pCGN empty vector or pCGN-pUL29/28 effector plasmid. For ISRE and NF-κB luciferase reporter plasmids, 100 ng of reporter was transfected into pUL38-expressing U-2 OS cells with either 500 ng of control or pUL29/28 effector plasmids. Luciferase activity was assayed 48 h posttransfection using a luciferase reporter assay system (Promega) and a Victor3 luminometer (Perkin-Elmer). Activity was measured by using equal protein amounts within each lysate and normalized to the luciferase activity from empty vector. Expression of pUL29/28 and pUL38 was assayed by Western blot analysis using the same lysates.
Results
Identification of HDAC-1 interacting proteins during HCMV infection
As discussed above, regulation of the viral chromatin state, including deacetylation of histones, plays a pivotal role in efficient HCMV infection. We were interested in determining what viral proteins might influence these events through interactions with HDACs. We focused on HDAC1, a 55 kDa nuclear protein. HDAC1 previously has been shown to influence herpesvirus replication as well as latency [30], [32], [55]–[57].
We constructed a retrovirus vector expressing the neomycin resistance gene and the HDAC1 open reading frame (ORF) with the enhanced green fluorescent protein (GFP) ORF fused at its C-terminus (pHDAC1gfp). For these studies, we used life-extended fibroblasts, which can be maintained for extended periods in culture and were demonstrated to support efficient HCMV replication [43]. Cells were transduced using the HDAC1-GFP retrovirus, cells containing the retrovirus were selected by treatment with G418, and a population expressing a moderate level of the fusion protein was isolated by FACS. We observed fluorescence within the nuclei of >99% of the pHDAC1gfp-expressing cells, which are termed HF-HD1GFP cells (Fig. 1A, left panels). Western blot analysis, performed on whole cell lysates from life-extended fibroblasts or HF-HD1GFP cells by using an antibody to HDAC1, demonstrated expression of a 55 kDa endogenous HDAC1 in both cell populations and an additional 75 kDa HDAC1-GFP exclusive to the transduced cells (Fig. 1A, right panel). The 75 kDa protein, pHDAC1gfp, was present at about half the level of endogenous protein, providing confidence that abnormal interactions due to overexpression of the fusion protein are not likely to occur.
Overexpression of HDAC proteins occurs in many cancers, and can affect cell proliferation [58]. To test whether constitutive pHDAC1-gfp expression altered the growth phenotype of the fibroblasts, we monitored their growth rate and found that the proliferation of HF-HD1GFP cells was indistinguishable from the parental HF cells (Fig. 1B, left panel). In addition, HF-HD1GFP cell growth was inhibited by trichostatin A (TSA), a class I and II HDAC inhibitor (Fig. 1B, left panel). To determine whether expression of HDAC1-GFP influenced HCMV replication, we completed a multistep growth analysis (Fig. 1B, right panel). Parental HF or HF-HD1GFP cells were infected at a multiplicity of 0.01 pfu/ml and medium was collected after various time intervals. A similar amount of infectious virus was released from the two cell lines indicating that expression of pHDAC1gfp does not affect HCMV replication. We conclude that HF-HD1GFP cells are a suitable cell line to identify HDAC1-interacting proteins during HCMV infection.
To identify cellular and viral proteins that interact with HDAC1, we infected HF-HD1GFP cells with wild-type HCMV (BADwt). At 24 hpi, pHDAC1gfp and associated proteins were isolated from cell extracts by using one-step immunoaffinity purification on magnetic beads coated with antibodies to GFP [36], [46], [47]. Isolated viral and host proteins were resolved by electrophoresis and identified by mass spectrometry. The major protein bands evident in the Coomassie blue-stained polyacrylamide gel were identified by employing sequential MALDI QqTOF MS and MALDI IT MS/MS analyses (Fig. 1C), and a complete listing of these proteins is presented in Table S1. Although their capture with pHDAC1gfp suggests they associate with HDAC1, confirmation is needed to definitively identify the proteins as interacting partners. Therefore, the proteins in table S1 must be considered as potential HDAC1-interacting proteins.
The HCMV proteins pUL29/28, pUL38, and pUL97 were identified in the capture experiment (Fig. 1C and Table S1). pUL97 is the 78 kDa viral kinase that is necessary for efficient replication and viral egress [59], [60]. pUL29/28 is a 79 kDa nuclear protein expressed from a spliced RNA containing the UL28 and UL29 ORFs, which is packaged in viral particles [61]. Disruption of pUL29/28 results in a virus that replicates poorly and expresses reduced levels of immediate-early genes [39], [40], [61]. pUL38 blocks cells from dying by apoptosis during infection [37]. It functions, in part, by regulating the unfolded protein response [38] and mTOR signaling pathways [36].
Our analysis also identified numerous cellular proteins predicted to interact with HDAC1 (Table S1). Analysis of infected fibroblasts expressing gfp alone (Fig. S1) and studies using different viral proteins tagged with gfp [62] did not detect these proteins, suggesting that they are unique to HDAC1gfp and not the gfp sequence itself. HDAC1 is found in several multi-protein complexes including NuRD [12], coREST [13], and Sin3 [14] complexes. Several proteins previously identified in these complexes were found in our capture experiment. We observed associations with components of the NuRD complex, including HDAC1, HDAC2, MTA1, MTA2, CHD3, CHD4, RBBP4 and LSD1. HDAC1, HDAC2, and RBBP4 are also elements of the Sin3 complex, and additional Sin3 components observed in our studies included SIN3A, SIN3B, and SDS3. HDAC1, HDAC2, and LSD1 are also elements of the coREST complex. In addition, putative interactions were observed with several cellular proteins previously not known to interact with HDAC1 (Table S1). Finally, we observed free GFP among the captured proteins, and this was not surprising since our past studies have demonstrated that loss of a portion of the epitope tag from fusion proteins occurs during HCMV infection [36], [62].
In a previous study we identified, but did not confirm, predicted associations of pUL38 with pUL29/28, HDAC1 and several additional components of the NuRD complex [36], and we have demonstrated that both pUL29/28 and pUL38 are required for efficient HCMV replication [37], [61]. With these earlier results in mind, we focused on the predicted association of HDAC1 with the viral proteins pUL29/28 and pUL38.
HCMV pUL29/28 and pUL38 interact with components of the NuRD complex
To validate the predicted interactions of pUL29/28 and pUL38 with HDAC1, we performed immunoprecipitation experiments from lysates of normal primary fibroblasts that did not express the HDAC1gfp fusion protein using an antibody to endogenous HDAC1. Because no antibody exists to HCMV pUL29/28, we infected fibroblasts with BADinUL29F, which expresses pUL29/28 with an amino-terminal FLAG epitope and replicates like wild-type virus [61]. Upon immunoprecipitation of endogenous HDAC1, we observed associations with both pUL29/28 and pUL38 at 24 hpi by Western blot assay using anti-FLAG and anti-pUL38 antibodies, respectively (Fig. 2A). Neither protein was detected in lysates from uninfected cells.

To test whether pUL29/28 and pUL38 associate with additional components of the NuRD complex, we repeated the analysis using an antibody to the MTA2 protein, a specific component of the complex [12]. Western blot analysis demonstrated that both pUL29/28 and pUL38 interact with MTA2 (Fig. 2A). We performed the reciprocal immunoprecipitation experiment to confirm the interactions (Fig. 2B, left panel). Fibroblasts were infected with BADinUL29F, pUL29/28 was immunoprecipitated through its FLAG epitope, and Western blot assays detected MTA2 and pUL38 in the precipitates. We also confirmed the interaction between pUL29/28 and HDAC1 using this approach (Fig. 2B, right panel). These interactions were not detected using an isotype-specific control antibody to the myc epitope, arguing for specificity of the immunoprecipitation. We conclude that HDAC1 and a second component of the NuRD complex, MTA2, interact either directly or indirectly with both HCMV pUL29/28 and pUL38 proteins during infection.
Our isolation of HDAC1-interacting proteins detected multiple components of both the NuRD and Sin3 complexes (Fig. 1 and Table S1). We next asked if pUL29/28 and pUL38 associate specifically with components of the NuRD complex or both HDAC1-containing complexes. Antibodies to SIN3A, a constituent of Sin3, or MTA2, a subunit of NuRD, were used to test for coprecipitation of the viral proteins. Both pUL29/28 and pUL38 precipitated with MTA2, while a low level of pUL29/28 and no detectable pUL38 were observed interacting with mSin3 (Fig. 2C, left panel). Thus, pUL29/28 interacts with HDAC1 in association with elements of at least two complexes, NuRD and Sin3. Its interaction with the NuRD constituent appears to be more robust than with the Sin3 element, although this could result in part from differences in performance of the two antibodies used for immunoprecipitation or from reduced accessibility of mSin3 to the antibody when pUL29/28 is present in the complex.
Next, we tested whether pUL29/28 binds to the TSC2 subunit of the tuberous sclerosis protein complex, because we previously demonstrated that pUL38 binds to TSC2 [36]. A TSC2-specific antibody coprecipitated pUL38 from infected cells, but very little pUL29/28, whereas the anti-MTA2 antibody clearly coprecipitated both pUL38 and pUL29/28 (Fig. 2C, right panel). Thus, the two viral proteins do not always track together, but HDAC1 clearly interacts with both pUL29/28 and pUL38 in the HDAC1-NuRD complex.
To further investigate the interaction of HDAC1 with pUL29/28 and pUL38, we performed an immunofluorescence microscopy analysis. Visual inspection of the resulting images using cells infected with the BADinUL29F virus showed that pUL29/28 and HDAC1 exhibit significant colocalization within the nucleus at 24 hpi (Fig. 3A, upper panels). No pUL29/28 signal was observed in an uninfected cells (data not shown) as well as an uninfected cell within the same image (Fig. 3A, upper panels), demonstrating specificity of the FLAG antibody to tagged pUL29/28. Quantitative measurement of images confirmed the colocalization [36], [63], demonstrating that Pearson's correlation (r) was greater for pUL29/28 and HDAC1 (r = 0.70) than for another HCMV nuclear protein, IE1, and HDAC1 (r = 0.47), which was concentrated near the edges of infected nuclei as described previously [52], [64]. Perfect colocalization of two proteins yields r = 1.0 [36]. We also observed colocalization between pUL29/28 and pUL38 within the nucleus (r = 0.77) (Fig. 3B, upper panels). This assay used the BADinUL38TAP virus expressing epitope tagged pUL38 [36] and primary fibroblasts expressing a myc-tagged pUL29/28. No signal for myc-tagged pUL29/28 was observed in a neighboring cell from an uninfected control sample (Fig. 3B, middle panels), demonstrating specificity of the antibody to pUL29/28. Finally, pUL38 and HDAC1 were colocalized (r = 0.79) at 24 hpi (Fig. 3B, lower panels). These correlation values are consistent with values observed for known protein-protein interactions [36]. Taken together, our results demonstrate that HDAC1 specifically interacts with both pUL29/28 and pUL38. This interaction likely results from an association of the viral proteins with the NuRD and Sin3 complexes.

pUL29/28 is necessary for pUL38 binding to HDAC1
pUL29/28 plus pUL38 were coprecipitated with antibodies against HDAC1, and pUL38 plus HDAC1 were precipitated with pUL29/28 antibody, arguing that these proteins exist within the same complex. We next tested whether pUL38 depends on pUL29/28 for interaction with HDAC1 by performing coimmunoprecipitations using uninfected fibroblasts expressing pUL38. Using a pUL38-specific antibody to assay by Western blot, we observed the viral protein in lysates of pUL38-expressing cells (Fig. 4A, lane 2–6). However, we did not detect an interaction between pUL38 and HDAC1 (Fig. 4A, lane 4), although in a control experiment, pUL38 was co-precipitated by using TSC2-specific antibody (Fig. 4A, lane 6). To determine the role of additional viral proteins in mediating this interaction, we infected pUL38-expressing cells with a pUL38-deficient virus [37]. We repeated the immunoprecipitation of HDAC1 and in this case we detected pUL38 by Western blot assay (Fig. 4A, lane 5). pUL38 is expressed in several isoforms of varying molecular weight due to the use of alternative starting methionines [36], and it appears that lower molecular weight forms preferentially interact with HDAC1 whereas higher molecular weight forms interact mainly with TSC2 (Fig. 4A, compare lanes 5 and 6). This data indicates that another viral protein or cellular protein induced during infection mediates the interaction between pUL38 and HDAC1.

Because pUL29/28 interacts with both pUL38 and HDAC1 (Fig. 2), we next asked whether pUL29/28 is necessary for the interaction between pUL38-HDAC1. We infected fibroblasts at a multiplicity of 3 pfu/cell with BADwt and two mutant viruses containing disruptions in either the UL28 (BADsubUL28) or UL29 (BADsubUL29) ORFs, both of which fail to express normal levels of immediate-early genes [39], [61]. pUL38 was coprecipitated with an HDAC1-specific antibody from whole cell lysates after infection with wild-type virus but not pUL29/28-deficient viruses (Fig. 4B). From these results, we conclude that pUL29/28 is necessary for the interaction between pUL38 and HDAC1 during infection. Next, we asked whether any additional viral proteins are needed for the interaction between HDAC1 and pUL29/28. For these studies, we used the smaller isoform of the protein since we were unable to isolate the full length protein from uninfected cells under conditions that were compatible with immunoprecipitation experiments. We transiently expressed pUL29 containing a hemagglutinin epitope tag in U-2 OS cells [41]. Under these conditions, HCMV pUL29 was co-immunoprecipitated with HDAC1 (Fig. 4C), while no band at the appropriate molecular weight was observed using the empty vector control (Fig. 4C). We conclude that no additional viral proteins are necessary to mediate the interaction of pUL29/28 and HDAC1, and that the critical region within the viral protein needed for the interaction is present within the UL29-coded domain.
pUL38 is exclusively nuclear at 8 hpi, but present in both the nucleus and cytoplasm at 24 hpi [36], [37]; in contrast pUL29/28 is expressed exclusively within the nucleus through 48 hpi and then in nucleus and cytoplasm at 72 hpi [61]. To determine whether the pUL29/28-pUL38-HDAC1 complex is formed during a defined time over the course of HCMV infection, i.e., when both viral constituents are clearly in the nucleus, we infected fibroblasts with BADinUL29F and collected whole cell lysates during the course of HCMV infection beginning at 6 hpi. Using an antibody against HDAC1, both pUL29/28 and pUL38 were co-precipitated beginning at 6 hpi and at all times tested through 72 hpi (Fig. 5, upper panel). We confirmed the presence of the complex at late times by using antibody to the MTA2 constituent of the NuRD complex (Fig. 5, lower panel). The pUL29/28-pUL38-HDAC1 complex forms very early during infection and exists through late times, presumably reflecting the fact that both viral proteins are present in the nucleus at all times after infection that were tested.

pUL29/28 and pUL38-deficient viruses are not rescued by inhibition of deacetylase activity
It is well established that inhibition of HDACs during cytomegalovirus infection stimulates immediate-early gene expression [20]–[24], [65]. Consequently, we reasoned that, if pUL29/28 or pUL38 interacts with HDAC1 to inhibit its activity, then an HDAC inhibitor might compensate for loss of pUL29/28 or pUL38 function. To test this possibility, fibroblasts were infected with BADwt, a pUL38-deficient (BADsubUL38) or pUL29/28-deficient (BADsubUL29) virus in the presence or absence of the HDAC antagonist, trichostatin A (TSA). Because we previously observed an abnormality in particle to PFU ratio for BADsubUL38 [37], we infected fibroblasts at a multiplicity of 0.1 pfu/cell for the wild-type virus and used an equivalent number of particles for the mutants. Culture medium was collected at 10 days post infection and viral titers were determined by quantifying the number of IE1-positive fibroblasts generated by a unit volume of virus (Fig. 6A). In the absence of drug, we observed an ∼102-fold decrease in virus yield for BADsubUL29 and an ∼104-fold decrease for BADsubUL38, consistent with our previous observations [37], [61], and the addition of TSA did not significantly rescue the growth of either mutant.

The absence of pUL29/28 results in reduced levels of immediate-early gene expression at very early times during infection [61]. To determine if inhibiting HDAC activity could rescue this phenotype, we infected cells pretreated with 500 nM of TSA [26], [66] with BADwt or BADsubUL29 and quantified IE1 RNA accumulation (Fig. 6B). The pUL29/28-deficient mutant generated reduced levels of RNA as compared to wild-type virus in the absence of TSA as previously seen [61]. Upon addition of TSA, a similar increase in IE1 levels was observed in both wild-type and BADsubUL29 infected cells and, importantly, TSA did not restore expression of the immediate-early RNA within mutant virus-infected cells to wild-type levels. Chemical inhibition of HDAC activity does not rescue the growth defect that occurs during replication in the absence of pUL29/28 or pUL38.
Multiple constituents of the NuRD complex are needed for efficient HCMV replication
Since inhibition of HDAC activity did not rescue the mutant viruses, we tested the possibility that NuRD components are needed by HCMV. The NuRD complex was disrupted by using short hairpin RNAs (shRNAs) targeting two specific components of the complex that were detected in the pHDAC1gfp capture experiment: CHD4 and RBBP4 (Fig. 1C). This approach has been successfully used by others to evaluate NuRD function [67], [68]. shRNAs specific for CHD4 or RBBP4 along with a non-specific scrambled sequence were introduced into primary human fibroblasts using a lentivirus vector containing the puromycin-resistance gene, and shRNA-expressing cells were selected by treating cultures with the drug. To demonstrate gene silencing, we assayed the steady state levels of CHD4 and RBBP4 RNA by quantitative RT-PCR (qRT-PCR) using two separate primer sets for each transcript. shRNA specific for CHD4 resulted in a ∼70% reduction in RNA levels as compared to the scrambled control, shRNA against RBBP4 resulted in an ∼85% reduction, and expression of the two shRNAs together generated a similar reduction (Fig. 7A).
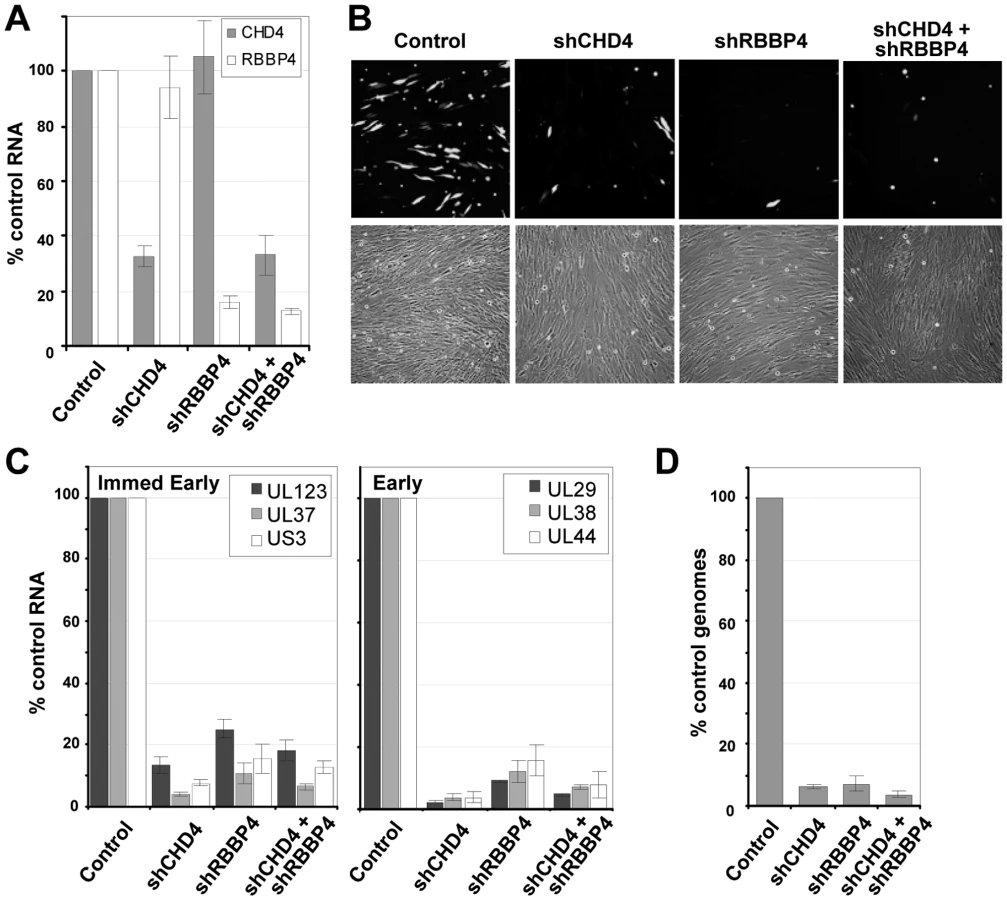
To test for a role of the NuRD subunits in HCMV replication, we infected fibroblasts expressing the scrambled control, CHD4, RBBP4 or both shRNAs with wild-type virus expressing a GFP marker gene at a multiplicity of 0.25 PFU/cell, and visualized GFP-positive cells 96 h later (Fig. 7B). The shRNAs against CHD4, RBBP4, or both components of the NuRD complex dramatically reduced the production of GFP-positive cells as compared to the control shRNA. To evaluate the impact on HCMV replication, we quantified both viral gene expression and genome replication during infection. Using qRT-PCR, we observed an ∼80% reduction in the accumulation of immediate-early RNAs (UL123, UL37 exon 1 and US3) and a >90% reduction in early RNAs (UL29, UL38 and UL44) at 10 hpi upon disruption of the NuRD complex as compared to control cells (Fig. 7C). These changes were accompanied by a >90% decrease in HCMV DNA accumulation at 96 hpi (Fig. 7D). We conclude that CHD4 and RBBP4, and by extension very likely a functional NuRD complex, are required for efficient HCMV replication.
pUL29/28 activates the major immediate-early promoter (MIEP) independently of pUL38
To confirm that pUL29/28 and/or pUL38 influence the accumulation of IE1 by modulating activity of the MIEP, which controls expression of UL123 mRNA, and to test whether the promoter was controlled by the viral proteins outside the context of the viral genome, we performed assays using a reporter in which luciferase expression is controlled by the MIEP. We have previously demonstrated that pUL29/28 alone can activate the MIEP in HEK-293T cells [41]. Increasing amounts of empty vector or a vector expressing HA epitope-tagged pUL29/28 were transfected together with the reporter into U-2 OS cells, and luciferase activity was determined 48 h later. Increasing amounts of pUL29/28 expression resulted in an ∼7-fold increase in luciferase activity relative to empty vector (Fig. 8A, left upper panel). Using the same lysates that were used to measure luciferase activity, we observed by Western blot analysis the expected increase in the levels of pUL29/28 with increasing input levels of the expression vector relative to cellular tubulin (Fig. 8A, left lower panel). To monitor the possible influence of pUL38 on MIEP activity, we repeated the experiment using U2OS-38 cells expressing pUL38 (Fig. 8A, right lower panel). Transfection of the MIEP luciferase reporter into pUL38-expressing cells resulted in similar levels of luciferase activity as observed in the control cells (Fig. 8A, left upper panel), arguing that pUL38 does not activate the MIEP. In a control experiment, increasing amounts of pUL29/28 increased MIEP activation in the U2OS-38 cells, but not to a greater extent than in cells lacking pUL38. Previously, we demonstrated that expression of pUL38 promotes active mTORC1 kinase and presumably protein translation [36]. To confirm that the increase in luciferase activity is the result of changes in RNA accumulation of the luciferase gene and not translation, we isolated RNA from pUL29/28-expressing U2OS or U2OS-38 cells and quantified the levels of UL29 and luciferase RNA by qRT-PCR. The similar ratios of the viral RNA relative to luciferase RNA (Fig. 8A, right upper panel) demonstrated that the luciferase assay reflected changes in RNA accumulation.

ISRE and NF-κB cis-acting elements contribute to full MIEP induction during infection [69], [70]. We tested whether pUL29/28 can activate expression of promoters comprised of these elements to determine whether it functions in part through these specific transcription factor binding sites, and more broadly to ask if the viral protein can influence expression from promoters other than the MIEP. Although it activated the MIEP reporter by a factor of 7, pUL29/28 had relatively little effect on reporter constructs containing promoters controlled by the ISRE or NF-κB elements (Fig. 8B). To confirm that pUL29/28-mediated activation of the MIEP reporter required NuRD components, the experiment was repeated in U2OS cells lacking either CHD4 or RBBP4. The shRNA-mediated knockdowns were confirmed by measuring RNA levels (Fig. 8C, left panel), and MIEP reporter activity in the presence of pUL29/28 in both knockdown cells was reduced to a modest extent but significant in only the CHD4 knockdown samples, as compared with normal U-2 OS cells (Fig. 8C, right panel).
To determine whether this activity could result from a direct interaction of pUL29/28 and elements of the NuRD complex at the MIEP, we performed chromatin immunoprecipitation (ChIP) experiments. Using the inUL29F virus, we infected primary fibroblasts and immunoprecipitated crosslinked DNA using an antibody to the MTA2 component of NuRD as well as to the FLAG epitope in pUL29/28. Both antibodies immunoprecipitated more MIEP DNA than a control antibody in three independent experiments (Fig. 8D).
We conclude that pUL29/28 works with components of the NuRD complex to activate the MIEP independently of pUL38, and both proteins interact with the MIEP during infection.
Discussion
Histone acetylation/deacetylation plays a pivotal role in the regulation of HCMV gene expression [20]–[24]. Analysis of proteins captured with pHDAC1gfp by mass spectrometry predicts that HDAC1 associates with a substantial number of cellular and viral proteins during infection (Fig. 1C and Table S1). HDAC1 exists in different complexes, and the capture assay identified putative interacting proteins that are found within the NuRD, coREST, and Sin3 complexes. We performed reciprocal co-immunoprecipitations to confirm the interaction of pUL38 and pUL29/28 with HDAC1 and to verify that the viral proteins interact with the MTA2 subunit of the NuRD complex (Fig. 2A and B). A relatively lower, but detectable, amount of pUL29/28 was coimmunoprecipitated with a component of the Sin3 complex (Fig. 2C), consistent with the view that the viral protein exists in multiple HDAC1-containing complexes within the infected cell, as predicted by the mass spectrometry analysis.
In addition to these HCMV proteins, we detected peptides corresponding to the viral kinase pUL97 in the HDAC1 capture experiment (Table S1). Although we did not validate this predicted interaction, other herpesvirus-coded kinases have been reported to hyperphosphorylate HDAC1, including the herpes simplex type 1 US3 kinase [56] and the varicella zoster virus ORF66p kinase [71]. Consequently, the possible HDAC1-pUL97 interaction could reflect a kinase-substrate interaction. We failed to detect IE1 or IE2 peptides in the HDAC1 capture experiment. These immediate-early proteins regulate HCMV gene expression, in part, by altering histone acetylation [10], [26], and IE2 has been previously reported to interact with HDAC1 [10] in transfected cells. Our failure to observe IE2 may be in part due to the isolation conditions (e.g. lysis buffer) or poor ionization of corresponding peptides during MS analysis, although we have detected IE2 in other MS analyses that were focused on the immediate-early protein (data not shown). Alternatively, the proteins might interact at the start of infection but not at 24 hpi, when we performed the capture assay.
We have previously shown that pUL29/28 and pUL38 are needed for efficient HCMV replication [37], [39], [41]. Our current results demonstrate that these proteins target the NuRD complex; but, rather than antagonize NuRD (Fig. 6), the viral proteins modulate its activity to support HCMV replication. We observed an ∼10-fold reduction in both HCMV RNA and DNA accumulation in cells expressing shRNAs directed against two different components of the NuRD complex, CHD4 and RBBP4 (Fig. 7C and D). A pUL29/28-deficient virus expressed ∼2-fold-reduced levels of immediate-early RNAs, but accumulated ∼10-fold less DNA when compared to wild-type virus [41]. The more severe effect on viral RNA accumulation observed for a NuRD as compared to a pUL29/28 deficiency suggests that NuRD likely plays a more complex role than simply regulating the MIEP in HCMV replication. The relatively small drop in pUL29/28-induced MIEP reporter activity upon disruption of NuRD components supports this view (Fig. 8C).
Our data demonstrate that components of the NuRD complex, with which pUL29/28 interacts, are needed for efficient replication. This is in contrast to the consequences of drug-mediated, global HDAC inhibition during infection, which enhances immediate-early gene expression [20]–[24], overcomes hDaxx-mediated repression on the MIEP [25] and rescues an IE1-deficient virus [26]. We observed similar increases in IE1 RNA expression in both pUL29-deficient and wild-type virus in the presence of TSA (Fig. 6B), suggesting that the mechanism to overcome hDaxx-mediated repression on the MIEP is still in place in the absence of pUL29/28.
pUL38 interacts with NuRD components only in the presence of pUL29/28 (Fig. 4); and pUL29/28, but not pUL38, can activate the MIEP in a reporter assay (Fig. 8A and B). The NuRD subunit-dependent (Fig. 8C) ability of pUL29/28 to modulate the MIEP in the absence of additional viral proteins argues that it plays a significant role in modulation of the NuRD complex during infection. Further, pUL29/28 failed to significantly activate reporters controlled by ISRE or NF-κB elements (Fig. 8B), supporting the interpretation that that pUL29/28 modulates NuRD activity to generate promoter-specific outcomes.
How does pUL29/28 modulate the NuRD complex to favor expression of the MIEP? The HCMV genome undergoes chromatin remodeling and changes in acetylation during the course of infection through unknown mechanisms [8], [9]. The NuRD complex contains both HDAC and nucleosome remodeling activity [12] and participates in both activating and repressing transcription [72]. As discussed above, global inhibition of HDAC activity enhances viral replication, raising the possibility that NuRD HDAC activity is not important for HCMV. Perhaps pUL29/28 modulates the activity of the MIEP and accumulation of immediate-early RNAs by influencing nucleosome positioning through its interaction with NuRD. In this regard, we observed an interaction of the MTA2 component of NuRD and pUL29/28 with the MIEP (Fig. 8D). There is precedent for a role of nucleosome remodeling in HCMV gene expression. The p150 subunit of the chromatin assembly factor 1 (CAF1) is localized to HCMV replication centers during viral infection [8], and it has been shown to influence MIEP activation [73]. CAF1 consists of three subunits, p150, p60 and p48 (RBBP4), and functions in nucleosome assembly [74] and p150 has been observed in remodeling complexes [75]. Alternatively, a complex of pUL29/28 and NuRD may act globally to alter the nuclear environment. The NuRD complex deacetylates both cellular proteins and histones at cellular promoters [12]. Consistent with a role at sites other than the HCMV genome, pUL29/28 and pUL38 are not restricted to viral replication centers during the course of infection (Fig. 3) [37], [41]. The NuRD complex regulates expression of several cellular genes, including snail [76], wnt4 [77], and brca1 [78]. Interestingly, expression of Snail and wnt4 are rapidly down regulated during HCMV infection [79]. Beyond histones, NuRD also has been demonstrated to directly deacetylate transcription factors, including p53 and HIF-1α [17], [18]. Casavant et al. [80] have argued that p53 is necessary for efficient HCMV replication, influencing viral gene expression [81].
Previously, we have demonstrated that HCMV pUL38 also interacts with and antagonizes the TSC complex during infection [36]. This association is clearly distinct from its association with the NuRD complex, because pUL29/28 is not associated with TSC2 (Fig. 2C). Further, a lower molecular weight isoform of pUL38 binds pUL29/28 and MTA2 than interacts with TSC2 (data not shown). The different forms of pUL38 have been attributed to alternative usage of ATG start codons [36]. In addition, the interaction of pUL38-pUL29/28 with NuRD subunits is evident by 6 hpi (Fig. 5) within the nucleus (Fig. 3), while pUL38-TSC complex is detected after 24 hpi and predominantly within the cytoplasm [36]. The consequences of the pUL38 interaction with NuRD subunits remains unclear. However, our studies suggest that the contribution of NuRD to replication is complex, and we speculate that pUL38 is important in regulating NuRD. Both TSC and NuRD complexes are targets of the human papillomavirus (HPV) E6 and E7 proteins [33], [82]. This conservation in cell targets among different viruses argues for their importance, and consequently for a central role of pUL29/28 and pUL38 in viral pathogenesis.
Supporting Information
{kind=link}
Zdroje
1. MocarskiE
ShenkT
PassRF
2007 Cytomegaloviruses. Fields Virology Lippincott Williams & Wilkins 2702 2772
2. StreblowDN
DumortierJ
MosesAV
OrloffSL
NelsonJA
2008 Mechanisms of cytomegalovirus-accelerated vascular disease: induction of paracrine factors that promote angiogenesis and wound healing. Curr Top Microbiol Immunol 325 397 415
3. WallerEC
DayE
SissonsJG
WillsMR
2008 Dynamics of T cell memory in human cytomegalovirus infection. Med Microbiol Immunol 197 83 96
4. CinatlJJr
NevelsM
PaulusC
MichaelisM
2009 Activation of telomerase in glioma cells by human cytomegalovirus: another piece of the puzzle. J Natl Cancer Inst 101 441 443
5. TerhuneSS
SchroerJ
ShenkT
2004 RNAs are packaged into human cytomegalovirus virions in proportion to their intracellular concentration. J Virol 78 10390 10398
6. BresnahanWA
ShenkT
2000 A subset of viral transcripts packaged within human cytomegalovirus particles. Science 288 2373 2376
7. ReevesMB
DaviesAA
McSharryBP
WilkinsonGW
SinclairJH
2007 Complex I binding by a virally encoded RNA regulates mitochondria-induced cell death. Science 316 1345 1348
8. NitzscheA
PaulusC
NevelsM
2008 Temporal Dynamics of Cytomegalovirus Chromatin Assembly in Productively Infected Human Cells. J Virol
9. Cuevas-BennettC
ShenkT
2008 Dynamic Histone H3 Acetylation and Methylation at Human Cytomegalovirus Promoters during Replication in Fibroblasts. J Virol 82 9525 9536
10. ReevesM
MurphyJ
GreavesR
FairleyJ
BrehmA
2006 Autorepression of the human cytomegalovirus major immediate-early promoter/enhancer at late times of infection is mediated by the recruitment of chromatin remodeling enzymes by IE86. J Virol 80 9998 10009
11. IoudinkovaE
ArcangelettiMC
RynditchA
De ContoF
MottaF
2006 Control of human cytomegalovirus gene expression by differential histone modifications during lytic and latent infection of a monocytic cell line. Gene 384 120 128
12. DenslowSA
WadePA
2007 The human Mi-2/NuRD complex and gene regulation. Oncogene 26 5433 5438
13. LakowskiB
RoelensI
JacobS
2006 CoREST-like complexes regulate chromatin modification and neuronal gene expression. J Mol Neurosci 29 227 239
14. SilversteinRA
EkwallK
2005 Sin3: a flexible regulator of global gene expression and genome stability. Curr Genet 47 1 17
15. WangY
ZhangH
ChenY
SunY
YangF
2009 LSD1 is a subunit of the NuRD complex and targets the metastasis programs in breast cancer. Cell 138 660 672
16. GuschinD
WadePA
KikyoN
WolffeAP
2000 ATP-Dependent histone octamer mobilization and histone deacetylation mediated by the Mi-2 chromatin remodeling complex. Biochemistry 39 5238 5245
17. LuoJ
SuF
ChenD
ShilohA
GuW
2000 Deacetylation of p53 modulates its effect on cell growth and apoptosis. Nature 408 377 381
18. YooYG
KongG
LeeMO
2006 Metastasis-associated protein 1 enhances stability of hypoxia-inducible factor-1alpha protein by recruiting histone deacetylase 1. EMBO J 25 1231 1241
19. ParkJJ
KimYE
PhamHT
KimET
ChungYH
2007 Functional interaction of the human cytomegalovirus IE2 protein with histone deacetylase 2 in infected human fibroblasts. J Gen Virol 88 3214 3223
20. Kuntz-SimonG
ObertG
1995 Sodium valproate, an anticonvulsant drug, stimulates human cytomegalovirus replication. J Gen Virol 76 (Pt 6) 1409 1415
21. TangQ
MaulGG
2003 Mouse cytomegalovirus immediate-early protein 1 binds with host cell repressors to relieve suppressive effects on viral transcription and replication during lytic infection. J Virol 77 1357 1367
22. SaffertRT
KalejtaRF
2006 Inactivating a cellular intrinsic immune defense mediated by Daxx is the mechanism through which the human cytomegalovirus pp71 protein stimulates viral immediate-early gene expression. J Virol 80 3863 3871
23. MichaelisM
SuhanT
ReinischA
ReisenauerA
FleckensteinC
2005 Increased replication of human cytomegalovirus in retinal pigment epithelial cells by valproic acid depends on histone deacetylase inhibition. Invest Ophthalmol Vis Sci 46 3451 3457
24. HummelM
YanS
LiZ
VargheseTK
AbecassisM
2007 Transcriptional reactivation of murine cytomegalovirus ie gene expression by 5-aza-2'-deoxycytidine and trichostatin A in latently infected cells despite lack of methylation of the major immediate-early promoter. J Gen Virol 88 1097 1102
25. WoodhallDL
GrovesIJ
ReevesMB
WilkinsonG
SinclairJH
2006 Human Daxx-mediated repression of human cytomegalovirus gene expression correlates with a repressive chromatin structure around the major immediate early promoter. J Biol Chem 281 37652 37660
26. NevelsM
PaulusC
ShenkT
2004 Human cytomegalovirus immediate-early 1 protein facilitates viral replication by antagonizing histone deacetylation. Proc Natl Acad Sci U S A 101 17234 17239
27. PaulusC
KraussS
NevelsM
2006 A human cytomegalovirus antagonist of type I IFN-dependent signal transducer and activator of transcription signaling. Proc Natl Acad Sci U S A 103 3840 3845
28. TaylorTJ
KnipeDM
2004 Proteomics of herpes simplex virus replication compartments: association of cellular DNA replication, repair, recombination, and chromatin remodeling proteins with ICP8. J Virol 78 5856 5866
29. LomonteP
ThomasJ
TexierP
CaronC
KhochbinS
2004 Functional interaction between class II histone deacetylases and ICP0 of herpes simplex virus type 1. J Virol 78 6744 6757
30. GuH
LiangY
MandelG
RoizmanB
2005 Components of the REST/CoREST/histone deacetylase repressor complex are disrupted, modified, and translocated in HSV-1-infected cells. Proc Natl Acad Sci U S A 102 7571 7576
31. RadkovSA
TouitouR
BrehmA
RoweM
WestM
1999 Epstein-Barr virus nuclear antigen 3C interacts with histone deacetylase to repress transcription. J Virol 73 5688 5697
32. KnightJS
LanK
SubramanianC
RobertsonES
2003 Epstein-Barr virus nuclear antigen 3C recruits histone deacetylase activity and associates with the corepressors mSin3A and NCoR in human B-cell lines. J Virol 77 4261 4272
33. BrehmA
NielsenSJ
MiskaEA
McCanceDJ
ReidJL
1999 The E7 oncoprotein associates with Mi2 and histone deacetylase activity to promote cell growth. EMBO J 18 2449 2458
34. LongworthMS
LaiminsLA
2004 The binding of histone deacetylases and the integrity of zinc finger-like motifs of the E7 protein are essential for the life cycle of human papillomavirus type 31. J Virol 78 3533 3541
35. LongworthMS
WilsonR
LaiminsLA
2005 HPV31 E7 facilitates replication by activating E2F2 transcription through its interaction with HDACs. EMBO J 24 1821 1830
36. MoormanNJ
CristeaIM
TerhuneSS
RoutMP
ChaitBT
2008 Human cytomegalovirus protein UL38 inhibits host cell stress responses by antagonizing the tuberous sclerosis protein complex. Cell Host Microbe 3 253 262
37. TerhuneS
TorigoiE
MoormanN
SilvaM
QianZ
2007 Human cytomegalovirus UL38 protein blocks apoptosis. J Virol 81 3109 3123
38. XuanB
QianZ
TorigoiE
YuD
2009 Human cytomegalovirus protein pUL38 induces ATF4 expression, inhibits persistent JNK phosphorylation, and suppresses endoplasmic reticulum stress-induced cell death. J Virol 83 3463 3474
39. YuD
SilvaMC
ShenkT
2003 Functional map of human cytomegalovirus AD169 defined by global mutational analysis. Proc Natl Acad Sci U S A 100 12396 12401
40. DunnW
ChouC
LiH
HaiR
PattersonD
2003 Functional profiling of a human cytomegalovirus genome. Proc Natl Acad Sci U S A 100 14223 14228
41. MitchellDP
SavarynJP
MoormanNJ
ShenkT
TerhuneSS
2009 Human cytomegalovirus UL28 and UL29 open reading frames encode a spliced mRNA and stimulate accumulation of immediate-early RNAs. J Virol 83 10187 10197
42. RomanowskiMJ
ShenkT
1997 Characterization of the human cytomegalovirus irs1 and trs1 genes: a second immediate-early transcription unit within irs1 whose product antagonizes transcriptional activation. J Virol 71 1485 1496
43. BresnahanWA
HultmanGE
ShenkT
2000 Replication of wild-type and mutant human cytomegalovirus in life-extended human diploid fibroblasts. J Virol 74 10816 10818
44. KinsellaTM
NolanGP
1996 Episomal vectors rapidly and stably produce high-titer recombinant retrovirus. Hum Gene Ther 7 1405 1413
45. YuD
SmithGA
EnquistLW
ShenkT
2002 Construction of a self-excisable bacterial artificial chromosome containing the human cytomegalovirus genome and mutagenesis of the diploid TRL/IRL13 gene. J Virol 76 2316 2328
46. CristeaIM
CarrollJW
RoutMP
RiceCM
ChaitBT
2006 Tracking and elucidating alphavirus-host protein interactions. J Biol Chem 281 30269 30278
47. CristeaIM
WilliamsR
ChaitBT
RoutMP
2005 Fluorescent proteins as proteomic probes. Mol Cell Proteomics 4 1933 1941
48. KrutchinskyAN
ZhangW
ChaitBT
2000 Rapidly switchable matrix-assisted laser desorption/ionization and electrospray quadrupole-time-of-flight mass spectrometry for protein identification. J Am Soc Mass Spectrom 11 493 504
49. KrutchinskyAN
KalkumM
ChaitBT
2001 Automatic identification of proteins with a MALDI-quadrupole ion trap mass spectrometer. Anal Chem 73 5066 5077
50. LuoY
LiT
YuF
KramerT
CristeaIM
2009 Resolving the Composition of Protein Complexes Using a MALDI LTQ Orbitrap. J Am Soc Mass Spectrom
51. ZhuH
ShenY
ShenkT
1995 Human cytomegalovirus IE1 and IE2 proteins block apoptosis. J Virol 69 7960 7970
52. FengX
SchroerJ
YuD
ShenkT
2006 Human cytomegalovirus pUS24 is a virion protein that functions very early in the replication cycle. J Virol 80 8371 8378
53. NaldiniL
BlomerU
GallayP
OryD
MulliganR
1996 In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 272 263 267
54. NaldiniL
BlomerU
GageFH
TronoD
VermaIM
1996 Efficient transfer, integration, and sustained long-term expression of the transgene in adult rat brains injected with a lentiviral vector. Proc Natl Acad Sci U S A 93 11382 11388
55. LuF
ZhouJ
WiedmerA
MaddenK
YuanY
2003 Chromatin remodeling of the Kaposi's sarcoma-associated herpesvirus ORF50 promoter correlates with reactivation from latency. J Virol 77 11425 11435
56. PoonAP
GuH
RoizmanB
2006 ICP0 and the US3 protein kinase of herpes simplex virus 1 independently block histone deacetylation to enable gene expression. Proc Natl Acad Sci U S A 103 9993 9998
57. ZhangY
JonesC
2001 The bovine herpesvirus 1 immediate-early protein (bICP0) associates with histone deacetylase 1 to activate transcription. J Virol 75 9571 9578
58. GlozakMA
SetoE
2007 Histone deacetylases and cancer. Oncogene 26 5420 5432
59. WolfDG
CourcelleCT
PrichardMN
MocarskiES
2001 Distinct and separate roles for herpesvirus-conserved UL97 kinase in cytomegalovirus DNA synthesis and encapsidation. Proc Natl Acad Sci U S A 98 1895 1900
60. KroskyPM
BaekMC
CoenDM
2003 The human cytomegalovirus UL97 protein kinase, an antiviral drug target, is required at the stage of nuclear egress. J Virol 77 905 914
61. MitchellDP
SavarynJP
MoormanNJ
ShenkT
TerhuneSS
2009 Human cytomegalovirus UL28 and UL29 open reading frames encode a spliced mRNA and stimulate accumulation of immediate-early RNAs. J Virol
62. MoormanNJ
Sharon-FrilingR
ShenkT
CristeaIM
2009 A targeted spatial-temporal proteomic approach implicates multiple cellular trafficking pathways in human cytomegalovirus virion maturation. Mol Cell Proteomics
63. CostesSV
DaelemansD
ChoEH
DobbinZ
PavlakisG
2004 Automatic and quantitative measurement of protein-protein colocalization in live cells. Biophys J 86 3993 4003
64. NevelsM
BruneW
ShenkT
2004 SUMOylation of the human cytomegalovirus 72-kilodalton IE1 protein facilitates expression of the 86-kilodalton IE2 protein and promotes viral replication. J Virol 78 7803 7812
65. GrovesI
ReevesM
SinclairJ
2009 Lytic infection of permissive cells with human cytomegalovirus is regulated by an intrinsic “pre-immediate early” repression of viral gene expression mediated by histone post-translational modification. J Gen Virol
66. GuoW
ShanB
KlingsbergRC
QinX
LaskyJA
2009 Abrogation of TGF-beta1-induced fibroblast-myofibroblast differentiation by histone deacetylase inhibition. Am J Physiol Lung Cell Mol Physiol 297 L864 870
67. PegoraroG
KubbenN
WickertU
GohlerH
HoffmannK
2009 Ageing-related chromatin defects through loss of the NURD complex. Nat Cell Biol 11 1261 1267
68. Ramirez-CarrozziVR
NazarianAA
LiCC
GoreSL
SridharanR
2006 Selective and antagonistic functions of SWI/SNF and Mi-2beta nucleosome remodeling complexes during an inflammatory response. Genes Dev 20 282 296
69. DeMerittIB
MilfordLE
YurochkoAD
2004 Activation of the NF-kappaB pathway in human cytomegalovirus-infected cells is necessary for efficient transactivation of the major immediate-early promoter. J Virol 78 4498 4507
70. NetterwaldJ
YangS
WangW
GhannyS
CodyM
2005 Two gamma interferon-activated site-like elements in the human cytomegalovirus major immediate-early promoter/enhancer are important for viral replication. J Virol 79 5035 5046
71. WaltersMS
ErazoA
KinchingtonPR
SilversteinS
2009 Histone Deacetylase 1 and 2 are Phosphorylated at Novel Sites During Varicella Zoster Virus Infection. J Virol
72. Miccio A, Wang Y, Hong W, Gregory GD, Wang H, et al. NuRD mediates activating and repressive functions of GATA-1 and FOG-1 during blood development. EMBO J 29 442 456
73. LeeSB
OuDS
LeeCF
JuanLJ
2009 Gene-specific transcriptional activation mediated by the p150 subunit of the chromatin assembly factor 1. J Biol Chem 284 14040 14049
74. TakamiY
OnoT
FukagawaT
ShibaharaK
NakayamaT
2007 Essential role of chromatin assembly factor-1-mediated rapid nucleosome assembly for DNA replication and cell division in vertebrate cells. Mol Biol Cell 18 129 141
75. KitagawaH
FujikiR
YoshimuraK
MezakiY
UematsuY
2003 The chromatin-remodeling complex WINAC targets a nuclear receptor to promoters and is impaired in Williams syndrome. Cell 113 905 917
76. FujitaN
JayeDL
KajitaM
GeigermanC
MorenoCS
2003 MTA3, a Mi-2/NuRD complex subunit, regulates an invasive growth pathway in breast cancer. Cell 113 207 219
77. ZhangH
SinghRR
TalukderAH
KumarR
2006 Metastatic tumor antigen 3 is a direct corepressor of the Wnt4 pathway. Genes Dev 20 2943 2948
78. MolliPR
SinghRR
LeeSW
KumarR
2008 MTA1-mediated transcriptional repression of BRCA1 tumor suppressor gene. Oncogene 27 1971 1980
79. BrowneEP
WingB
ColemanD
ShenkT
2001 Altered cellular mRNA levels in human cytomegalovirus-infected fibroblasts: viral block to the accumulation of antiviral mRNAs. J Virol 75 12319 12330
80. CasavantNC
LuoMH
RosenkeK
WinegardnerT
ZurawskaA
2006 Potential role for p53 in the permissive life cycle of human cytomegalovirus. J Virol 80 8390 8401
81. HannemannH
RosenkeK
O'DowdJM
FortunatoEA
2009 The presence of p53 influences the expression of multiple human cytomegalovirus genes at early times postinfection. J Virol 83 4316 4325
82. LuZ
HuX
LiY
ZhengL
ZhouY
2004 Human papillomavirus 16 E6 oncoprotein interferences with insulin signaling pathway by binding to tuberin. J Biol Chem 279 35664 35670
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2010 Číslo 6
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Requirement of NOX2 and Reactive Oxygen Species for Efficient RIG-I-Mediated Antiviral Response through Regulation of MAVS Expression
- Formation of Complexes at Plasmodesmata for Potyvirus Intercellular Movement Is Mediated by the Viral Protein P3N-PIPO
- Insight into the Mechanisms of Adenovirus Capsid Disassembly from Studies of Defensin Neutralization
- Two Novel Point Mutations in Clinical Reduce Linezolid Susceptibility and Switch on the Stringent Response to Promote Persistent Infection
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy