
Host Cell Invasion and Virulence in Sepsis Is Facilitated by the Multiple Repeats within FnBPA
Entry of Staphylococcus aureus into the bloodstream can lead to metastatic abscess formation and infective endocarditis. Crucial to the development of both these conditions is the interaction of S. aureus with endothelial cells. In vivo and in vitro studies have shown that the staphylococcal invasin FnBPA triggers bacterial invasion of endothelial cells via a process that involves fibronectin (Fn) bridging to α5β1 integrins. The Fn-binding region of FnBPA usually contains 11 non-identical repeats (FnBRs) with differing affinities for Fn, which facilitate the binding of multiple Fn molecules and may promote integrin clustering. We thus hypothesized that multiple repeats are necessary to trigger the invasion of endothelial cells by S. aureus. To test this we constructed variants of fnbA containing various combinations of FnBRs. In vitro assays revealed that endothelial cell invasion can be facilitated by a single high-affinity, but not low-affinity FnBR. Studies using a nisin-inducible system that controlled surface expression of FnBPA revealed that variants encoding fewer FnBRs required higher levels of surface expression to mediate invasion. High expression levels of FnBPA bearing a single low affinity FnBR bound Fn but did not invade, suggesting that FnBPA affinity for Fn is crucial for triggering internalization. In addition, multiple FnBRs increased the speed of internalization, as did higher expression levels of FnBPA, without altering the uptake mechanism. The relevance of these findings to pathogenesis was demonstrated using a murine sepsis model, which showed that multiple FnBRs were required for virulence. In conclusion, multiple FnBRs within FnBPA facilitate efficient Fn adhesion, trigger rapid bacterial uptake and are required for pathogenesis.
Published in the journal:
. PLoS Pathog 6(6): e32767. doi:10.1371/journal.ppat.1000964
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1000964
Summary
Entry of Staphylococcus aureus into the bloodstream can lead to metastatic abscess formation and infective endocarditis. Crucial to the development of both these conditions is the interaction of S. aureus with endothelial cells. In vivo and in vitro studies have shown that the staphylococcal invasin FnBPA triggers bacterial invasion of endothelial cells via a process that involves fibronectin (Fn) bridging to α5β1 integrins. The Fn-binding region of FnBPA usually contains 11 non-identical repeats (FnBRs) with differing affinities for Fn, which facilitate the binding of multiple Fn molecules and may promote integrin clustering. We thus hypothesized that multiple repeats are necessary to trigger the invasion of endothelial cells by S. aureus. To test this we constructed variants of fnbA containing various combinations of FnBRs. In vitro assays revealed that endothelial cell invasion can be facilitated by a single high-affinity, but not low-affinity FnBR. Studies using a nisin-inducible system that controlled surface expression of FnBPA revealed that variants encoding fewer FnBRs required higher levels of surface expression to mediate invasion. High expression levels of FnBPA bearing a single low affinity FnBR bound Fn but did not invade, suggesting that FnBPA affinity for Fn is crucial for triggering internalization. In addition, multiple FnBRs increased the speed of internalization, as did higher expression levels of FnBPA, without altering the uptake mechanism. The relevance of these findings to pathogenesis was demonstrated using a murine sepsis model, which showed that multiple FnBRs were required for virulence. In conclusion, multiple FnBRs within FnBPA facilitate efficient Fn adhesion, trigger rapid bacterial uptake and are required for pathogenesis.
Introduction
Staphylococcus aureus is a major human pathogen and the continuous emergence and spread of antibiotic resistant strains (e.g. MRSA, VRSA) mean treatment options are often severely limited [1], [2]. Despite its normal role as a commensal organism living asymptomatically in the nasal cavities of a large proportion of the human population [3], S. aureus is also responsible for a raft of different infections that range in both anatomical site and severity. These infections are facilitated by a vast array of different virulence factors such as adhesins, invasins, toxins and modulins, which not only enable evasion of host immune responses [4], [5], but also contribute to colonization, dissemination, tissue damage and transmission [1].
Whilst some infections are superficial and self-limiting, S. aureus is also responsible for serious invasive diseases. Indeed, S. aureus is a leading cause of sepsis and infective endocarditis [1], [6]–[8]. Colonization of the heart and subsequent formation of vegetations involves a number of complex interactions [9]–[11]. Animal studies have shown that staphylococcal fibronectin-binding protein A (FnBPA) is able to support the colonization of heart valves by otherwise non-pathogenic Lactococcus lactis, as well as promote dissemination into the spleen [12], [13]. In addition, FnBPs were significantly associated with systemic inflammation, severe weight loss and mortality in a murine sepsis model [14]. The role of FnBPA in virulence is supported by in vitro work showing that L. lactis expressing FnBPA is able to activate endothelial cells, inducing inflammatory and pro-coagulant responses [15], [16]. In addition to binding fibronectin (Fn) and fibrinogen, FnBPA promotes attachment to the endothelium and triggers the uptake of S. aureus by endothelial cells, which is believed to facilitate bacterial persistence and the establishment of secondary (metastatic) infections [17]–[21].
In common with several other bacterial pathogens, S. aureus invades endothelial cells via cell surface integrins [22]–[24]. The bacterium binds Fn, which is attached to the endothelial cell by α5β1 integrins [18], [19]. This triggers bacterial uptake via a host-cell driven process involving actin remodelling, focal adhesion kinase and Src family kinases [25], [26]. In vitro studies suggest that Fn on the surface of endothelial cells is sufficient for S. aureus-integrin bridging and thus binding of exogenous Fn (e.g. from plasma) is not required [19], [26].
In addition to FnBPA, many strains express a second, related, Fn-binding protein (FnBPB). Indeed, the majority of clinical isolates screened encode both (77%), whilst the remainder encode just FnBPA (22%) or, rarely, just FnBPB (1%) [27]. Both proteins have N-terminal domains that bind fibrinogen and elastin [28], [29] followed by a region containing 11 (FnBPA) or 10 (FnBPB) non-identical repeats that are responsible for binding F1 modules in the N-terminal domain of Fn [30], [31]. Studies using synthesized peptides have demonstrated that some FnBRs (1, 4, 5, 9, 10 and 11) have a high affinity for Fn, whilst the others have a much lower affinity [30], [31]. The repeat region is intrinsically disordered, allowing the relatively small FnBRs (27–39 aa) to bind the much larger Fn molecule with high affinity [31]. It has been hypothesized that this arrangement enables a single FnBPA protein to bind multiple Fn molecules, leading to integrin clustering and bacterial uptake [30], [32], [33]. Previous work, using an old organizational scheme for FnBPA based solely on amino acid sequence similarity demonstrated that partial deletions in the repeat region did not detectably diminish the ability of bacteria to bind Fn or invade endothelial cells [13], [19]. However, because it is difficult to relate this work to the new domain organization based on structural data [30], [31], it is not clear how many FnBRs are necessary for, or how the affinity of a given repeat for Fn affects invasion of host cells. Furthermore, it is unknown how the bacterium benefits from such apparent redundancy. This study addresses these questions by using bacteria expressing FnBPA variants containing various combinations of FnBRs to dissect their role in Fn-binding, the invasion of endothelial cells and virulence in a murine sepsis model.
Results
Construction of FnBPA variants
FnBPA contains 11 non-identical repeats with either high or low affinity for fibronectin (Fig. 1A). FnBPA variants were generated using a technique based on the circular PCR approach described by Massey et al. [13] (Fig. 1A, B and Table 1). Primers were modified to include 5′ phosphate, which facilitated simple blunt-ended ligation of PCR products to form a plasmid encoding fnbA variants lacking DNA encoding various repeats (Fig. 1B). Certain constructs were themselves used as templates to produce further fnbA variants (Table 1). Previous work (using the old domain organizational scheme (Fig. 1A)), suggested that only a few repeats were needed to confer binding to fibronectin and invasion of endothelial cells [13], [19] and we therefore produced FnBPA variants containing various combinations of 1–3 repeats. As such, 3 different types of construct were produced; those containing a mixture of high - and low-affinity repeats, those containing just high-affinity repeats and those containing just low-affinity repeats (Fig. 1C). Western immunoblots confirmed expression of each of the constructs, which appeared as bands of the expected size (Fig. 1D). DNA sequencing ensured the integrity of each construct (data not shown).


Surface expression levels of FnBPA are equivalent between variants
To verify that each of the FnBPA variants were expressed at equivalent concentrations we employed an ELISA approach using antibodies raised to the N-terminal region of FnBPA that is conserved between our variants. Standard plots (Fig. S1) indicated that this would allow us to accurately measure FnBPA expression levels and detect any differences between variants. Because S. aureus expresses protein A, we used a S. aureus ΔfnbA/B (Δfnb) strain (DU5883) as the blank in our ELISA assay, meaning that values relate purely to FnBPA expression levels. As expected from our Western immunoblot data (Fig. 1D), FnBPA expression levels in the 8325.4 wild-type (WT) strain were low (Fig. 2A). These levels were higher amongst the bacteria expressing plasmid-encoded FnBPA, which were equivalent between variants (Fig. 2A).

We also measured FnBPA expression by quantifying adhesion of S. aureus expressing each FnBPA variant to immobilized fibrinogen (Fig. 2B). Fibrinogen binding is conferred by the N-terminal region that is constant in all of the variants [28]. Attachment to fibrinogen was maintained in the 8325.4 Δfnb strain due to the presence of fibrinogen-binding ClfA and ClfB proteins. However, binding of S. aureus expressing plasmid-encoded FnBPA was significantly enhanced above that of the 8325.4 WT or Δfnb strains, reflecting the higher levels of plasmid-encoded FnBPA, which was equal between variants indicating equivalent expression levels (Fig. 2B).
A single high-affinity FnBR is sufficient for FnBPA-mediated adhesion to Fn and invasion of endothelial cells
Previous work using synthesized peptides [31] determined that some FnBRs have a high affinity for Fn, whilst others bind more weakly (Fig. 1A). To examine how the composition of the Fn-binding region of FnBPA affects both adhesion to Fn and endothelial cell invasion, various combinations of FnBPA repeats were expressed on the surface of a S. aureus 8325.4 mutant strain lacking both FnBPA and FnBPB [34]. In keeping with previous work [19], [34], S. aureus lacking FnBPs exhibited significantly reduced binding to Fn (Fig. 3A) and invasion of endothelial cells (Fig. 3B; Fig. S2). S. aureus expressing FnBPA lacking the entire Fn-binding repeat region (FnBPR0) did not adhere to Fn or invade endothelial cells above the levels of the FnBP-deficient mutant (Fig. 3A, B). Expression of FnBPA from the plasmid was higher than that of WT bacteria (Fig. 2A, B) and accordingly S. aureus FnBPR1-11 bound Fn more strongly than WT 8325.4 (Fig. 3A). Bacteria expressing FnBPA variants with increasing numbers of high-affinity repeats bound Fn incrementally, where three repeats were sufficient to confer the same level of binding to Fn as S. aureus expressing the full length protein (or for consistency in nomenclature here FnBPR1-11) (Fig. 3A). Despite binding Fn at a lower level, a single high-affinity repeat (FnBPR1 or FnBPR11) was sufficient to confer equivalent invasion of endothelial cells when compared with the full length protein. A single low-affinity repeat (FnBPR2 or FnBPR8) did not confer any binding to Fn above that observed for the knockout strain, but the addition of an extra low-affinity repeat (FnBPR7,8) did increase this, albeit at a significantly lower level when compared with the high-affinity repeats (Fig. 3A). Three low-affinity repeats (FnBPR6–8) were needed before adhesion to Fn or invasion of endothelial cells was equivalent to that conferred by the full length protein (Fig. 3A, B).

Selected constructs were also expressed in the mouse-derived S. aureus strain LS-1. Initial experiments were performed to assess the expression levels of each of the constructs by ELISA using anti-FnBPA antibodies (Fig. 4A) and fibrinogen binding (Fig. 4B), as described above for 8325.4. As for 8325.4, LS-1 WT expressed FnBPA at lower levels than the plasmid-encoded gene. Expression levels were equivalent between variants (Fig. 4A). LS-1 WT and FnBPA-defective strains both bound fibrinogen (due to the presence of fibrinogen-binding proteins ClfA and ClfB [14]) but LS-1 strains expressing each of the FnBPA variants bound in 2-fold greater numbers (Fig. 4B), reflecting the higher levels of FnBPA expression that occurs when the gene is located on the plasmid. There were no differences in binding levels between plasmid-encoded FnBPA variants (Fig. 4B). Binding to either human or murine Fn was dependant on the presence of at least 1 FnBR (Fig. 4C, D). Similarly, invasion of EA. hy926 cells was dependent on the presence of at least a single high-affinity FnBR (Fig. 4E).

A nisin-inducible system facilitates controlled surface expression of FnBPA variants
Although FnBPA variants expressing a single or very few high-affinity repeats were able to mediate adhesion and invasion at the same level as full length FnBPA, we considered the possibility that the relatively high levels of protein expression of the constructs (Fig. 2A, B) might mask important differences between full length FnBPA and variant forms. As such we chose the following constructs for further study; FnBPR1-11 (full-length protein), FnBPR1 (single high-affinity repeat), FnBPR2 (single low-affinity repeat) and FnBPR1, 10, 11 (3 high-affinity repeats and binds and invades equivalently to FnBPR1-11). FnBPR0 was included as a control as it wouldn't be expected to bind or invade at any expression level. To assess how selected FnBPA variants might affect adhesion and internalization at lower expression levels an inducible expression system was employed [35]. This and similar systems have been used previously to vary expression levels of several Gram-positive proteins on the surface of L. lactis [35]–[37]. Surface expression of FnBPA variants was quantified by ELISA using antibodies to the A domain of FnBPA (which is common to all variants). Analysis of L. lactis expressing FnBPR1-11 grown in a range of nisin concentrations (0–200 ng ml−1) revealed that, in keeping with previous reports [35], [36], low-level expression occurred in the absence of nisin (Fig. 5A). A dose-dependent increase in FnBPR1-11 expression was seen with increasing concentrations of nisin up to 200 ng ml−1 (Fig. 5A), beyond which bacterial growth was inhibited (data not shown). Expression levels of each of the FnBPA variants were equivalent when identical nisin concentrations were used (Fig. 5A). In addition to the ELISA we employed a fibrinogen-binding assay to assess FnBPA function as the A domain, which is constant in all our constructs, is known to bind fibrinogen (Fig. 5B). In the absence of nisin L. lactis expressing each FnBPA variant bound fibrinogen at very low levels, which increased in a dose-dependent manner and were equal at nisin concentrations >10 ng ml−1 (Fig. 5B). Adhesion to fibrinogen at lower surface expression levels (<10 ng ml−1) was variable and, in keeping with recent work [13], suggested that FnBR1 is required for optimal binding to this ligand.
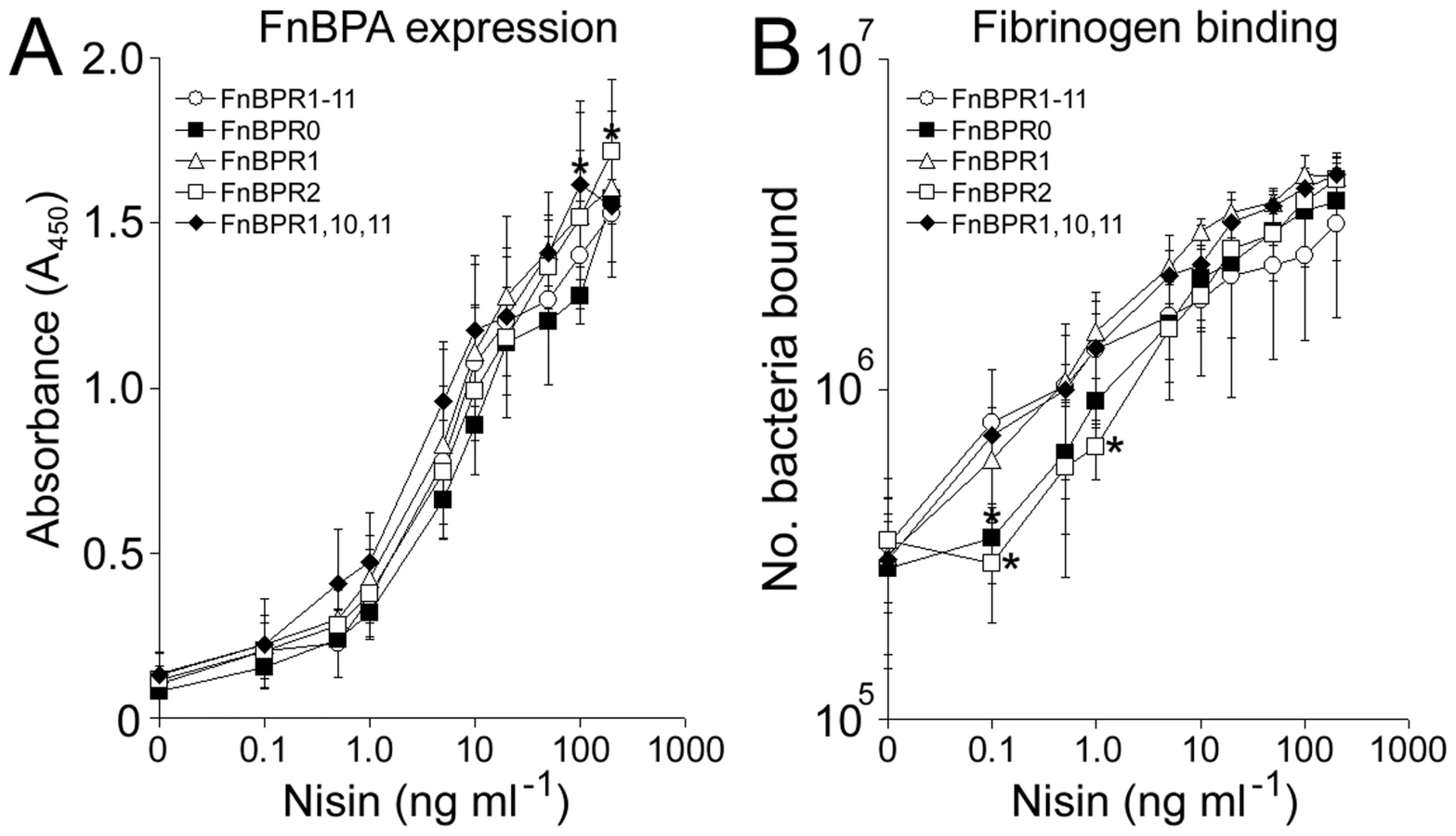
Multiple FnBRs within FnBPA support Fn adhesion and trigger bacterial internalization at low levels of surface expression
The abilities of five FnBPA variants to adhere to Fn and to trigger bacterial internalization were compared over a range of expression levels. In the absence of nisin, L. lactis expressing FnBPR1-11 adhered strongly to Fn (Fig. 6A). Increasing FnBPR1-11 expression (0–10 ng ml−1 nisin) promoted adhesion 2.5-fold before reaching saturation (Fig. 6A). These results mirrored those seen for bacterial internalization, with high levels occurring at basal (absence of nisin) FnBPA expression levels, which increased steadily (>10-fold) before reaching a plateau, also at 10 ng ml−1 nisin (Fig. 6B). By contrast, binding to Fn or invasion of endothelial cells by L. lactis expressing FnBPR0 (no Fn-binding repeats) was not enhanced at any level of expression (Fig. 6). At basal expression levels L. lactis expressing FnBPR1 (containing a single high-affinity repeat) bound to Fn at a low level and was not internalized (relative to FnBPR0, Fig. 6). However, both adhesion and invasion levels increased dramatically (20-fold and 63–fold respectively) with increasing levels of expression, eventually reaching similar levels to that of FnBPR1-11 at saturation (Fig. 6). Adhesion of L. lactis expressing FnBPR2 (containing a single low-affinity repeat) to Fn occurred only at levels of induction >1 ng ml−1 nisin and increased in a dose-dependent manner, eventually reaching levels similar to that supported by FnBPR1 at 100 ng ml−1 nisin (Fig. 6A). However, FnBPR2 did not trigger bacterial internalization at any expression level (Fig. 6B).

The adhesion profile of L. lactis expressing FnBPR1,10,11 (containing 3 high-affinity repeats) was very similar to that of FnBPR1-11 (Fig. 6A). At the basal-level of expression L. lactis FnBR1,10,11 triggered invasion at levels 3.5-fold lower than full length FnBPR1-11 but 3-fold higher than FnBPR1. Invasion levels increased with increasing nisin concentration, reaching the same level as full length FnBPR1-11 at 10 ng ml−1 (Fig. 6B).
Together these findings suggest that although a single high affinity repeat can confer wild-type levels of adhesion and invasion, its expression needs to reach a high density before it can do so. The expression of multiple repeats within a single molecule allows invasion of endothelial cells at low FnBPA surface density.
The rate of invasion of endothelial cells is directly associated with the density of binding repeats expressed on the surface of bacteria
Fn-binding proteins of pathogenic bacteria are believed to cause clustering of cell-surface integrins, leading to cell signalling events that trigger uptake of the bacterium [24]. We hypothesized that multiple FnBRs might trigger uptake in a more efficient manner than a single repeat, leading to an increased rate of bacterial internalization. To assess this we determined the number of adherent and internalized bacteria at various time points from 15 to 60 minutes post-incubation with endothelial cells. Figures 7A and 7B show that adhesion of FnBPA-expressing bacteria to the endothelial cell surfaces was consistent between variants over time. Indeed, adhesion to this cell line does not appear to depend on fibronectin binding since S. aureus FnBPR0 binds as efficiently as FnBPR1-11 (data not shown). S. aureus expressing full length FnBPR1-11 showed a continual increase in the number of internalized bacteria over this time period, reaching a plateau at 45–60 minutes (Fig. 7C). S. aureus expressing only a single repeat (FnBPR1) had approximately 20-fold fewer internalized bacteria after 15 minutes compared with the full length protein but this difference became less pronounced with increasing incubation time, and there was no significant difference in the number of internalized bacteria after 60 minutes (Fig. 7C). S. aureus expressing three high affinity repeats invaded more quickly than those expressing a single repeat but less quickly that those expressing all 11 repeats, supporting the hypothesis that multiple repeats increase the rate at which S. aureus can invade cells. These results were verified by examining invasion mediated by full length FnBPA on the surface of L. lactis at varying densities (Fig. 7D). After 60 minutes' incubation of bacteria and endothelial cells L. lactis expressing all densities of FnBPA had invaded at similar levels, but at 15 and 30 minutes incubation a clear association between surface density and bacterial uptake could be seen (Fig. 7D).

Uptake mechanisms do not differ with varying densities of binding repeats
Although triggered by FnBPA, staphylococcal entry into endothelial cells involves host cell processes [18], [20]. There appear to be two major mechanisms of invasion of human cells by pathogenic bacteria, the first involving integrin-triggered remodelling of actin and the second making use of host cell structures known as caveolae [24]. In keeping with previous reports [24]–[26], the invasion of the cells used in this study by S. aureus appears to occur via an actin remodelling mechanism (Fig. S2). As the rate of bacterial internalization was affected by FnBPA composition and surface density, we hypothesized that this variation might reflect the triggering of different cellular uptake mechanisms. To examine this we investigated the mode of entry of S. aureus expressing FnBPR1-11 and FnBPR1, as well as L. lactis expressing FnBPR1-11 at high or low surface densities, using inhibitors of cellular processes. Consistent with wild-type S. aureus 8325.4 (Fig. S2D), internalization of S. aureus expressing either FnBPR1-11 or FnBPR1 was unaffected by genistein or methyl-β-cyclodextrin but was inhibited by Wortmannin and cytochalasin D (Fig. 8A). Although uptake of both S. aureus expressing FnBPR1-11 and FnBPR1 was slightly reduced by the Src kinase inhibitor PP2, it was only significant for FnBPR1 (p = <0.05) (Fig. 8A). L. lactis expressing FnBPR1-11 was similarly affected, regardless of expression level (Fig. 8B). Internalization of bacteria weakly (absence of nisin) or strongly (200 ng ml−1 nisin) expressing FnBPR1-11 was inhibited by wortmannin and cytochalasin D but not genistein or methyl-β-cyclodextrin (Fig. 8B). Inhibition of L. lactis uptake by PP2 was much more pronounced than for S. aureus, particularly at low expression levels (Fig. 8). These findings verify that the density of Fn-binding repeats does not affect the uptake mechanism exploited by the bacteria.

Full-length FnBPA facilitates bacterial adhesion to immobilized Fn in the presence of soluble Fn
Blood contains high levels of soluble (plasma) Fn. Although FnBPA has been shown to bind fluid-phase Fn [20], we hypothesized that to bind the endothelium in vivo S. aureus must be able to bind preferentially to immobilized (cell bound) Fn. To test this we measured the adhesion of bacteria expressing FnBPA variants to immobilized fibronectin in the presence of increasing concentrations of soluble fibronectin. Adhesion of S. aureus expressing FnBPR1-11 to immobilized Fn was unaffected by the presence of soluble Fn except at the highest concentrations used (Fig. 9). By contrast, Fn-binding by S. aureus expressing FnBPA variants containing one or three repeats was significantly inhibited at low concentrations of soluble Fn (Fig. 9). This suggests that full-length FnBPA is likely to be significantly more efficient at conferring adhesion to the endothelium under in vivo conditions.

Fibronectin-binding does not affect S. aureus survival or modulate TNFα production in whole human blood
S. aureus has a number of mechanisms that aid evasion of the host immune system including surface adhesins such as Clf and Spa that inhibit phagocytosis [4]. Work with Streptococcus pyogenes suggested that the Fn-binding protein PrtF, which is structurally similar to FnBPA, inhibits phagocytosis in a whole blood model [38]. As such, we assessed whether the FnBR region of FnBPA might perform a similar function. Using a similar whole blood model (see Materials and Methods section), we determined the survival of S. aureus strains 8325.4 and LS-1 expressing full length FnBPA (FnBPR1-11) or the FnBR-deficient variant FnBPR0. Over a period of up to 6 h there were no significant differences in bacterial CFU between strains expressing FnBPR1-11 or the FnBR-deficient FnBPR0 (Fig. 10A), indicating that fibronectin binding does not affect S. aureus survival.

In addition, we assessed whether Fn-binding might affect the inflammatory response of whole blood to S. aureus by determining the production of TNFα triggered by each strain. All bacteria examined elicited TNFα expression that was significantly greater than that of whole blood incubated without bacteria (Fig. 10B). Although there was a significant difference between strains, the FnBR-region of FnBPA had no effect on TNFα production by whole blood (Fig. 10B).
Multiple FnBRs are essential for full virulence in a murine sepsis model
We hypothesized from our in vitro data that bacteria expressing FnBPA variants with multiple repeats would be able to bind to and invade endothelium more efficiently in vivo than bacteria expressing FnBPA variants with few or no repeats, causing greater levels of mortality. To test this we employed a murine sepsis model to assess the virulence of S. aureus LS-1 (a mouse infective strain) expressing FnBPA variants. S. aureus LS-1 Δfnb expressing FnBPR0, FnBPR1, FnBPR1,10,11 or FnBPR1-11 were phenotypically similar to 8325.4 Δfnb expressing the same constructs with respect to Fn-binding and invasion of endothelial cells (Fig. 3). Mice were injected intravenously with one of these four S. aureus LS-1 strains and survival and weight loss monitored over 14 days. By day 9, S. aureus expressing full length FnBPR1-11 had killed >60% of mice, which was significantly greater than any of the three other strains (p = <0.007, Fig. 11A). Those that survived continued to suffer significant weight loss of approximately 30%, suggesting severe infection (Fig. 11B). By contrast, no significant difference in mortality was observed between S. aureus expressing FnBPR0, FnBPR1 or FnBPR1,10,11, which killed up to 20% of the mice (Fig. 11A). The pattern of weight loss was also similar between these three strains, although mice infected with S. aureus expressing FnBPR1 and FnBPR1,10,11 showed significantly different weight changes between days 4 and 13 after inoculation (p = 0.02, Fig. 11B).

In addition to measurements of mortality and weight loss we also assessed bacterial numbers (CFU) in the kidneys 3 days after inoculation (Fig. 11C). This time point was chosen because previous experiments (Fig. 11A, B) indicated that it was close to the initiation of mortality (indeed, one mouse in the FnBPR1-11 group died before sacrifice). As expected from the data in Figures 11A and 11B, the kidneys of mice inoculated with S. aureus FnBPR1-11 contained significantly more CFU than the kidneys of mice infected with S. aureus FnBPR0 (23-fold greater, p = 0.0022). S. aureus FnBPR1-11 was also approximately 6-fold more abundant than S. aureus expressing either FnBPR1 or FnBPR1,10,11 (p = 0.024). There was no significant difference in the bacterial load between S. aureus FnBPR0, FnBPR1 or FnBPR1,10,11. We also used the bacterial colonies from the kidneys to determine loss of the FnBPA-encoding plasmids (Fig. 11D). Plasmid loss was highly variable between animals but was not significantly different between FnBPA variants and did not correlate with bacterial load (data not shown). The difference between S. aureus expressing FnBPR0, FnBPR1 or FnBPR1,10,11 when compared with those expressing full length FnBPR1-11 demonstrates that full length FnBPA is required for full pathogenicity in this model, most likely during the early stages of infection.
Discussion
Bacterial invasion of host cells is a process common to many pathogens. It likely aids evasion of extracellular immune factors and provides a protected niche from antibiotics, and can therefore allow bacteria to cause persistent and recurrent infections [39], [40]. Additionally, bacterial invasion of the endothelium can lead to inflammation, endocarditis and may lead to traversal of blood vessels and subsequent formation of metastatic infections [22]. As such it is crucial that we gain a greater understanding of how this pathogen interacts with the host to identify means of blocking these infections [1].
There is clear and compelling evidence that FnBPA alone is sufficient to trigger S. aureus invasion of cells via Fn bridging to α5β1 integrins [18], [20]. Previous work revealed that staphylococcal attachment to Fn and invasion of host cells is maintained even when substantial portions of the Fn-binding region have been deleted [13], [19]. As such, it was unclear what benefits are conferred by multiple FnBRs. As there is evidence that a single FnBPA molecule is able to bind up to nine Fn molecules [33], we hypothesized that a minimum number of repeats in close proximity would be required to trigger Fn clustering and subsequent integrin clustering, which leads to cell invasion [32], [33]. Furthermore, we hypothesized that the composition of FnBPA variants with respect to high - and low-affinity repeats would affect adhesion and internalization. Here we show that high surface densities of repeats are required for adhesion and internalization in vitro. However, it is not important to have multiple repeats within a single FnBPA molecule because high level expression of individual FnBPA variants containing a single high-affinity binding repeat results in invasion at WT levels (Fig. 6). In addition to demonstrating that high surface density of FnBRs promotes cell invasion, we show that speed of uptake is modulated by composition and expression level of FnBPA (Fig. 7).
Although the data presented here suggest that Fn binding is indicative of the potential to trigger invasion, our studies using low-affinity repeats suggest a more complicated situation. The inability of the FnBPA variant containing a single low-affinity repeat (FnBPR2) to trigger internalization at high surface expression levels, despite supporting adhesion to Fn, suggests that invasin-receptor affinity is crucial (Fig. 6). Three tandem low-affinity repeats however support invasion (Fig. 3). It is therefore possible that certain FnBPA-Fn interactions induce a conformational change in the glycoprotein, which triggers integrin signalling and bacterial uptake. In vivo, S. aureus will not only encounter cell-bound Fn but also the soluble form. Although adhesion of S. aureus expressing FnBPR1 and FnBPR1,10,11 to immobilized Fn was significantly inhibited at even low concentrations of soluble Fn, S. aureus FnBPR1-11 was only affected at the highest concentration used (Fig. 9). This suggests that the three other high-affinity repeats (4, 5 and 9) and perhaps also the five low-affinity repeats (2, 3, 6, 7 and 8) are important in overcoming the presence of soluble ligand.
Although our data suggest that, in tandem, multiple low-affinity repeats can perform almost as well as single high-affinity repeats, it is not clear what advantage the bacteria gain from maintaining these repeats. The answer may lie in studies of the immune response to FnBPA during staphylococcal infection. Analysis of a panel of antisera from different patients demonstrated that antibodies recognizing the Fn-binding region do not inhibit bacterial binding of Fn [41]. Furthermore, the binding of these antibodies to FnBPA was enhanced in the presence of Fn, presumably due to the creation of ligand-induced binding sites [41]. A more recent study revealed that despite strong antibody recognition of high-affinity repeats when complexed with Fn, there was significantly lower recognition of low-affinity repeats either in the presence or absence of the glycoprotein [31]. As such, the lack of antibody recognition of low-affinity repeats may allow the bacteria to adhere to host tissues even in the presence of anti-FnBPA antibodies.
A role for FnBPA in sepsis has been previously established [14], although it was not clear whether the fibrinogen - and elastin-binding region or the Fn-binding region was important. Using the same model of sepsis we show that the FnBRs are required for virulence (Fig. 11). We also show that increased efficiency of the full length protein (FnBPR1-11) at mediating binding to Fn and cell invasion translates into a difference in virulence. Despite the ability to bind Fn and invade cells in vitro, neither S. aureus expressing FnBPR1 nor FnBPR1,10,11 were any more virulent than that expressing FnBPR0. The enhanced pathogenesis conferred by multiple FnBRs does not appear to be due to elevated survival in blood or the stimulation of pro-inflammatory TNFα. However, the greater bacterial load in the kidneys of mice infected with S. aureus FnBPR1-11, compared with FnBPR0 or FnBPR1, suggests that multiple repeats significantly enhance systemic invasion.
The demonstration of the role of FnBPA in septic death does not rule out the involvement of other staphylococcal factors in the progression of infection. Indeed, a role for clumping factor in sepsis using this model and strain has been shown previously [41] and it is likely that different factors will be required at different stages of the infective process [1]. Experiments examining the loss of FnBPA-encoding plasmids from infecting bacteria suggest that this protein acts early in the infectious process and its retention is not required for the latter stages of infection. Significant plasmid loss at day 3 is unsurprising since the bacterial load for S. aureus FnBPR1-11 is 10 times greater than the inoculum, indicating significant replication within this organ. These data indicate that the FnBRs within FnBPA significantly enhance bacterial exit from the blood and colonization of the kidneys soon after entry into the vascular system. This is supported by previous in vivo experiments that showed S. aureus attachment to the endothelium is dependent on FnBPA and occurs within 5 minutes of inoculation [21]. Once colonization is established, S. aureus is likely to employ other virulence factors (e.g. other surface proteins, toxins and proteases), which together with host factors, promote progression to abscess formation [1], leading to weight loss and mortality. However, early extravasation, mediated by FnBPA containing multiple FnBRs, appears to be essential for full virulence.
In summary, the presence of multiple FnBRs within FnBPA results in a high surface density of Fn-binding sites. This triggers a rapid invasion of endothelial cells even at low surface expression levels and enables binding in the presence of soluble ligand. This appears to be particularly relevant to pathogenesis and the development of systemic infection.
Materials and Methods
Ethics statement
Approval for experiments using human blood was granted by the Bath Research Ethics Committee (NHS National Research Ethics Services, reference 08/H0101/18). Donors gave informed consent in writing prior to the commencement of any procedures.
Approval for animal experiments was granted by the Göteborg Animal Experimentation Ethics board and their ethical and husbandry guidelines followed for all experiments. NMRI mice were obtained from Charles River (Sulzfeld, Germany) and were maintained in the animal facility of the Department of Rheumatology, University of Göteborg, Sweden. Mice were housed up to 10 animals per cage with a 12 h light-dark cycle, and were fed standard laboratory chow and water ad libitum. The animals were 8 weeks old at the start of the experiments.
The overall condition of each mouse was examined by assessing signs of systemic inflammation, i.e. weight decrease, reduced alertness, and ruffled coat. In keeping with approved husbandry standards, in cases of severe systemic infection, when a mouse was judged too ill to survive another 24 h, it was killed by cervical dislocation and considered dead due to sepsis.
Bacterial strains and growth conditions
A detailed list of the strains used in this study is presented in Table 2. S. aureus strains were cultured for 16 h in Brain-Heart Infusion (BHI) broth at 37°C in air with shaking. S. aureus CFU were quantified on Tryptic Soy Agar (TSA) plates incubated overnight at 37°C in air. L. lactis strains were cultured in M17 broth (supplemented with 0.5% w/v glucose) for 16 h at 30°C in air (with the appropriate concentration of nisin where necessary). Escherichia coli was grown in Luria broth at 37°C with shaking. Where appropriate, bacteria were incubated in the presence of the following antibiotics: ampicillin 100 µg ml−1, chloramphenicol 10 µg ml−1 and erythromycin 5 µg ml−1 (L. lactis) or 250 µg ml−1 (E. coli).

Endothelial cell culture
The cell line EA. hy926, established by the fusion of human umbilical endothelial cells (HUVEC) and the permanent lung epithelial carcinoma cell line A549 [42], was cultured in Dulbecco's Modified Eagle's medium (DMEM; Invitrogen) supplemented with foetal bovine serum (FBS; 10%) and L-glutamine (2 mM) at 37°C and 5% CO2. Pooled primary HUVECs were purchased from Lonza (Basel, Switzerland) and cultured in endothelial basal medium supplemented with 2% fetal bovine serum, bovine brain extract (including heparin), human endothelial growth factor, hydrocortisone and GA-1000 (Gentamicin & Amphotericin B) at 37°C and 5% CO2 according to manufacturer's instructions (Lonza).
Endothelial cells were cultured in T75 flasks to approximately 95% confluency, liberated with trypsin-EDTA, resuspended in the relevant culture medium and added to 24-well plates containing thermanox glass coverslips [19]. Plates were incubated for 48 h as described above before the coverslips were removed, dip washed in PBS and added to new 24-well plates containing fresh medium and bacteria [19]. In experiments using metabolic inhibitors, these were incubated with the cells 1 h prior to the addition of bacteria and concentrations maintained during the assay; genistein (200 µM), wortmannin (20 nM), cytochalasin D (50 µM), PP2 (10 µM) and methyl-β-cyclodextrin (2 mM).
Construction of FnBPA variants
FnBPA constructs containing various combinations of Fn-binding repeats were produced using a circular PCR system based on that described by Massey et al. [19]. Primers were designed to various repeats or DNA just outside of the fnbA repeat region and were synthesized with 5′ phosphorylation in order to allow self-ligation of the PCR product. Primer sequences and the combinations used to produce the various products can be found in Table 1. Plasmid pFnBA4 [34] was used as a template for the amplification of DNA lacking in various repeats. PCR was performed using Phusion high-fidelity DNA polymerase (New England Biolabs (NEB) Ipswich, MA). Template DNA was digested with DpnI and products cleaned using a PCR purification kit or gel extracted (Qiagen, Hilden, Germany). Linear DNA was self-ligated using T4 DNA ligase (NEB) to produce plasmids containing DNA encoding the fnbA gene lacking various combinations of FnBRs (Table 2). Ligated PCR products were transformed into CaCl2 competent E. coli [43] and plated onto LB agar containing 100 µg ml−1 ampicillin. Selected colonies were grown overnight in LB broth (5 ml, 100 µg ml−1 Ampicillin), bacteria pelleted by centrifugation and plasmids recovered using a miniprep kit (Qiagen). Constructs were checked by sequencing (Geneservice, London, UK) and transformed into S. aureus RN4220 by electroporation. Plasmids were recovered using a combination of lysostaphin and the Qiagen miniprep kit [19] and re-transformed into S. aureus 8325.4 Δfnb.
Construction and controlled expression of FnBPA variants in L. lactis
Controlled expression of FnBPA variants was achieved using a nisin-inducible system previously used to control the expression of the enterococcal surface protein PrgB [35]. The prgB gene was excised from plasmid pMSP7517 using NcoI and XhoI and the remaining plasmid recovered by gel excision. pFnBA4 and associated constructs were used as templates for PCR reactions using primers designed to amplify the entire fnbA gene (and repeat-region variants) and contained NcoI (AAACCATGGAGGAGGTATTATAGTGAAAAACAATCTTAGG) and XhoI (AAACTCGAGCTAACTTTATCTCTCAGTTCGTTATC) sites respectively (underlined). Digested PCR products were ligated into digested pMSP7517 and transformed into CaCl2-treated E. coli K12 ER2925, which was more suitable for antibiotic selection using erythomycin than E. coli DH5α (data not shown). Plasmids were recovered, checked by sequencing and electro-transformed into L. lactis NZ9800 [36]. Expression was induced by culture (16 h, 30°C) of L. lactis containing FnBPA variant constructs (Table 2) in the presence of various concentrations of nisin (range 0–200 ng ml−1). Expression levels were determined by ELISA and binding to immobilized fibrinogen (see below).
Determination of surface protein expression by Western immunoblot
S. aureus was cultured as described and the bacteria pelleted by centrifugation (5,000 x g, 8 min). Bacteria were washed 3 times in PBS, resuspended in spheroplasting buffer (30% raffinose, Tris-HCl pH 7.5) [27] containing 100 µg ml−1 lysostaphin and incubated for 1 h at 37°C. Spheroplasts were pelleted by centrifugation and the supernatant containing surface proteins recovered. Aliquots (40 µl) were mixed with 5X concentrated sample buffer (10 µl; 50% glycerol containing 50 mM Tris-HCl pH 6.8, 10% SDS and 10 mM 2-mercaptoethanol) and heated at 100°C for 5 min before being subjected to SDS-PAGE (7.5% acrylamide). Separated proteins were electroblotted onto nitrocellulose membrane using a BioRad (Hercules, Ca) semi-dry blotter (25V, 1 h).
Membranes were blocked by incubation in 3% BSA (1 h) and probed with antibodies to the N-terminal domain of FnBPA [19] for 1 h with agitation. Membranes were washed 4 times with PBS and incubated with HRP-conjugated goat anti-rabbit antibodies (1 h with agitation). Membranes were subsequently washed 4 times with PBS, rinsed with ddH2O and reactive bands detected using the Opti-4CN detection kit (BioRad).
Determination of surface protein expression by ELISA
S. aureus strains were cultured as described above, washed with PBS, adjusted to OD600 = 0.1 and aliquots (50 µl) immobilized onto triplicate plastic wells (Nunc Maxisorp) by incubation at 4°C for 16 h. Wells were blocked with BSA (1 h, 4°C), washed once with PBS and then incubated with antibodies (50 µl, 1∶500 in PBS for S. aureus 8325.4 or 1∶2000 for S. aureus LS-1) raised to the A region of FnBPA [19] for 1 h at 37°C. Wells were washed 4 times with PBS and incubated with HRP-conjugated protein A (50 µl, 1∶5000, Sigma) for 30 min at 37°C. Wells were washed a further 4 times with PBS and bound antibody detected and quantified with 100 µl TMB (3,3′,5,5′-tetramethylbenzidine) ELISA detection substrate (Sigma) for 10 min at room temperature. The reaction was stopped with 100 µl 2M HCl and product measured at 450 nm using a microplate reader.
L. lactis expressing FnBPA variants were washed in PBS as described above, diluted 1∶16 in PBS and 50 µl aliquots added to duplicate wells of a microtitre plate as described for S. aureus. Wells were blocked as described above and immobilized bacteria were incubated with anti-FnBPA antibodies (50 µl, 1∶1000, 1 h, 37°C) before washing three times with PBS. Bacteria were then incubated with HRP-conjugated anti-rabbit antibodies (50 µl, 1∶5000, 1 h, 37°C) and quantified using TMB as described above.
Solid-phase Fn and fibrinogen binding assay
The adhesion of bacteria to human Fn (Sigma), murine Fn (Biopur, Bubendorf, Switzerland) or fibrinogen (Fn-depleted, Enzyme Research Labs, Swansea, UK) was assessed using a protocol based on those of Jakubovics et al. and Peacock et al. [27], [44]. Fn or fibrinogen (1 µg protein in 100 µl PBS [pH 7.4] per well) was immobilized onto plastic Nunc Maxisorp Immuno modules by incubation at 4°C for 16 h and remaining binding sites were blocked with 300 µl 3% BSA (in PBS) at 25°C for 1 h. Blocked wells were washed with PBS and incubated with 100 µl bacteria (approx. 1×108 S. aureus or 5×108 L. lactis in PBS) at 37°C for 1 h. Non-adherent bacteria were removed by 3 rounds of washing with PBS and adherent bacteria fixed with 0.25% paraformaldehyde (5 min, 25°C). Wells were washed with PBS a further 2 times. Fixed bacteria were stained with crystal violet (0.5%, 2 min) before a further three rounds of washing, as described above. Adherent, fixed bacteria were enumerated by solubilization of crystal violet with 100 µl 7% acetic acid. The contents of each well was quantified by measurement at A595 using a microplate reader. For both Fn and fibrinogen binding, readings were blanked against bacteria binding to wells coated only with BSA. Absorbance measurements were converted to bacterial numbers by the use of standard plots of known bacterial numbers against A595 readings (Figure S3). These showed that the relationship of A595 to bacterial numbers is linear (Figure S3). Each experiment was performed at least three times. Since S. aureus was stained more strongly with crystal violet than L. lactis (Figure S3), different amounts of bacteria were used in assays in order to allow bacterial quantification over as wide a range of adhesion levels as possible.
Endothelial adhesion and invasion assays
Cultured cells were dissociated from plastic flasks using trypsin-EDTA solution (Invitrogen) and approximately 5×105 (in 0.5 ml medium) were seeded into each well of 24-well plates (Nunc) containing 13 mm plastic Thermanox cover slips (Fisher) and allowed to attach for 48 h (37°C, 5% CO2). Coverslips were dip-washed once in PBS and placed in the well of a new 24-well plate containing 450 µl of DMEM containing 10% FBS.
To each well, 50 µl of washed bacteria were added (approximately 1×107 CFU S. aureus or 5×107 L. lactis) and incubated for 5–90 minutes at 37°C in 5% CO2.
To measure the total number of bacteria associated with the cells (adherent and internalized), coverslips were dip-washed 3 times in PBS and added to wells containing 500 µl 0.5% Triton X-100. Wells containing coverslips were agitated by pipetting to fully lyse the cells and CFU were enumerated by serial dilution and plating onto TSA agar plates and incubated overnight at 37°C in 5% CO2.
For invasion assays, the bacterial suspension was removed and replaced with 500 µl DMEM/10% FBS supplemented with 200 µg ml−1 gentamicin and incubated at 37°C in 5% CO2 for 60 min. Coverslips were washed 3 times in PBS, lysed and plated for CFU as described for the adhesion assay above. In assays where metabolic inhibitors were used, these were added to cell monolayers for 60 min prior to the experiment and concentrations maintained during incubation with bacteria.
Statistics
For adhesion and invasion assays, statistical analyses were performed with Student's t test by using the Bonferroni correction for multiple comparisons. Values that were statistically significantly different from control values are indicated by asterices in the figures. Error bars indicate the mean average ± standard deviation of multiple independent experiments (indicated in the figure legend).
Whole human blood model of infection
The ability of S. aureus to survive in human blood and generate pro-inflammatory cytokine release was assessed using a whole human blood model. Blood from a healthy male was drawn into heparinised tubes and used immediately. Bacteria were washed and diluted in PBS and 10 µl (containing approximately 20,000 CFU) was added to 800 µl blood and incubated at 37°C for 1, 2 or 6 h with mixing. Aliquots of the blood/bacteria mixture (5 µl) were mixed with PBS (95 µl) and plated onto tryptic soy agar to enumerate CFU. In addition to plating for CFU, blood at the final time point (6 h) was centrifuged (5,000 x g, 5 min) and 100 µl serum recovered for analysis of TNFα production using a BD OptEIA ELISA kit (BD Biosciences, San Diego, USA).
Murine sepsis model
Groups of 15 mice were infected by intravenous injection with S. aureus strain LS-1 Δfnb [14] expressing FnBPR1-11 (1.1×107 CFU), FnBPR0 (1.1×107 CFU), FnBPR1 (1.0×107 CFU) or FnBPR1,10,11 (1.2×107 CFU) and mortality and weight change were followed until day 14. The clinical evaluation was performed in a blinded manner. The overall condition of each mouse was examined by assessing signs of systemic inflammation, i.e. weight decrease, reduced alertness, and ruffled coat. In keeping with approved husbandry standards, in cases of severe systemic infection, when a mouse was judged too ill to survive another 24 h, it was killed by cervical dislocation and considered dead due to sepsis. The bacterial load in kidneys was assessed as described previously [14]. Two independent groups of 4 mice were inoculated with S. aureus (as described above) and sacrificed 3 days later. Kidney pairs were recovered and CFU enumerated on TSA plates. Retention of the FnBPA-encoding plasmid was determined by plating onto TSA with and without chloramphenicol. Statistical evaluation was done by using the Kruskal-Wallis test with a following post-hoc analysis, or the Logrank (Mantel-Cox) test at survival analysis. P = <0.05 was considered to be significant. No correction for multiple comparisons was employed. Data are reported as medians, interquartile ranges, and 80% central ranges, unless otherwise mentioned.
Gene accession information
FnBPA variants were derived from the fnbA gene of S. aureus 8325.4, Swissprot accession SAOUHSC_02803.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. LowyFD
1998 Staphylococcus aureus infections. N Engl J Med 339 520 532
2. MenichettiF
2005 Current and emerging serious Gram-positive infections. Clin Microbiol Infect 3 22 28
3. PeacockSJ
de SilvaI
LowyFD
2001 What determines nasal carriage of Staphylococcus aureus? Trends Microbiol 9 605 610
4. FosterTJ
2005 Immune evasion by Staphylococci. Nat Rev Microbiol 3 948 958
5. RooijakkersSH
van KesselKP
van StrijpJA
2005 Staphylococcal innate immune evasion. Trends Microbiol 13 596 601
6. PettiCA
FowlerVGJr
2002 Staphylococcus aureus bacteremia and endocarditis. Infect Dis Clin North Am 16 413 435
7. FowlerVGJr
MiroJM
HoenB
CabellCH
AbrutynE
2005 Staphylococcus aureus endocarditis: a consequence of medical progress. JAMA 293 3012 3021
8. BeynonRP
BahlVK
PrendergastBD
2006 Infective endocarditis. BMJ 333 334 339
9. HillEE
HerijgersP
HerregodsMC
PeetermansWE
2006 Evolving trends in infective endocarditis. Clin Microbiol Infect 12 5 12
10. BashoreTM
CabellC
FowlerVJr
2006 Update on infective endocarditis. Curr Probl Cardiol 31 274 352
11. ChorianopoulosE
BeaF
KatusHA
FreyN
2009 The role of endothelial cell biology in endocarditis. Cell Tissue Res 335 153 163
12. QueYA
HaefligerJA
PirothL
FrançoisP
WidmerE
2005 Fibrinogen and fibronectin-binding cooperate for valve infection and invasion in Staphylococcus aureus experimental endocarditis. J Exp Med 201 1627 1635
13. PirothL
QueYA
WidmerE
PanchaudA
PiuS
2008 The fibrinogen - and fibronectin-binding domains of Staphylococcus aureus fibronectin-binding protein A synergistically promote endothelial invasion and experimental endocarditis. Infect Immun 76 3824 3831
14. PalmqvistN
FosterT
FitzgeraldJR
JosefssonE
TarkowskiA
2005 Fibronectin-binding proteins and fibrinogen-binding clumping factors play distinct roles in staphylococcal arthitis and systemic inflammation. J Infect Dis 191 791 798
15. HeyingR
van de GevelJ
QueYA
MoreillonP
BeekhuizenH
2007 Fibronectin-binding proteins and clumping factor A in Staphylococcus aureus experimental endocarditis: FnBpA is sufficient to activate human endothelial cells. Thomb Haemost 97 617 626
16. HeyingR
van de GevelJ
QueYA
PirothL
MoreillonP
2009 Contribution of (sub)domains of Staphylococcus aureus fibronectin-binding protein to the proinflammatory and procoagulant response of human vascular endothelial cells. Thomb Haemost 101 495 504
17. PeacockSJ
FosterTJ
CameronBJ
BerendtAR
1999 Bacterial fibronectin-binding proteins and endothelial cell surface fibronectin mediate adherence of Staphylococcus aureus to resting human endothelial cells. Microbiology 145 3477 3486
18. SinhaB
FrançoisPP
NüsseO
FotiM
HartfordOM
1999 Fibronectin-binding protein acts as Staphylococcus aureus invasin via fibronectin bridging to integrin α5β1. Cell Microbiol 1 101 117
19. MasseyRC
KantzanouMN
FowlerT
DayNP
SchofieldK
2001 Fibronectin-binding protein A of Staphylococcus aureus has multiple, substituting, binding regions that mediate adherence to Fn and invasion of endothelial cells. Cell Microbiol 3 839 851
20. SinhaB
FrancoisP
QueYA
HussainM
HeilmannC
2000 Heterologously expressed Staphylococcus aureus fibronectin-binding proteins are sufficient for invasion of host cells. Infect Immun 68 6871 6878
21. KerdudouS
LaschkeMW
SinhaB
PreissnerKT
MengerMD
2006 Fibronectin-binding proteins contribute to the adherence of Staphylococcus aureus to intact endothelium in vivo. Thomb Haemost 96 183 189
22. SinhaB
HerrmannM
2005 Mechanism and consequences of invasion of endothelial cells by Staphylococcus aureus. Thomb Haemost 94 266 277
23. IsbergRR
Tran Van NhieuG
1994 Binding and internalization of microorganisms by integrin receptors. Trends Microbiol 2 10 14
24. Nitsche-SchmitzDP
RohdeM
ChhatwalGS
2007 Invasion mechanisms of Gram-positive pathogenic cocci. Thomb Haemost 98 488 496
25. AgererF
LuxS
MichelA
RohdeM
OhlsenK
2005 Cellular invasion by Staphylococcus aureus reveals a functional link between focal adhesion kinase and cortactin in integrin-mediated internalization. J Cell Sci 118 2189 2200
26. SchöderA
SchöderB
RoppenserB
LinderS
Sinha
2006 Staphylococcus aureus fibronectin-binding protein-A induces motile attachment sites and complex actin remodeling in living endothelial cells. Mol Biol Cell 17 5198 5210
27. PeacockSJ
DayNP
ThomasMG
BerendtAR
FosterTJ
2000 Clinical isolates of Staphylococcus aureus exhibit diversity in fnb genes and adhesion to human fibronectin. J Infect 41 23 31
28. WannER
GurusiddappaS
HookMJ
2000 The Fn-binding MSCRAMM FnbpA of Staphylococcus aureus is a bifunctional protein that also binds to fibrinogen. Biol Chem 275 13863 13871
29. RocheFM
DownerR
KeaneF
SpezialeP
ParkPW
2004 The N-terminal A domain of fibronectin-binding proteins A and B promotes adhesion of Staphylococcus aureus to elastin. J Biol Chem 279 38433 38440
30. Schwarz-LinekU
WernerJM
PickfordAR
GurusiddappaS
KimJH
2003 Pathogenic bacteria attach to human fibronectin though a tandem beta-zipper. Nature 423 177 181
31. MeenanNA
VisaiL
ValtulinaV
Schwarz-LinekU
NorrisNC
2007 The tandem beta-zipper model defines high affinity fibronectin-binding repeats within Staphylococcus aureus FnBPA. J Biol Chem 282 25893 25902
32. FrömanG
SwitalskiLM
SpezialeP
HöökMJ
1987 Isolation and characterization of a fibronectin receptor from Staphylococcus aureus. Biol Chem 262 6564 6571
33. BinghamRJ
Rudiño-PiñeraE
MeenanNA
Schwarz-LinekU
TurkenburgJP
2008 Crystal structures of fibronectin-binding sites from Staphylococcus aureus FnBPA in complex with Fn domains. Proc Natl Acad Sci U S A 105 12254 12258
34. GreeneC
McDevittD
FrancoisP
VaudauxPE
LewDP
1995 Adhesion properties of mutants of Staphylococcus aureus defective in fibronectin-binding proteins and studies on the expression of fnb genes. Mol Microbiol 17 1143 1152
35. HirtH
ErlandsenSL
DunnyGM
2000 Heterologous inducible expression of Enterococcus faecalis pCF10 aggregation substance asc10 in Lactococcus lactis and Streptococcus gordonii contributes to cell hydrophobicity and adhesion to fibrin. J Bacteriol 182 2299 2306
36. BryanEM
BaeT
KleerebezemM
DunnyGM
2000 Improved vectors for nisin-controlled expression in gram-positive bacteria. Plasmid 44 183 190
37. LoughmanA
FitzgeraldJR
BrennanMP
HigginsJ
DownerR
2005 Roles for fibrinogen, immunoglobulin and complement in platelet activation promoted by Staphylococcus aureus clumping factor A. Mol Microbiol 57 804 818
38. HylandKA
WangB
ClearyPP
2007 Protein F1 and Streptococcus pyogenes resistance to phagocytosis. Infect Immun 75 3188 3191
39. GarzoniC
KelleyWL
2009 Staphylococcus aureus: new evidence for intracellular persistence. Trends Microbiol 17 59 65
40. SendiP
ProctorRA
2009 Staphylococcus aureus as an intracellular pathogen: the role of small colony variants. Trends Microbiol 17 54 58
41. JosefssonE
HigginsJ
FosterTJ
TarkowskiA
2008 Fibrinogen binding sites P336 and Y338 of clumping factor A are crucial for Staphylococcus aureus virulence. PLoS ONE 3 e2206
42. EdgellCJ
McDonaldCC
GrahamJB
1983 Permanent cell line expressing human factor VIII-related antigen established by hybridization. Proc Natl Acad Sci U S A 80 3734 3737
43. SambrookJ
FritschEF
ManiatisT
1989 Molecular Cloning: a Laboratory Manual, 2nd edn. Cold Spring Harbor,, NY Cold Spring Harbor Laboratory
44. JakubovicsNS
StrömbergN
van DolleweerdCJ
KellyCG
JenkinsonHF
2005 Differential binding specificities of oral streptococcal antigen I/II family adhesins for human or bacterial ligands. Mol Microbiol 55 1591 1605
45. KreiswirthBN
LöfdahlS
BetleyMJ
O'ReillyM
SchlievertPM
1983 The toxic shock syndrome exotoxin structural gene is not detectably transmitted by a prophage. Nature 305 709 712
46. NovickR
1967 Properties of a cryptic high-frequency transducing phage in Staphylococcus aureus. Virology 33 155 166
47. HartleibJ
KöhlerN
DickinsonRB
ChhatwalGS
SixmaJJ
2000 Protein A is the von Willebrand factor binding protein on Staphylococcus aureus. Blood 96 2149 2156
48. BremellT
LangeS
YacoubA
RydénC
TarkowskiA
1991 Experimental Staphylococcus aureus arthitis in mice. Infect Immun 59 2615 2623
49. HartfordO
O'BrienL
SchofieldK
WellsJ
FosterTJ
2001 The Fbe (SdrG) protein of Staphylococcus epidermidis HB promotes bacterial adherence to fibrinogen. Microbiology 147 2545 2552
50. KuipersOP
BeerthuyzenMM
SiezenRJ
De VosWM
1993 Characterization of the nisin gene cluster nisABTCIPR of Lactococcus lactis. Requirement of expression of the nisA and nisI genes for development of immunity. Eur J Biochem 216 281 291
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2010 Číslo 6
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Requirement of NOX2 and Reactive Oxygen Species for Efficient RIG-I-Mediated Antiviral Response through Regulation of MAVS Expression
- Formation of Complexes at Plasmodesmata for Potyvirus Intercellular Movement Is Mediated by the Viral Protein P3N-PIPO
- Insight into the Mechanisms of Adenovirus Capsid Disassembly from Studies of Defensin Neutralization
- Two Novel Point Mutations in Clinical Reduce Linezolid Susceptibility and Switch on the Stringent Response to Promote Persistent Infection
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy