
Inhibition of Host Vacuolar H-ATPase Activity by a Effector
Legionella pneumophila is an intracellular pathogen responsible for Legionnaires' disease. This bacterium uses the Dot/Icm type IV secretion system to inject a large number of bacterial proteins into host cells to facilitate the biogenesis of a phagosome permissive for its intracellular growth. Like many highly adapted intravacuolar pathogens, L. pneumophila is able to maintain a neutral pH in the lumen of its phagosome, particularly in the early phase of infection. However, in all cases, the molecular mechanisms underlying this observation remain unknown. In this report, we describe the identification and characterization of a Legionella protein termed SidK that specifically targets host v-ATPase, the multi-subunit machinery primarily responsible for organelle acidification in eukaryotic cells. Our results indicate that after being injected into infected cells by the Dot/Icm secretion system, SidK interacts with VatA, a key component of the proton pump. Such binding leads to the inhibition of ATP hydrolysis and proton translocation. When delivered into macrophages, SidK inhibits vacuole acidification and impairs the ability of the cells to digest non-pathogenic E. coli. We also show that a domain located in the N-terminal portion of SidK is responsible for its interactions with VatA. Furthermore, expression of sidK is highly induced when bacteria begin to enter new growth cycle, correlating well with the potential temporal requirement of its activity during infection. Our results indicate that direct targeting of v-ATPase by secreted proteins constitutes a virulence strategy for L. pneumophila, a vacuolar pathogen of macrophages and amoebae.
Published in the journal:
. PLoS Pathog 6(3): e32767. doi:10.1371/journal.ppat.1000822
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1000822
Summary
Legionella pneumophila is an intracellular pathogen responsible for Legionnaires' disease. This bacterium uses the Dot/Icm type IV secretion system to inject a large number of bacterial proteins into host cells to facilitate the biogenesis of a phagosome permissive for its intracellular growth. Like many highly adapted intravacuolar pathogens, L. pneumophila is able to maintain a neutral pH in the lumen of its phagosome, particularly in the early phase of infection. However, in all cases, the molecular mechanisms underlying this observation remain unknown. In this report, we describe the identification and characterization of a Legionella protein termed SidK that specifically targets host v-ATPase, the multi-subunit machinery primarily responsible for organelle acidification in eukaryotic cells. Our results indicate that after being injected into infected cells by the Dot/Icm secretion system, SidK interacts with VatA, a key component of the proton pump. Such binding leads to the inhibition of ATP hydrolysis and proton translocation. When delivered into macrophages, SidK inhibits vacuole acidification and impairs the ability of the cells to digest non-pathogenic E. coli. We also show that a domain located in the N-terminal portion of SidK is responsible for its interactions with VatA. Furthermore, expression of sidK is highly induced when bacteria begin to enter new growth cycle, correlating well with the potential temporal requirement of its activity during infection. Our results indicate that direct targeting of v-ATPase by secreted proteins constitutes a virulence strategy for L. pneumophila, a vacuolar pathogen of macrophages and amoebae.
Introduction
The delivery of newly formed phagosomes to the lysosomal system by the endocytic pathway is essential for the digestion of phagocytosed materials. To evade such destruction, successful intracellular bacterial pathogens have evolved various mechanisms, including inhibition of phagolysosomal fusion, resistance to lysosomal digestion or the escape to the host cell cytosol. For intravacuolar pathogens, active modification of lipid and protein composition of phagosomal membrane is critical for their survival and replication. Moreover, since lysosomal enzymes often are active only in an acidic environment, regulation of pH in the phagosomal lumen is one common strategy employed by pathogens to avoid lysosomal killing [1],[2].
Legionella pneumophila is a facultative intracellular pathogen responsible for Legionnaires' disease. Upon being phagocytosed, this bacterium orchestrates various cellular processes to initiate a unique trafficking pathway that eventually leads to the formation of a phagosome permissive for its multiplication [3]. The biogenesis and maintenance of the bacterial replicative vacuole is mediated by protein substrates of the Dot/Icm type IV secretion system [4],[5]. For example, RalF activates and recruits the small GTPase Arf1 to the bacterial vacuole [6]. Similarly, another small GTPase Rab1 is recruited to the bacterial vacuole by SidM/DrrA, which with LepB [7], completely hijacks the activity of this important regulatory molecule in membrane trafficking [8],[9]. Whereas SidM/DrrA functions to release Rab1 from its GDI and activates the protein by loading it with GTP, LepB promotes the GTPase activity [10]. These proteins, along with other effectors such as SidJ that is involved in the recruitment of endoplasmic reticulum (ER) proteins to the bacterial vacuole [11], are thought to be responsible for the transformation of the nascent phagosome into a vacuole derived from the ER that resembles an immature autophagosome [12],[13],[14]. L. pneumophila also actively modulates cell death pathways of infected macrophages, presumably to ensure the well being of the host cell for a complete infection cycle. Inhibition of cell death is mediated through the activation of an NF-κB-dependent induction of antiapoptotic genes and by effectors such as SidF that directly antagonize proapoptotic BNIP3 and Bcl-rambo [15],[16],[17], and SdhA, an effector of unknown mechanism of action [18]. Effectors that modulate other cellular processes, including protein synthesis, ubiquitination and lipid metabolism have also been identified, but how the bacterium benefits from the functions of these virulence factors is less clear [19],[20],[21],[22]. Finally, a recent study indicated that the effector AnkB contributes significantly to bacterial intracellular growth but does not affect any of the above host cellular processes, suggesting the targeting of yet unidentified host pathways by L. pneumophila [23].
The yeast Saccharomyces cerevisiae has been widely used to study bacterial effectors, largely due to its genetic manipulability and the conservation of many eukaryotic cellular processes [24]. A large number of L. pneumophila effectors have been identified by their ability to kill yeast cells [20],[25] or to interfere with its vesicle trafficking processes [26],[27]. In eukaryotic cells, the pH of intracellular compartments is an intricately regulated parameter that is crucial for many biological processes, including membrane trafficking, protein degradation and coupled transport of small molecules [28]. Organellar acidification primarily is mediated by ATP-dependent proton transporters known as the vacuolar H+-ATPases or v-ATPases, which is a large multisubunit complex with an approximate molecular mass of 103 kDa [28]. The structure of v-ATPases can be divided into two major functional domains: a 570-kDa peripheral subcomplex, known as V1, that binds and hydrolyzes ATP, and an integral membrane subcomplex, termed V0, that serves as the pore through which protons traverse the membrane bilayer [28]. L. pneumophila is able to maintain a neutral luminal pH during infection, particularly within the first 6 hrs after uptake [29],[30], a hallmark shared by many intravacuolar pathogens. One such example is Mycobacterium avium whose vacuoles fail to acidify below pH 6.3, probably by selectively inhibiting fusion with v-ATPase-containing vesicles or by rapidly removing the complex from its phagosomes [31]. Interestingly, a recent organelle proteomic study reveals that, in the soil amoebae host Dictyostelium discoideum, v-ATPase is associated with the Legionella containing vacuole (LCV) even in early phase of infection [32]. This finding is contradictory to the well-established notion that in macrophages the bacterial phagosome maintains a neutral luminal pH for several hours, suggesting that the pathogen may initially antagonize the activity of the proton transporter.
Proton transporters from different orders of eukaryotes are highly conserved in structure and function, and some genes of mammalian or plant v-ATPase components can complement the corresponding yeast mutants [33],[34]. However, whereas in mammals mutations eliminating subunits of v-ATPase generate various phenotypes, ranging from the absence of any severe phenotype to lethality to embryonic development [35],[36], yeast v-ATPase mutants are viable but only in acidic medium [37]. In this study, we have taken the advantage of this conditional phenotype of yeast vma mutants to identify L. pneumophila proteins that may target the host v-ATPases. Here we report one such protein that inhibits v-ATPase by directly interacting with one component of the proton transporter.
Results
Identification of SidK, a Legionella protein that affects growth of yeast cells in neutral pH medium
One of the prominent phenotypes associated with yeast v-ATPase mutants is their inability to grow in neutral pH medium [37]. We reasoned that if L. pneumophila codes for proteins that inhibit v-ATPase activity, expression of such proteins in a yeast strain would impair its ability to grow in neutral pH medium. To this end, we cloned individual L. pneumophila hypothetical genes into pGBKT7 (Clontech) [20]. Yeast strains harboring each of the plasmids were tested for their ability to grow in medium with a pH of 7.5. Of the first 97 genes screened (Table S1), one gene that consistently interferes with yeast growth under this condition was obtained (Fig. 1A and B). This gene (lpg0968), designated SidK is predicted to code for a protein of approximately 65 kDa. It is present in the genomes of all sequenced strains of L. pneumophila but has no detectable homology to proteins in the database, nor does it contain predictable domains or motifs suggestive of known biochemical activities. Interestingly, this gene is divergently transcribed from lpg0969, a gene that appears to inhibit yeast growth by interfering with unknown host functions [27].

Since the original construct was made by fusing the testing gene to the DNA binding domain of the Gal4 protein on pGBKT7 (Clontech), we attempted to eliminate the potential effect of protein fusion by expressing untagged sidK in yeast strain BY4741 [38]. To this end, we used a series of vectors that differ in copy number and promoter strength (Table S3 and ref. [39]). The expression of sidK on these vectors is proportional to the strength of the promoter, with the GPD (glyceraldehyde-3-phosphate dehydrogenase) promoter giving the highest protein level (Fig. 1C). Consistent with the protein levels, in 18 hrs after the establishment of subcultures of identical cell density, strains in which SidK was expressed from the GPD promoter almost completely lost the ability to grow in neutral pH medium (Fig. 1D). On the other hand, only a marginal growth defect was observed when the gene was expressed from the moderate ADH promoter (Fig. 1C and D, strain 1), indicating that the effect of SidK on yeast growth under this condition is dose-dependent. Taken together, these data indicate that we have identified a L. pneumophila gene that affects yeast growth in neutral pH medium, probably by interfering with its v-ATPase activity directly or with activities relevant to the proton transporter.
SidK is a substrate of the Dot/Icm system that is delivered into host cytosol during infection
To exert an effect on its cellular targets, a bacterial virulence factor must first reach the host cytosol via specialized secretion systems. We thus examined whether SidK is a substrate of the Dot/Icm secretion system. We first employed the Cya assay [40] by fusing SidK to the carboxyl end of the catalytic domain of the Bordetella pertusis cyclic AMP synthetase. Infection of macrophages with a L. pneumophila strain expressing Cya fused to the known effector SidJ led to production of high-level cAMP in a Dot/Icm-dependent manner (Fig. 2A). Importantly, although the Cya-SidK fusion expressed similarly in the wild type and the dotA mutant, high levels of cAMP were only detected in infections using the wild type strain (Fig. 2A), indicating that SidK contains signals recognizable by the Dot/Icm system. Similar results were obtained with the SidC staining assay [41], in which fusion to SidK restores the translocation of the transfer deficient SidCΔC100 mutant to wild type levels (Fig. 2B–D). These results indicate that SidK is a substrate of the Dot/Icm system.

To determine whether SidK is injected into host cells by L. pneumophila during infection, we attempted to detect SidK in lysates of infected cells generated by saponin fractionation [41]. Despite considerable effort, we were unable to detect this protein in the soluble fraction of lysates of cells infected by L. pneumophila for up to 3 hrs (data not shown). Considering the possibility that the amount of translocated SidK is beyond detection by this method; we used a SidK specific antibody to enrich the protein. After immunoprecipitation, SidK protein was detected in lysates of cells infected with wild-type strain but not with the Dot/Icm deficient mutant or a sidK deletion mutant (Fig. 2E, lanes 2–3). Expression of SidK from a plasmid in the sidK deletion mutant restored the delivery of this protein into infected cells (Fig. 2E, lane 4). Collectively, these results indicate that SidK is a substrate of the Dot/Icm system and is injected into infected cells by L. pneumophila during infection. Furthermore, we cannot readily detect SidK in concentrated lysates of infected cells (data not shown), suggesting that the amount of translocated protein is low.
Next, we examined the potential role of sidK in L. pneumophila infection by constructing an in-frame deletion mutant and tested its intracellular growth. The mutant did not exhibit detectable growth defect in either mouse bone marrow-derived macrophages or D. discoideum (Fig. S1). These observations extend the list of L. pneumophila effectors not essential for its intracellular growth in standard infection models, thus adding another layer to the remarkable potential functional redundancy among substrates of the Dot/Icm system [5].
Since wild type L. pneumophila maintains a neutral pH in its vacuole and SidK appears to interfere with the functions of v-ATPase, we analyzed whether deletion of sidK affects luminal pH of LCVs in mouse macrophages. Relevant L. pneumophila strains labeled with 5(6)-carboxyfluorescein-N-hydroxysuccinamide ester (FLUOS, Fluka) were used to infect macrophages and images obtained from individual phagosomes were used to calculate intravacuolar pH against a standard curve as described [30]. As expected, vacuoles containing heat-killed bacteria quickly acidified to pH values of about 4, whereas phagosomes harboring wild type L. pneumophila maintain a neutral pH at the time points examined (Fig. S2-A). Furthermore, although the dotA mutant was not lysed by the macrophages in the experimental duration (Fig. S2-B), its vacuoles were also acidified, indicating that the Dot/Icm system is required for the biogenesis of a bacterial phagosome of neutral pH. Interestingly, vacuoles containing the sidK deletion mutant still are able to block their acidification, thus maintaining a neutral luminal pH in the experimental duration (Fig. S2-A). Given the proficient intracellular growth displayed by the mutant (Fig. S1), this result was not unexpected. L. pneumophila mutants lacking a single effector gene rarely exhibit detectable intracellular growth defect, possibly due to functional redundancy among bacterial and/or host factors [5],[42].
Expression of sidK is repressed at stationary phase and is induced within one hour after transition into fresh medium
Consistent with the observation that L. pneumophila grown at post-exponential phase are more infectious, many substrates of the Dot/Icm system are highly induced when bacterial cultures enter this growth phase [5]. That the luminal pH of LCVs is neutral in the early phase of infection points to the requirement of bacterial factors that target v-ATPase in this period of infection. We thus examined the protein level of SidK at different time points throughout the L. pneumophila growth cycle in broth. In contrast to many substrates of the Dot/Icm transporter whose expression is induced at post-exponential phase, very little SidK was present in L. pneumophila grown at this stage (Fig. 3). Rather, accumulation of SidK was apparent within 1 hr after diluting saturated cultures into fresh medium, and reached the peak 2 hrs after dilution (Fig. 3B). When the bacterium begins to replicate (approximately 4–5 hrs after dilution), protein level of SidK begins to decrease and became difficult to detect throughout the rest of the growth cycle (Fig. 3A–B), a pattern consistent with the slow progression of LCVs to lysosomal organelles [30]. We also determined the kinetics of SidK translocation during infection by saponin fractionation. Translocated SidK was not detectable until 3 hrs after infection and the protein is present in the soluble fraction of infected cells in the first 12 hrs of infection (Fig. 3C).

SidK interacts with the host vacuolar ATPase
Since mutations affecting various yeast genes not directly involved in v-ATPase function can result in mutants sensitive to neutral pH medium [43], we furthered our study on the mechanism of action of SidK by identifying its cellular targets with the unbiased affinity chromatograph method. We incubated Affigel beads (Bio-Rad) coated with purified SidK (Fig. S3) with PBS soluble fraction of U937 cell lysates. After removing unbound proteins by washing with PBS, proteins retained on the beads were separated by SDS-PAGE and were visualized by silver staining. At least three proteins with molecular weights ranging between 20 kDa and 75 kDa were retained by beads coated with SidK, but not by uncoated beads (Fig. 4A, lane 3). By mass spectrometry analysis, we identified these proteins as three components of the mammalian vacuolar H+-ATPase: VatA, the ubiquitous VatB2 subunit and VatE (Fig. 4A, lane 3). These results indicate v-ATPase is the potential target of SidK.

To further investigate the interactions between v-ATPase and SidK, we transfected mammalian cells with combinations of plasmids that direct the expression of GFP-SidK or Flag-VatA, one essential component of the proton transporter [28]. Lysates of transfected cells were subjected to co-immunoprecipitation (co-IP) with an anti-Flag antibody to detect possible SidK/VatA complexes. GFP-SidK was detected in immunoprecipitates only from cells coexpressing Flag-VatA (Fig. 4B). No signals were detected in untransfected samples or in samples transfected with plasmids expressing only Flag-VatA or GFP-SidK, indicating that the interactions were specific. Similar results were obtained in reciprocal experiments using the anti-GFP antibody (Fig. 4C). Furthermore, VatA was detected in precipitates from cells that was transfected only with the plasmid expressing SidK, indicating that this protein interacts with endogenous VatA (Fig. 4C, lane 3).
The structure and function of v-ATPase from mammals and yeast are highly similar [28]. We thus examined whether SidK interacts with yeast v-ATPase. When lysates of yeast cells expressing GFP or GFP-SidK were immunoprecipitated with a GFP-specific antibody, Vma1 (equivalent of VatA) was detected in precipitates, again only in samples expressing SidK (Fig. 4D, lane 1 in left panel). Similar results were obtained in reciprocal immumoprecipitation using a Vma1 specific antibody (data not shown). Taken together, these data establish that v-ATPase is the cellular target of SidK.
SidK directly interacts with the VatA (Vma1) subunit of the v-ATPase
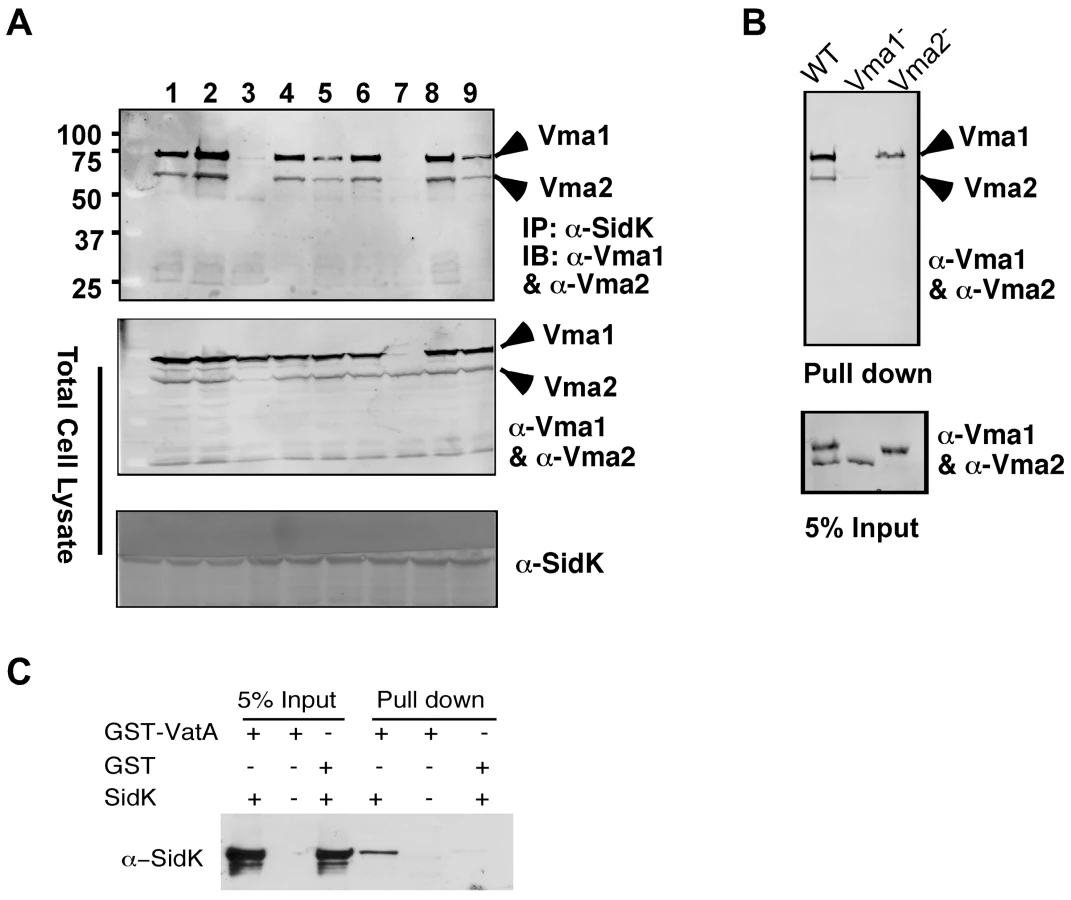
Under normal physiological condition, components of the V1 domain of v-ATPases form a stable complex [28]. In agreement with this notion, in immunoprecipitation experiments aiming at detecting interactions between SidK and components of the V1 complex, positive interactions were observed in many if not all components (data not shown). Thus, we further investigated which subunit of the V1 domain is directly targeted by SidK. Because some V1 components are recalcitrant to purification in their soluble form, we used yeast mutants that lack individual V1 component genes [38] to identify the subunit that directly interacts with SidK. We first examined the formation of protein complexes between SidK and two V1 subunits, Vma1 and Vma2 in these mutants. Vma1 and Vma2 can be coimmunoprecipitated by the SidK antibody in mutants lacking vma4, 5, 7, 8, 10 or 13, indicating that none of these subunits is required for the formation of protein complexes between SidK and Vma1 or Vma2 (Fig. 5A). However, in the absence of Vma1, no interactions between SidK and Vma2 or any other V1 components were detected (Fig. 5A, lane 7 and data not shown). Importantly, although at a low level, Vma1 was detected in precipitates obtained by the SidK antibody in the vma2 mutant (Fig. 5A, lane 3). Furthermore, when beads coated with SidK were incubated with lysates of different vma mutants, Vma1 from the lysates of the vam2 mutant was retained (Fig. 5B, lane 3). Under the same condition, SidK coated beads did not retain Vma2 or other V1 components expressed in the vma1 mutant (Fig. 5B, lane 2 and data not shown). Collectively, these results point to Vma1 as the direct target of SidK. To confirm this conclusion, we purified recombinant mammalian VatA as a GST tagged protein (GST-VatA) and tested its interaction with SidK. As expected, formation of SidK/GST-VatA complexes can be captured by GST beads (Fig. 5C). From these results, we conclude that SidK targets the v-ATPase by directly interacting with the VatA (Vma1) subunit.
A N-terminal domain of SidK is important for binding to Vma1
We extended our analysis of the interactions between SidK and VatA by determining the region on SidK important for target binding. To this end, we constructed a series of SidK deletion mutants (Fig. 6A). To eliminate the potential discrepancy that may arise from the loss of epitopes in these mutants when the polyclonal anti-SidK antibody is used in subsequent experiments, we expressed GFP fusions of these mutants in mammalian cells. Deletion of 30 amino acids from the N-terminus of SidK did not detectably affect its binding with VatA (Fig. 6B, lane 2). However, although it was expressed at a high level, a mutant missing the first 94 amino acids no longer detectably bound VatA (Fig. 6B, lane 3). On the other hand, the VatA binding activity of SidK is far more tolerant to deletions in its carboxyl end. A mutant lacking as many as 382 amino acids from this end of SidK still co-precipitated with VatA at a level similar to that of the full-length protein (Fig. 6B, lane 8). Similar results were obtained in yeast, with the exception of sidKΔC382, which did not bind Vma1, probably due to the different binding affinity of SidK to v-ATPases from these two organisms (Fig. S4). Collectively, these results indicate that a domain that lies within residue 30 to 191 of SidK is important for interacting with VatA.

To determine whether any of the SidK deletion mutants are still active in inhibiting v-ATPase functions, we tested their ability to inhibit yeast growth in neutral pH medium. Our results indicate that under this condition, SidKΔN30 consistently inhibits yeast growth, whereas the binding inactive mutant SidKΔN94 exhibits very little effect (Fig. 6C). Similarly, the binding of SidKΔC98 and SidKΔ286 to VatA is comparable to that of full-length protein and both mutants exhibit detectable inhibition in yeast growth (Fig. 6C). Although these mutants can be stably expressed in yeast from the GPD promoter, their expression levels are considerably lower than that of full-length SidK (Fig. S5). Since high protein level is required for SidK to exert full inhibitory effect on yeast growth (Fig. 1), the low activity exhibited by these mutants may result from the their low protein levels in yeast.
SidK inhibits v-ATPase-mediated ATP hydrolysis and proton translocation
The V1 domain of v-ATPase provides the energy required for proton translocation across membranes by binding and hydrolyzing ATP via subunit Vma2 and Vma1, respectively [44]. Our observation that SidK directly binds Vma1 prompted us to examine the effect of such binding on ATP hydrolysis. Thus, we followed a standard procedure [45] to prepare yeast membrane and examined the effect of SidK on its ATPase activity. In this assay, exogenous ATP was added to purified yeast membranes and the release of free phosphate was measured by malachite green [46]. In samples receiving 2 µM BSA, the level of free phosphate steadily increased throughout the experimental duration (Fig. 7A, open triangles). On the other hand, the addition of 1 µM bafilomycin A1 (Baf A1), a commonly used inhibitor of v-ATPase [47] led to strong inhibition of ATP hydrolysis, thus low level of free phosphate (Fig. 7A, diamonds). Importantly, we found that recombinant SidK inhibited v-ATPase activity in a dose-dependent manner. Significant inhibition was observed by 0.1 µM SidK, and a higher amount of protein caused more severe inhibition (Fig. 7A, closed triangles); 1 µM of SidK exerted inhibition at a level similar to that of Baf A1 (Fig. 7A, squares). Next, we probed the mechanism of action of SidK by adding its antibody to the reactions. The antibody did not detectably affect SidK activity even when it was two-fold in excess (Fig. S6).

To confirm that the observed inhibition of ATP hydrolysis was indeed a result of blocking v-ATPase activity, we did similar experiments with membranes prepared from the vma1 mutant. For the same amount of membranes, the overall ATP hydrolysis activity of the mutant markedly decreased (compare 3rd bar of mutant and 1st bar of WT in Fig. 7B). Moreover, the ATP hydrolysis activity of membranes from the mutant is resistant to Baf A1 or SidK, but not to EDTA and vanadate, two general inhibitors for ATPases [48] (Fig. 7B, mutant 4th & 5th bar). On the other hand, ATPase activity in membranes prepared from wild type yeast consistently exhibited sensitivity to SidK, again in a dose-dependent manner (Fig. 7B, wild type 2nd-5th bar). We also tested the effect of SidK on the ATP hydrolysis activity of mammalian Hsp70, an ATPase structurally distant from the v-ATPase [49]. EDTA but not SidK abolished ATP hydrolysis by this heat shock protein; vanadate also exhibited inhibitory effect, but to a lesser extent (Fig. S7). Taken together, these results indicate that binding of SidK to Vma1 leads to specific inhibition of the ATP hydrolysis activity of the proton transporter.
Because v-ATPase-mediated ATP hydrolysis is coupled with proton translocation, and thus the acidification of vesicles [50], inhibition of v-ATPase activity by SidK would block vesicle acidification. To test this hypothesis, we determined the effect of SidK on the sequestration of the lipophilic amine acridine orange (AO) by yeast vesicles. Nonprotonated AO permeates membranes, and, if the pH of the vesicles drops as a result of v-ATPase-mediated proton translocation, it becomes protonated and sequestered, leading to quenching of its fluorescence [51]. In samples receiving the solvent DMSO that does not affect v-ATPase activity, more AO was trapped in the vesicles as proton translocation was initiated by adding ATP, leading to quenching of fluorescence at 525 nm (Fig. 7C, diamonds). On the other hand, inclusion of 1 µM SidK to the reaction blocked such quenching during the entire experimental duration (Fig. 7C, squares). The effect of SidK at this concentration is comparable to that of Baf A1, which almost completely blocked AO fluorescence quenching (Fig. 7C, triangles). From these observations, we conclude that inhibition of ATP hydrolysis activity of v-ATPase by SidK prevents proton translocation.
SidK interferes with phagosome acidification and phagolysosomal digestion of bacteria
To determine whether SidK affects the functions of v-ATPase in vivo, we delivered His6-SidK into mouse bone marrow-derived macrophages by syringe loading [52] and examined the acidification of phagosomes by using the dextran-coupled pH sensitive fluorescein, whose fluorescence drops very sharply at pH values below 5.5 [53]. The pH insensitive Cascade Blue dextran was included in the feeding mixture as a loading control. Macrophages in all samples emitted blue fluorescence signals at similar intensity, indicating that the dyes were equally loaded into the cells (Fig. 8A, lower panel). Importantly, compared to macrophages receiving BSA, cells loaded with SidK gave significantly stronger green fluorescence signals (Fig. 8A, upper panel, the first two images). Similarly, compared to cells loaded with BSA, cells treated with the v-ATPase inhibitor Baf A1 also emitted stronger fluorescence signals (Fig. 8A, upper panel, the right image). These results indicate that SidK interferes with efficient acidification of phagosomes, thus inhibiting the decrease of their luminal pH values. To substantiate this observation, we used LysoRed, which accumulates in acidified organelles to stain protein-loaded macrophages that have been fed with E. coli cells expressing GFP for 10 hrs. Strong red fluorescence signals were readily detected in cells loaded with BSA, but not in cells receiving SidK (Fig. 8B, left panel), further supporting the notion that SidK inhibits phagosomal acidification. No effect was detected in additional controls with two other Legionella effector proteins (data not shown). Moreover, at this time point, we observed that the number of E. coli cells in macrophages loaded with SidK was significantly higher than that of cells containing BSA (Fig. 8B, middle panel), suggesting that inhibition of phagosomal acidification by SidK impaired the lysosomal digestion of internalized bacteria.

Since macrophages from mice lacking a functional a3 subunit exhibit delayed digestion of bacteria [54], we set to more thoroughly examine the effect of SidK on macrophage-mediated lysis of E. coli cells. We first determined the survival of E. coli in macrophages loaded with different proteins. Macrophages loaded with His6-SidK or BSA are capable of killing phagocytosed bacteria, but cells containing SidK were less efficient in the clearance of the bacteria, and such differences became significant 6 hrs after adding the bacteria (Fig. 8C). Similar results were obtained when macrophages harboring one or more intact E. coli cells were scored. In cells receiving His6-SidK, more than 90% of the cells harbor intact bacterial cells throughout the 24 hrs experiment duration (Fig. 8D). However, 8 hrs after adding the bacteria, less than 40% of the macrophages loaded with BSA contained intact bacterial cells and the ratio of such cell population dropped to less than 10% at the12-hr time point (Fig. 8D and E). Taken together, these results indicate that SidK can inhibit v-ATPase activity in vivo and such inhibition leads to defects in phagosomal acidification and impairment in lysosomal digestion of bacteria by macrophages.
Discussion
Many intracellular bacterial pathogens reside and replicate in phagosomes of unique physiological and biochemical properties. One such property is an actively regulated pH homeostasis important for successful infection of these pathogens. Since the vacuolar ATPase is the primary cellular machinery involved in controlling vacuolar pH [50], it is believed that pathogens capable of maintaining a neutral phagosomal pH encode specific traits to inhibit the accumulation of the proton transporters on their vacuolar membranes. However, although the importance of maintaining proper phagosomal pH, presumably by actively regulating the activity of v-ATPase, is generally recognized, almost nothing is known concerning the molecular mechanisms responsible for such regulation. Using a screening strategy based on the sensitivity of yeast v-ATPase mutants to neutral pH medium, we have identified SidK, a L. pneumophila protein that targets the proton transporter.
A pathogen can employ at least two mechanisms to maintain a neutral luminal pH in its phagosomes: By preventing the accumulation of v-ATPases on the phagosomal membranes or by inhibiting the activity of acquired v-ATPases. Although we have not been able to consistently detect SidK on LCVs, probably because of low protein level and/or poor antibody quality (Fig. 2 and data not shown), the presence of v-ATPases on LCVs [32] strongly suggests that SidK targets the proton pumps on the bacterial phagosomes. This feature differs from vacuoles of Mycobacterium ovium that do not contain detectable v-ATPases [31]. However, these two mechanisms are not mutually exclusive, because in addition to blocking its acquisition, the pathogen may need to antagonize v-ATPases that accidentally associate with its phagosomes. It is worth noting that detecting the association of v-ATPase with specific organelles can be complicated by low abundance of this protein complex on the membranes. For example, only a few v-ATPases were detected on a phagosome containing a latex bead [55]. Similarly, v-ATPases associated with LCVs can be detected by the sensitive mass spectrometry but not by standard immunostaining (ref. [32] and data not shown). Thus, direct targeting of v-ATPase by specific virulence factors could be a mechanism shared by many intravacuolar pathogens.
Our data showed that SidK interacts with v-ATPases by directly binding to the VatA subunit (Fig. 5). Moreover, this protein appears to have a higher affinity for VatA in the presence of VatB (Fig. 5A). Such differences may result from the conformation assumed by VatA when it is associated with VatB [28]. Preferably binding to the VatA/VatB and/or the fully assembled v-ATPase complex clearly will result in higher inhibitory efficiency for SidK, as its effect can be diluted by free VatA if these two proteins interact similarly regardless of their statuses. Reversible assembly of the V1 and V0 domain is important in regulating v-ATPase activity under different physiological conditions [28]. Whether binding of SidK to v-ATPase causes disassembly of the transporter, block of its rotary movement or other functional mechanisms of the proton transporter remains to be determined. Our biochemical studies indicate that SidK blocks organelle acidification during infection. Two lines of evidence indicate that SidK inhibits v-ATPase in vivo. First, macrophages harboring physically delivered recombinant SidK failed to block emission of fluorescence signals by the pH sensitive fluorescein (Fig. 8A). Second, cells loaded with SidK sequester significantly lower levels of LysoRed, a fluorescence dye that prefers to accumulate in acidic environments (Fig. 8B). Moreover, similar to macrophages from mice lacking a subunit of the v-ATPase [54], cells loaded with SidK displayed a significant delay in the digestion of phagocytosed bacteria (Fig. 8C–E). Thus, it is clear that by binding to VatA, SidK is able to block phagosomal acidification, thus contributing to the protection of internalized L. pneumophila during infection.
Bacterial effectors often enzymatically modify their targets to divert the cellular processes in ways beneficial to the survival of the pathogens [42]. However, despite considerable effort, we were unable to detect novel post-translational modifications on VatA co-purified with SidK from yeast (data not shown). Moreover, our attempt to determine the mechanism of action of SidK by its antibody is not conclusive because the antibody is able to immunoprecipitate Vma1 or VatA, indicating that it can form a stable complex with these two proteins (Fig. 5A and data not shown). Although SidK-mediated modifications of VatA or other v-ATPase subunits could substantiate its effect, two lines of evidence indicate the importance of physical binding in the activity of SidK. First, in contrast to other highly effective L. pneumophila effectors, such as those involved in inhibiting host protein synthesis or membrane trafficking [5], a much higher level of SidK is required to significantly inhibit yeast growth in neutral pH medium, a condition that is completely unable to support growth of yeast vma mutants (Fig. 1). For instance, if the effect of SidK was mediated by a highly catalytic mechanism, one would expect more severe inhibition when expressed from the ADH (alcohol dehydrogenase) promoter (Fig. 1C). Similarly, a considerable amount of SidK is needed to inhibit v-ATPase activity in yeast membranes (Fig. 7). Second, some deletion mutants capable of binding VatA still are able to exert inhibitory effect on yeast growth in neutral pH medium (Fig. 6C). Given the requirement of high-level SidK for full growth inhibition, the loss of inhibitory effect by deletion mutants still competent for binding VatA very likely is a result of lower protein levels (Fig. S5). Alternatively, SidK may need other L. pneumophila proteins to exert its full activity or our experimental conditions are not optimal for its activity.
In A/J mouse macrophages, vacuoles containing L. pneumophila become acidified during the replication phase of infection. Delayed maturation of the LCV promotes intracellular growth, since Baf A1 treatment blocks acidification and acquisition of lysosomal markers and also reduces the bacterial yield [30]. Consistent with the observation that expression of sidK peaks early in the lag phase before declining to almost undetectable levels as the bacteria enter the post-exponential phase, translocated SidK did not become detectable until 3 hrs after bacterial uptake. That translocated SidK is still detectable 12 hrs after infection suggests a delayed inhibition of SidK expression during infection or that SidK expresses differently in intracellular bacteria. In D. discoideum, the association of v-ATPases with the LCV is detectable from 15 min to 14 hrs after bacterial uptake [32]. The presence of this transporter in the early phase of infection suggests that the undetected SidK and/or other effectors antagonize its activity. Given the important roles of v-ATPase in vesicular trafficking, particularly in the endocytic pathway [56] that was recently shown to be involved in remodeling the membranes of the LCVs in D. discoideum [32], prolonged biochemical modifications of the v-ATPase may interfere with the ability of the bacteria within phagosomes to efficiently acquire nutrients and materials from certain membrane trafficking pathways. Thus, L. pneumophila appears to use a combination of strategies to modulate the activities of v-ATPase at different phases of infection for its benefit.
L. pneumophila appears to acquire many of its genes important for its interactions with host by horizontal gene transfer, which may account for at least in part the high plasticity of its genomes [57],[58]. Although the distinct biochemical activities of these genes can interfere with host cellular processes, a single gene often plays only a small incremental role in its evolution to parasitize its hosts, which may explain the remarkable functional redundancy among effectors of the Dot/Icm system [5],[42]. For example, at least four proteins are involved in inhibiting host protein synthesis by targeting the elongation factor eEF1A [20],[59]. Consistent with this notion, with a few exceptions, deletion of one or more Dot/Icm substrate genes did not cause detectable defect in intracellular growth [5],[42],[60]. Thus, our observation that deletion of sidK did not impair intracellular growth of L. pneumophila or its phagosomal pH is not completely unexpected. It is very likely that multiple Dot/Icm substrates are involved in the modulation of v-ATPase activity. Identification and elucidation of activities of such proteins should pave the way toward further understanding of the mechanisms underlying organelle acidification and of the means whereby it can be disrupted by pathogens.
Materials and Methods
All animal use procedures were in strict accordance with the NIH Guide for the Care and Use of Laboratory Animals and were approved by the Purdue Animal Care and Use Committee (PACUC).
Bacterial strains and growth conditions
Bacterial strains used in this study are listed in Table S2. Strains of E. coli were grown in LB and the medium was supplemented with the appropriate antibiotics when necessary. The L. pneumophila strain Philadelphia-1 strain Lp02 [61] was the parent of all derivatives used in this study. L. pneumophila was grown and maintained on CYE medium as previously described [62]. When necessary antibiotics were included as described [62]. To construct the sidK in-frame deletion mutant ZL114, we constructed plasmid pZL886 by cloning two DNA fragments generated by primers PL192/PL193 and PL194/PL195 (Table S4) into SacI/SalI digested pSR47s [61]. The primers were designed so that after deletion, only the first and last 15 amino acids are left in the mutant. pZL886 was introduced into Lp02 and the deletion mutant was obtained by following the standard allelic exchange method [63]. To complement the mutation, we inserted the coding region of sidK into pJB908 [40]. In complementation experiments, the vector used for expressing the gene of interest was introduced into the wild type strain or mutants and the bacterial cultures grown to the post-exponential phase as determined by optical density of the cultures (OD600 = 3.3-3.8) as well as an increase of bacterial motility.
Plasmid construction
The plasmids used in this study are listed in Table S3 and the sequences of all primers are in Table S4. Plasmids harboring individual full-length L. pneumophila hypothetical genes were in Table S1. The open reading frame of sidK and its derivatives were cloned into pEGFPC1 (Clontech) for expression in mammalian cells. A number of vectors, including pGBKT7 (Clontech), p415ADH, p415TEF, p425TEF and p425GPD [39] were used to express sidK in yeast either as an untagged form or as GFP fusions (see text for details). To express His6-SidK in L. pneumophila, we first amplified the multiple cloning site region of pQE30 (Qiagen) with primers QE5′EcoRV/QE3′XbaI and inserted it into Ecl136II/XbaI digested pJB908 [40]. to generate pZL507. The sidK gene was then inserted into pZL507 as a BamHI/XhoI fragment to give pZL1333. cDNA clones coding for relevant subunits of the v-ATPase were amplified from a human kidney cDNA library (Clontech) or from clones purchased from the ATCC. For expression in mammalian cells, sidK or each of these genes was inserted into pEGFPC1 (Clontech) or pFlag-CMV (Sigma). The vatH gene (pEF-HA-NBP1) was a gift from Dr. M. Peterlin of University of California, San Francisco. The integrity of all genes was verified by sequencing analysis.
Yeast manipulation and screening of L. pneumophila proteins that inhibits yeast growth in neutral pH medium
Yeast strains used were PJ69-4A [64], BY4741 [38] and their derivatives (Table S2). Yeast was grown in YPD medium or in appropriate amino acid dropout minimal media at 30°C. Using a standard protocol [65], we transformed plasmids carrying full-length hypothetical L. pneumophila genes [20] into yeast strain PJ69-4A [64] and grew the resulting strains over night in minimal medium of pH 5.5. After diluting at 1∶40 into medium of pH 7.5 buffered with 50 mM MES and 50 mM MOP, the cultures were incubated with vigorous shaking for 24 hrs. Cultures that did not grow to high density were retained for further analysis. For quantitative study of growth, yeast subcultures of 2×106 cells/ml were made in appropriate Dropout medium and cell growth was monitored by measuring the OD600 at indicated time points.
To prepare cell lysates for protein analysis, cells from 5 ml overnight cultures were first lysed with a cracking buffer (40 mM Tris-Cl [pH 6.8], 5% SDS, 0.1 mM EDTA, 8 M urea, bromothymol Blue 0.4 mg/ml) with glass beads. Samples were resolved by SDS-PAGE after adding Laemmli buffer.
Cell culture and transfection
Mouse macrophages were prepared from bone marrow of female A/J mice of 6–10 weeks of age following published protocols [12]. U937 cells were cultured in RPMI medium supplemented with 10% fetal bovine/calf serum (FBS) and 5 mM glutamate, and if needed, the cells were differentiated into macrophages with 50 ng/ml phorbol myristic acid (PMA) as described [12]. 293T cells were cultured in Dulbecco's modified minimum Eagle's medium (DMEM) supplemented with 10% FBS. For transfection, we grew cells to about 80% confluence and transfected them with Lipofectamine 2000 (Invitrogen) following manufacturer's instructions. For growth curve experiments, macrophages were plated into 24-well plates at 2×105 cell per well. For immunoprecipitation and fractionation, about 2×107 cells plated on standard petri dishes were used. Infection was performed at the indicated MOIs as required by the particular experiments.
Protein expression and purification
To purify GST-SidK, we inserted the predicted sidK orf into pGEX-4T-1 (Qiagen) to generate pZL797. E. coli strain XL1Blue containing pZL797 was grown in 1 liter LB to OD600 of 0.7. After inducing with 0.2 mM IPTG for 6 hrs at 25°C, harvested cells were lysed by passing through a French press twice at 1,500 psi. Cleared supernatant was incubated with glutathione beads (Qiagen) for 2 hrs at 4°C and the beads were washed with 40X bed volume of PBS buffer containing 0.5% Triton X-100. GST-SidK was eluted with PBS containing 10 mM reduced glutathione. When needed, GST tag was removed by Thrombin, a protease that was subsequently removed by benzamidine-Sepharose beads (GE). GST-VatA was purified with a similar procedure.
To purify His6-SidK from L. pneumophila, we introduced pZL1333 into the non-virulent strain Lp03 [61]. A 50 ml of saturated culture was diluted into 1 liter AYE broth, when the culture reached exponential growth phase (OD600 = 0.5), expression of the gene was induced with 0.2 mM IPTG for 16 hrs. Cleared cell lysates were incubated with Ni2+-Agrose beads for 2 hrs at 4°C and the beads were washed with 40 times of the bed volume of TBS buffer (50 mM Tris-HCl, 150 mM NaCl, pH 7.4) containing 10 mM imidazole. The protein was eluted with 200 mM imidazole. After dialyzing against TBS to remove imidazole, the protein was further purified by gel filtration with an FPLC system using a Superdex 200 10/300 GL column (GE Healthcare). TBST buffer (50 mM Tris-Cl, 150 mM NaCl, 0.1% Triton-X100, pH 7.4) was used as eluent and the flow rate was set at 0.4 ml/min. The single peak corresponding to the protein was collected, dialyzed in the appropriate buffer for subsequent use. His6-Hsp70 was similarly purified from E. coli. Protein concentrations were determined by the Bradford assay; the purity of all proteins was more than 95% as assessed by SDS-PAGE followed by Coomassie bright blue staining (Figs. S3 and S7).
Affinity chromatograph from cell lysates and in vitro protein binding
The procedure for affinity pulldown was described elsewhere [20]. Briefly, U937 cells collected from 500 ml culture suspended in 3.0 ml PBS containing 5 mM DTT and protease inhibitors (Roche) were lysed with a glass homogenizer (Wheaton). The lysates were subjected to centrifugation at 10,000×g for 10 min at 4°C to remove unbroken cells and nuclei, the post-nuclear supernatant was added to Affigel beads coated with SidK and incubated for 14 hrs at 4°C. We then washed the beads five times with PBS and dissolved bound proteins with SDS sample buffer. After SDS-PAGE, proteins were visualized by silver staining (Bio-Rad). Individual protein bands retained by beads coated by SidK but not by GST were excised, digested with trypsin, and analyzed by matrix-assisted laser desorption/ionization/mass spectrometry (MALDI/MS) (Taplin Biological Mass Spectrometry Facility, Harvard Medical School).
For GST pull down experiments, 10 µg purified GST or GST-VatA was mixed with 2 µg His6-SidK in PBS for 4 hrs at 4°C. After adding 40 µl of 50% pre-washed glutathione beads slurry, binding was allowed to proceed for 1 hr. The beads were then washed 5 times with PBS containing 500 mM NaCl. Retained proteins were detected by immunoblot after SDS-PAGE.
Coimmunoprecipitation
Twenty-four hrs after transfection, cells were collected and lysed in a lysis buffer (0.2% of NP-40, 50 mM Tris-HCl pH = 7.5, 150 mM NaCl, 1 mM EDTA, 15% glycerol, and protease inhibitors (1 mM Na3VO4, 1 mM PMSF, 10 µg/ml Aprotinin, 2 µg/ml Leupeptin, 0.7 µg/ml Pepstatin)). After removing debris by centrifugation at 10,000 g for 10 min at 4°C, 2 mg protein (approximately 1 ml) was used for immunoprecipitation by adding the appropriate antibody and 30 µl of 40% protein G-sepharose beads (GE Healthcare). After incubating at 4°C on a rotary shaker for 4 hrs, the beads were washed 5 times with the lysis buffer before being dissolved in Laemmli buffer.
For immunoprecipitation with yeast lysates, cells harvested from 50 ml mid-log phase cultures were digested with Zymolyase, and the resulting spheroplasts were lysed with the lysis buffer used for mammalian cells. The lysates containing approximately 2 mg proteins were incubated with appropriate antibody and protein G sepharose for 16 hrs at 4°C. The beads were removed by washing 5 times with the lysis buffer. In both cases, protein associated with beads were dissolved in Laemmli buffer and resolved by SDS-PAGE. Proteins transferred to nitrocellulose membranes were detected by immunoblot.
Antibodies and Western blot
Antisera against Legionella, ICDH (isocitrate dehydrogenase) were described in an early study [4]. SidK cleaved from GST-SidK were used as an antigen to produce a specific antibody following a standard protocol (Pocono Rabbit Farm and Laboratory, Canadensis, PA). GFP antibody was prepared similarly with purified His6-GFP. When necessary, antibodies were affinity-purified against the antigens covalently coupled to an Affigel matrix (Bio-Rad) using standard protocols [66]. Monoclonal or polyclonal antibodies against Flag, Vma1, Vma2, PGK (3-phosphoglycerate kinase), VatA and Hsp70 were purchased from Sigma, Invitrogen, Abcam and Santa Cruz Biotechnology (sc-65521), respectively.
For Western blots, samples resolved by SDS-PAGE were transferred onto nitrocellulose membranes. After blocking with 4% milk in PBS buffer containing 0.2% Tween 20, membranes were incubated with the appropriate primary antibody: anti-SidK, 1∶2,500; anti-VatA, 1∶10,000; anti-Vma1, 1∶1,000; anti-Vma2, 1∶1,000; anti-GFP, 1∶50,000; anti-PGK, 1∶ 2000; anti-ICDH, 1∶5,000; anti-Hsp70, 1∶2000. Horseradish peroxidase conjugated secondary antibodies and enhanced bioluminescence reagents were used to detect the signals (Pierce, Rockford, IL). Alternatively, membranes were incubated with an appropriate IRDye infrared secondary antibody (Li-Cor's Biosciences, Lincoln, Nebraska, USA) and the signals were detected, and if necessary, the intensity of the bands are quantitiated by using the Odyssey infrared imaging system.
Preparation of yeast vacuolar membrane vesicles
Yeast vacuolar membrane vesicles were prepared according to the standard method [45] with some modification. Briefly, exponentially growing yeast cells (O.D. = 0.6) were harvested, washed twice with distilled water and digested with zymolyase at 30°C for 90 min in 1 M sorbitol. The spheroplasts were resuspended in 7 volumes of Buffer A (10 mM MES/Tris (pH 6.9), 12% Ficoll 400, and 0.1 mM MgCl2), homogenized in a loosely fitting Dounce homogenizer (Wheaton) with 20 strokes, and centrifuged in a swinging bucket rotor at 4,500 g for 10 min. The supernatants were transferred to new centrifuge tubes, and buffer A was layered on the top. After centrifugation at 51,900×g for 40 min, the white layer on the top was collected and resuspended in Buffer A with a homogenizer, and Buffer B (10 mM MES/Tris (pH 6.9), 8% Ficoll 400, and 0.5 mM MgCl2) was layered on the top. After similar centrifugation, vacuoles free from lipid granules or other membranous organelles were collected from the top of the tube. Vacuolar membrane vesicles were prepared by diluting the purified vacuoles in a vesicle buffer (10 mM MES/Tris (pH 6.9), 5 mM MgCl2, and 25 mM KC1).
V-ATPase-mediated ATP hydrolysis assay
Equal amount of vacuolar membranes in ATPase buffer (10 mM HEPES, 5 mM MgCl2, 125 mM KCl, pH = 7.0) were preincubated for 40 min at room temperature with or without the testing chemicals or proteins. To test the effect of antibody, purified antibody was added to the reactions for 20 min before the addition of ATP. BSA dissolved in the same buffer as that of SidK was used as a negative control. The reaction was initiated by adding 1 mM of ATP, and samples were withdrawn at indicated time points to measure the production of inorganic phosphate using the malachite green method [46]. Briefly, the malachite green reagent was made of 2 volumes of 0.0812% malachite green, 1 volume of 5.72% ammonium molybdate dissolved in 6 M HCl, 1 volume of 2.32% polyvinyl alcohol and 2 volumes of distilled water. 90 µl of the malachite green reagent was added to 10 µl samples withdrawn at indicated time points. The reactions were allowed to proceed for 2 min and were terminated with 1/10 volume of 34% sodium citrate. After incubation for another 20 min, absorbance at OD620 nm was measured. A standard curve simultaneously obtained with a series of phosphate solutions of known concentrations was used to determine the amount of phosphate released by the membranes.
Proton translocation activity
The ATP-driven proton transport activity was assayed by measuring the uptake of proton in the yeast membrane vesicles using acridine orange (AO) quenching assay [67]. Purified vacuolar membrane vesicles were diluted in AO buffer (5 mM HEPES, pH = 7.0, 5 mM MgCl2, 150 mM KCl, 6 µM AO), and preincubated for 40 min at room temperature with or without the testing chemicals or proteins. The reaction was initiated by adding 2 mM of ATP and quickly mixed. The quenching of acridine orange was monitored by the Spex FluoroMax 3 spectrofluorometer (Jobin Yvon) with excitation at 493 nm and emission at 525 nm.
Measurement of vacuolar pH
The pH of L. pneumophila-containing phagosomes was determined by fluorescence ratio imaging using 5(6)-carboxyfluorescein-N-hydroxysuccinamide ester (FLUOS, Fluka) stained L. pneumophila as previously described [30] with the modifications detailed below. For labeling, L. pneumophila were cultured to the post-exponential phase, defined by motility and OD600 = 3.6 - 4.6, washed once with 100 mM potassium phosphate buffer, pH 8.0, and then incubated for 20 min at room temperature with 0.8 mg/ml FLUOS in 4% DMSO in 100 mM potassium phosphate, pH 8.0. This treatment did not affect viability of bacteria as determined by quantifying colony formation.
We infected macrophages plated on 24 mm coverslips with an MOI of ∼10. Infections were synchronized by washing infected cells 4 times with RPMI/FBS 60 minutes after uptake. Following an additional 70–180 minutes of incubation, we washed the monolayers 3 times with 37°C Ringers Buffer (RB; 55 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 2 mM NaH2PO4, 10 mM HEPES, and 10 mM glucose, pH 7.2), and placed the samples at a 37°C chamber with 1 ml RB and visualized on an Olympus IX70 inverted microscope. Images were acquired from the attached CoolSNAP HQ2 14-bit CCD camera (Photometrics) and subsequently analyzed using Metamorph Premier v6.3 software (Molecular Devices).
Fluorescence images were obtained at excitation wavelengths of 492 nm and 436 nm and corrected for bias, shading, and background. Individual bacteria were masked by manual thresholding using an addition image of both wavelengths. The mask was then applied to each original corrected image, and the average fluorescence intensity over the masked area of each particle was determined at each wavelength. The pH of each L. pneumophila-containing phagosome was calculated from the ratio of the fluorescence intensity at an excitation wavelength of 492 nm to the intensity at excitation 436 nm. The fluorescence intensity ratios from two independent experiments were converted to pH using a single standard curve of quartic function. The standard curve was established using FLUOS-labeled bacteria immobilized on a coverslip coated with poly-(L)-lysine. Samples were processed as above, with greater than 130 bacteria analyzed per pH at 10 incremental values ranging from pH 3.5 to 8.5 in clamping buffer (130 mM KCl, 1 mM MgCl2, 15 mM HEPES, 15 mM MES).
Only intact rod-shaped bacteria were evaluated, with greater than 50 bacteria analyzed per coverslip in each of two independent experiments. To verify that sidK did not affect lysosomal degradation, the fraction of wild-type and sidK mutant bacteria that were degraded was quantified 1 h post-infection by fixed immunofluorescence microscopy as previously described [68] (Fig. S2). Heat killed (80°C 20 minutes) wild-type bacteria served as a control for particles that trafficked to an acidic compartment [69].
Protein loading, lysosomal digestion of bacteria and phagosomal pH evaluation
We delivered recombinant proteins into macrophages by the syringe loading method [52] with some modifications: Briefly, cells were washed and collected in ice cold Dulbecco's PBS (DPBS) (Cellgro) by centrifugation (200 g 5 min). We then washed the cells twice with 37°C DPBS containing 1.2 mM CaCl2 before adding 200 µl warm loading solutions (DPBS containing 1.2 mM CaCl2 and the protein to be loaded at 0.4 mg/ml) to cell pellet containing 5×106 cells. A P-200 micropipettor (Rainin) set at 100 µl was used to pipett the cell suspension for 100 times at 37°C. After pipetting, the mixture was incubated at 37°C for 2 min. After washing twice with warm DPBS containing 1.2 mM CaCl2, cells were seeded onto glass coverslips in 24-well plates with a density of 4×105 per well and were incubated at 37°C for 12–16 hrs.
The bactericidal activity of macrophages loaded with different proteins was measured according to a published method [54] with minor modifications. Cells of E. coli strain XL1-Blue expressing the mCherry RFP or GFP were added to macrophages at an MOI of 10 for 1 hr at 37°C. The culture supernatant was replaced with fresh tissue culture medium containing 100 µg/ml gentamicin to kill extracellular bacteria. After 1 hr of incubation, the medium was replaced with fresh medium containing 10 µg/ml gentamicin. At indicated time points, cells were washed extensively (5x) with warm PBS and lysed with 0.02% saponin. The lysates were plated on LB plates and colonies were counted after overnight incubation at 37°C.
For the staining with fluorescein dextran, 10 kD fluorescent dextran (Invitrogen) was added to the cells to 0.2 mg/ml; the pH insensitive 10 kD Cascade Blue dextran (Invitrogen) was added at 0.2 mg/ml as a loading control. After incubating at 37°C for1 hr, the cells were washed 5 times and incubated at 37°C for an additional 4 hrs before being imaged. For the staining with LysoRed (LysoTracker Red DND-99, Invitrogen), cells of an E. coli strain expressing GFP were added to macrophage monolayer at an MOI of 20 for 1 hr at 37°C. After incubating with a medium containing 100 µg/ml gentamicin for 1 hr and then a medium containing 10 µg/ml gentamicin for an additional 8 hrs, a medium containing 50 nM LysoRed was added to the samples for 15 min. Cells were washed 3 times with fresh medium and subjected to imaging analysis immediately under an Olympus X-81 fluorescence microscope. All images were acquired with identical digital imaging parameters (objectives, exposure duration, contrast ratios, etc.) and were similarly processed using the IPlab software package (BD Biosciences).
Gene accession number
The gene described in this manuscript is lpg0968 with an accession number of YP_095002 in the Genebank.
Supporting Information
Zdroje
1. HuynhKK
GrinsteinS
2007 Regulation of vacuolar pH and its modulation by some microbial species. Microbiol Mol Biol Rev 71 452 462
2. OhkumaS
PooleB
1978 Fluorescence probe measurement of the intralysosomal pH in living cells and the perturbation of pH by various agents. Proc Natl Acad Sci U S A 75 3327 3331
3. ShinS
RoyCR
2008 Host cell processes that influence the intracellular survival of Legionella pneumophila. Cell Microbiol 10 1209 1220
4. LiuY
GaoP
BangaS
LuoZQ
2008 An in vivo gene deletion system for determining temporal requirement of bacterial virulence factors. Proc Natl Acad Sci U S A 105 9385 9390
5. IsbergRR
O'ConnorTJ
HeidtmanM
2009 The Legionella pneumophila replication vacuole: making a cosy niche inside host cells. Nat Rev Microbiol 7 13 24
6. NagaiH
KaganJC
ZhuX
KahnRA
RoyCR
2002 A bacterial guanine nucleotide exchange factor activates ARF on Legionella phagosomes. Science 295 679 682
7. ChenJ
de FelipeKS
ClarkeM
LuH
AndersonOR
2004 Legionella effectors that promote nonlytic release from protozoa. Science 303 1358 1361
8. MachnerMP
IsbergRR
2006 Targeting of host Rab GTPase function by the intravacuolar pathogen Legionella pneumophila. Dev Cell 11 47 56
9. MurataT
DelpratoA
IngmundsonA
ToomreDK
LambrightDG
2006 The Legionella pneumophila effector protein DrrA is a Rab1 guanine nucleotide-exchange factor. Nat Cell Biol 8 971 977
10. IngmundsonA
DelpratoA
LambrightDG
RoyCR
2007 Legionella pneumophila proteins that regulate Rab1 membrane cycling. Nature 450 365 369
11. LiuY
LuoZQ
2007 The Legionella pneumophila effector SidJ is required for efficient recruitment of endoplasmic reticulum proteins to the bacterial phagosome. Infect Immun 75 592 603
12. SwansonMS
IsbergRR
1995 Association of Legionella pneumophila with the macrophage endoplasmic reticulum. Infect Immun 63 3609 3620
13. TilneyLG
HarbOS
ConnellyPS
RobinsonCG
RoyCR
2001 How the parasitic bacterium Legionella pneumophila modifies its phagosome and transforms it into rough ER: implications for conversion of plasma membrane to the ER membrane. J Cell Sci 114 4637 4650
14. AmerAO
SwansonMS
2005 Autophagy is an immediate macrophage response to Legionella pneumophila. Cell Microbiol 7 765 778
15. BangaS
GaoP
ShenX
FiscusV
ZongWX
2007 Legionella pneumophila inhibits macrophage apoptosis by targeting pro-death members of the Bcl2 protein family. Proc Natl Acad Sci U S A 104 5121 5126
16. LosickVP
IsbergRR
2006 NF-kappaB translocation prevents host cell death after low-dose challenge by Legionella pneumophila. J Exp Med 203 2177 2189
17. Abu-ZantA
JonesS
AsareR
SuttlesJ
PriceC
2007 Anti-apoptotic signalling by the Dot/Icm secretion system of L. pneumophila. Cell Microbiol 9 246 264
18. LagunaRK
CreaseyEA
LiZ
ValtzN
IsbergRR
2006 A Legionella pneumophila-translocated substrate that is required for growth within macrophages and protection from host cell death. Proc Natl Acad Sci U S A 103 18745 18750
19. BelyiY
NiggewegR
OpitzB
VogelsgesangM
HippenstielS
2006 Legionella pneumophila glucosyltransferase inhibits host elongation factor 1A. Proc Natl Acad Sci U S A 103 16953 16958
20. ShenX
BangaS
LiuY
XuL
GaoP
2009 Targeting eEF1A by a Legionella pneumophila effector leads to inhibition of protein synthesis and induction of host stress response. Cell Microbiol 11 911 926
21. WeberSS
RagazC
ReusK
NyfelerY
HilbiH
2006 Legionella pneumophila exploits PI(4)P to anchor secreted effector proteins to the replicative vacuole. PLoS Pathog 2 e46 doi:10.1371/journal.ppat.0020046
22. KuboriT
HyakutakeA
NagaiH
2008 Legionella translocates an E3 ubiquitin ligase that has multiple U-boxes with distinct functions. Mol Microbiol 67 1307 1319
23. Al-KhodorS
PriceCT
HabyarimanaF
KaliaA
Abu KwaikY
2008 A Dot/Icm-translocated ankyrin protein of Legionella pneumophila is required for intracellular proliferation within human macrophages and protozoa. Mol Microbiol 70 908 923
24. SiggersKA
LesserCF
2008 The Yeast Saccharomyces cerevisiae: a versatile model system for the identification and characterization of bacterial virulence proteins. Cell Host Microbe 4 8 15
25. CampodonicoEM
ChesnelL
RoyCR
2005 A yeast genetic system for the identification and characterization of substrate proteins transferred into host cells by the Legionella pneumophila Dot/Icm system. Mol Microbiol 56 918 933
26. ShohdyN
EfeJA
EmrSD
ShumanHA
2005 Pathogen effector protein screening in yeast identifies Legionella factors that interfere with membrane trafficking. Proc Natl Acad Sci U S A 102 4866 4871
27. HeidtmanM
ChenEJ
MoyMY
IsbergRR
2008 Large-scale identification of Legionella pneumophila Dot/Icm substrates that modulate host cell vesicle trafficking pathways. Cell Microbiol
28. ForgacM
2007 Vacuolar ATPases: rotary proton pumps in physiology and pathophysiology. Nat Rev Mol Cell Biol 8 917 929
29. HorwitzMA
1983 The Legionnaires' disease bacterium (Legionella pneumophila) inhibits phagosome-lysosome fusion in human monocytes. J Exp Med 158 2108 2126
30. Sturgill-KoszyckiS
SwansonMS
2000 Legionella pneumophila replication vacuoles mature into acidic, endocytic organelles. J Exp Med 192 1261 1272
31. Sturgill-KoszyckiS
SchlesingerPH
ChakrabortyP
HaddixPL
CollinsHL
1994 Lack of acidification in Mycobacterium phagosomes produced by exclusion of the vesicular proton-ATPase. Science 263 678 681
32. UrwylerS
NyfelerY
RagazC
LeeH
MuellerLN
2009 Proteome analysis of Legionella vacuoles purified by magnetic immunoseparation reveals secretory and endosomal GTPases. Traffic 10 76 87
33. LuX
YuH
LiuSH
BrodskyFM
PeterlinBM
1998 Interactions between HIV1 Nef and vacuolar ATPase facilitate the internalization of CD4. Immunity 8 647 656
34. KimW
WanCY
WilkinsTA
1999 Functional complementation of yeast vma1 delta cells by a plant subunit A homolog rescues the mutant phenotype and partially restores vacuolar H(+)-ATPase activity. Plant J 17 501 510
35. InoueH
NoumiT
NagataM
MurakamiH
KanazawaH
1999 Targeted disruption of the gene encoding the proteolipid subunit of mouse vacuolar H(+)-ATPase leads to early embryonic lethality. Biochim Biophys Acta 1413 130 138
36. FinbergKE
WagnerCA
BaileyMA
PaunescuTG
BretonS
2005 The B1-subunit of the H(+) ATPase is required for maximal urinary acidification. Proc Natl Acad Sci U S A 102 13616 13621
37. NelsonH
NelsonN
1990 Disruption of genes encoding subunits of yeast vacuolar H(+)-ATPase causes conditional lethality. Proc Natl Acad Sci U S A 87 3503 3507
38. WinzelerEA
ShoemakerDD
AstromoffA
LiangH
AndersonK
1999 Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science 285 901 906
39. MumbergD
MullerR
FunkM
1995 Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene 156 119 122
40. BardillJP
MillerJL
VogelJP
2005 IcmS-dependent translocation of SdeA into macrophages by the Legionella pneumophila type IV secretion system. Mol Microbiol 56 90 103
41. VanRheenenSM
LuoZQ
O'ConnorT
IsbergRR
2006 Members of a Legionella pneumophila family of proteins with ExoU (phospholipase A) active sites are translocated to target cells. Infect Immun 74 3597 3606
42. NinioS
RoyCR
2007 Effector proteins translocated by Legionella pneumophila: strength in numbers. Trends Microbiol 15 372 380
43. SambadeM
AlbaM
SmardonAM
WestRW
KanePM
2005 A genomic screen for yeast vacuolar membrane ATPase mutants. Genetics 170 1539 1551
44. StevensTH
ForgacM
1997 Structure, function and regulation of the vacuolar (H+)-ATPase. Annu Rev Cell Dev Biol 13 779 808
45. UchidaE
OhsumiY
AnrakuY
1985 Purification and properties of H+-translocating, Mg2+-adenosine triphosphatase from vacuolar membranes of Saccharomyces cerevisiae. J Biol Chem 260 1090 1095
46. YimL
Martinez-VicenteM
VillarroyaM
AguadoC
KnechtE
2003 The GTPase activity and C-terminal cysteine of the Escherichia coli MnmE protein are essential for its tRNA modifying function. J Biol Chem 278 28378 28387
47. HussM
WieczorekH
2009 Inhibitors of V-ATPases: old and new players. J Exp Biol 212 341 346
48. HenebergP
2009 Use of protein tyrosine phosphatase inhibitors as promising targeted therapeutic drugs. Curr Med Chem 16 706 733
49. SousaR
LaferEM
2006 Keep the traffic moving: mechanism of the Hsp70 motor. Traffic 7 1596 1603
50. NishiT
ForgacM
2002 The vacuolar (H+)-ATPases–nature's most versatile proton pumps. Nat Rev Mol Cell Biol 3 94 103
51. PalmgrenMG
1991 Acridine orange as a probe for measuring pH gradients across membranes: mechanism and limitations. Anal Biochem 192 316 321
52. McNeilPL
2001 Direct introduction of molecules into cells. Curr Protoc Cell Biol Chapter 20 Unit 20 21
53. HanJ
BurgessK
2009 Fluorescent Indicators for Intracellular pH. Chem Rev PMID 19831417
54. Sun-WadaGH
TabataH
KawamuraN
AoyamaM
WadaY
2009 Direct recruitment of H+-ATPase from lysosomes for phagosomal acidification. J Cell Sci 122 2504 2513
55. KinchenJM
RavichandranKS
2008 Phagosome maturation: going through the acid test. Nat Rev Mol Cell Biol 9 781 795
56. MarshanskyV
FutaiM
2008 The V-type H+-ATPase in vesicular trafficking: targeting, regulation and function. Curr Opin Cell Biol 20 415 426
57. CazaletC
RusniokC
BruggemannH
ZidaneN
MagnierA
2004 Evidence in the Legionella pneumophila genome for exploitation of host cell functions and high genome plasticity. Nat Genet 36 1165 1173
58. de FelipeKS
PampouS
JovanovicOS
PericoneCD
YeSF
2005 Evidence for acquisition of Legionella type IV secretion substrates via interdomain horizontal gene transfer. J Bacteriol 187 7716 7726
59. BelyiY
TabakovaI
StahlM
AktoriesK
2008 Lgt: a family of cytotoxic glucosyltransferases produced by Legionella pneumophila. J Bacteriol 190 3026 3035
60. LuoZQ
IsbergRR
2004 Multiple substrates of the Legionella pneumophila Dot/Icm system identified by interbacterial protein transfer. Proc Natl Acad Sci U S A 101 841 846
61. BergerKH
IsbergRR
1993 Two distinct defects in intracellular growth complemented by a single genetic locus in Legionella pneumophila. Mol Microbiol 7 7 19
62. ConoverGM
DerreI
VogelJP
IsbergRR
2003 The Legionella pneumophila LidA protein: a translocated substrate of the Dot/Icm system associated with maintenance of bacterial integrity. Mol Microbiol 48 305 321
63. MerriamJJ
MathurR
Maxfield-BoumilR
IsbergRR
1997 Analysis of the Legionella pneumophila fliI gene: intracellular growth of a defined mutant defective for flagellum biosynthesis. Infect Immun 65 2497 2501
64. JamesP
HalladayJ
CraigEA
1996 Genomic libraries and a host strain designed for highly efficient two-hybrid selection in yeast. Genetics 144 1425 1436
65. GietzRD
SchiestlRH
WillemsAR
WoodsRA
1995 Studies on the transformation of intact yeast cells by the LiAc/SS-DNA/PEG procedure. Yeast 11 355 360
66. DumenilG
IsbergRR
2001 The Legionella pneumophila IcmR protein exhibits chaperone activity for IcmQ by preventing its participation in high-molecular-weight complexes. Mol Microbiol 40 1113 1127
67. ShiraishiY
NagaiJ
MurakamiT
TakanoM
2000 Effect of cisplatin on H+ transport by H+ -ATPase and Na+/H+ exchanger in rat renal brush-border membrane. Life Sci 67 1047 1058
68. DalebrouxZD
EdwardsRL
SwansonMS
2009 SpoT governs Legionella pneumophila differentiation in host macrophages. Mol Microbiol 71 640 658
69. JoshiAD
Sturgill-KoszyckiS
SwansonMS
2001 Evidence that Dot-dependent and -independent factors isolate the Legionella pneumophila phagosome from the endocytic network in mouse macrophages. Cell Microbiol 3 99 114
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2010 Číslo 3
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Kaposi's Sarcoma-Associated Herpesvirus ORF57 Protein Binds and Protects a Nuclear Noncoding RNA from Cellular RNA Decay Pathways
- Endocytosis of the Anthrax Toxin Is Mediated by Clathrin, Actin and Unconventional Adaptors
- Perforin and IL-2 Upregulation Define Qualitative Differences among Highly Functional Virus-Specific Human CD8 T Cells
- Exoerythrocytic Parasites Secrete a Cysteine Protease Inhibitor Involved in Sporozoite Invasion and Capable of Blocking Cell Death of Host Hepatocytes
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy