
Antagonistic Cross-Regulation between Sox9 and Sox10 Controls an Anti-tumorigenic Program in Melanoma
For the development of future cancer therapies it is imperative to understand the molecular processes underlying tumor initiation and expansion. Many key factors involved in these processes have been identified based on cell culture and transplantation experiments, but their relevance for tumor formation and disease progression in the living organism is often unclear. Therefore, genetically modified mice spontaneously developing tumors present indispensable models for cancer research. Here, we address this issue by studying the formation of melanoma, the most fatal skin tumor in industrialized countries. To this end, we use a transgenic mouse model to elucidate cellular and molecular mechanisms regulating congenital nevus and melanoma initiation. We show that a transcription factor called SOX10 promotes melanoma formation by repressing an anti-tumorigenic program involving the activity of a related factor, SOX9. When SOX10 is inactivated, SOX9 becomes upregulated and induces cell cycle arrest and death in melanoma cells. Furthermore, upon experimental elevation of SOX9 levels, SOX10 activity is suppressed, revealing an antagonistic relationship between SOX9 and SOX10 in melanoma initiation. Knowledge of how an anti-tumorigenic program can be stimulated by modulating the activities of these key factors might help to design novel therapeutic strategies.
Published in the journal:
. PLoS Genet 11(1): e32767. doi:10.1371/journal.pgen.1004877
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004877
Summary
For the development of future cancer therapies it is imperative to understand the molecular processes underlying tumor initiation and expansion. Many key factors involved in these processes have been identified based on cell culture and transplantation experiments, but their relevance for tumor formation and disease progression in the living organism is often unclear. Therefore, genetically modified mice spontaneously developing tumors present indispensable models for cancer research. Here, we address this issue by studying the formation of melanoma, the most fatal skin tumor in industrialized countries. To this end, we use a transgenic mouse model to elucidate cellular and molecular mechanisms regulating congenital nevus and melanoma initiation. We show that a transcription factor called SOX10 promotes melanoma formation by repressing an anti-tumorigenic program involving the activity of a related factor, SOX9. When SOX10 is inactivated, SOX9 becomes upregulated and induces cell cycle arrest and death in melanoma cells. Furthermore, upon experimental elevation of SOX9 levels, SOX10 activity is suppressed, revealing an antagonistic relationship between SOX9 and SOX10 in melanoma initiation. Knowledge of how an anti-tumorigenic program can be stimulated by modulating the activities of these key factors might help to design novel therapeutic strategies.
Introduction
Sox (Sry (sex determining region Y)-related HMG box) genes encode a family of transcription factors that are characterized by a conserved high-mobility group (HMG) domain mediating their binding to DNA in a sequence-specific manner [1–3]. While the majority of Sox proteins functions as transcriptional activators, some members of the Sox family including Sox9 and Sox10 may also act as transcriptional repressors [4–6]. Sox genes play key roles in embryonic development and are major determinants of stem cell behavior, regulating cell fate decisions and maintaining cellular identity [3]. Their crucial role in normal tissue formation and homeostasis is evident from the fact that several mutations in Sox genes are causative for developmental diseases, and accumulating evidence demonstrates the important functional role of Sox family proteins in a variety of cancers [7–10].
A common feature of SoxE group proteins, which includes Sox9 and Sox10, is their expression in neural crest (NC) cells during embryonic development [2, 11]. NC cells are a transient embryonic cell population that gives rise to most of the peripheral nervous system, chondrocytes and osteoblasts of craniofacial structures, smooth muscle cells of the cardiovascular system, and melanocytes, the pigmented cells of the skin [12]. While Sox9 is expressed in premigratory NC cells and in the pharyngeal apparatus, Sox10 is found in NC cells at the time of their emigration and is essential for their self-renewal and survival [12–16]. Loss of Sox10 results in absence of most NC derivatives, whereas Sox10 haploinsufficiency causes Waardenburg Hirschsprung syndrome, characterized by aganglionic megacolon, pigmentary abnormalities and often deafness due to loss of sensory innervation [13, 17–20]. In the melanocytic lineage, Sox10 is expressed during all stages of development as well as in the adult and is required in different species for the generation and homeostasis of embryonic and adult melanocytes in vitro and in vivo [13, 21–25].
In contrast, loss of Sox9 in the NC does not lead to general defects in NC-derived structures, but specifically affects the development of mesectodermal derivatives, such as smooth muscle cells and craniofacial bones and cartilage [11, 26–28]. Furthermore, heterozygous mutations in Sox9 in both mice and humans, result in campomelic dysplasia, a syndrome associated with dwarfism, skeletal malformations, cleft palate, XY sex reversal and often hermaphroditism [28–30]. However, data on Sox9 expression in melanocytes are inconsistent, and a functional implication of Sox9 in melanocyte formation has not been provided so far [23].
Based on the assumption that mechanisms of tumor formation might be related to those underlying the generation of the cell type, from which the tumor develops, we and others have recently addressed the function of Sox10 in melanoma. These studies demonstrated a crucial role of Sox10 in the pathogenesis of giant congenital naevi and melanoma in both mice and humans by regulating proliferation and survival of melanocytic cells and maintenance of their cellular identity [9; 31]. However, the precise molecular mechanisms mediating Sox10 function in melanoma remain to be investigated.
Here we show that in contrast to Sox10, Sox9 appears to be expressed at very low levels only and is functionally not required in melanocyte stem cells, committed melanocytes, and melanoma cells. However, Sox9 expression is elevated upon Sox10 deletion in mouse and human melanoma cells [9], and critically contributes to the anti-tumorigenic effects observed upon Sox10 inactivation in giant congenital naevus and melanoma.
Results
Expression analysis of SOXE factors in human skin melanocytes, giant congenital naevi, and primary melanoma in vivo
While SOX10 expression and function has been well established in adult melanocytes, naevi, and melanoma tissue in human and mice [9; 23], studies on SOX9 expression in melanocytic cells are controversial. SOX9 was reported to be expressed in cultured human melanocytes in vitro [32], human melanocytes in vivo [33], and in human melanoma [34–36]. Other reports, however, failed to reveal Sox9 mRNA and protein expression in melanoblasts and differentiated melanocytes during development and postnatally [21,37]. Given the close relationship between SoxE factors, one conceivable explanation for these discrepancies might be that antibodies raised against a given SoxE protein fail to discriminate between SOX9 and SOX10 epitopes. Indeed, when we performed immunohistochemistry on murine skin to test the specificity of various anti-SOX9 antibodies, several of them recognized both melanocytes and epithelial cells in the outer root sheet (a region in hair follicles known to express and functionally require Sox9; [37–38] (S1 Fig.). In contrast, the antibody sc-20095 exclusively detected protein expression in epithelial cells but not in melanocytes. Of note, in human melanoma cell lines in vitro, all antibodies tested but sc-20095 not only recognized SOX9, but also a protein of the molecular weight of SOX10 and detected by a SOX10-specific antibody (S2A-S2D Fig.).
To further investigate the specificity of anti-SOX9 antibodies, we performed SOX10 knockdown in human melanoma cell lines in vitro and analyzed SOX10 and SOX9 expression using Western blot analysis (S2E-S2K Fig.). As shown in S2E-S2K Fig., different anti-SOX9 antibodies used in earlier studies detected SOX10 protein expression, which was lost upon SOX10 knock-down. The only anti-SOX9 antibody, which did not display cross-reactivity with SOX10 protein, was sc-20095.
Therefore, we chose to reassess SOX9 expression in human melanocytes and melanocytic skin lesions using the specific SOX9 antibody sc-20095. Double immunostaining for SOX9 and MITF (Microphthalmia-associated transcription factor), an established marker of melanocytes [39] revealed no detectable SOX9 expression in human skin melanocytes in vivo (Fig. 1A-G). In contrast, SOX10 was readily detectable in human melanocytes (S3A-S3B Fig.). Moreover, while SOX10 was expressed in 100% of human giant congenital naevi, SOX9 expression was not detected in the same set of patient samples (n = 17; (Fig. 1H; S3D Fig.); [9]). Likewise, all samples of a melanoma tissue microarray composed of 56 primary melanoma biopsies revealed strong SOX10 expression (Fig. 1I; [9]). SOX9 expression, however, was found in only 41% (23/56) of the primary melanoma samples, in which it was expressed in a few scattered cells accounting for less than 10% of all melanoma cells (Fig. 1I). In contrast, expression of SOX9 was readily detectable in the epithelial lineage of normal skin as well as in basal cell carcinoma, an epithelial skin cancer (Fig. 1E, F; S3C Fig.; [37,40]). To investigate the mRNA expression of SOX10 and SOX9 in a large set of human melanoma samples, we used of the TCGA (The Cancer Genome Atlas) database. Interestingly, the vast majority of human melanoma samples displayed much higher SOX10 than SOX9 expression (Fig. 1J) and only very few samples were characterized by a SOX9 high / SOX10 low expression pattern (Fig. 1K). Thus, SOX10 but not SOX9 is prominently expressed in normal human melanocytes, human giant congenital naevi, and primary melanoma.

SOX10 and SOX9 exhibit divergent functional roles in murine melanocyte stem cells and hair pigmentation
To corroborate our findings in an experimentally amenable system, we extended the analysis of Sox9 expression to mouse melanocytes, taking advantage of a previously described iDct-GFP mouse line (Fig. 2A,B; [41]). Doxycycline-induced GFP-labelled melanocytes were isolated via fluorescence-activated cell sorting (FACS) and subjected to RNA-Seq analysis (Fig. 2A). While Sox10, Mitf and Tyr were expressed at high levels (Sox10 reads were 1292, 1372, 1776 and 2488 at E15.5, E17.5, P1 and P7, respectively), the expression of Sox9 was extremely low (Sox9 reads were 68, 65, 105 and 128 at E15.5, E17.5, P1 and P7, respectively). These data are in accordance with earlier studies on Sox9 mRNA and protein expression in murine melanoblasts and melanocytes [21,37] and suggest that in contrast to Sox10, Sox9 expression is virtually absent in the melanocytic lineage during mouse embryogenesis and postnatally.

Melanocyte stem cells are found in a specialized region of the hair follicle called bulge and give rise to melanocyte progenitors and differentiated melanocytes [42]. The latter are located in the lower hair follicle portion termed bulb, where they transfer pigment to the growing hair. When melanocyte stem cells are functionally impaired, they fail to generate melanocytes, which results in hair graying [43]. To further investigate the expression of Sox10 and Sox9 in the melanocytic lineage of the mouse skin, we made use of Dct::LacZ transgenic mouse line expressing LacZ driven by the dopachrome tautomerase (Dct) promoter that allows genetic tracking of melanocyte stem cells and their derivatives in the hair follicle (Fig. 2C; [44]). Sox10 expression was detected in X-Gal-positive melanocyte stem cells located in the bulge region (Fig. 2E, upper panels) as well as in differentiated melanocytes in the hair follicular bulb (Fig. 2E, lower panels). Similarly to the situation in human melanocytes, immunostaining with the specific anti-Sox9 antibody sc-20095 demonstrated absence of Sox9 expression in X-Gal-positive melanocyte stem cells and their progeny (Fig. 2D, upper and lower panels). Sox9 expression was restricted to cells of the epithelial lineage, namely the outer root sheath and the epithelial stem cell compartment in the bulge area (Fig. 2D; S4 Fig.), in agreement with previous reports [37,38,45].
To address the function of Sox10 and Sox9 in the melanocytic lineage in vivo, we conditionally ablated either Sox10 (Fig. 2H) or Sox9 (Fig. 2F) using Tyr-CreERT2 transgenic mice [46] carrying floxed alleles of Sox10 [47] and Sox9 [27], respectively. Tamoxifen-induced homozygous deletion of Sox10 in Sox10fl/fl Tyr-CreERT2 mice resulted in progressive hair graying, revealing an essential function of Sox10 for melanocyte stem cell homeostasis (Fig. 2I, [23]). In contrast, homozygous deletion of Sox9 in the melanocytic lineage did not cause hair graying even after a prolonged period after tamoxifen-induced gene deletion (Fig. 2G). Thus, these two closely related genes are not only differentially expressed but also play distinct roles in the biology of melanocytes in vivo.
Sox10 and Sox9 are differentially expressed in mouse giant congenital naevi and melanoma and exhibit functionally distinct roles in tumor initiation
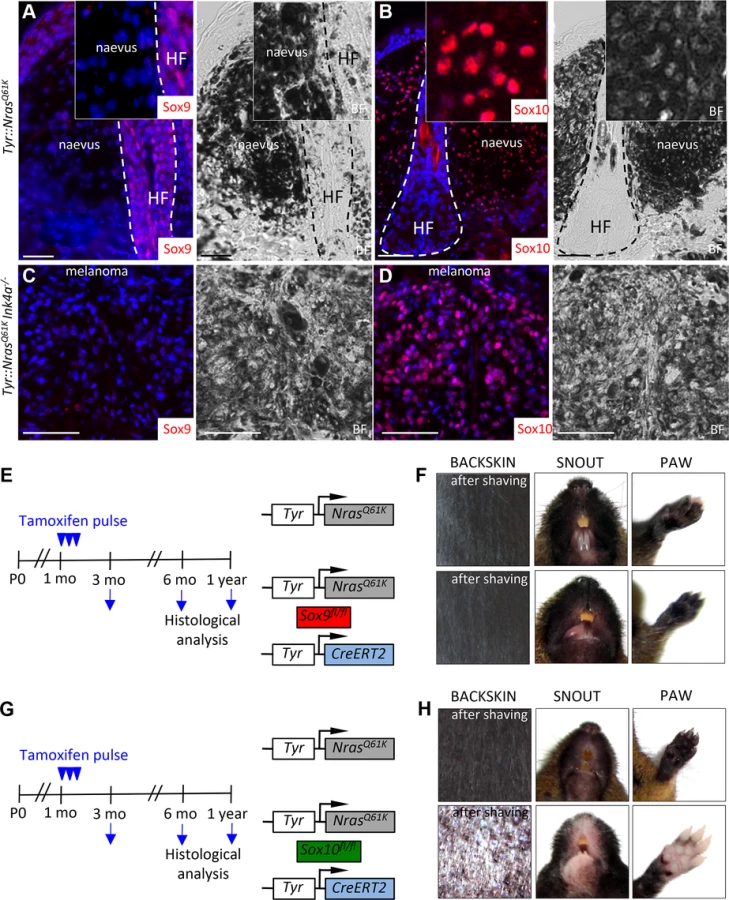
To functionally assess the role of SoxE factors in melanomagenesis, we first performed immunohistochemical staining for Sox9 and Sox10 of giant congenital naevi formed in Tyr::NrasQ61K mice and in melanoma derived from giant congenital naevi in Tyr::NrasQ61KInk4a−/− mice [9,48]. In contrast to the widespread expression of Sox10 displayed by mouse naevi and melanoma tissue (Fig. 3B,D, [9]), immunostaining of both naevi and primary melanoma sections did not reveal any detectable expression of Sox9 protein (Fig. 3A,C; S4B Fig.), consistent with the data that we have obtained for human giant congenital naevi and melanoma (Fig. 1H-I). Despite the lack of detectable Sox9 expression, low levels of Sox9 might be functionally implicated in the formation of melanocytic lesions arising in Tyr::NrasQ61K mice. To address this issue, we generated Tyr::NrasQ61K Sox9fl/fl Tyr-CreERT2 mice and conditionally deleted both Sox9 alleles by tamoxifen treatment of the mice (Fig. 3E). However, skin hyperpigmentation induced by oncogenic NRas was not affected in Tyr::NrasQ61K Sox9fl/fl Tyr-CreERT2 mice and was comparable to that presented by their control Tyr::NrasQ61K littermates (Fig. 3F). These data reveal that in contrast to Sox10 (Fig. 3G,H}, Sox9 is not required for the formation of melanocytic lesions.
Deletion of SOX10 in giant congenital naevus cells results in the induction of SOX9 expression
To gain insight into the possible interplay between SOX10 and SOX9 during tumor progression, we quantified the expression levels of SOX10 and SOX9 in normal human melanocytes, in cells from giant congenital naevi, and in a melanoma cell line (M010817; [49]) using quantitative RT-PCR analysis (Fig. 4A-C). Notably, NRASQ61K-associated tumor progression was associated with an increase in SOX10 expression (Fig. 4B). Cells from giant congenital naevi showed a 5-fold increase in SOX10 expression when compared to normal melanocytes, while M010817 melanoma cells displayed a 10-fold increase in SOX10 expression when compared to normal melanocytes (Fig. 4B). In striking contrast to the increase in SOX10 expression, SOX9 expression levels were low in human melanocytes and further decreased with melanoma progression (Fig. 4C).

To assess whether the disparate expression of SOX9 and SOX10 is a general feature of human melanoma samples, we analyzed the endogenous expression of these SOXE proteins in a large set of human melanoma cell lines previously categorized into cells with proliferative and invasive signatures, respectively [49]. Of note, all cell lines with a proliferative signature were characterized by high SOX10/low SOX9 mRNA expression (Fig. 4D). However, many cell lines with an invasive signature displayed the opposite expression pattern and showed low SOX10/high SOX9 mRNA expression. These date were confirmed on the protein level by Western blot analysis of several proliferative and invasive cell lines (Fig. 4E).
Interestingly, an inverse correlation between SOX10 and SOX9 expression has previously been observed in several systems, including cultured human melanocytes, where upon the induction of differentiation, SOX10 levels were reduced concomitantly with an increase in SOX9 levels [32]. Thus, high expression of a given SoxE transcription factor might be causative for reduced expression of the related SoxE factor. We therefore asked whether deregulation of SOX10 leads to changes in SOX9 expression and found a significant upregulation of SOX9 mRNA expression upon SOX10 knockdown in human giant congenital naevus cells (Fig. 4F-H). This is in analogy to the upregulation of SOX9 mRNA previously observed in melanoma cells upon SOX10 knockdown [9]. Moreover, SOX10 knockdown also resulted in upregulation of SOX9 protein levels in human melanoma cells (S5A Fig.). The combined data indicate that SOX10 normally suppresses SOX9 expression in cells from melanocytic lesions.
To address whether the findings in human cells in vitro also apply to the in vivo situation in mice, we isolated melanocytic progenitors using fluorescence-activated cell sorting (FACS) from the skin of Tyr::NrasQ61K and Tyr::NrasQ61K Sox10LacZ/+ mice that lack one allele of Sox10 (Fig. 4I) and subsequently measured Sox9 expression levels using quantitative RT-PCR (Fig. 4K, L). Elevated Sox10 levels mediated by oncogenic NRas in melanocytic cells from Tyr::NrasQ61K mice [9] were associated with decreased Sox9 mRNA expression as compared to normal melanoblasts wild-type for NRas. However, reduced Sox10 levels brought about by Sox10 heterozygosity resulted in upregulation of Sox9 expression, as revealed by comparing cells from Tyr::NrasQ61K with cells from Tyr::NrasQ61K Sox10LacZ/+ mice. Thus, in cells derived from both human and mouse melanocytic lesions, reduced Sox10 levels were accompanied by elevated Sox9 expression.
SOX10 and SOX9 display antagonistic functions in melanoma cells
Based on our findings it is conceivable that increased levels of SOX9 might mediate at least some of the anti-tumorigenic effects observed upon SOX10 loss-of-function in melanoma. To address this hypothesis, we overexpressed SOX9 in human M010817 melanoma cells in vitro and compared the gene expression profile of these cells with that of SOX10 knock-down melanoma cells, using the parental M010817 cell line as control [9]. Unsupervised hierarchical clustering revealed that overexpression of SOX9 led to a gene expression profile closely resembling the SOX10 knockdown-signature, which included in both conditions regulators of cell cycle progression, apoptosis, and melanocytic and mesectodermal differentiation (Fig. 5A). Among others, these data suggest that while SOX10 acts as an inhibitor of apoptosis in melanoma cells, SOX9 elicits a proapoptotic response. Similarly, SOX10 and SOX9 appear to play antagonistic functions in the regulation of the cell cycle, melanocytic differentiation, and mesectodermal differentiation (Fig. 5A).

Notably, SOX9 overexpression resulted in decreased expression of a number of genes associated with melanocytic differentiation, such as MLANA, MITF, DCT, TYR, and importantly SOX10. To confirm the downregulation of SOX10 upon SOX9 overexpression observed in microarray analysis (Fig. 5A) also on the protein level, we performed Western blot analysis and observed a pronounced downregulation of SOX10 protein upon SOX9 overexpression in human melanoma cell lines (Fig. 5B). Moreover, chromatin immunoprecipitation assays in human melanoma M010817 cells indicated that SOX9 binds to the promoter of SOX10, suggesting a direct regulation of the SOX10 gene by SOX9 (Fig. 5C). Thus, whereas SOX10 loss-of-function leads to increased SOX9 expression (Fig. 4), high levels of SOX9 suppress SOX10 expression, revealing cross-regulatory interactions between these two transcription factors.
Next, we addressed whether the cross-regulation between SOX10 and SOX9 is functionally relevant in human melanoma cells. To this end, we performed RNAi experiments to test whether interfering with SOX9 overexpression upon SOX10 knockdown could rescue M010817 melanoma cells (Fig. 5D-F). Using two independent sets of siRNAs, the elevated SOX9 levels observed in SOX10 knockdown cells were reverted below control levels by means of concomitant SOX9 knockdown (Fig. 5E). Importantly, both SOX9 gain-of-function and SOX10 knockdown promoted apoptosis (Fig. 5F; Fig. S5B). However, SOX10 knockdown cells were rescued when SOX9 expression was simultaneously downregulated, resulting in numbers of apoptotic cells comparable to those found in control cells (number of Annexin V-positive apoptotic M010817 cells: control, 9.95±0.9%; SOX10 siRNA #1, 19.96±0.13%; SOX10 siRNA #2 18.8±0.49%; combination of SOX10 siRNA #1/SOX9 siRNA, 9.45±0.79%; combination of SOX10 siRNA #2/SOX9 siRNA. 10.75±0.4%) (Fig. 5F; S5B Fig.). These data indicate that at least in vitro, SOX9 plays a key role in mediating the cellular phenotype obtained in human melanoma cells upon suppression of SOX10.
Deletion of Sox9 rescues the phenotype of Tyr::NrasQ61KSox10fl/+Tyr-CreERT2 mice, restoring NrasQ61K-mediated naevus formation
As in human melanoma cells in vitro, reducing Sox10 levels in vivo elicits an anti-tumorigenic effect, preventing melanocytic hyperplasia in Tyr::NrasQ61KSox10fl/+Tyr-CreERT2 mice [9]. To functionally test whether upregulation of Sox9 is the key factor accountable for the lack of hyperplasia in these mice in vivo, we performed simultaneous conditional ablation of both Sox10 and Sox9 genes in the Tyr::NrasQ61K mice (Fig. 6A, B). In agreement with our previous observations [9], the skin of the snout, paws and the back was noticeably lighter in color in Tyr::NrasQ61KSox10fl/+Tyr-CreERT2 mice as compared to their control Tyr::NrasQ61K littermates (Fig. 6A, left and middle panels). In contrast, macroscopic examination of the skin of Tyr::NrasQ61KSox10fl/+Sox9fl/flTyr-CreERT2 animals showed pronounced hyperpigmentation, a hallmark of the skin phenotype found in Tyr::NrasQ61K mice (Fig. 6A, right panels). Thus, conditional deletion of Sox9 rescued the effect of Sox10 haploinsufficiency in Tyr::NrasQ61K mice.

As shown in Fig. 2, conditional ablation of both alleles of Sox10 in the normal melanocytic lineage using Tyr-CreERT2 resulted in loss of functional melanocytes and hair graying (Fig. 2I). Likewise, homozygous deletion of Sox10 in Tyr::NrasQ61K mice not only prevented NrasQ61K-stimulated skin hyperpigmentation, but also led to hair greying in these mice (Fig. 6B, left panels). To determine whether these phenotypes involve Sox9 activity and whether, therefore, loss of Sox9 could rescue the effects of homozygous Sox10 deletion, we generated Tyr::NrasQ61KSox10fl/flSox9fl/flTyr-CreERT2 animals. Surprisingly, loss of Sox9 in Sox10 homozygous conditional knock-out animals efficiently restored skin hyperpigmentation as seen on the snout and paws of Tyr::NrasQ61KSox10fl/flSox9fl/flTyr-CreERT2 animals when compared to mice lacking Sox10 but not Sox9 in the melanocytic lineage (Fig. 6B, right panels).
We have previously demonstrated that skin hyperpigmentation in Tyr::NrasQ61K mice is due to hyperplasia of ectopically located pigment cells emerging in the dermis, around the upper part of the hair follicle [9]. To measure the degree of hyperpigmentation in Tyr::NrasQ61K mice with conditional Sox10 deletion (heterozygous and homozygous) versus mice simultaneously lacking both Sox9 alleles and one or both Sox10 alleles in the melanocytic lineage, we quantified the percentage of hair follicles associated with ectopically located melanocytic cells in the back skin of these mouse lines. In Tyr::NrasQ61K mice, more than 90% of all hair follicles displayed ectopic pigment cells (Fig. 6D). In contrast, in the absence of one or both alleles of Sox10, there were almost no hair follicles with ectopic pigment cells, despite NrasQ61K expression (Fig. 6C, D). Strikingly, however, the percentage of hair follicles associated with ectopic melanoblasts in Tyr::NrasQ61KSox10fl/+Sox9fl/flTyr-CreERT2 animals was reverted to numbers similar to those found in Tyr::NrasQ61K mice (93±1.8% and 96±1%, respectively) (Fig. 6C, D). Moreover, even in the absence of both Sox10 alleles, loss of Sox9 rescued the NrasQ61K–dependent appearance of melanocytic cells found outside hair follicles (93.5±3%) (Fig. 6D). These data reveal a key role of Sox9 in preventing melanoma initiation and provide novel insights into the functional interplay between Sox10 and Sox9 during melanoma formation.
Discussion
Our study identifies the structurally related transcription factors SOX10 and SOX9 as functionally antagonistic regulators of postnatal melanocyte and melanoma development. Although we did not find SOX9 to be expressed in the melanocytic lineage when SOX10 is present, SOX9 expression becomes evident upon SOX10 inactivation in naevus and melanoma cells. In this context, SOX9 appears to promote the major cellular processes induced by SOX10 loss-of-function, namely stop of proliferation and apoptosis. Intriguingly, SOX9 and SOX10 are engaged in a cross-regulatory feedback loop whereby SOX9, which is induced upon SOX10 inactivation, itself suppresses SOX10, thus strengthening an anti-tumorigenic program.
In many cell lineages and tissues, SOX10 and SOX9 are co-expressed and functionally redundant [50]. We propose that this is not the case in melanocytic cells and that SOX9, unlike SOX10, is neither required for normal melanocyte stem cell homeostasis nor for formation of congenital nevi and primary melanoma. Our findings disagree with some previously published studies reporting SOX9 expression in the normal and tumor-associated melanocytic lineage [32–34,36,51]. However, as we demonstrate here, most previously used anti-SOX9 antibodies display cross-reactivity with SOX10, owing to the close relationship between these two SoxE factors. Having identified anti-SOX10 and anti-SOX9 specific antibodies, we reveal virtually exclusive expression patterns of these transcription factors in the normal human skin and in a large set of melanoma biopsies and cell lines. While SOX10 expression is restricted to neural crest derivatives, including melanoblasts, differentiated melanocytes, and virtually all human naevus and melanoma biopsies tested (Fig. 1; S3 Fig.; [9], SOX9 expression in melanocytic cells was restricted to few scattered cells in a subset of melanoma biopsies. In contrast, SOX9 was strongly expressed in epithelial cells of the hair follicle, which are devoid of SOX10 expression.
In support of these data, Sox10 protein expression in the mouse skin is detected in vivo throughout all stages of melanocyte development from stem cells to differentiated melanocytes in the hair follicular bulb (Fig. 2). Mice lacking Sox10 in the melanocyte lineage display hair graying, indicating that Sox10 is necessary for maintenance of melanocyte stem cells and committed melanoblasts [23] (Fig. 2). Likewise, Sox10 is required for the establishment of giant congenital naevi as well as melanoma [9]. In contrast, murine Sox9 appeared not to be expressed in melanocytic cells of the normal skin, nevi, and primary melanoma, while it was readily detectable in epithelial cells in accordance with previous reports [33,37,38]. Importantly, loss of function analyses failed to reveal a crucial role of Sox9 in normal melanocytes, as conditional deletion of Sox9 did not affect generation and long-term maintenance of melanocytes in vivo and did not result in hair graying, a phenotype characteristic for the loss of Sox10. Likewise, lack of Sox9 did not prevent emergence of melanocytic lesions induced by oncogenic NRasQ61K (Fig. 3). These data demonstrate that Sox10 and Sox9 are not only expressed in different cellular compartments in the skin, but also play distinct roles in normal and transformed melanocytes.
In cell types other than melanocytes Sox9 and Sox10 can act redundantly. For instance, in oligodendroglial progenitors, concomitant expression of Sox9 can compensate for the loss of Sox10 [52,53]. Similarly, in avian and Xenopus embryos, Sox9 and Sox10 are co-expressed in premigratory neural crest cells and are both able to induce ectopic neural crest cell formation upon forced expression in chicken neural tube [11,24,54]. In addition, the two factors are able to cross-regulate each other at this early stage of neural crest formation. Interfering with Sox10 function leads to inhibition of Sox9 expression [55], suggesting that Sox10 is required for the expression of Sox9 in pre-migratory neural crest. On the other hand, Sox9 overexpression in Xenopus embryos leads to upregulation of Sox10 expression [24], suggesting that Sox9 can also act upstream of Sox10. As development proceeds, however, Sox10 expression persists in the trunk neural crest and is downregulated in cranial neural crest cells giving rise to mesectodermal structures, while Sox9 expression is absent in trunk neural crest cells but present in the cranial neural crest [24,26]. These divergent expression patterns are established by signaling pathways differentially regulating transcription of Sox9 and Sox10, respectively. In particular, TGFβ (transforming growth factor β) simultaneously triggers induction of Sox9 and reduction of Sox10 expression [56]. Accordingly, mice lacking Sox10 display phenotypes that are distinct from those obtained upon loss of Sox9 [13,54,56–58]. In particular, Sox10 but not Sox9 is expressed in and required for the generation of melanoblasts during mouse embryogenesis [13, 21]. Finally, in agreement with our expression studies on human skin, humans carrying mutations in SOX9 display campomelic dysplasia affecting the skeleton and reproductive system but not melanocytes, whereas patients with mutations in SOX10 often exhibit pigmentary anomalies [20,50,59].
However, the divergent functions of Sox10 and Sox9 in the skin appear not to be simply due to their differential expression patterns. Depending on the cellular context, these two transcription factors can also elicit different responses in one and the same cell lineage rather than playing redundant roles. Studies in Xenopus embryos demonstrated that while expression of Sox10 at the two-cell stage was sufficient to activate the expression of Trp-2 (Dct) and the induction of melanocytic precursors, the expression of Sox9 failed to do so [24]. In mice, loss of Sox9 promotes apoptosis and other phenotypes in neural crest cells, but Sox10 is maintained in these cells and cannot rescue the Sox9-mutant phenotype [54]. Moreover, while Sox9 activates the expression of genes involved in the induction of osteochondrogenesis in neural crest cells in pharyngeal arches [26,28,60], Sox10 is involved in the specification of a glial and melanocytic gene expression program [13]. In this context, Sox10 and Sox9 play antagonistic roles, in that Sox9 promotes cells cycle exit and mesenchymal fates, while Sox10 activates proliferation and suppresses mesenchymal fate acquisition [56]. Accordingly, Sox10 inactivation results in induction of Sox9-dependent fates in postmigratory neural crest cells. This interplay between Sox10 and Sox9 functions during normal neural crest development is highly reminiscent of our findings in melanocytic lesions, where Sox10 also promotes proliferation and survival, while Sox9 counteracts these cellular processes. Indeed, in human melanoma cells, loss of SOX10 not only resulted in upregulation of SOX9 expression, but also in global transcriptional changes highly similar to the changes observed upon SOX9 overexpression (Fig. 5), indicating that these factors appear to play opposing functions in melanoma. Interestingly, a study by Passeron et al. revealed that overexpression of SOX9 prevents melanoma formation [35] by increasing the expression of the CDK inhibitor p21 and subsequent cell cycle arrest. Likewise, a recent report by Pavan and colleagues established that the expression of p21 and p27 were increased upon SOX10 knockdown [31]. Thus, our data might provide one explanation for the anti-tumorigenic effect of SOX9, namely by downregulation of SOX10. This is in accordance with our previously published results on the essential role of SOX10 for melanoma initiation and progression [9]. Of note, the anti-tumorigenic effect elicited by suppressing SOX10 was abolished by concomitant SOX9 inactivation both in human melanoma cells as well as in mice. Thus, antagonistic SOX10/SOX9 constitutes a key node in the genetic network underlying melanomagenesis. Nonetheless, it is conceivable that further cues mediate SOX10-pro - and SOX9-anti-tumorigenic effects, respectively. Moreover, our data do not exclude a role of SOX9 at later stages of melanoma disease progression, in particular during metastasis formation by invasive cells. Indeed, while we could attribute a SOX10 high/SOX9 low signature to proliferative human melanoma cell lines and to all human and murine melanoma tissues analyzed, several human melanoma cell lines reported to display invasive features [49] exhibited SOX10 low/SOX9 high expression. Although this remains to be shown, these invasive cell lines with SOX10 low/SOX9 high expression might have been established by capturing or inducing invasive tumor cells that appear to be rather rare in biopsies of bulk tumor tissue. Likewise, apart from experimentally reducing SOX10 levels, other stimuli such as UV exposure might also lead to upregulation of SOX9 [51]. In any case, our discovery of the antagonistic interaction between SOX10 and SOX9, together with the further characterization of their mode of action in melanoma cells, might not only provide new mechanistic insights into how SoxE group proteins are regulated and act in the context of melanoma initiation and maintenance, but might also point to novel strategies for melanoma therapies.
Materials and Methods
Human specimens
All analyses involving human skin, giant congenital naevi and melanoma tissue were performed in accordance with the ethical committee in canton Zurich, Switzerland. TMA containing melanoma tissue was constructed as previously described [61].
Mice
Tyr::NrasQ61K [48] were provided by F. Beermann (EPFL Lausanne, Switzerland). Dct-LacZ mice were described previously [44]. Sox10fl/fl mice were described previously [47]. Sox9fl/fl mice [27] were a kind gift from G. Scherer (Institute of Human Genetics, Freiburg, Germany). Tyr-CreERT2 line [46] was provided by L. Chin (The University of Texas MD Anderson Cancer Center, Houston, Texas, USA). Rosa26-lacZ mice were obtained from Jackson laboratory. All animal experiments were performed in accordance with Swiss law and have been approved by the veterinary authorities of Zurich.
Tamoxifen injections
Mice were subjected to intraperitoneal injections of tamoxifen (T5648, Sigma), diluted with the mixture of ethanol and sunflower oil (1 : 9 ratio). Tamoxifen was injected for 5 consecutive days.
Histological analysis and immunohistochemistry
Immunohistochemistry on paraffin sections was performed as previously described [9]. Briefly, skin samples were fixed in 4% buffered paraformaldehyde and embedded in paraffin. For immunohistochemistry, antigen retrieval was performed in citrate buffer (pH 6.0) for 10 minutes at 110°C in HistoPro (Rapid Microwave Histoprocessor, Milestone, USA). The following primary antibodies were used: anti-Sox10 (goat, 1 : 200, Santa Cruz Biotechnology, Santa Cruz, CA), anti-Sox10 (mouse, 1 : 200, R&D), anti-Sox9 (rabbit, 1 : 100, sc-20095, Santa Cruz Biotechnology, Santa Cruz, CA), anti-Sox9 (rabbit, 1 : 100, ab36748, Abcam), anti-Sox9 (M00006662, Abnova), anti-Sox9 (AB5535, Millipore), anti-Sox9 (GTX 109661, GenTex), anti-MITF (mouse, clone 6D3, 1 : 500) was a kind gift from Heinz Arnheiter (NIH, USA). Images were captured with a Leica DMI 6000B Microscope and using LAS AF (Leica Application Suite Advanced Fluorescence) software. For whole mount X-Gal staining, skin samples were fixed with 4% buffered paraformaldehyde, washed with PBS and subjected to X-Gal staining solution overnight at 37°C. After several washing steps, tissue was paraffin embedded and sectioned. 5 μm thick sections were further counterstained with eosin solution and mounted.
RNA isolation, reverse transcription and quantitative PCR
Total RNA was isolated using Trizol according to manufacturer’s instructions (Invitrogen). 1 μg aliquots of RNA were reverse transcribed with Reverse Transcription System (Promega) according to the manufacturer’s instructions. Data collection and analysis were performed by ABI Viia7 Fast Real-Time PCR Systems (Applied Biosystems). Gene expression values of averaged triplicate reactions were normalized to RPL28 expression levels. RPL28 primers are as follows: 5’-GCAATTGGTTCCGCTACAAC-3’ and 5’-TGTTCTTGCGGATCATGTGT-3’. The expression of SOX10 and SOX9 was measured using primers purchased from QIAGEN: SOX10 (Hs_SOX10_1_SG); SOX9 (Hs_SOX9_1_SG).
Sequencing
Cells derived from patients with giant congenital naevi were sequenced for NRAS. Primers for sequencing for Exon 1 (mutation G12) and Exon 2 (mutation Q61K) of NRAS gene were as follows: NRAS_1F 5’-ATAGAAAGCTTTAAAGTACTG-3’ and NRAS_1R 5’-TTCCTTTAATACAGAATATGG-3’, NRAS_2F 5’-CCCCTTACCCTCCACAC-3’ and NRAS_2R 5’-AACCTAAAACCAACTCTTCCCA-3’.
Cell culture and transfection assays
Silencing RNA (siRNA) transfection was carried out using INTERFERin transfection solution according to the manufacturer’s protocol (Polyplus-transfection, Illkirch, France). Cells were transfected with 10 nM of siRNA (Qiagen) for 96 hours before RNA was extracted or used for FACS analysis. As control siRNA, the All-Star negative siRNA sequence (Qiagen) was used, and gene-specific siRNAs targeting siSOX10 (SI00729414, SI00729421) and siSOX9 (SI00007595, SI00007609) were obtained from Qiagen. Transfection of DNA was carried using JetPEI transfection solution according to the manufacturer’s protocol (Polyplus-transfection, Illkirch, France). Cells were transfected with 1 ug of pCMV6-SOX9 (Origene SC321884) or empty vector for 96 hours before RNA was extracted or used for FACS analysis.
Melanocyte FACS and RNA seq
Melanocytes were purified by FACS from doxycycline-treated iDct-GFP mice as previously described [41]. Total RNA was prepared from FACS-sorted cell fractions containing GFP-positive melanoblasts/melanocytes according to standard Illumina RNA-Seq paired-end protocol and sequenced on the Illumina GAIIx to 80 bp per read.
Microarray analysis
Total RNA was isolated from melanoma cell cultures using TRIzol according to the manufacturer’s instructions (Invitrogen). Total RNA was amplified and biotin-labelled using the Message Amp II-Biotin Enhanced aRNA Amplification Kit (Ambion, Austin, TX, USA). Biotin-labelled RNA was hybridized to Affymetrix HG-U133 plus 2.0 oligonucleotide microarrays following the manufacturer’s protocol (Affymetrix, Santa Clara, CA, USA). After hybridization, microarrays were washed and stained using a GeneChip Fluidics Station 450 (Affymetrix) and then scanned using a GeneChip Scanner 7G (Affymetrix). Raw data was processed by R using the affycoretools package (RMA). Gene expression datasets for SOX10 knockdown were obtained from NCBI GEO GSE37059. Gene expression analysis was performed by R using the limma package. P-values were adjusted by FDR p-value adjustment. For melanoma cell lines analysis (proliferative vs invasive): Normalized expression values were downloaded from GSE4840 containing microarray data for twenty three melanoma cell cultures. Pearson’s product moment correlation (r) was calculated for the SOX10 and SOX9 expression values across all twenty three samples. P-value was determined from the t statistic calculated from r.
Flow cytometry and cell sorting
Skin tissue (from back skin) was digested with a mixture of Dispase (Roche) and Collagenase I (Worthington) for 1 hour at 37°C and enzymatic reaction was stopped by addition of DMEM media supplemented with 10%FCS as previously described [9]. Subsequently, single cell suspension was filtered through 40 μm strainers (BD). For cell cycle analysis, Click-iT EdU Alexa Fluor 647 Flow Cytometry Assay Kit (Invitrogen) was used. Cells were labeled with PI according to manufacturer’s protocol and DNA content was measured using a BD FACSCanto II flow cytometer (BD Biosciences) and a BD FACSDiva software (BD Biosciences). For measurement of apoptosis, Annexin V-PE Apoptosis Detection Kit I (BD Pharmingen, 559763) was used. FACSAria sorter and FACS DiVa software (BD Biosciences) were used for cell sorting.
Chromatin immunoprecipitation
ChIP analysis was performed as previously described [62]. Sox9 antibody was from Santa Cruz Biotechnology (sc-20095, Santa Cruz Biotechnology). SOX10 promoter sequences were amplified with forward primer (5’-CCTCTGCCTCGTGTGACTAC-3’) and reverse primer (5’-TCCTGTCTGGAGTGGGCTG-3’).
Supporting Information
Zdroje
1. Wilson M., and Koopman P. (2002) Matching SOX: partner proteins and co-factors of the SOX family of transcriptional regulators. Curr Opin Genet Dev 12 : 441–6. 12100890
2. Wegner M. (1999) From head to toes: the multiple facets of Sox proteins. Nucleic Acids Res 27 : 1409–20. 10037800
3. Sarkar A., and Hochedlinger K. (2013) The sox family of transcription factors: versatile regulators of stem and progenitor cell fate. Cell Stem Cell 12 : 15–30. 23290134
4. Girard M., and Goossens M. (2006) Sumoylation of the SOX10 transcription factor regulates its transcriptional activity. FEBS Lett 580 : 1635–41. 16494873
5. Uchikawa M., Kamachi Y., and Kondoh H. (1999) Two distinct subgroups of Group B Sox genes for transcriptional activators and repressors: their expression during embryonic organogenesis of the chicken. Mech Dev 84 : 103–20. 10473124
6. Lee P.C., Taylor-Jaffe K.M., Nordin K.M., Prasad M.S., Lander R.M., et al. (2012) SUMOylated SoxE factors recruit Grg4 and function as transcriptional repressors in the neural crest. J Cell Biol 198 : 799–813. doi: 10.1083/jcb.201204161 22927467
7. Rudin C.M., Durinck S., Stawiski E.W., Poirier J.T., Modrusan Z., et al. (2012) Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer. Nat Genet 44 : 1111–6. doi: 10.1038/ng.2405 22941189
8. Castillo S.D., Matheu A., Mariani N., Carretero J., Lopez-Rios F., et al. (2012) Novel transcriptional targets of the SRY-HMG box transcription factor SOX4 link its expression to the development of small cell lung cancer. Cancer Res 72 : 176–86. doi: 10.1158/0008-5472.CAN-11-3506 22084397
9. Shakhova O., Zingg D., Schaefer S.M., Hari L., Civenni G., et al. (2012) Sox10 promotes the formation and maintenance of giant congenital naevi and melanoma. Nat Cell Biol 14 : 882–90. doi: 10.1038/ncb2535 22772081
10. Vervoort S.J., van Boxtel R., and Coffer P.J. (2013) The role of SRY-related HMG box transcription factor 4 (SOX4) in tumorigenesis and metastasis: friend or foe? Oncogene 32 : 3397–409.
11. Cheung M., and Briscoe J. (2003) Neural crest development is regulated by the transcription factor Sox9. Development 130 : 5681–93. 14522876
12. O. Shakhova, and L. Sommer (2010) Neural crest-derived stem cells. Stem Book.
13. Britsch S., Goerich D.E., Riethmacher D., Peirano R.I., Rossner M., et al. (2001) The transcription factor Sox10 is a key regulator of peripheral glial development. Genes Dev 15 : 66–78. 11156606
14. Paratore C., Eichenberger C., Suter U., and Sommer L. (2002) Sox10 haploinsufficiency affects maintenance of progenitor cells in a mouse model of Hirschsprung disease. Hum Mol Genet 11 : 3075–85. 12417529
15. Kim J., Lo L., Dormand E., and Anderson D.J. (2003) SOX10 maintains multipotency and inhibits neuronal differentiation of neural crest stem cells. Neuron 38 : 17–31. 12691661
16. Mollaaghababa R., and Pavan W.J. (2003) The importance of having your SOX on: role of SOX10 in the development of neural crest-derived melanocytes and glia. Oncogene 22 : 3024–34. 12789277
17. Bondurand N., Dastot-Le Moal F., Stanchina L., Collot N., Baral V., et al. (2007) Deletions at the SOX10 gene locus cause Waardenburg syndrome types 2 and 4. Am J Hum Genet 81 : 1169–85.
18. Kuhlbrodt K., Schmidt C., Sock E., Pingault V., Bondurand N., et al. (1998) Functional analysis of Sox10 mutations found in human Waardenburg-Hirschsprung patients. J Biol Chem 273 : 23033–8. 9722528
19. Herbarth B., Pingault V., Bondurand N., Kuhlbrodt K., Hermans-Borgmeyer I., et al. (1998) Mutation of the Sry-related Sox10 gene in Dominant megacolon, a mouse model for human Hirschsprung disease. Proc Natl Acad Sci U S A 95 : 5161–5. 9560246
20. Pingault V., Bondurand N., Kuhlbrodt K., Goerich D.E., Prehu M.O., et al. (1998) SOX10 mutations in patients with Waardenburg-Hirschsprung disease. Nat Genet 18 : 171–3. 9462749
21. Stolt C.C., Lommes P., Hillgartner S., and Wegner M. (2008) The transcription factor Sox5 modulates Sox10 function during melanocyte development. Nucleic Acids Res 36 : 5427–40. doi: 10.1093/nar/gkn527 18703590
22. Potterf S.B., Mollaaghababa R., Hou L., Southard-Smith E.M., Hornyak T.J., et al. (2001) Analysis of SOX10 function in neural crest-derived melanocyte development: SOX10-dependent transcriptional control of dopachrome tautomerase. Dev Biol 237 : 245–57. 11543611
23. Harris M.L., Buac K., Shakhova O., Hakami R.M., Wegner M., et al. (2013) A Dual Role for SOX10 in the Maintenance of the Postnatal Melanocyte Lineage and the Differentiation of Melanocyte Stem Cell Progenitors. PLoS Genet 9:e1003644. doi: 10.1371/journal.pgen.1003644 23935512
24. Aoki Y., Saint-Germain N., Gyda M., Magner-Fink E., Lee Y.H., et al. (2003) Sox10 regulates the development of neural crest-derived melanocytes in Xenopus. Dev Biol 259 : 19–33. 12812785
25. Nonaka D., Chiriboga L., and Rubin B.P. (2008) Sox10: a pan-schwannian and melanocytic marker. Am J Surg Pathol 32 : 1291–8. doi: 10.1097/PAS.0b013e3181658c14 18636017
26. Spokony R.F., Aoki Y., Saint-Germain N., Magner-Fink E., and Saint-Jeannet J.P. (2002) The transcription factor Sox9 is required for cranial neural crest development in Xenopus. Development 129 : 421–32. 11807034
27. Kist R., Schrewe H., Balling R., and Scherer G. (2002) Conditional inactivation of Sox9: a mouse model for campomelic dysplasia. Genesis 32 : 121–3. 11857796
28. Bell D.M., Leung K.K., Wheatley S.C., Ng L.J., Zhou S., et al. (1997) SOX9 directly regulates the type-II collagen gene. Nat Genet 16 : 174–8. 9171829
29. Kent J., Wheatley S.C., Andrews J.E., Sinclair A.H., and Koopman P. (1996) A male-specific role for SOX9 in vertebrate sex determination. Development 122 : 2813–22. 8787755
30. Morais da Silva S., Hacker A., Harley V., Goodfellow P., Swain A., et al. (1996) Sox9 expression during gonadal development implies a conserved role for the gene in testis differentiation in mammals and birds. Nat Genet 14 : 62–8. 8782821
31. Cronin J.C., Watkins-Chow D.E., Incao A.A., Hasskamp J.H., Schonewolf N., et al. (2013) SOX10 ablation arrests the cell cycle, induces senescence and suppresses melanomagenesis. Cancer Res 73 : 5709–18. doi: 10.1158/0008-5472.CAN-12-4620 23913827
32. Cook A.L., Smith A.G., Smit D.J., Leonard J.H., and Sturm R.A. (2005) Co-expression of SOX9 and SOX10 during melanocytic differentiation in vitro. Exp Cell Res 308 : 222–35. 15896776
33. Krahl D., and Sellheyer K. (2010) Sox9, more than a marker of the outer root sheath: spatiotemporal expression pattern during human cutaneous embryogenesis. J Cutan Pathol 37 : 350–6. doi: 10.1111/j.1600-0560.2009.01369.x 19614725
34. Flammiger A., Besch R., Cook A.L., Maier T., Sturm R.A., et al. (2009) SOX9 and SOX10 but not BRN2 are required for nestin expression in human melanoma cells. J Invest Dermatol 129 : 945–53. 18923447
35. Passeron T., Valencia J.C., Namiki T., Vieira W.D., Passeron H., et al. (2009) Upregulation of SOX9 inhibits the growth of human and mouse melanomas and restores their sensitivity to retinoic acid. J Clin Invest 119 : 954–63. doi: 10.1172/JCI34015 19273910
36. Bakos R.M., Maier T., Besch R., Mestel D.S., Ruzicka T., et al. (2009) Nestin and SOX9 and SOX10 transcription factors are coexpressed in melanoma. Exp Dermatol 19:e89–94. doi: 10.1111/j.1600-0625.2009.00991.x 19845757
37. Nowak J.A., Polak L., Pasolli H.A., and Fuchs E. (2008) Hair follicle stem cells are specified and function in early skin morphogenesis. Cell Stem Cell 3 : 33–43. doi: 10.1016/j.stem.2008.05.009 18593557
38. Vidal V.P., Chaboissier M.C., Lutzkendorf S., Cotsarelis G., Mill P., et al. (2005) Sox9 is essential for outer root sheath differentiation and the formation of the hair stem cell compartment. Curr Biol 15 : 1340–51. 16085486
39. Goding C.R. (2000) Mitf from neural crest to melanoma: signal transduction and transcription in the melanocyte lineage. Genes Dev 14 : 1712–28. 10898786
40. Vidal V.P., Ortonne N., and Schedl A. (2008) SOX9 expression is a general marker of basal cell carcinoma and adnexal-related neoplasms. J Cutan Pathol 35 : 373–9. doi: 10.1111/j.1600-0560.2007.00815.x 18333897
41. Zaidi M.R., Davis S., Noonan F.P., Graff-Cherry C., Hawley T.S., et al. (2011) Interferon-gamma links ultraviolet radiation to melanomagenesis in mice. Nature 469 : 548–53. doi: 10.1038/nature09666 21248750
42. Nishimura E.K., Jordan S.A., Oshima H., Yoshida H., Osawa M., et al. (2002) Dominant role of the niche in melanocyte stem-cell fate determination. Nature 416 : 854–60. 11976685
43. Nishimura E.K., Granter S.R., and Fisher D.E. (2005) Mechanisms of hair graying: incomplete melanocyte stem cell maintenance in the niche. Science 307 : 720–4. 15618488
44. Mackenzie M.A., Jordan S.A., Budd P.S., and Jackson I.J. (1997) Activation of the receptor tyrosine kinase Kit is required for the proliferation of melanoblasts in the mouse embryo. Dev Biol 192 : 99–107. 9405100
45. Fantauzzo K.A., Kurban M., Levy B., and Christiano A.M. (2012) Trps1 and its target gene Sox9 regulate epithelial proliferation in the developing hair follicle and are associated with hypertrichosis. PLoS Genet 8: e1003002. doi: 10.1371/journal.pgen.1003002 23133399
46. Bosenberg M., Muthusamy V., Curley D.P., Wang Z., Hobbs C., et al. (2006) Characterization of melanocyte-specific inducible Cre recombinase transgenic mice. Genesis 44 : 262–7. 16676322
47. Finzsch M., Schreiner S., Kichko T., Reeh P., Tamm E.R., et al. (2010) Sox10 is required for Schwann cell identity and progression beyond the immature Schwann cell stage. J Cell Biol 189 : 701–12. doi: 10.1083/jcb.200912142 20457761
48. Ackermann J., Frutschi M., Kaloulis K., McKee T., Trumpp A., et al. (2005) Metastasizing melanoma formation caused by expression of activated N-RasQ61K on an INK4a-deficient background. Cancer Res 65 : 4005–11. 15899789
49. Hoek K.S., Eichhoff O.M., Schlegel N.C., Dobbeling U., Kobert N., et al. (2008) In vivo switching of human melanoma cells between proliferative and invasive states. Cancer Res 68 : 650–6. doi: 10.1158/0008-5472.CAN-07-2491 18245463
50. Stolt C.C., and Wegner M. (2010) SoxE function in vertebrate nervous system development. Int J Biochem Cell Biol 42 : 437–40. 19647093
51. Passeron T., Valencia J.C., Bertolotto C., Hoashi T., Le Pape E., et al. (2007) SOX9 is a key player in ultraviolet B-induced melanocyte differentiation and pigmentation. Proc Natl Acad Sci U S A 104 : 13984–9. 17702866
52. Stolt C.C., Schmitt S., Lommes P., Sock E., and Wegner M. (2005) Impact of transcription factor Sox8 on oligodendrocyte specification in the mouse embryonic spinal cord. Dev Biol 281 : 309–17. 15893981
53. Finzsch M., Stolt C.C., Lommes P., and Wegner M. (2008) Sox9 and Sox10 influence survival and migration of oligodendrocyte precursors in the spinal cord by regulating PDGF receptor alpha expression. Development 135 : 637–46. doi: 10.1242/dev.010454 18184726
54. Cheung M., Chaboissier M.C., Mynett A., Hirst E., Schedl A., et al. (2005) The transcriptional control of trunk neural crest induction, survival, and delamination. Dev Cell 8 : 179–92. 15691760
55. Honore S.M., Aybar M.J., and Mayor R. (2003) Sox10 is required for the early development of the prospective neural crest in Xenopus embryos. Dev Biol 260 : 79–96.
56. John N., Cinelli P., Wegner M., and Sommer L. (2011) Transforming growth factor beta-mediated Sox10 suppression controls mesenchymal progenitor generation in neural crest stem cells. Stem Cells 29 : 689–99. doi: 10.1002/stem.607 21308864
57. Paratore C., Goerich D.E., Suter U., Wegner M., and Sommer L. (2001) Survival and glial fate acquisition of neural crest cells are regulated by an interplay between the transcription factor Sox10 and extrinsic combinatorial signaling. Development 128 : 3949–61. 11641219
58. Reiprich S., Stolt C.C., Schreiner S., Parlato R., and Wegner M. (2008) SoxE proteins are differentially required in mouse adrenal gland development. Mol Biol Cell 19 : 1575–86. doi: 10.1091/mbc.E07-08-0782 18272785
59. Mansour S., Hall C.M., Pembrey M.E., and Young I.D. (1995) A clinical and genetic study of campomelic dysplasia. J Med Genet 32 : 415–20. 7666392
60. Lefebvre V., Dumitriu B., Penzo-Mendez A., Han Y., and Pallavi B. (2007) Control of cell fate and differentiation by Sry-related high-mobility-group box (Sox) transcription factors. Int J Biochem Cell Biol 39 : 2195–214. 17625949
61. Mihic-Probst D., Kuster A., Kilgus S., Bode-Lesniewska B., Ingold-Heppner B., et al. (2007) Consistent expression of the stem cell renewal factor BMI-1 in primary and metastatic melanoma. Int J Cancer 121 : 1764–70. 17597110
62. Santoro R. (2014) Analysis of chromatin composition of repetitive sequences: the ChIP-Chop assay. Methods Mol Biol 1094 : 319–28. doi: 10.1007/978-1-62703-706-8_25 24162999
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2015 Číslo 1
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- The Global Regulatory Architecture of Transcription during the Cell Cycle
- A Truncated NLR Protein, TIR-NBS2, Is Required for Activated Defense Responses in the Mutant
- Proteasomes, Sir2, and Hxk2 Form an Interconnected Aging Network That Impinges on the AMPK/Snf1-Regulated Transcriptional Repressor Mig1
- The SWI2/SNF2 Chromatin Remodeler BRAHMA Regulates Polycomb Function during Vegetative Development and Directly Activates the Flowering Repressor Gene
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy